

Introduction
Carbapenems can be an effective treatment of infections with multidrug-resistant Gram-negative bacteria such as Pseudomonas aeruginosa,1 Acinetobacter spp.,2 Klebsiella pneumoniae,3 and other Enterobacteriaceae.4 They are semi-synthetic or synthetic β-lactam compounds that are distinguished from other β-lactam compounds such as penicillins and cephalosporins by the absence of a sulfur atom in the bicyclic core and a different stereochemistry at Cα of the β-lactam ring (in penicillins and carbapenems, this atom is usually referred to as C6; in cephalosporins as C7) (Figure 1). The most popular carbapenem antibiotics are imipenem5, 6 (Merck, 1985), meropenem7, 8 (Sumitomo Pharmaceuticals and AstraZeneca, 1996), ertapenem9, 10 (Merck, 2005), and doripenem11, 12 (Shionogi Co. and Johnson & Johnson, 2005) (Figure 2). All of these broad-spectrum drugs are used intravenously. Carbapenems are considered to be drugs of last resort due to the fact that they are not inactivated by and effectively inhibit many β-lactamases (most Ambler class A and C β-lactamases13), while these enzymes efficiently hydrolyze penicillins and cephalosporins. β-Lactamases hydrolyze the β-lactam ring of β-lactam antibiotics blocking peptidyltransferase (also referred to as penicillin binding protein or PBP) activity that is critical for the peptidoglycan biosynthesis of the bacterial cell wall.14 β-Lactam antibiotics inhibit peptidyltransferase by forming a stable acyl-enzyme intermediate after an active-site serine of pepdityltransferase cleaves the β-lactam ring through a nucleophilic attack.15 Similar to peptidyltransferase, most β-lactamases contain an active site serine, which exerts a nucleophilic attack on and cleaves the β-lactam ring, resulting in turnover by the enzyme. These enzymes are referred to as serine β-lactamases (SBLs) and, based on sequence and structural homology, have been grouped into classes A, C, and D by Ambler.13 CTX-M β-lactamases are a group of class A SBLs expressed by Enterobacteriaceae that confer resistance toward the third-generation cephalosporin cefotaxime (Figure 3).16 As a consequence, carbapenems are frequently used to treat infections with Enterobacteriaceae expressing these enzymes. The increased use of carbapenems drives the emergence of carbapenem resistance mechanisms.
Figure 1.

Chemical structures of the bicyclic cores of different classes of β-lactam antibiotics. The penem core is found in penicillins and consists of a β-lactam ring fused with a tetrahydrodrothiazole ring. The cephem core is found in cephalosporins and consists of a β-lactam ring fused with a dihydrothiazine ring. The carbapenem core consists of a β-lactam ring fused with a dihydropyrrole ring. Heavy atoms of the bicyclic systems are numbered according to common use rather than according to the IUPAC nomenclature to facilitate comparisons between the different antibiotics. Note that the numbering of the R groups is arbitrary; here we started labeling R groups from the ones attached to the core atoms with the lowest number.
Figure 2.

Chemical structures of four commonly prescribed carbapenems: imipenem ((5R,6S)-3-[2-(aminomethylideneamino)ethylsulfanyl]-6-(1-hydroxyethyl)-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid), meropenem (3-[5-(dimethylcarbamoyl)pyrrolidin-2-yl] sulfanyl-6- (1-hydroxyethyl)-4-methyl-7-oxo- 1-azabicyclo[3.2.0] hept-2-ene-2-carboxylic acid); ertapenem ((4R,5S,6S)-3-[(3S,5S)-5-[(3-carboxyphenyl)carbamoyl]pyrrolidin-3-yl]sulfanyl-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid); and doripenem ((4R,5S,6S)-6-(1-hydroxyethyl)-4-methyl-7-oxo-3-[(3S,5S)-5-[(sulfamoylamino)methyl]pyrrolidin-3-yl]sulfanyl-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid).
Figure 3.

Chemical structures of selected non-carbapenem β-lactam antibiotics in clinical use: oxacillin ((2S,5R,6R)-3,3-dimethyl-6-[(5-methyl-3-phenyl-1,2-oxazole-4-carbonyl)amino]-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid), a penicillin; cefotaxime ((6R,7R,Z)-3-(acetoxymethyl)-7-(2-(2-aminothiazol-4-yl)-2-(methoxyimino)acetamido)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid), a third generation cephalosporin; ceftazidime ((6R,7R,Z)-7-(2-(2-aminothiazol-4-yl)-2-(2-carboxypropan-2-yloxyimino)acetamido)-8-oxo-3-(pyridinium-1-ylmethyl)-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylate), another third generation cephalosporin; and aztreonam (2-(([(1Z)-1-(2-amino-1,3-thiazol-4-yl)-2-([(2S,3S)-2-methyl-4-oxo-1-sulfoazetidin-3-yl]amino)-2-oxoethylidene]amino)oxy)-2-methylpropanoic acid), a monocyclic β-lactam or monobactam.
An increasing number of recent reports indicate that some β-lactamases can efficiently hydrolyze carbapenems. This alarming situation is made worse by the lack of new antibiotics at or near clinic that are active against resistant Gram-negative organisms, particularly nonfermenters such as Pseudomonas aeruginosa and Acinetobacter baumannii. Among SBLs, the most notable carbapenemases are variants of the OXA group (class D) and Klebsiella pneumoniae carbapenemases (KPCs, class A). Metallo-β-lactamases (MBLs), a separate class of enzymes (Ambler class B13), employ a water/hydroxide ion nucleophile activated by coordination to one or two Zn(II) ions and recognize a broad spectrum of β-lactams, including carbapenems. This perspective focuses on the MBLs of the IMP type (IMPs efficiently hydrolyze imipenem) and the VIM type (VIM represents Verona integron-borne metallo-β-lactamase), since these enzymes seem to be the clinically most important MBLs.17
We will (1) give a summary of these four important groups of carbapenemases, OXA, KPC, IMP, and VIM, including their epidemiology, structure, mechanism, and substrate specificity, (2) summarize approaches that have been undertaken to develop MBL inhibitors to reverse antibiotic resistance (potent SBL inhibitors such as clavulanic acid18 are already in clinical use), and (3) propose a novel approach to efficiently screen for such drugs using the Shape Signatures algorithm.
Clinically Important Carbapenemases
The carbapenemases of the OXA, KPC, IMP, and VIM types are clinically important enzymes. They are all encoded on mobile genetic elements, located on plasmids or chromosomes, and are frequently isolated from patients suffering from antibiotic resistant infections.
OXA β-Lactamases
OXA β-lactamases are classified by a preference for the β-lactam antibiotic oxacillin (Figure 3). These enzymes are class D SBLs of about 28 kDa molecular weight19 and exhibit an α/β protein fold. Several distinct lineages within the very divergent OXA group of enzymes have acquired the ability to hydrolyze carbapenems. Although relatively weak toward most carbapenem substrates compared to the KPC, IMP, and VIM enzymes discussed below, the activity of these enzymes is sufficient to confer carbapenem resistance. OXA carbapenemases are frequently found in Acinetobacter spp., in particular, in Acinetobacter baumannii. These Gram-negative bacteria colonize and infect patients, especially in intensive care units.20 The first OXA β-lactamase variant with carbapenemase activity, OXA-23, was isolated from a patient in Edinburgh, UK, in 1985.21 It was originally called ARI-1 and later renamed OXA-23 due to its sequence similarity to other OXA enzymes.22 Today, this enzyme has been isolated in other European countries, but also in Africa, Asia, South America, and North America (See Supporting Information S2 for more details). More recently, the emergence of OXA-48 in Enterobacteriaceae has given rise to concern.23 The OXA genes are very diverse; some are encoded on plasmids and some are chromosomal. Based on phylogenetic analysis, Barlow and Hall have suggested that they have been transferred from chromosomes to plasmids several times millions of years ago.24
To date, 162 variants of OXA β-lactamases have been documented, including a few redundant assignments.25 Several of these also possess the ability to inactivate carbapenems and have been found world wide (See Supporting Information S2 for more details). An extensive review of this group including phylogenetic trees and activity profiles has been published by Walther-Rasmussen and Hoiby.26 At the time of publication of that review (2006), 121 different OXA enzymes had been reported, of which 45 were known to have carbapenemase activity.
OXA carbapenemases confer only moderate resistance levels to pathogenic bacteria and their action is often complemented by other resistance mechanisms such as porin deficiencies and efflux pump overexpression.26 Other recent reviews on carbapenemases also include more detail on OXA β-lactamases.27, 28 Overall, the diversity, global presence, and the fact that transfer to plasmids has probably occurred long ago suggest that the OXA enzymes are an evolutionarily established group of SBLs.
Klebsiella Pneumoniae Carbapenemases (KPCs)
While there are several class A SBLs with carbapenemase activity, Klebsiella pneumoniae carbapenemases (KPCs) are by far the most important in the clinic. These are enzymes of about 28.5 kDa molecular weight (calculated29 for the mature proteins missing the N-terminal 24 residues) that also exhibit an α/β protein fold. Although the name suggests that they are specific to Klebsiella pneumoniae and foremost carbapenemases, enzymes of this group have also been found in other pathogenic bacteria, such as Pseudomonas aeruginosa,30 Serratia marcescens,31 and Enterobacter spp.,32 and they can also inactivate cephalosporins such as cefotaxime (Figure 3).27 The first KPC (originally named KPC-1) was found in a clinical isolate of Klebsiella pneumoniae in North Carolina in 1996.33 Currently, nine KPC variants have been reported25 and isolated world wide, most frequently in the United States and Israel (Figure 4 and Supporting Information S2-S3). The sequences of KPC-1 and KPC-2 (a point mutant of KPC-1) have been found to be identical after resequencing,34 and we will refer to this enzyme as KPC-2. The other eight variants are labeled KPC-3 through KPC-10. All known KPCs deviate from KPC-2 by only up to a few amino acid substitutions (Figure 5), suggesting that they may be direct descendents of KPC-2 (See Supporting Information S2-S3 for more details).
Figure 4.

World map illustrating the global spread of KPC enzymes. A blank world map was obtained from http://upload.wikimedia.org/ and countries with KPC occurences were colored in different opacities of red (symbolizing SBLs) according to the number of publications found on PubMed at http://www.ncbi.nlm.nih.gov/. Publications were retrieved using search strings such as “KPC-* United States” and titles and abstracts were checked for content. Only articles reporting occurences of KPCs were included, while review articles and reports restricted to computational and/or in vitro studies were excluded. Countries, for which ten or more publications with KPC reports were found, were colored in red with 100% opacity; those with fewer publications with lower opacities: 7-9 publications, 80%; 4-6 publications, 60%; 1-3 publications, 40%; no publications, white (see color code in the Figure). For more details see Supporting Information S2-S3.
Figure 5.

Radial phylogenetic tree of currently known KPC enzymes. Amino acid sequences of KPC enzymes including the leader sequence were retrieved from GenBank at http://www.ncbi.nlm.nih.gov/and aligned using Clustal X Version 2.0.9129 using default parameters. The phylogenetic tree was visualized using TreeView.130 The bar at the lower left corner gives a measure for amino acid sequence diversity. For instance, two enzymes differing by only one of 293 amino acid residues share 99.66% sequence identity and differ by 0.34% (0.0034). The KPC-9 sequence was missing five and four residues at the N- and C-termini, respectively. Since these residues are 100% conserved in the other enzymes, we added the missing residues accordingly. For more details see Supporting Information S2-S3.
In a review article published in 2007, Walther-Rasmussen and Hoiby included a section on KPC enzymes; at that time only four KPC variants were known.35 The fact that KPC enzymes have spread and evolved to this degree in only thirteen years is alarming.
Metallo-β-Lactamases (MBLs)
β-Lactamases that employ one or two active site Zn(II) ions to catalyze the cleavage of β-lactams belong to the class B or metallo-β-lactamases (MBLs),13 enzymes of about 25 kDa molecular weight. They all exhibit an αββα fold,36 which includes a compact core of two β-sheets sandwiched by α-helices on either side and is now known as the metallo-β-lactamase fold.37 The active site containing one or two Zn(II) ions is located at the edge of the two β sheets.36 MBLs have been further divided into three subgroups, B1, B2, and B3, based on sequence, structure, and activity similarities.38, 39 MBLs from subclass B1 and B3 possess a binuclear active site, which requires one or two Zn(II) ions for full activity. The Zn1 site is formed by three histidine residues, whereas the Zn2 site is formed by aspartic acid, cysteine/histidine, and a histidine residue. Subclass B2 enzymes are catalytically active with one Zn(II) ion binding to the Zn2 site. Enzymes of subclass B2 selectively hydrolyze carbapenems, whereas enzymes of subclasses B1 and B3 also hydrolyze penicillins and cephalosporins.40 MBLs cannot inactivate aztreonam (Figure 3). MBLs are a relatively novel class of enzymes and new types and variants of known types are isolated from the environment and clinical isolates at an alarming pace. We will focus here on the clinically important IMP and VIM enzymes, which belong to subclass B1. The Zn1 binding site of B1 enzymes consists of His116, His118, and His196; the Zn2 binding site consists of Asp120, Cys221, and His263 (we use the standard numbering scheme for MBLs38, 39 throughout this article). Mutations at position 120 have shown that the Zn2 binding site and with it the Zn1-Zn2 distance are quite flexible.41
IMP β-Lactamases
Together with other MBLs such as BcII (Bacillus cereus β-lactamase II),42, 43 CcrA from Bacteroides fragilis,44, 45 BlaB from Chryseobacterium meningosepticum,46 VIM enzymes (see below), and SPM-1 (Sao Paulo metallo-β-lactamase 1) found in Pseudomonas aeruginosa,47, 48 IMP enzymes form subclass B1. IMP-1 was originally isolated in 1991 from a patient with a Serratia marcescens infection in Japan and characterized as a protein of 246 amino acids in length and molecular weight of about 30 kDa (25.1 kDa as determined by electrospray ionization mass spectrometry49 and calculation29), encoded by the chromosomal blaIMP gene of 39.4% GC content.50 This GC content is much lower than the usual GC content of Serratia marcescens genes (56.2 to 58.4%)51 and an extraneous origin of the blaIMP gene has been suggested.50 IMP-1 has also been isolated from Pseudomonas aeruginosa, Klebsiella pneumoniae, Pseudomonas putida, Alcaligenes xylosoxidans,52 Acinetobacter junii,53 Providencia rettgeri,54 Acinetobacter baumannii,55 and Enterobacter aerogenes,56 usually on a mobile gene cassette inserted into an integron, either on the chromosome or on a plasmid.57 After being isolated first in Japan,50, 52 IMP-1 was isolated in several other Asian countries, in Europe, and in South America (See Supporting Information S3-S5 for more details). The global spread of IMP-1 and other IMP enzymes is depicted in Figure 6.
Figure 6.

World map illustrating the global spread of IMP enzymes. The map was prepared as described for Figure 4 except that IMP-specific search strings were used to retrieve articles and that countries with IMP occurence were colored in green, symbolizing MBLs. In some cases, where no published articles were available, GenBank entries were taken into account, e.g., for Thailand. For more details see Supporting Information S3-S5.
Crystal structures of IMP-141, 58-61 and the two D120A and D120E mutants41 have been reported. They confirm the general structure of MBLs described above. Besides the Zn(II)-ligating residues, a characteristic residue present in all IMP enzymes and in most subclass B1 enzymes (not VIM enzymes) is Lys224, which interacts with the carboxylate group of a co-crystallized mercaptocarboxylate inhibitor58 and supposedly the carboxylate at C3/C4 of penicillins and carbapenems/cephalosporins. A mechanism has been proposed by Wang et al. for nitrocefin hydrolysis by CcrA,62 the active site of which is virtually identical to that of IMP-158, 63 and which shares 35.9% sequence identity with IMP-1.50 According to this mechanism, the Zn(II) ions are coordinated by a bridging water/hydroxide molecule that performs a nucleophilic attack on the carbonyl carbon in the β-lactam ring of the substrate, resulting in cleavage of the amide bond and deactivation of the antibiotic. Relevant for inhibitor design, this mechanism differs from the one catalyzed by SBLs in that it lacks a covalent enzyme-substrate intermediate.64
To date, twenty six IMP variants have been isolated in countries across the globe, representing every continent except Africa and Antarctica (Figure 6). The phylogenetic relationship between these enzymes is shown in Figure 7. In the following, we will point out a few landmark enzymes; for a detailed account of all variants, refer to Supporting Information S3-S5.
Figure 7.

Radial phylogenetic tree of currently known IMP enzymes, generated as described for Figure 5. Two clusters of closely related enzymes, the “IMP-1 cluster” and “IMP-2 cluster” are shown as insets (note the different scale of the sequence diversity measures). For more details see Supporting Information S3-S5.
IMP-2 was first isolated from Acinetobacter baumannii in Italy in 1997.65 Its relatively low amino acid sequence identity (85%) to IMP-1, the difference between the gene cassettes carrying the IMP-1 and IMP-2 encoding genes, and the different geographic origins suggest different phylogenetic origins of IMP-1 and IMP-2.65 The substrate spectrum of IMP-2 was overall similar to that of IMP-1 (penicillins, cephalosporins, and carbapenems, but not aztreonam), however, with significantly lower catalytic efficiencies toward ampcillin and cephaloridine and significantly higher catalytic efficiencies toward carbenicillin and meropenem65 relative to IMP-1.57 The phylogenetic tree in Figure 7 shows that IMP-1 and IMP-2 belong to two distinct groups of closely related enzymes (discussed in detail in the Supporting Information S3-S4).
IMP-4 was the first MBL to be discovered on the continent of Australia.66 The first IMP enzyme to be discovered in the Americas was IMP-7, which was isolated from Pseudomonas aeruginosa in Canada in 1995 and 1996.67 IMP-15 and IMP-18 are variants that were isolated in the United States. IMP-15 was recently isolated from Pseudomonas aeruginosa in Kentucky, USA, and it was likely imported from Mexico, where the patient was injured and initially treated.68 IMP-18 was isolated from Pseudomonas aeruginosa in the United States,69 Mexico,70 and Puerto Rico.71
In summary, the group of IMP enzymes exhibits great diversity and an almost explosive discovery of new variants all around the globe. This group of enzymes is certainly one of the most concerning threats to state-of-the-art antimicrobial chemotherapy. Some attempts to predict novel IMP variants that might evolve in the future have been undertaken. For instance, point mutants of IMP-1 harboring a N233A72 and a F218Y73 mutation, respectively, were found to exhibit enhanced catalytic efficiencies compared to IMP-1.
VIM β-Lactamases
VIM β-lactamases are another group of MBLs of subclass B139 of about 25 kDa molecular weight. The first enzyme of this group reported was isolated from Pseudomonas aeruginosa found in a patient at the Verona University Hospital in Northern Italy in 1997.74 It was found that the gene encoding this enzyme was borne on an integron, which gave rise to its name Verona integron-borne metallo-β-lactamase 1 or VIM-1.74 Apart from Pseudomonas aeruginosa in Italy, VIM-1 has been isolated from other organisms in other European countries and in Turkey, but to our knowledge not outside these countries (See Supporting Infromation S6 for more details). The global spread of VIM-1 and other VIM enzymes is depicted in Figure 8.
Figure 8.

World map illustrating the global spread of VIM enzymes. The map was prepared as described for Figure 6 except that VIM-specific search strings were used to retrieve articles. For more details see Supporting Information S6-S8.
Twenty three VIM variants have been reported to date.25 Their phylogenetic relationships are shown in Figure 9. VIM-2, the second VIM enzyme to be reported, was actually isolated prior to VIM-1 in 1996 in France, also from Pseudomonas aeruginosa.75 Its amino acid sequence is 90% identical to that of VIM-1 and it also hydrolyzes all tested β-lactam antibiotics except the monobactam aztreonam.75 Its activity toward cephalosporins is similar to and that toward penicillins and carbapenems higher than that of VIM-1.76 Geographically, VIM-2 is more widely spread than VIM-1. Interestingly, and similar to the IMP enzymes, VIM-1 and VIM-2 also belong to clusters of closely related enzymes (Figure 9). VIM-2 is the only enzyme of the VIM group that has been crystallized, in complex with a mercaptocarboxylate inhibitor77 and as the free enzyme both with an oxidized and reduced Cys221.78 VIM-7 is the first VIM enzyme to be isolated in the United States. Its gene was found on an integron in a Pseudomonas aeruginosa isolate in 2001.79 VIM-2 was not isolated there until 2003.80 VIM-7 has less sequence identity (77%) to VIM-1 than any of the other variants up to VIM-6 (89 to 99%). It has therefore been suggested that VIM-7 has arisen independently in the United States rather than having been imported from Europe or Asia.79 VIM-2, on the other hand, may have been disseminated to the United States from Jordan.81 VIM-7 effectively hydrolyzes all tested β-lactam antibiotics except aztreonam.76 Its activity toward cephalosporins is lower and that toward penicillins higher than that of VIM-2.76 Its activity toward the two carbapenems imipenem and meropenem is similar to, but that toward ertapenem significantly higher than that of VIM-2.76 It has been suggested that some of the changes in catalytic efficiencies may be due to a Y218F mutation.76 Details on the occurence and relationship of all VIM variants can be found in the Supporting Information S6-S8.
Figure 9.

Radial phylogenetic tree of currently known VIM enzymes, generated as described for Figure 5. For more details see Supporting Information S6-S8.
As for the other carbapenemase groups described above, the occurrence of VIMs in different organisms, their global spread and the continued discovery of new variants in just a little more than a decade demonstrates their rapid dissemination and evolution. Figure 8 indicates that the VIM enzymes are so far the most globally spread MBLs.
Recent Approaches to Finding MBL Inhibitors
The structural complexity and heterogeneity of MBLs have posed challenges to finding a general inhibitor to treat infections caused by MBL-producing strains. A number of structures have been determined by X-ray crystallography for enzymes from all three subclasses B1-B3, summarized in Table 1, which provide important insights for understanding the catalytic mechanism, substrate selectivity and for guiding rational (computer-aided) inhibitor design.
Table 1.
Summary of different MBLs, organisms from which they were isolated, their subclass, metal ions bound in the crystal structure, and the corresponding PDB entry codes.
| MBL | Organism | Subclass | Metal ions | PDB Entry Codes |
|---|---|---|---|---|
| BcII | Bacillus cereus | B1 | 2 Zn | 2UYX, 2BFK, 2BFL, 2BFZ, 2BG2, 2BG6, 2BG7, 2BG8, 2BGA, 3FCZ, 1BC2, 1BVT |
| 2 Cd | 1MQO | |||
| 1 Zn | 2NZE, 2NZF, 2NXA, 2NYP, 1DXK, 2BC2, 3BC2, 1BMC |
|||
|
| ||||
| CcrA | Bacteroides fragilis | B1 | 2 Zn | 1A8T, 2BMI, 1A7T, 1ZNB, 1KR3,1HLK |
| 2 Cd | 2ZNB | |||
| 1 Zn, 1 Hg | 3ZNB | |||
|
| ||||
| IMP-1 | Serratia marcescens | B1 | 2 Zn | 2DOO, 1VGN, 1WUO, 1WUP |
| Pseudomonas aeruginosa | 1JJE, 1JJT, 1DDK, 1DD6 |
|||
|
| ||||
| BlaB |
Elizabethkingia meningospetica |
B1 | 2 Zn | 1M2X |
|
| ||||
| SPM-1 | Pseudomonas aeruginosa | B1 | 1 Zn | 2FHX |
|
| ||||
| VIM-2 | Pseudomonas aeruginosa | B1 | 2 Zn | 2YZ3, 1KO2, 1KO3 |
|
| ||||
| CphA | Aeromonas hydrophila | B2 | 2 Zn | 3F9O, 3FAI |
| 1 Zn | 1X8G, 1X8H, 1X8I, 2GKL, 2QDS |
|||
|
| ||||
| L1 |
Stenotrophomonas maltophilia |
B3 | 2 Zn | 2QDT, 2QDS, 2AIO, 2FU8, 2FU9, 2FM6, 2QIN, 2HB9, 2GFJ, 2GFK, 1SML |
| 2 Cu | 2FU7 | |||
| 1 Zn | 2H6A | |||
| 2FU6 | ||||
|
| ||||
| FEZ-1 | Fluoribacter gormanii | B3 | 2 Zn | 1L9Y, 1JT1, 1K07 |
|
| ||||
| BJP-1 | Bradyrhizobium japonicum | B3 | 2 Zn | 2GMN |
A superposition of crystal structures of MBLs from all three subclasses is shown in Figure 10. Selected enzymes include VIM-2 (subclass B1, blue, PDB code 2YZ377), IMP-1 (subclass B1, cyan, PDB code 1DD658), CphA (subclass B2, orange, PDB code 2QDS82), and L1 (subclass B3, red, PDB code 2AIO83). This representation reveals that in addition to the metal binding sites, the MBL active site is characterized by two hypervariable and flexible loops. The first loop (loop1) connects β-strands 3 and 4, while the second loop (loop2) is positioned opposite to loop1 connecting β-strand 11 and alpha-helix 4. The X-ray crystal structures of MBLs co-complexed with hydrolyzed substrate/inhibitors depict extensive interactions with these loops, suggesting that they may play a key role in substrate recognition, binding, and catalysis. It is logical to infer that these loops would also bear equal significance in the binding affinity and specificity of small-molecule inhibitors. Interestingly, the amino acid side chains of Trp64, Val67, and Lys224 play key roles in enzyme-inhibitor interactions in co-crystals of the MBL IMP-1 and a succinic acid.59 The residues Phe61 and Arg228 in VIM-2, Val67 and Lys224 in CphA, and Leu68 and Ser223 (Leu69 and Ser225 according to Garau et al.39) in L1 are anticipated to serve as important contact points to guide medicinal chemistry efforts toward the design of pan MBL inhibitors.
Figure 10.

Superimposition of crystal structures of MBLs from three subclasses. Selected enzymes include VIM-2 (subclass B1, blue, PDB code 2YZ3), IMP-1 (subclass B1, cyan, PDB code, 1DD6), CphA (subclass B2, orange, PDB code 2QDS), and L1 (subclass B3, red, PDB code 2AIO). Two flexible loops are highlighted by green circles. Important residues in the two loops are depicted and colored by atom type (carbon, grey; oxygen, red; nitrogen, blue). Clearly, residues in loop1 are hydrophobic, while those in loop2 possess mostly positively charged side chains.
A wide array of computational methods such as molecular dynamics simulations, quantum mechanics calculations, and virtual ligand-receptor screening (“docking and scoring”) have shown utility in discerning of the catalytic mechanism of MBLs and modeling the structural and dynamic effects of substrate/inhibitor binding at the atomic level. Biologically relevant phenomena that are not easily observed experimentally, including the protonation state of the zinc-bound water/hydroxide or ligand, the active site hydrogen bonding network, the Zn1-Zn2 distance, and the catalytic reaction pathway, have been the focus of molecular modeling studies on MBLs with either one or two active-site Zn(II) ions.84-93 For some binuclear B1 and B3 MBLs, the second Zn(II) ion in the Zn2 binding site serves to stabilize the build up of negative charge on the anionic N atom during hydrolysis of the peptide bond, thereby decreasing the activation free energy for nucleophilic attack. As a result, the overall catalytic efficiency of the binuclear enzyme is enhanced over the mononuclear variant. Results from these simulations suggested that separate catalytic mechanisms might apply to MBLs depending on differences in Zn(II) utilization, which is in agreement with experimental reports.62
There is growing concern about the rapid emergence of MBL expressing strains that exhibit a pan-resistant phenotype, particularly in the event that MBLs evolve into more efficient enzymes through mutation. Complementing experimental techniques, molecular modeling approaches have been employed to predict MBL evolution in order to effectively tackle MBL-conferred resistance to antibiotics.94, 95 Using rankings derived from molecular dynamics simulations, the variations in observed catalytic efficiencies of two wild-type enzymes (IMP-1 and IMP-6) to four cephalosporins were successfully reproduced.96 Later, the same approach was applied to computationally predict novel MBL point mutants that may cause problems if they evolve naturally. Five predicted variants from this study were characterized through in vitro experiments.49 As predicted, a variant of IMP-6 with only a single-site mutation converted the tested antibiotics more efficiently than IMP-6. This case represents one example by which computational approaches have advanced our understanding of crucial aspects of MBL pharmacology.
To address the emerging threat to public health posed by MBL-conferred resistance to antibiotics, new and more effective MBL inhibitors are urgently needed. Several classes of MBL inhibitors have been reported in the literature, including biphenyl tetrazoles,97 trifluoromethyl alcohol, ketone derivatives of L- and D-alanine,98 thiols,82, 99-102 thioester derivatives,99, 103, 104 hydroxamates,105, 106 cysteinyl peptides,107 mercaptocarboxylates,58, 99, 100, 108 sulfonylhydrazones,109 2,3-disubstituted succinic acids,59 phthalic acid derivatives,110 pyridine dicarboxylates,111 and tricyclic natural compounds.112 Representative compounds from each class are shown in Figure 11. Most of these compounds exhibit appreciable inhibitory activity only on a limited number of MBLs; consequently, they are unsuitable for treating infections caused by pan-resistant bacterial strains. To our knowledge, there are no MBL inhibitors currently in late-stage clinical development. In the private sector, this drought in the antibiotic pipeline is largely driven by economic pressures. Antibiotics are considered financially unattractive. They are generally used for short periods of time and for a specific indication, and they are subject to antibiotic resistance due to overuse. Encouragingly, the situation is beginning to improve. Thanks largely to efforts by organizations such as the Bill & Melinda Gates Foundation, public-private partnerships, and the NIH, today major funding initiatives are directed toward antibiotic drug development. Clearly, sustained awareness and adequate financial resources will be essential to mount an appropriate response.
Figure 11.

Chemical structures of selected MBL inhibitors.
Studies on the X-ray crystal structures of inhibitor-MBL co-complexes are providing critical information on the binding modes of the inhibitors and their specific interactions with the active-site residues. The collection of those MBL inhibitors is summarized in Table 2 and they bind in a similar fashion within the MBL active site. In general, inhibitors of MBLs feature two functional groups: hydrophobic moieties which bind in a predominantly hydrophobic pocket around loop1, and a metal-ligating group that interacts with the Zn(II) ion(s). In some cases, interactions between inhibitors and MBLs are stabilized by hydrogen bonds or electrostatic interactions with conserved residues such as Lys224 in loop2. Insights gleaned from exploring the X-ray crystal structures of MBLs from the three subclasses with various inhibitors present opportunities to formulate new strategies for the rational design of potent, broad-spectrum inhibitors of MBLs.
Table 2.
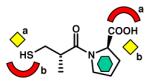
MBL inhibitors that were co-crystalized with the enzymes. Binding mode for each inhibitor is illustrated by three symbols (yellow diamond, metal-ligating group; green hexagon, hydrophobic group; red arch, electrostatically interacting group). a and b designate two different binding modes that were observed for the same inhibitor, depending on the different subclass of interacting MBLs.
| Structural Class | Chemical Structure | MBL [subclass] (PDB entry code) |
|---|---|---|
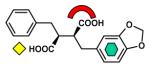
| Biphenyl tetrazole |

|
CcrA [B1] (1A8T) |
| Tricyclic compounds |
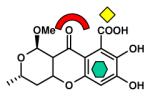
|
CcrA [B1] (1KR3, 1HLK) |
| Dicarboxylic acid |
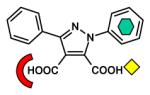
|
L1 [B3] (2GFJ) |
| Dicarboxylic acid |
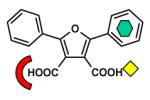
|
L1 [B3] (2GFK) |
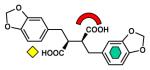
| Dicarboxylic acid |
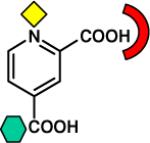
|
CphA [B2] (2GKL) |
| Dicarboxylic acid |
|
IMP-1 [B1] (1JJE) |
| Dicarboxylic acid |
|
IMP-1 [B1] (1JJT) |
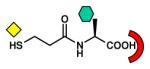
| Thiol |
|
L1 [B3] (2QDT) |
| Thiol |
|
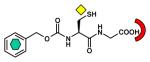
CphA [B2] (2QDS) L1 [B3] (2FU8) BlaB [B1] (1M2X) FEZ-1 [B3] (1JT1) |
| Thiol |
|
L1 [B3] (2FU9) |
| Thiol |
|
IMP-1 [B1] (2DOO) |
| Thiol |
|
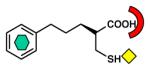
VIM-2 [B1] (2YZ3) |
| Thiol |
|
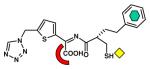
IMP-1 [B1] (1DD6) |
| Thiol |
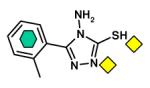
|
L1 [B3] (2HB9) |
Combining X-ray crystallographic and molecular modeling studies, Lienard and coworkers recently showed that several thiols provide broad-spectrum inhibition of MBLs with values of the inhibition constant in the sub-micromolar range against all three MBL subclasses.82 These represent the first published MBL inhibitors that may serve as templates for further development toward clinically useful drugs with inhibitory activities against MBLs spanning all three subclasses. Interestingly, their analysis revealed that the same compound may adopt differential binding modes when interacting with MBLs from different subclasses. For example, Compound 2 utilized a thiol to coordinate the Zn(II) ion in IMP-1 from subclass B1 and the carboxylate group to form strong electrostatic interactions with the conserved Lys224 residue. On the contrary, in CphA (subclass B2), the thiol is hydrogen bonded with the side chains of neighboring residues while the carboxylate group is now interacting with the Zn(II) ion. These findings, together with structural data on new thiol inhibitors, provide an excellent basis for the design and development of more potent MBL inhibitors with broad-spectrum activities.
Virtual screening (VS) has been an effective computational approach that has become the mainstay in early-stage drug discovery at major pharmaceutical companies. The process involves the rapid in silico assessment of binding potentials for large libraries of chemical structures to biological macromolecular targets of interest, typically a protein receptor or enzyme. VS of ligand-receptor pairs requires three basic components: databases of chemical structures for screening; the structure of the target protein; and a computational algorithm to explore and assess (i.e., score) the binding mode(s) of the ligand within the protein's binding pocket. Commonly termed ligand-receptor “docking and scoring” by specialists in the field, VS has been widely adopted and adapted for multiple purposes including for prediction of ligand-protein affinities and interatomic interactions and for rapid screening of extensive chemical databases in search of prospective drug leads for many protein targets.97 Computational modeling of the cationic Zn(II) metal center poses multiple challenges, including considerations of solvation effects, geometric structure, and charge parameterization particularly in the vicinity of metal ions.113 To account for charge transfer in Zn(II) centers in a docking study with CcrA, an empirical decrease of the formal charge of +2 of the Zn(II) ion by 0.2 per Zn(II) ligand in combination with a balancing change of charge of the ligands has been employed.114 To account for distribution of the positive charge of the Zn(II) ion into the electron-deficient sp3 orbitals, a cationic dummy atom approach115 has been used in molecular dynamics simulations of IMP-1.92 A thorough discussion of these issues is beyond the scope of this Perspective.
Olsen et al.116 presented a successful application of docking and scoring to MBLs. In their study, the GOLD docking program provided the best overall performance in terms of low RMSDs between experimental and docked structures and good statistical correlations between the GOLD score and the corresponding experimental inhibitor activities. In a similar study, structure-based VS techniques were deployed against several metalloenzymes including CcrA. This process yielded five inhibitors which were later confirmed experimentally to possess low micromolar activities against this MBL from Bacteroides fragilis.114 The results of these studies maintained that the docking and scoring approach is a reliable method for identification of potential new MBL inhibitors.
Shape Signatures: a Novel Approach to Finding MBL Inhibitors
Our particular interest is focused on finding MBL inhibitor templates that can be developed into broad-spectrum inhibitors for class B β-lactamases. Therefore, an appropriate VS scheme would require screening of the prospective inhibitors against a select array of MBL enzymes that includes at least one MBL from each subclass (e.g. IMP-1 and/or VIM-2 for B1, CphA for B2, and L1 for B3). The size, composition, and structural diversity of chemical databases for screening are important factors that influence the final outcome of a VS study. Recently, a fragment-based screening approach was proposed to prioritize a series of inhibitor fragments for AmpC, a Class A β-lactamase.117 In this study, the workers observed that fragment-based screening explored a broader region of chemical space than screening of lead-like and drug-like databases. This study highlights the importance of input compound library preparation in the screening process.
To date there are few reports of VS studies targeting the class B β-lactamases (MBLs).114 We propose a VS scheme, incorporating both traditional and novel computational approaches for the design and filtering of compound libraries, toward discovery of MBL inhibitors exhibiting broad-spectrum inhibitory activities against class B β-lactamases (Figure 12). In the process of designing libraries of MBL-directed compounds, we introduce a novel computational method, Shape Signatures, which rapidly compares molecules based on similarity in shape, polarity, and other bio-relevant properties.118-124 The degree of similarity between a pair of molecules can be assessed by comparing their 1D Signatures (shape only) or 2D Shape Signatures (shape and surface charge distribution). This process is fast and efficient, and it eliminates tedious and subjective atom-based alignment of the molecules required in many traditional molecular modeling approaches. Unlike traditional quantitative structure-activity relationship (QSAR)-based approaches that require the user to compute and select hundreds of discrete descriptors for each molecule, the Shape Signature can be viewed as a very compact descriptor that encodes molecular shape and electrostatics in a single entity. Large Shape Signature databases of small-molecule compounds can be screened easily and rapidly using simple metrics to identify small molecules that are similar in shape and polarity, but not necessarily similar in chemical structure. Likewise, the Shape Signatures of small molecules will identify complementary protein receptor sites. Consequently, it is well-suited for diverse applications in virtual screening, drug discovery, and predictive toxicology. The Shape Signatures method excels at scaffold hopping (crossing chemical families); therefore, it is more likely to discover structurally diverse molecules that may show attractive bioactivity for the three subtypes of MBLs.
Figure 12.

Proposed VS scheme for the discovery of MBL inhibitors with broad-spectrum activities against B1, B2, and B3 subclasses.
Shape Signatures for a small-molecule compound are probability distributions, expressed as histograms, derived from a ray-trace of the volume enclosed by the solvent-accessible surface of the molecule. They are rotationally-invariant descriptors that can be used to rapidly compare molecules in terms of shape similarity, and the method can easily accommodate additional biorelevant properties defined on the molecular surface, such as electrostatic charge. Shape Signatures is computationally fast, easy to use in that it avoids specification of complex structural queries or molecular alignment schemes, and can accommodate an almost limitless number of chemical compounds. The method thus lends itself to rapid comparison of large databases of chemical compounds with each other or with a known bioactive molecule of interest (the query compound).
Commonly used computational approaches include ligand-based drug design with pharmacophores, structure-based drug design (drug-receptor docking), QSARs, and quantitative structure-property relationships (QSPRs). Regulatory agencies as well as the pharmaceutical industry are actively involved in development of computational tools that will improve the speed and efficiency of drug discovery and development, decrease the use of animals, increase predictability, and decrease uncertainty.125
In the present case, our query compounds are the thiols which have already exhibited broad-spectrum inhibitory activity for the three MBL subclasses.82 The initial aim is to prepare a balanced chemical database that is structurally diverse yet incorporates our current understanding of the structure-activity relationships of known MBL ligands. The database should reflect the key features and commonalities of existing MBL inhibitors as well as the valuable information presented by the available crystal structures of MBL-inhibitor complexes. This can be achieved by filtering the initial chemical database using MBL-binding features incorporated in separate pharmacophores derived from known inhibitors of each of the three subclasses. Pharmacophores represent the geometric arrangement of the minimal structural elements of ligands (hydrogen bond donors/acceptors, metal ligating moieties, and hydrophobic regions) that are deemed essential for binding to the target protein(s). Compounds matching at least one of the subclass pharmacophores would proceed to the next step for docking and scoring into the structural models of representative B1, B2, and B3 enzymes. The Zn(II) centers could be represented similar to the approaches mentioned above. The “hits” from this VS scheme would be ranked and prioritized based on their binding modes and docking scores, after which a subset is selected for further consideration in the drug discovery process. Adopting this screening process in concert with medicinal chemistry, structural analysis, methods in biochemistry and molecular biology, and pre-clinical evaluation, the authors have embarked on a strategy to discover pan MBL inhibitors that show promise for eventual applications in the clinic.
Concluding Remarks
There is an urgent need for pan MBL small molecule inhibitors that can be used in combination with currently approved antibiotics.126 Extensive progress has been made in the discovery of specific, potent MBL inhibitors against individual enzymes; however, there is a paucity of inhibitors with clinical potential in the treatment of infections caused by a broad spectrum of bacterial pathogens. One major challenge is to find good inhibitors that can permeate through the bacterial envelope. Identifying inhibitors that act on MBLs and SBLs is an even bigger challenge due to the different catalytic mechanisms and active site shapes. Shape Signatures could assist the design of such inhibitors or at least point to whether such an inhibitor design is feasible or not. Human microflora itself has been recently shown to harbor a reservoir of antibiotic resistance genes,127 which could be caused by constant exposure to low levels of antibiotics in food and drinking water.128 This Perspectives article provides new tools for the medicinal chemist for the design of β-lactamase inhibitors using a combination of virtual screening and molecular epidemiology.
Supplementary Material
Acknowledgments
This research was supported by awards from Research Corporation for Science Advancement (to P.O. and J.H.T.), Kean University President Farahi (to J.H.T.), the Provost's Teacher-Scholar Program at Cal Poly Pomona (to P.O.), a Howard Hughes Medical Institute Research Apprenticeship (to K.T.D.), and an NIH R21-GM081394 grant from the National Institute of General Medical Sciences (to W.J.W.).
Abbreviations
- SBL
serine β-lactamase
- MBL
metallo-β-lactamase
- OXA
oxacillinase
- KPC
Klebsiella pneumoniae carbapenemase
- IMP
imipenemase
- VIM
Verona integron-borne metallo-β-lactamase
- VS
virtual screening
- QSAR
quantitative structure-activity relationship
- QSPR
quantitative structure-property relationship
Biographies
Peter Oelschlaeger studied Biology and Chemistry at the University of Hohenheim, where he received an M.S. (Diplom) in Biology and a teaching degree in Biology and Chemistry in 1999. He obtained his Ph.D. (Dr. rer. nat.) in Biology from the University of Stuttgart in 2002. After postdoctoral work with Stephen L. Mayo at the California Institute of Technology and with Arieh Warshel at the University of Southern California and teaching in the Biology Department at Occidental College, he started his independent academic career in 2007 as an Assistant Professor in the Chemistry Department at California State Polytechnic University, Pomona (Cal Poly Pomona). His research employs a combined computational/experimental approach to study the mechanism and evolution of metallo-β-lactamases and their role in antibiotic resistance.
Ni Ai earned her B.S. degree in Chemical Physics from University of Science and Technology of China in 2000 and her Ph.D. degree in Cellular and Molecular Pharmacology from University of Medicine and Dentistry of New Jersey (UMDNJ) in 2005. She currently is a research specialist in the academic laboratory of Professor William Welsh at UMDNJ-Robert Wood Johnson Medical School. Her research focuses on the development and application of computational approaches for the study of ligand-protein interactions, particularly as they relate to drug discovery.
Kevin DuPrez began his undergraduate studies at Cal Poly Pomona in Fall 2004. During Summer 2007, he was a participant in the Eugene and Ruth Roberts Summer Student Academy at City of Hope, Duarte, CA. He joined the Oelschlaeger Laboratory at Cal Poly Pomona in January 2008, studying for his senior thesis entitled “Inhibition studies of metallo-β-lactamase variants”. He graduated from Cal Poly Pomona in June 2009 with B.S. degrees in Biotechnology and Chemistry and is now pursuing a Ph.D. degree in the Graduate Program in Biochemistry and Molecular Biology at the University of California, Riverside.
William J. Welsh occupies the Norman H. Edelman Endowed Professorship in Bioinformatics in the Department of Pharmacology at the Robert Wood Johnson Medical School (RWJMS) in Piscataway, NJ, University of Medicine and Dentistry of New Jersey (UMDNJ). Dr. Welsh serves as Director of the Informatics Institute of UMDNJ and the Environmental Bioinformatics & Computational Toxicology Center. Dr. Welsh earned his B.S. degree in Chemistry from St. Joseph's University (Phila., PA) in 1969 and his Ph.D. degree in Theoretical Physical Chemistry from the University of Pennsylvania (Phila., PA) in 1975. He also conducted postdoctoral studies at the University of Cincinnati (Cinti., OH) and the National Institutes of Health. Dr. Welsh's laboratory specializes in the development and application of computational tools for drug discovery and predictive toxicology.
Jeffrey H. Toney's career has spanned both the pharmaceutical industry and academia. His academic training is in Chemistry (B.S., University of Virginia; M.S. and Ph.D., Northwestern University) and included research experience as a postdoctoral fellow in Molecular Biology (Dana Farber Cancer Institute, Harvard Medical School) and in Chemical Biology (Massachusetts Institute of Technology). His current scholarship is focused on drug discovery using an interdisciplinary approach. As a Senior Research Fellow at Merck Research Laboratories, he studied a variety of therapeutic targets. He has held the Herman and Margaret Sokol Professorship in Chemistry at Montclair State University and served as Department Chairperson of Chemistry and Biochemistry. He is currently serving as Dean of the College of Natural, Applied and Health Sciences at Kean University.
Footnotes
Supporting Information Available: Additional Information on the Epidemiology of Clinically Important Carbapenemases. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rossolini GM, Mantengoli E. Treatment and control of severe infections caused by multiresistant Pseudomonas aeruginosan. Clin. Microbiol. Infect. 2005;11(Suppl 4):17–32. doi: 10.1111/j.1469-0691.2005.01161.x. [DOI] [PubMed] [Google Scholar]
- 2.Karageorgopoulos DE, Falagas ME. Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect. Dis. 2008;8:751–762. doi: 10.1016/S1473-3099(08)70279-2. [DOI] [PubMed] [Google Scholar]
- 3.Ramphal R, Ambrose PG. Extended-spectrum β-lactamases and clinical outcomes: current data. Clin. Infect. Dis. 2006;42(Suppl 4):S164–172. doi: 10.1086/500663. [DOI] [PubMed] [Google Scholar]
- 4.Isturiz R. Global resistance trends and the potential impact on empirical therapy. Int. J. Antimicrob. Agents. 2008;32(Suppl 4):S201–206. doi: 10.1016/S0924-8579(09)70003-2. [DOI] [PubMed] [Google Scholar]
- 5.Barza M. Imipenem: first of a new class of β-lactam antibiotics. Ann. Intern. Med. 1985;103:552–560. doi: 10.7326/0003-4819-103-4-552. [DOI] [PubMed] [Google Scholar]
- 6.Rodloff AC, Goldstein EJ, Torres A. Two decades of imipenem therapy. J. Antimicrob. Chemother. 2006;58:916–929. doi: 10.1093/jac/dkl354. [DOI] [PubMed] [Google Scholar]
- 7.Mohr JF., 3rd. Update on the efficacy and tolerability of meropenem in the treatment of serious bacterial infections. Clin. Infect. Dis. 2008;47(Suppl 1):S41–51. doi: 10.1086/590065. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin CM, Lyseng-Williamson KA, Keam SJ. Meropenem: a review of its use in the treatment of serious bacterial infections. Drugs. 2008;68:803–838. doi: 10.2165/00003495-200868060-00006. [DOI] [PubMed] [Google Scholar]
- 9.Odenholt I. Ertapenem: a new carbapenem. Expert Opin. Investig. Drugs. 2001;10:1157–1166. doi: 10.1517/13543784.10.6.1157. [DOI] [PubMed] [Google Scholar]
- 10.Keating GM, Perry CM. Ertapenem: a review of its use in the treatment of bacterial infections. Drugs. 2005;65:2151–2178. doi: 10.2165/00003495-200565150-00013. [DOI] [PubMed] [Google Scholar]
- 11.Keam SJ. Doripenem: a review of its use in the treatment of bacterial infections. Drugs. 2008;68:2021–2057. doi: 10.2165/00003495-200868140-00007. [DOI] [PubMed] [Google Scholar]
- 12.Matthews SJ, Lancaster JW. Doripenem monohydrate, a broad-spectrum carbapenem antibiotic. Clin. Ther. 2009;31:42–63. doi: 10.1016/j.clinthera.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Ambler RP. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 14.Popham DL, Young KD. Role of penicillin-binding proteins in bacterial cell morphogenesis. Curr. Opin. Microbiol. 2003;6:594–599. doi: 10.1016/j.mib.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Sauvage E, Powell AJ, Heilemann J, Josephine HR, Charlier P, Davies C, Pratt RF. Crystal structures of complexes of bacterial DD-peptidases with peptidoglycan-mimetic ligands: the substrate specificity puzzle. J. Mol. Biol. 2008;381:383–393. doi: 10.1016/j.jmb.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawkey PM, Jones AM. The changing epidemiology of resistance. J. Antimicrob. Chemother. 2009;64(Suppl 1):i3–10. doi: 10.1093/jac/dkp256. [DOI] [PubMed] [Google Scholar]
- 17.Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-β-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 2005;18:306–325. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finlay J, Miller L, Poupard JA. A review of the antimicrobial activity of clavulanate. J. Antimicrob. Chemother. 2003;52:18–23. doi: 10.1093/jac/dkg286. [DOI] [PubMed] [Google Scholar]
- 19.Bethel CR, Distler AM, Ruszczycky MW, Carey MP, Carey PR, Hujer AM, Taracila M, Helfand MS, Thomson JM, Kalp M, Anderson VE, Leonard DA, Hujer KM, Abe T, Venkatesan AM, Mansour TS, Bonomo RA. Inhibition of OXA-1 β-lactamase by penems. Antimicrob. Agents Chemother. 2008;52:3135–3143. doi: 10.1128/AAC.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergogne-Berezin E, Towner KJ. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 1996;9:148–165. doi: 10.1128/cmr.9.2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paton R, Miles RS, Hood J, Amyes SG. ARI 1: β-lactamase-mediated imipenem resistance in Acinetobacter baumannii. Int. J. Antimicrob. Agents. 1993;2:81–87. doi: 10.1016/0924-8579(93)90045-7. [DOI] [PubMed] [Google Scholar]
- 22.Donald HM, Scaife W, Amyes SG, Young HK. Sequence analysis of ARI-1, a novel OXA β-lactamase, responsible for imipenem resistance in Acinetobacter baumannii 6B92. Antimicrob. Agents Chemother. 2000;44:196–199. doi: 10.1128/aac.44.1.196-199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livermore DM. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009;64(Suppl 1):i29–36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 24.Barlow M, Hall BG. Phylogenetic analysis shows that the OXA β-lactamase genes have been on plasmids for millions of years. J. Mol. Evol. 2002;55:314–321. doi: 10.1007/s00239-002-2328-y. [DOI] [PubMed] [Google Scholar]
- 25.Jacoby G, Bush K. Amino Acid Sequences for TEM, SHV and OXA Extended-Spectrum and Inhibitor Resistant ß-Lactamases. http://www.lahey.org/Studies/other.asp#table1.
- 26.Walther-Rasmussen J, Hoiby N. OXA-type carbapenemases. J. Antimicrob. Chemother. 2006;57:373–383. doi: 10.1093/jac/dki482. [DOI] [PubMed] [Google Scholar]
- 27.Queenan AM, Bush K. Carbapenemases: the versatile β-lactamases. Clin. Microbiol. Rev. 2007;20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walsh TR. Clinically significant carbapenemases: an update. Curr. Opin. Infect. Dis. 2008;21:367–371. doi: 10.1097/QCO.0b013e328303670b. [DOI] [PubMed] [Google Scholar]
- 29.Putnam C. Protein Calculator v3.3. http://www.scripps.edu/~cdputnam/protcalc.html.
- 30.Villegas MV, Kattan JN, Correa A, Lolans K, Guzman AM, Woodford N, Livermore D, Quinn JP. Dissemination of Acinetobacter baumannii clones with OXA-23 Carbapenemase in Colombian hospitals. Antimicrob. Agents Chemother. 2007;51:2001–2004. doi: 10.1128/AAC.00226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang R, Zhou HW, Cai JC, Chen GX. Plasmid-mediated carbapenem-hydrolysing β-lactamase KPC-2 in carbapenem-resistant Serratia marcescens isolates from Hangzhou, China. J. Antimicrob. Chemother. 2007;59:574–576. doi: 10.1093/jac/dkl541. [DOI] [PubMed] [Google Scholar]
- 32.Hossain A, Ferraro MJ, Pino RM, Dew RB, 3rd, Moland ES, Lockhart TJ, Thomson KS, Goering RV, Hanson ND. Plasmid-mediated carbapenem-hydrolyzing enzyme KPC-2 in an Enterobacter sp. Antimicrob. Agents Chemother. 2004;48:4438–4440. doi: 10.1128/AAC.48.11.4438-4440.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, Alberti S, Bush K, Tenover FC. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001;45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, Alberti S, Bush K, Tenover FC. Author's Correction. Antimicrob. Agents Chemother. 2008;52:809. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walther-Rasmussen J, Hoiby N. Class A carbapenemases. J. Antimicrob. Chemother. 2007;60:470–482. doi: 10.1093/jac/dkm226. [DOI] [PubMed] [Google Scholar]
- 36.Carfi A, Pares S, Duee E, Galleni M, Duez C, Frere JM, Dideberg O. The 3-D structure of a zinc metallo-β-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J. 1995;14:4914–4921. doi: 10.1002/j.1460-2075.1995.tb00174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aravind L. An evolutionary classification of the metallo-β-lactamase fold proteins. In Silico Biol. 1999;1:69–91. [PubMed] [Google Scholar]
- 38.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM. Standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 2001;45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garau G, Garcia-Saez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frere JM, Dideberg O. Update of the standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 2004;48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi Y, Kuroki T, Yasuzawa H, Higashi T, Jin W, Kawanami A, Yamagata Y, Arakawa Y, Goto M, Kurosaki H. Probing the role of Asp-120(81) of metallo-β-lactamase (IMP-1) by site-directed mutagenesis, kinetic studies, and X-ray crystallography. J. Biol. Chem. 2005;280:20824–20832. doi: 10.1074/jbc.M414314200. [DOI] [PubMed] [Google Scholar]
- 42.Sabath LD, Abraham EP. Zinc as a cofactor for cephalosporinase from Bacillus cereus 569. Biochem. J. 1966;98:11C–13C. doi: 10.1042/bj0980011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davies RB, Abraham EP. Separation, purification and properties of β-lactamase I and β-lactamase II from Bacillus cereus 569/H/9. Biochem. J. 1974;143:115–127. doi: 10.1042/bj1430115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuchural GJ, Jr., Malamy MH, Tally FP. β-Lactamase-mediated imipenem resistance in Bacteroides fragilis. Antimicrob. Agents Chemother. 1986;30:645–648. doi: 10.1128/aac.30.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Rasmussen BA, Bush K. Biochemical characterization of the metallo-β-lactamase CcrA from Bacteroides fragilis TAL3636. Antimicrob. Agents Chemother. 1992;36:1155–1157. doi: 10.1128/aac.36.5.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossolini GM, Franceschini N, Riccio ML, Mercuri PS, Perilli M, Galleni M, Frere JM, Amicosante G. Characterization and sequence of the Chryseobacterium (Flavobacterium) meningosepticum carbapenemase: a new molecular class B β-lactamase showing a broad substrate profile. Biochem. J. 1998;332(Pt 1):145–152. doi: 10.1042/bj3320145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy TA, Simm AM, Toleman MA, Jones RN, Walsh TR. Biochemical characterization of the acquired metallo-β-lactamase SPM-1 from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2003;47:582–587. doi: 10.1128/AAC.47.2.582-587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toleman MA, Simm AM, Murphy TA, Gales AC, Biedenbach DJ, Jones RN, Walsh TR. Molecular characterization of SPM-1, a novel metallo-β-lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J. Antimicrob. Chemother. 2002;50:673–679. doi: 10.1093/jac/dkf210. [DOI] [PubMed] [Google Scholar]
- 49.Oelschlaeger P, Mayo SL, Pleiss J. Impact of remote mutations on metallo-β-lactamase substrate specificity: implications for the evolution of antibiotic resistance. Protein Sci. 2005;14:765–774. doi: 10.1110/ps.041093405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osano E, Arakawa Y, Wacharotayankun R, Ohta M, Horii T, Ito H, Yoshimura F, Kato N. Molecular characterization of an enterobacterial metallo β-lactamase found in a clinical isolate of Serratia marcescens that shows imipenem resistance. Antimicrob. Agents Chemother. 1994;38:71–78. doi: 10.1128/aac.38.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colwell RR, Mandel M. Adansonian Analysis and Deoxyribonucleic Acid Base Composition of Serratia Marcescens. J. Bacteriol. 1965;89:454–461. doi: 10.1128/jb.89.2.454-461.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senda K, Arakawa Y, Ichiyama S, Nakashima K, Ito H, Ohsuka S, Shimokata K, Kato N, Ohta M. PCR detection of metallo-β-lactamase gene (blaIMP) in gram-negative rods resistant to broad-spectrum β-lactams. J. Clin. Microbiol. 1996;34:2909–2913. doi: 10.1128/jcm.34.12.2909-2913.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tysall L, Stockdale MW, Chadwick PR, Palepou MF, Towner KJ, Livermore DM, Woodford N. IMP-1 carbapenemase detected in an Acinetobacter clinical isolate from the UK. J. Antimicrob. Chemother. 2002;49:217–218. doi: 10.1093/jac/49.1.217. [DOI] [PubMed] [Google Scholar]
- 54.Shiroto K, Ishii Y, Kimura S, Alba J, Watanabe K, Matsushima Y, Yamaguchi K. Metallo-β-lactamase IMP-1 in Providencia rettgeri from two different hospitals in Japan. J. Med. Microbiol. 2005;54:1065–1070. doi: 10.1099/jmm.0.46194-0. [DOI] [PubMed] [Google Scholar]
- 55.Liu SY, Lin JY, Chu C, Su LH, Lin TY, Chiu CH. Integron-associated imipenem resistance in Acinetobacter baumannii isolated from a regional hospital in Taiwan. Int. J. Antimicrob. Agents. 2006;27:81–84. doi: 10.1016/j.ijantimicag.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Biendo M, Canarelli B, Thomas D, Rousseau F, Hamdad F, Adjide C, Laurans G, Eb F. Successive emergence of extended-spectrum β-lactamase-producing and carbapenemase-producing Enterobacter aerogenes isolates in a university hospital. J. Clin. Microbiol. 2008;46:1037–1044. doi: 10.1128/JCM.00197-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laraki N, Galleni M, Thamm I, Riccio ML, Amicosante G, Frere JM, Rossolini GM. Structure of In31, a blaIMP-containing Pseudomonas aeruginosa integron phyletically related to In5, which carries an unusual array of gene cassettes. Antimicrob. Agents Chemother. 1999;43:890–901. doi: 10.1128/aac.43.4.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry. 2000;39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 59.Toney JH, Hammond GG, Fitzgerald PM, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao YD. Succinic acids as potent inhibitors of plasmid-borne IMP-1 metallo-β-lactamase. J. Biol. Chem. 2001;276:31913–31918. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 60.Kurosaki H, Yamaguchi Y, Higashi T, Soga K, Matsueda S, Yumoto H, Misumi S, Yamagata Y, Arakawa Y, Goto M. Irreversible inhibition of metallo-β-lactamase (IMP-1) by 3-(3-mercaptopropionylsulfanyl)propionic acid pentafluorophenyl ester. Angew. Chem. Int. Ed. Engl. 2005;44:3861–3864. doi: 10.1002/anie.200500835. [DOI] [PubMed] [Google Scholar]
- 61.Kurosaki H, Yamaguchi Y, Yasuzawa H, Jin W, Yamagata Y, Arakawa Y. Probing, inhibition, and crystallographic characterization of metallo-β-lactamase (IMP-1) with fluorescent agents containing dansyl and thiol groups. ChemMedChem. 2006;1:969–972. doi: 10.1002/cmdc.200600115. [DOI] [PubMed] [Google Scholar]
- 62.Wang Z, Fast W, Benkovic SJ. On the mechanism of the metallo-β-lactamase from Bacteroides fragilis. Biochemistry. 1999;38:10013–10023. doi: 10.1021/bi990356r. [DOI] [PubMed] [Google Scholar]
- 63.Concha NO, Rasmussen BA, Bush K, Herzberg O. Crystal structure of the wide-spectrum binuclear zinc β-lactamase from Bacteroides fragilis. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 64.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 65.Riccio ML, Franceschini N, Boschi L, Caravelli B, Cornaglia G, Fontana R, Amicosante G, Rossolini GM. Characterization of the metallo-β-lactamase determinant of Acinetobacter baumannii AC-54/97 reveals the existence of bla(IMP) allelic variants carried by gene cassettes of different phylogeny. Antimicrob. Agents Chemother. 2000;44:1229–1235. doi: 10.1128/aac.44.5.1229-1235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peleg AY, Franklin C, Bell J, Spelman DW. Emergence of IMP-4 metallo-β-lactamase in a clinical isolate from Australia. J. Antimicrob. Chemother. 2004;54:699–700. doi: 10.1093/jac/dkh398. [DOI] [PubMed] [Google Scholar]
- 67.Gibb AP, Tribuddharat C, Moore RA, Louie TJ, Krulicki W, Livermore DM, Palepou MF, Woodford N. Nosocomial outbreak of carbapenem-resistant Pseudomonas aeruginosa with a new bla(IMP) allele, bla(IMP-7) Antimicrob. Agents Chemother. 2002;46:255–258. doi: 10.1128/AAC.46.1.255-258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martin CA, Morita K, Ribes JA, Deshpande LM, Sader HS, Castanheira M. IMP-15-producing Pseudomonas aeruginosa strain isolated in a U.S. medical center: a recent arrival from Mexico. Antimicrob. Agents Chemother. 2008;52:2289–2290. doi: 10.1128/AAC.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanson ND, Hossain A, Buck L, Moland ES, Thomson KS. First occurrence of a Pseudomonas aeruginosa isolate in the United States producing an IMP metallo-β-lactamase, IMP-18. Antimicrob. Agents Chemother. 2006;50:2272–2273. doi: 10.1128/AAC.01440-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garza-Ramos U, Tinoco P, Silva-Sanchez J, Morfin-Otero R, Rodriguez-Noriega E, Leon-Garnica G, Sader HS, Jones RN. Metallo-β-lactamase IMP-18 is located in a class 1 integron (In96) in a clinical isolate of Pseudomonas aeruginosa from Mexico. Int. J. Antimicrob. Agents. 2008;31:78–80. doi: 10.1016/j.ijantimicag.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 71.Wolter DJ, Khalaf N, Robledo IE, Vazquez GJ, Sante MI, Aquino EE, Goering RV, Hanson ND. Surveillance of carbapenem-resistant Pseudomonas aeruginosa isolates from Puerto Rican Medical Center Hospitals: dissemination of KPC and IMP-18 β-lactamases. Antimicrob. Agents Chemother. 2009;53:1660–1664. doi: 10.1128/AAC.01172-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Materon IC, Palzkill T. Identification of residues critical for metallo-β-lactamase function by codon randomization and selection. Protein Sci. 2001;10:2556–2565. doi: 10.1110/ps.40884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oelschlaeger P, Mayo SL. Hydroxyl groups in the ββ sandwich of metallo-β-lactamases favor enzyme activity: a computational protein design study. J. Mol. Biol. 2005;350:395–401. doi: 10.1016/j.jmb.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 74.Lauretti L, Riccio ML, Mazzariol A, Cornaglia G, Amicosante G, Fontana R, Rossolini GM. Cloning and characterization of blaVIM, a new integron-borne metallo-β-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 1999;43:1584–1590. doi: 10.1128/aac.43.7.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poirel L, Naas T, Nicolas D, Collet L, Bellais S, Cavallo JD, Nordmann P. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob. Agents Chemother. 2000;44:891–897. doi: 10.1128/aac.44.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Samuelsen O, Castanheira M, Walsh TR, Spencer J. Kinetic characterization of VIM-7, a divergent member of the VIM metallo-β-lactamase family. Antimicrob. Agents Chemother. 2008;52:2905–2908. doi: 10.1128/AAC.00166-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y, Kurosaki H. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J. Med. Chem. 2007;50:6647–6653. doi: 10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- 78.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J. Mol. Biol. 2008;375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 79.Toleman MA, Rolston K, Jones RN, Walsh TR. blaVIM-7, an evolutionarily distinct metallo-β-lactamase gene in a Pseudomonas aeruginosa isolate from the United States. Antimicrob. Agents Chemother. 2004;48:329–332. doi: 10.1128/AAC.48.1.329-332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lolans K, Queenan AM, Bush K, Sahud A, Quinn JP. First nosocomial outbreak of Pseudomonas aeruginosa producing an integron-borne metallo-β-lactamase (VIM-2) in the United States. Antimicrob. Agents Chemother. 2005;49:3538–3540. doi: 10.1128/AAC.49.8.3538-3540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aboufaycal H, Sader HS, Rolston K, Deshpande LM, Toleman M, Bodey G, Raad I, Jones RN. blaVIM-2 and blaVIM-7 carbapenemase-producing Pseudomonas aeruginosa isolates detected in a tertiary care medical center in the United States: report from the MYSTIC program. J. Clin. Microbiol. 2007;45:614–615. doi: 10.1128/JCM.01351-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lienard BM, Garau G, Horsfall L, Karsisiotis AI, Damblon C, Lassaux P, Papamicael C, Roberts GC, Galleni M, Dideberg O, Frere JM, Schofield CJ. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org. Biomol. Chem. 2008;6:2282–2294. doi: 10.1039/b802311e. [DOI] [PubMed] [Google Scholar]
- 83.Spencer J, Read J, Sessions RB, Howell S, Blackburn GM, Gamblin SJ. Antibiotic recognition by binuclear metallo-β-lactamases revealed by X-ray crystallography. J. Am. Chem. Soc. 2005;127:14439–14444. doi: 10.1021/ja0536062. [DOI] [PubMed] [Google Scholar]
- 84.Park H, Brothers EN, Merz KM., Jr. Hybrid QM/MM and DFT investigations of the catalytic mechanism and inhibition of the dinuclear zinc metallo-β-lactamase CcrA from Bacteroides fragilis. J. Am. Chem. Soc. 2005;127:4232–4241. doi: 10.1021/ja042607b. [DOI] [PubMed] [Google Scholar]
- 85.Suarez D, Merz KM., Jr. Molecular dynamics simulations of the mononuclear zinc-β-lactamase from Bacillus cereus. J. Am. Chem. Soc. 2001;123:3759–3770. doi: 10.1021/ja003796a. [DOI] [PubMed] [Google Scholar]
- 86.Diaz N, Suarez D, Merz KM., Jr. Molecular dynamics simulations of the mononuclear zinc-β-lactamase from Bacillus cereus complexed with benzylpenicillin and a quantum chemical study of the reaction mechanism. J. Am. Chem. Soc. 2001;123:9867–9879. doi: 10.1021/ja0113246. [DOI] [PubMed] [Google Scholar]
- 87.Suarez D, Brothers EN, Merz KM., Jr. Insights into the structure and dynamics of the dinuclear zinc β-lactamase site from Bacteroides fragilis. Biochemistry. 2002;41:6615–6630. doi: 10.1021/bi0121860. [DOI] [PubMed] [Google Scholar]
- 88.Dal Peraro M, Llarrull LI, Rothlisberger U, Vila AJ, Carloni P. Water-assisted reaction mechanism of monozinc β-lactamases. J. Am. Chem. Soc. 2004;126:12661–12668. doi: 10.1021/ja048071b. [DOI] [PubMed] [Google Scholar]
- 89.Dal Peraro M, Vila AJ, Carloni P. Structural determinants and hydrogen-bond network of the mononuclear zinc(II)-β-lactamase active site. J. Biol. Inorg. Chem. 2002;7:704–712. doi: 10.1007/s00775-002-0346-2. [DOI] [PubMed] [Google Scholar]
- 90.Dal Peraro M, Vila AJ, Carloni P. Protonation state of Asp120 in the binuclear active site of the metallo-β-lactamase from Bacteroides fragilis. Inorg. Chem. 2003;42:4245–4247. doi: 10.1021/ic026059j. [DOI] [PubMed] [Google Scholar]
- 91.Dal Peraro M, Vila AJ, Carloni P, Klein ML. Role of zinc content on the catalytic efficiency of B1 metallo β-lactamases. J. Am. Chem. Soc. 2007;129:2808–2816. doi: 10.1021/ja0657556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oelschlaeger P, Schmid RD, Pleiss J. Insight into the mechanism of the IMP-1 metallo-β-lactamase by molecular dynamics simulations. Protein Eng. 2003;16:341–350. doi: 10.1093/protein/gzg049. [DOI] [PubMed] [Google Scholar]
- 93.Wang C, Guo H. Inhibitor binding by metallo-β-lactamase IMP-1 from Pseudomonas aeruginosa: quantum mechanical/molecular mechanical simulations. J. Phys. Chem. B. 2007;111:9986–9992. doi: 10.1021/jp073864g. [DOI] [PubMed] [Google Scholar]
- 94.Oelschlaeger P, Pleiss J. Hydroxyl groups in the ββ sandwich of metallo-β-lactamases favor enzyme activity: Tyr218 and Ser262 pull down the lid. J. Mol. Biol. 2007;366:316–329. doi: 10.1016/j.jmb.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 95.Oelschlaeger P. Outsmarting metallo-β-lactamases by mimicking their natural evolution. J. Inorg. Biochem. 2008;102:2043–2051. doi: 10.1016/j.jinorgbio.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 96.Oelschlaeger P, Schmid RD, Pleiss J. Modeling domino effects in enzymes: molecular basis of the substrate specificity of the bacterial metallo-β-lactamases IMP-1 and IMP-6. Biochemistry. 2003;42:8945–8956. doi: 10.1021/bi0300332. [DOI] [PubMed] [Google Scholar]
- 97.Toney JH, Fitzgerald PM, Grover-Sharma N, Olson SH, May WJ, Sundelof JG, Vanderwall DE, Cleary KA, Grant SK, Wu JK, Kozarich JW, Pompliano DL, Hammond GG. Antibiotic sensitization using biphenyl tetrazoles as potent inhibitors of Bacteroides fragilis metallo-β-lactamase. Chem. Biol. 1998;5:185–196. doi: 10.1016/s1074-5521(98)90632-9. [DOI] [PubMed] [Google Scholar]
- 98.Walter M, Felici A, Galleni M, Soto R, RM A, JE B, JM F, M G, CJ S. Trifluoromethyl alcohol and ketone inhibitors of metallo-β-lactamases. Bioorg. Med. Chem. Lett. 1996;6:2455–2458. [Google Scholar]
- 99.Mollard C, Moali C, Papamicael C, Damblon C, Vessilier S, Amicosante G, Schofield CJ, Galleni M, Frere JM, Roberts GC. Thiomandelic acid, a broad spectrum inhibitor of zinc β-lactamases: kinetic and spectroscopic studies. J. Biol. Chem. 2001;276:45015–45023. doi: 10.1074/jbc.M107054200. [DOI] [PubMed] [Google Scholar]
- 100.Siemann S, Clarke AJ, Viswanatha T, Dmitrienko GI. Thiols as classical and slow-binding inhibitors of IMP-1 and other binuclear metallo-β-lactamases. Biochemistry. 2003;42:1673–1683. doi: 10.1021/bi027072i. [DOI] [PubMed] [Google Scholar]
- 101.Kurosaki H, Yasuzawa H, Yamaguchi Y, Jin W, Arakawa Y, Goto M. Detection of a metallo-β-lactamase (IMP-1) by fluorescent probes having dansyl and thiol groups. Org. Biomol. Chem. 2003;1:17–20. doi: 10.1039/b209086d. [DOI] [PubMed] [Google Scholar]
- 102.Jin W, Arakawa Y, Yasuzawa H, Taki T, Hashiguchi R, Mitsutani K, Shoga A, Yamaguchi Y, Kurosaki H, Shibata N, Ohta M, Goto M. Comparative study of the inhibition of metallo-β-lactamases (IMP-1 and VIM-2) by thiol compounds that contain a hydrophobic group. Biol. Pharm. Bull. 2004;27:851–856. doi: 10.1248/bpb.27.851. [DOI] [PubMed] [Google Scholar]
- 103.Yang KW, Crowder MW. Inhibition studies on the metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Arch. Biochem. Biophys. 1999;368:1–6. doi: 10.1006/abbi.1999.1293. [DOI] [PubMed] [Google Scholar]
- 104.Payne DJ, Bateson JH, Gasson BC, Khushi T, Proctor D, Pearson SC, Reid R. Inhibition of metallo-β-lactamases by a series of thiol ester derivatives of mercaptophenylacetic acid. FEMS Microbiol. Lett. 1997;157:171–175. doi: 10.1111/j.1574-6968.1997.tb12769.x. [DOI] [PubMed] [Google Scholar]
- 105.Lienard BM, Horsfall LE, Galleni M, Frere JM, Schofield CJ. Inhibitors of the FEZ-1 metallo-β-lactamase. Bioorg. Med. Chem. Lett. 2007;17:964–968. doi: 10.1016/j.bmcl.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 106.Walter M, MH V, RM A, G A, JE B, JM F, Galleni M, Rossolini GM, Schofield CJ. Hydroxamate inhibitors of Aeromonas hydrophila AE036 metallo-β-lactamase. Bioorg. Chem. 1999;27:35–40. [Google Scholar]
- 107.Bounaga S, Galleni M, Laws AP, Page MI. Cysteinyl peptide inhibitors of Bacillus cereus zinc β-lactamase. Bioorg. Med. Chem. 2001;9:503–510. doi: 10.1016/s0968-0896(00)00257-1. [DOI] [PubMed] [Google Scholar]
- 108.Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. Coordination geometries of metal ions in D- or L-captopril-inhibited metallo-β-lactamases. J. Biol. Chem. 2003;278:20659–20666. doi: 10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]
- 109.Siemann S, Evanoff DP, Marrone L, Clarke AJ, Viswanatha T, Dmitrienko GI. N-arylsulfonyl hydrazones as inhibitors of IMP-1 metallo-β-lactamase. Antimicrob. Agents Chemother. 2002;46:2450–2457. doi: 10.1128/AAC.46.8.2450-2457.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hiraiwa Y, Morinaka A, Fukushima T, Kudo T. Metallo-β-lactamase inhibitory activity of phthalic acid derivatives. Bioorg. Med. Chem. Lett. 2009;19:5162–5165. doi: 10.1016/j.bmcl.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 111.Horsfall LE, Garau G, Lienard BM, Dideberg O, Schofield CJ, Frere JM, Galleni M. Competitive inhibitors of the CphA metallo-β-lactamase from Aeromonas hydrophila. Antimicrob. Agents Chemother. 2007;51:2136–2142. doi: 10.1128/AAC.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Payne DJ, Hueso-Rodriguez JA, Boyd H, Concha NO, Janson CA, Gilpin M, Bateson JH, Cheever C, Niconovich NL, Pearson S, Rittenhouse S, Tew D, Diez E, Perez P, De La Fuente J, Rees M, Rivera-Sagredo A. Identification of a series of tricyclic natural products as potent broad-spectrum inhibitors of metallo-β-lactamases. Antimicrob. Agents Chemother. 2002;46:1880–1886. doi: 10.1128/AAC.46.6.1880-1886.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fernandez JR, Welsh WJ, Firestein BL. Structural characterization of the zinc binding domain in cytosolic PSD-95 interactor (cypin): Role of zinc binding in guanine deamination and dendrite branching. Proteins. 2008;70:873–881. doi: 10.1002/prot.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 115.Pang YP, Xu K, Yazal JE, Prendergas FG. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000;9:1857–1865. [PMC free article] [PubMed] [Google Scholar]
- 116.Olsen L, Pettersson I, Hemmingsen L, Adolph HW, Jorgensen FS. Docking and scoring of metallo-β-lactamases inhibitors. J. Comput. Aided Mol. Des. 2004;18:287–302. doi: 10.1023/b:jcam.0000046821.15502.71. [DOI] [PubMed] [Google Scholar]
- 117.Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for fragment inhibitors of AmpC β-lactamase. Proc Natl Acad Sci U S A. 2009;106:7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J. Med. Chem. 2003;46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 119.Meek PJ, Liu Z, Tian L, Wang CY, Welsh WJ, Zauhar RJ. Shape Signatures: speeding up computer aided drug discovery. Drug Discov. Today. 2006;11:895–904. doi: 10.1016/j.drudis.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 120.Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L, Welsh WJ, Bachmann K, Mani S. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol. Pharmacol. 2008;74:662–672. doi: 10.1124/mol.108.049437. [DOI] [PubMed] [Google Scholar]
- 121.Chekmarev DS, Kholodovych V, Balakin KV, Ivanenkov Y, Ekins S, Welsh WJ. Shape signatures: new descriptors for predicting cardiotoxicity in silico. Chem. Res. Toxicol. 2008;21:1304–1314. doi: 10.1021/tx800063r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kortagere S, Chekmarev D, Welsh WJ, Ekins S. New predictive models for blood-brain barrier permeability of drug-like molecules. Pharm. Res. 2008;25:1836–1845. doi: 10.1007/s11095-008-9584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang CY, Ai N, Arora S, Erenrich E, Nagarajan K, Zauhar R, Young D, Welsh WJ. Identification of previously unrecognized antiestrogenic chemicals using a novel virtual screening approach. Chem. Res. Toxicol. 2006;19:1595–1601. doi: 10.1021/tx060218k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kortagere S, Welsh WJ. Development and application of hybrid structure based method for efficient screening of ligands binding to G-protein coupled receptors. J. Comput. Aided Mol. Des. 2006;20:789–802. doi: 10.1007/s10822-006-9077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem. Biol. Interact. 2008;171:165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Oelschlaeger P, Toney JH. Are Human Microflora Influenced by Environmental Exposure to Antibiotics? Science. 2009 October 13; (E-Letter) [Google Scholar]
- 129.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 130.Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.