

Abstract
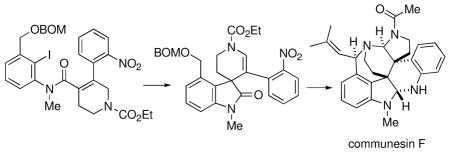
A new synthetic strategy for construction of the heptacyclic marine fungal alkaloid (±)-communesin F has been devised. Key reactions include an intramolecular Heck cyclization of a tetrasubstituted alkene to generate a tetracyclic enamide bearing one of the quaternary carbon centers (C7) of the alkaloid, an intramolecular reductive cyclization of an N-Boc aniline onto the oxindole moiety to form a pentacyclic framework containing the southern aminal, a stereoselective N-Boc-lactam enolate C-allylation to introduce the second quaternary carbon center (C8), and an azide reduction/N-Boc-lactam-opening cascade leading to the northern aminal.
Introduction and Background
Marine microbes are a relatively new source of unique natural products, in contrast to terrestrial microorganisms, which have been studied extensively for many decades. In 1993 Numata and coworkers reported two structurally unique polycyclic alkaloids, communesin A (1) and B (5) which were isolated from a Penicillium fungus growing on the marine alga Enteromorpha intestinalis (Figure 1).1 Communesins A (1) and B (5) show cytotoxicity against P-388 lymphocytic leukemia cells with moderate to potent activity (ED50 = 3.5 μg/mL and 0.45 μg/mL, respectively). The structures of these alkaloids were elucidated by extensive spectroscopic analysis. Thus, the communesins have a novel heptacyclic skeleton bearing two aminal functional groups, two vicinal quaternary carbon centers (C7, C8) and an epoxide moiety. The relative stereochemistry of the communesins (except for C21) was determined by NMR nOe studies, although the absolute configurations of 1 and 5 could not be established at that time.
FIGURE 1.

The Communesins and Perophoramidine
In 2003, Hemscheidt and coworkers described an alkaloid, nomofungin (9), which was isolated from the fermentation broth of an unidentified fungus derived from the bark of Ficus microcarpa L., growing on the Manoa campus of the University of Hawaii.2 The name, nomofungin, was chosen because the fungus producing this alkaloid was lost after isolation of the metabolite. Although nomofungin and communesin B (5) have identical 1H and 13C NMR spectral data, the metabolite was initially proposed to have structure 9, which has a lower N,O-acetal rather than the aminal of communesin B. However, it was later found that this assignment was incorrect, and that nomofungin is actually communesin B (5). It should be noted that the Hemscheidt studies did establish both the configuration at C21 of 5 using Murata’s J-based NMR method, as well as the absolute configuration of the molecule using the exciton chirality method as being 6R, 7R, 8R, 9S, 11S, 21R as shown. Hemscheidt also found that communesin B has cytotoxic activity against LoVo and KB cells (MIC 2.0 μg/mL, 4.5 μg/mL, respectively), which was shown to be due to the ability of the metabolite to cause microfilament disruption.
Recently, several other communesins have been isolated, including communesin C (6), D (7), E (2), F (8), G (3) and H (4).3 These metabolites differ only in the substituents at N15 and N16, except for communesin F, which has a double bond instead of an epoxide at C21,22. Communesins C and D, along with communesin B, were isolated from a Penicillium species growing on the sponge Axinella verrucosa.3a All three compounds exhibit moderate antiproliferative activity against a series of leukemia cell lines. Communesins A, B, D, E and F were also isolated from the fermentation broth of okara (the insoluble residue of whole soybeans) with Penicillium espansum Link MK-57.3b,4 These communesins show insecticidal activity against the third instar larvae of silkworms (LD50 = 150, 5, 80, 300 and 80 μg/g for communesins A, B, D, E and F, respectively). Communesins G and H, which were isolated from Penicillium rivulum Frisvad,3c were found to be inactive in antimicrobial, antiviral, and anticancer assays. Several additional communesin derivatives have recently been detected by mass spectrometry in a marine Penicillium, although the complete structures of these new metabolites have not yet been determined.3d,5
In 2002, Ireland et al. reported the isolation of a related metabolite, perophoramidine (10), from the tropical colonial ascidian Perophora namei collected in the Philippines.6 Extensive spectroscopic analysis showed that the compound has a hexacyclic ring system (i.e. one less ring than the heptacyclic skeleton of the communesins due to the absence of the azepine-G-ring). Perophoramidine has bis-amidine rather than the bis-aminal functionality present in the communesins, as well as three halogen atoms in the aromatic rings. Perophoramidine also has adjacent quaternary carbons (C4, C20) similar to the communesins, but the relative stereochemistry of these two stereogenic centers was found to be opposite that of the communesins. The absolute stereochemistry of perophoramidine has not yet been established. Perophoramidine has cytotoxicity against the HCT116 colon carcinoma cell line (IC50 = 60 μM) due to induction of apoptosis by poly(adenosine-5′-diphosphateribose)polymerase (PARP) cleavage.
Although there is relatively little in the way of experimental evidence to date for the biogenesis of these metabolites, it is clear that they are derived from two tryptamines units, along with an isoprenoid moiety in the case of the communesins.7 Structurally, the communesins and perophoramidine closely resemble members of the calycanthaceous family of plant alkaloids, whose biogenesis was suggested via an oxidative dimerization of tryptamine many years ago.8 More recently, Stoltz, et al. have proposed an alternative pathway for formation of the communesins via a hetero Diels Alder step.9
The novel structures of the communesins and perophoramidine, as well as the promising biological activity of these alkaloids, have caught the attention of a number of synthetic chemists. To date, however, few successful total syntheses of these complex metabolites have been reported, perhaps due in part to difficulties in the stereoselective formation of the adjacent quaternary carbon centers. Funk and Fuchs made a breakthrough on these molecules by completing an elegant total synthesis of racemic perophoramidine utilizing a key biomimetically patterned hetero Diels-Alder reaction.10 Unnatural dehaloperophoramidine has also been synthesized by Rainier and coworkers through a quite different strategy.11 More recently, Qin, et al. have published the first total synthesis of racemic communesin F.12 In addition, a few other preliminary synthetic approaches to the core ring system of the communesins and perophoramidine have been reported.13–15 Herein we report a new approach to the communesins, culminating in a total synthesis of racemic communesin F (8).16,17
Results and Discussion
Our initial unified approach to synthesis of both the communesins and perophoramidine was to rely on a tandem intramolecular Heck reaction/carbonylation of an acrylamide substrate such as 11 to form a lactam ester like 12 (Scheme 1). This compound would then be transformed to a tetracycle 13, where the lactone carbonyl would become C9 of the communesins (C24 of perophoramidine) and provide a handle to establish the quaternary carbon at C8. In a series of publications, we have described the implementation of this basic strategy.14 However, we have found that the success of the key Heck/carbonylation step was highly dependent upon the substituents in the two aromatic rings of 11, and even in the optimal cases reproducibility often was an issue, particularly on large scale reactions. As a result, we decided to investigate a new strategy which would bypass this problematic step.
SCHEME 1.

Thus, we considered the possibility of effecting a pivotal intramolecular Heck reaction18 of a system such as 14 to form a tetracyclic enamine derivative 15 having the C7 quaternary carbon (Scheme 2). In this case, a carbonylation is not necessary since the tetrahydropiperidine ring incorporates a carbon which will become C9 of the communesins. Moreover, the cyclization product 15 has the enamine functionality in a position to facilitate introduction of the second (C8) quaternary center. It should be noted that we approached this transformation with some trepidation, since examples of intramolecular Heck reactions of tetrasubstituted double bonds are relatively uncommon.19,20 In the work described below, we have investigated three series of Heck substrates 14 which differ in the substituent (X) at C12a in the F-ring. These substituents were intended to provide suitable functionality for eventual construction of the azepine-G-ring and also allow some flexibility with regard to protecting group compatibility during various steps in the synthesis.
SCHEME 2.

In order to prepare the requisite substrates 14 for the Heck reaction, we employed the three different F-ring anilines shown in Scheme 3. Thus, known nitro benzyl alcohol 1621 was converted to the BOM ether 17a and the TBS ether 17b via standard procedures in good yields. The corresponding anilines 18a and 18b were then prepared conveniently by iron-promoted reduction of the nitro compounds. The third aniline required, 18c, is a known compound which was accessed by the literature procedure.22
SCHEME 3.

The B/C-ring component required for synthesis of the Heck substrates 14 was easily prepared from commercially available materials. Therefore, known enol triflate 19,23 readily accessible from the corresponding β-ketoester, was coupled in a Suzuki-Miyaura reaction with o-nitrobenzeneboronic acid to afford β-arylated-α,β-unsaturated ester 20 in excellent yield (Scheme 4).24 This ester was converted to acid 21 by basic hydrolysis and then to the corresponding acid chloride 22. Without purification, 22 was combined with anilines 18a–c in the presence of Hunig’s base to cleanly afford amides 23a–c, respectively. It was found most convenient to replace the N-benzyl group of these intermediates at this point with a carbamate protecting group. Thus, exposure of 23a–c to ethyl chloroformate in methylene chloride at room temperature resulted in high yields of ethyl carbamates 24a–c.25 All three compounds could be transformed to the N-methylamides 25a–c in excellent yields with sodium hydride and methyl iodide.
SCHEME 4.

With the requisite substrates in hand, we proceded to explore the key intramolecular Heck cyclizations. We were pleased to find that both O-protected hydroxymethyl-substituted systems 25a and 25b underwent rapid, high yielding Heck reactions under the conditions shown in Scheme 4 to afford the desired spirocyclic enamides 26a and 26b, respectively. On the other hand, the cyclization of the C12a bromine-substituted substrate 25c proved to be much more problematic.
Therefore, when compound 25c was exposed to the same experimental conditions as used successfully for 25b, a 1:1 mixture of the desired Heck product 26c along with pentacyclic enamide 27 was formed in 74% total yield (Scheme 5). The latter compound presumably arises from a subsequent Heck arylation of bromide 26c. However, after some experimentation, it was found that by lowering the reaction temperature to 100 °C, and allowing the reaction to proceed for one hour produced a mixture of 26c and 27 in a 6.7:1 ratio in 51% total yield, along with 40% of recovered starting material 25c which could be recycled. Since compounds 26c and 27 were not easily separable by silica gel chromatography, the crude mixture was reduced with iron/concentrated HCl to afford aniline 28 which could then be separated from the amine derived from 27 (84% isolated yield based on the amount of nitro compound 26c in the mixture). This material was next converted to the Boc derivative 29c in high yield.
SCHEME 5.

The nitro groups of the two other Heck products 26a and 26b were also converted to the corresponding amines, although each required a different reduction procedure. The BOM-protected system 26a was reduced by catalytic hydrogenation at 40 atm using 5% Pt/C to afford the corresponding unstable aniline (without BOM hydrogenolysis), which was immediately transformed to the Boc derivative 29a in 87% yield for the two steps (Scheme 6). In the case of the TBS-protected compound 26b, catalytic hydrogenation was best effected using 10% Pd/C at 1 atm, followed by Boc protection to produce carbamate 29b in 85% overall yield.26
SCHEME 6.

The next objective in the synthesis was to construct the southern aminal functionality and the attendant D-ring of the communesins via a partial reduction of the lactam functionality of our three intermediates. After screening several hydride reagents,27 it was discovered that alane-dimethylethylamine complex effected lactam reduction/cyclization of 29a–c in one operation to directly generate the desired pentacyclic Boc-protected aminals 30a–c in good yields.
At this stage, we turned to introduction of the quaternary carbon at C8 with the desired stereochemistry, along with subsequently forming the upper aminal moiety. Initial studies were carried out with the TBS-protected enamide 30b, which was treated with an excess of butyllithium at low temperature, followed by addition of allyl iodide to afford the desired C-allylated imine 32 as the major product along with a small amount of the N-allyl enamine 33 (Scheme 7). Reaction of the enamide 30b with the alkyllithium reagent undoubtedly produces the intermediate lithio enamine 31,28 which undergoes alkylation from the less congested convex face to form 32 having the required vicinal quaternary carbon stereochemistry of the communesins. The structure and stereochemistry of this intermediate were confirmed by HMQC, HMBC and NOESY-NMR experiments. It might also be noted that we have been unable to alkylate 31 and related systems (Cf. 40, 50) with any two carbon electrophiles such as nitroethylene, ethyl iodoacetate, iodoacetonitrile, 2-iodoethylazide, N-nosylaziridine, ethylene oxide, etc.17 In addition, all attempted cyclopropanations of the enamide also failed.29,30
SCHEME 7.

Since it seemed possible that the C-allylation product 32 could arise via an aza-Cope rearrangement from the N-allyl compound 33, we briefly probed this possibility in a related series where the TBS group of 32 and 33 had been replaced by a p-methoxyphenyl (PMP) protecting group.17a Thus, both allyl pentacycles were first reexposed to the allylation conditions, but no change was detected. In refluxing toluene (bp 110 °C) both compounds were stable and no isomerization occurred. At a higher temperature in refluxing o-xylene (bp 143–145 °C), the N-allyl pentacycle began to decompose but none of the C-allyl compound was detected. However, at this same temperature the C-allyl pentacycle was slowly converted to the N-allyl product, and after heating for 17 hours, the ratio of C-allyl/N-allyl compounds in the mixture was found to be 1:0.6. This unexpected conversion can be rationalized by invoking a 1-aza-Cope rearrangement, which is usually unfavorable relative to its counterpart, a 3-aza-Cope rearrangement. However, in this specific system, relief of ring strain or of unfavorable steric interactions could be the driving force for the 1-aza-Cope rearrangement.31
With intermediate imine 32 now in hand, our plan for completion of the synthesis was to next construct the northern aminal functionality of the metabolites containing the A/B-ring system, and then finally establish the azepine-G-ring. Towards this end, it was necessary to oxidatively cleave the allyl group, although this transformation could not be directly effected on 32 due to the presence of the imine functionality. In an attempt to protect the imine, compound 32 was treated with diethyl pyrocarbonate in EtOH,32 which provided the desired α-ethoxycarbamate 34 as a mixture of diastereomers, but to our surprise a significant amount of aldehyde 35 was also produced (Scheme 8). The N,O acetal 34 was also found to be rather sensitive to handling and tended to rearrange to 35. Formation of this aldehyde can be rationalized as occurring via an intermediate N-acyliminium ion generated from the imine 32 and ethyl pyrocarbonate. As can be seen from models, there is a close proximity between the siloxymethyl group at C12a and the electrophilic N-acyliminium ion functionality which facilitates an intramolecular hydride transfer.33 In an attempt to decrease the amount of 35 formed from the imine, the reaction conditions were varied, but neither low temperature (−78 °C) nor addition of bases (NEt3, K2CO3) caused any significant improvement. It might also be noted that the PMP-protected variant of silyl ether 32 was even more prone to undergo this hydride migration.
SCHEME 8.

We therefore decided to explore manipulating the terminal olefin moiety of the crude 34/35 product mixture. It was found that by three sequential reactions (i.e. dihydroxylation, oxidative cleavage and reduction), the desired alcohol 36 could be obtained by chromatography as a 2:1 mixture of diastereomers in 45% yield based on imine 32, along with diol 37 in 30% yield, which is derived from aldehyde 35.
In an attempt to continue the synthesis, alcohol 36 was first converted to the corresponding azide under Mitsunobu conditions (Scheme 9). This azide was then reduced by catalytic hydrogenation and the resulting amine was protected in a one-pot reaction to afford N-Boc compound 38. Unfortunately, we were unable to cyclize 38 to the desired northern aminal 39, but rather under acidic conditions only a rearranged aldehyde was observed which results via a hydride migration from the C11 methylene group.
SCHEME 9.

Since the primary difficulty with the sequence involving the siloxymethyl series of compounds was due to the undesired hydride migrations outlined above, we decided to eliminate any possibility of this rearrangement by replacement of the C12a carbon substituent with a bromine. It might also be noted that a bromine substituent was used as a C12a functional handle to construct the azepine-G-ring in Qin’s total synthesis of communesin F.12b We therefore returned to brominated tetracyclic enamide 30c. Conversion of this compound to the corresponding lithio enamine using the butyllithium procedure described above (Cf. Scheme 7) was problematic since some halogen-metal exchange occurred under these conditions. However, the enamide could be tranformed by basic hydrolysis to the unstable NH enamine 40, which without purification was treated with LDA and allyl iodide to form the C-allyl imine 41 in 58% isolated yield along with 29% of the corresponding N-allyl enamine.
With alkylated pentacycle 41 in hand, we subsequently investigated the oxidative cleavage of the allyl group. As we had hoped, exposure of imine 41 to diethyl pyrocarbonate in ethanol cleanly produced α-ethoxycarbamate 42 (Scheme 10). However, much to our surprise dihydroxylation of 42 followed by oxidative cleavage with periodate gave hexacyclic acetal 43 (42% overall yield for three steps from 41) as a mixture of epimers instead of the desired aldehyde. The structure of the cyclic acetal 43 was confirmed by 2D NMR studies (HMQC, HMBC, and NOESY). Cyclic acetal 43 could then be hydrolyzed with aqueous acid to the hemiacetal 44 in good yield. With the hope that there might be some aldehyde in equilibrium with this hemiacetal, attempts were made at reductive aminations of 44, but to no avail. In view of these problems in forming the northern aminal, along with some of the poor reaction yields observed in the C12a bromine series of intermediates, the synthetic strategy was revised again.
SCHEME 10.

Because the many problems encountered in attempting to form the upper aminal primarily involved an imine, it was decided to avoid such functionality and instead utilize a B-ring lactam. Moreover, we elected to adopt an approach analogous to that of Qin,12b where the azepine-G-ring was positioned prior to forming the A/B-ring northern aminal. To investigate this new strategy, enamide 30a in the BOM-protected series was first hydrolyzed to the unstable NH enamine 45, which without purification was treated with cyanogen azide at room temperature to afford the N-cyanoamidine 47 (Scheme 11).34 This transformation is believed to occur via an initial [3+2]-dipolar cycloaddition of the enamine to afford triazole 46, which then rearranges with concomitant loss of nitrogen to form product 47. It was then found that basic hydrolysis of this amidine gave the lactam 48 as a single stereoisomer. Subsequent base mediated N-acylation of lactam 48 led to N-Boc lactam 49, but also caused isomerization to a 3:1 mixture of epimers at C8, which was of no consequence to the synthesis.
SCHEME 11.

Gratifyingly, alkylation of the mixture of N-Boc lactams 49 could be effected with potassium t-butoxide and allyl iodide to afford the desired product 51 as a single C8 stereoisomer in excellent yield. This alkylation proceeds via attack of the iodide on the N-Boc lactam enolate 50 from the least congested convex face, analogous to the stereoselective alkylations of the lithio enamines done previously (vide supra). To continue the synthesis it was found best to selectively remove the Boc group on the lactam at this point by basic cleavage to produce 52.
In a straightforward series of reactions, the allyl group of 52 was oxidatively cleaved to the aldehyde which without purification was reduced to the corresponding alcohol. This alcohol could then be transformed into the mesylate 53 in good overall yield for the three steps (Scheme 12). Removal of the BOM group of 53 was effected by hydrogenolysis with Pearlman’s catalyst and the resulting benzyl alcohol was immediately oxidized with the Dess-Martin periodinane to produce aldehyde 54. Displacement of the mesylate with sodium azide in DMF afforded azido aldehyde 55.
SCHEME 12.

We next investigated homologation of this compound to the α,β-unsaturated ketone 56, but to our surprise the aldehyde was found to be unreactive in Wadsworth-Emmons-Horner or Wittig condensations, perhaps for steric reasons. However, it was discovered that aldehyde 55 underwent a clean cross aldol reaction with acetone in the presence of aqueous sodium hydroxide to produce the desired (E)-unsaturated ketone 56 in excellent yield. In order to activate the δ-lactam for rearrangement in the next step, a Boc group was installed to form 57. With enone 57 in hand, our initial plan was to effect a one-pot tandem azide reduction, lactam opening and subsequent aza-Michael addition of the resulting N-Boc carbamate to the α,β-unsaturated ketone moiety to sequentially form the A-ring and the azepine-G-ring. Therefore, N-Boc-lactam azide 57 was reduced with trimethylphosphine in THF/water which led to the γ-lactam 58 in good yield, but this procedure did not promote the desired conjugate addition to the enone. All attempts to subsequently effect an aza-Michael addition of the N-Boc carbamate in 58 to the α,β-unsaturated ketone moiety to form the azepine ring under a variety of acidic or basic conditions unfortunately also failed.
We therefore decided to generate the azepine ring of communesin F using the strategy of Qin, et al.12a,35 Thus, exposure of ketone 58 to methyllithium in THF at low temperature led to the (E)-allylic alcohol 59 in 73% yield (Scheme 13). Subsequent treatment of this alcohol with PPTS at room temperature promoted a stereoselective allylic substitution reaction which produced the hexacyclic G-ring product 61 having the communesin configuration at C11 in 62% isolated yield. In addition, a 24% yield of the diene 60 resulting from dehydration of starting alcohol 59 was formed, similar to what was observed in the Qin system. The stereochemical outcome of this cyclization is not surprising since inspection of models indicates that the conformation shown in 59 is preferred in order to minimize steric interactions with the γ-lactam Bring. Thus, an acid catalyzed allylic substitution by the N-Boc group via this conformation produces the requisite communesin configuration at C11 in 61.
SCHEME 13.

To complete the total synthesis, it was now necessary to form the northern aminal. Towards this end, γ-lactam 61 was treated with commercially available triethyloxonium fluoroborate and DIEA in methylene chloride in order to form the corresponding ethyl imidate, similar to the conditions reported by Qin et al.12b Although this reaction produced some of the desired compound, a substantial amount of an unidentified byproduct was formed. Alternatively, the use of trimethyloxonium fluoroborate in the presence of Hunig’s base cleanly generated the desired methyl imidate 62.
Since the upper Boc protecting group proved to be much more labile towards acid than the one on the southern aminal, it could be selectively removed with 5% trifluoroacetic acid in methylene chloride. Upon neutralization of the resulting amine TFA salt with aqueous sodium bicarbonate, cyclization occurred spontaneously to form the heptacyclic amidine 63.36 Reduction of this amidine with sodium borohydride in acetic acid containing acetic anhydride occurred stereoselectively from the less hindered face to give the N-acetyl aminal 64. Finally, removal of the Boc protecting group on the lower aminal could then be effected with 40% TFA in methylene chloride to afford racemic communesin F (8). This material, which exists as a 2.6:1 mixture of acetamide rotamers in CDCl3, had proton and carbon NMR spectral data identical to that reported by the Qin group for the alkaloid.12b
In conclusion, we have achieved a stereoselective total synthesis of the heptacyclic fungal alkaloid (±)-communesin F (8) in about 30 steps from readily prepared enol triflate 19 and commercially available o-nitrobenzeneboronic acid. Notable reactions in the sequence include a rare example of an intramolecular Heck cyclization of a tetrasubstituted alkene to generate a spiro-tetracycle with the C7 quaternary carbon center of the metabolite, a one-pot reductive cyclization of an N-Boc aniline onto an oxindole moiety to form a pentacyclic system incorporating the southern aminal, a stereoselective N-Boc-lactam enolate C-allylation to introduce the C8 quaternary carbon center, and an azide reduction/N-Boc-δ-lactam opening cascade eventually leading to the northern aminal. In principle, it should be possible to effect the key intramolecular Heck reaction of substrate 25a enantioselectively to ultimately produce the natural (−)-enantiomer of communesin F.37 We hope to explore this transformation in future work.
Experimental Section
Synthesis of BOM Ether 17a
To a solution of (2-iodo-3-nitrophenyl)methanol21 (16, 263 mg, 0.94 mmol) in THF (5.0 mL) was added TBAI (70 mg, 0.19 mmol), DIPEA (0.25 mL, 1.41 mmol) and BOMCl (0.13 mL, 0.94 mmol). The reaction mixture was heated at 70 °C for 14 h and quenched by the addition of saturated aqueous NaHCO3. The solution was then extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/4) to give the BOM ether 17a (232 mg, 62%). 1H NMR (300 MHz, CDCl3) δ 7.67 (d, J = 7.6 Hz, 1H), 7.58 (dd, J = 1.5, 7.9 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.45-7.32 (m, 5H), 4.98 (s, 2H), 4.74 (s, 2H), 4.72 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 154.7, 143.8, 137.4, 131.0, 128.8, 128.4, 127.8, 127.7, 123.3, 94.4, 88.7, 73.9, 69.9; HRMS-ES (m/z): [M + H]+ calcd for C15H15NO4I, 400.0046; found, 400.0039.
Synthesis of Aniline 18a
To a solution of nitro compound 17a (3.16 g, 7.92 mmol) in EtOH (50.0 mL) was added iron powder (2.21 g, 39.59 mmol) and AcOH (6.8 mL). The reaction mixture was heated at 60 °C for 12 h and then cooled to rt. The mixture was filtered and the filtrate was concentrated. The residue was diluted with EtOAc and H2O and basified with solid Na2CO3. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 9/1) to provide the iodoaniline 18a (2.22 g, 76%). 1H NMR (300 MHz, CDCl3) δ 7.49-7.31 (m, 5H), 7.16 (br m, 1H), 6.89 (d, J = 7.4 Hz, 1H), 6.77 (br s, 1H), 4.94 (s, 2H), 4.74 (s, 2H), 4.69 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 146.5, 141.0, 137.7, 128.8, 128.4, 127.9, 127.6, 119.2, 114.3, 94.2, 88.4, 74.2, 69.6; HRMS-ES (m/z): [M + H]+ calcd for C15H17NO2I, 370.0304; found, 370.0295.
Synthesis of Ester 20
To a solution of enol triflate 1923 (763 mg, 1.94 mmol) and o-nitrobenzeneboronic acid (356 mg, 2.13 mmol) in DME (11.0 mL) and water (3.7 mL) were added Pd(PPh3)4 (45 mg, 0.039 mmol) and Na2CO3 (617 mg, 5.82 mmol). The reaction mixture was stirred at 80 °C for 1 h and then cooled to rt. The solution was diluted with H2O and extracted with EtOAc. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/3) to give arylated compound 20 (697 mg, 98%). 1H NMR (300 MHz, CDCl3) δ 8.03 (dd, J = 1.2, 8.2 Hz, 1H), 7.57 (ddd, J = 0.8, 7.6, 7.6 Hz, 1H), 7.46-7.39 (m, 3H), 7.36-7.26 (m, 3H), 7.19 (dd, J = 1.3, 7.6 Hz, 1H), 3.87 (q, J = 7.2 Hz, 2H), 3.73 (q, J =12.7 Hz, 2H), 3.48, 3.22 (ABq, J = 17.9 Hz, 2H), 2.83 (t, J = 5.8 Hz, 2H), 2.60 (m, 2H), 0.88 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 166.6, 148.3, 144.7, 138.2, 137.1, 133.5, 130.1, 129.4, 128.8, 128.4, 127.7, 125.1, 124.6, 61. 8, 60.7, 58.6, 49.2, 26.0, 14.0; HRMS-ES (m/z): [M + H]+ calcd for C21H23N2O4, 367.1658; found, 367.1651.
Synthesis Carboxylic Acid 21
To a solution of ester 20 (18.23 g, 49.75 mmol) in MeOH (260 mL) and water (108 mL) was added LiOH·H2O (10.45 g, 249.05 mmol). The reaction mixture was stirred at 50 °C for 12 h before MeOH was removed under reduced pressure. The resulting aqueous solution was acidified with 1 N HCl to pH 5–6 and extracted with EtOAc. The combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (MeOH/CH2Cl2, 1/10) to give the carboxylic acid 21 (16.48 g, 86%). 1H NMR (300 MHz, CDCl3) δ 12.6 (br s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.1 Hz, 1H), 7.33-7.26 (m, 6H), 7.13 (d, J = 7.2 Hz, 1H), 4.03-3.90 (m, 2H), 3.73-3.51 (m, 2H), 3.00 (br s, 1H), 2.78 (br s, 1H), 2.47 (br s, 2H); 13C NMR (75 MHz, CDCl3) δ 169.4, 148.5, 136.6, 135.6, 133.8, 131.8, 130.9, 130.7, 129.4, 129.3, 128.7, 127.3, 124.2, 58.6, 54.5, 46.6, 23.3; HRMS-ES (m/z): [M+H]+ calcd for C19H19N2O4, 339.1345; found, 339.1325.
Synthesis of Amide 23a
The acid 21 (11.33 g, 38.49 mmol) was dissolved in SOCl2 (25.0 mL) and the solution was refluxed for 3 h. Excess SOCl2 was distilled off and the resulting residue was dried under high vacuum and then dissolved in CH2Cl2 (30.0 mL). To a stirred solution of aniline 18a (9.16 g, 24.80 mmol) and DIPEA (17.3 mL, 99.31 mmol) in CH2Cl2 (50.0 mL) was added the above acid chloride 22 solution dropwise at rt. The reaction mixture was further stirred at rt for 12 h, and was diluted with CH2Cl2 and saturated aqueous NaHCO3. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 7/3) to give the amide 23a (14.88 g, 87% based on aniline 18a). 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.85 (s, 1H), 7.56 (t, J = 7.5 Hz, 1H), 7.47-7.31 (m, 12H), 7.28-7.18 (m, 2H), 4.91 (s, 2H), 4.70 (s, 2H), 4.63 (s, 2H), 3.76 (br d, J = 5.5 Hz, 2H), 3.36, 3.14 (ABq, J = 16.9 Hz, 2H), 2.97-2.77 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 166.1, 147.6, 140.7, 137.9, 137.4, 137.3, 134.8, 133.5, 130.6, 129.3, 128.65, 128.59, 128.3, 128.13, 128.09, 127.6, 127.4, 127.0, 124.8, 124.4, 121.4, 94.01, 93.94, 73.9, 69.4, 61.1, 56.6, 48.5, 26.2; HRMS-ES (m/z): [M + H]+ calcd for C34H33N3O5I, 690.1465; found, 690.1472.
Synthesis of Ethyl Carbamate 24a
To a stirred solution of amide 23a (3.13 g, 4.54 mmol) in CH2Cl2 (35.0 mL) was added ClCO2Et (0.52 mL, 5.45 mmol) dropwise at 0 °C. After the addition was complete, the ice bath was removed and the mixture was stirred at rt for 12 h before saturated aqueous NaHCO3 was added. The layers were separated. The aqueous layer was extracted with CH2Cl2 and the combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc/CH2Cl2, 5/3/2) to provide carbamate 24a (2.92 g, 96%). 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 7.9 Hz, 1H), 7.81 (dd, J = 1.3, 7.8 Hz, 1H), 7.73 (s, 1H), 7.63-7.58 (m, 1H), 7.47-7.27 (m, 7H), 7.24-7.14 (m, 2H), 4.86 (s, 2H), 4.65 (s, 2H), 4.58 (s, 2H), 4.41- 4.09 (br m, 2H), 4.22 (q, J = 7.0 Hz, 2H), 3.87-3.75 (br m, 2H), 2.85-2.68 (br m, 2H), 1.45-1.29 (br m, 3H); 13C NMR (75 MHz, CDCl3) δ 165.7, 154.9, 147.6, 140.8, 137.7, 137.3, 133.8, 133.6, 130.7, 129.1, 128.4, 128.2, 127.6, 127.5, 125.1, 124.7, 121.5, 94.3, 94.0, 73.9, 69.4, 61.4, 47.1, 39.5, 25.8, 14.4; HRMS-ES (m/z): [M + H]+ calcd for C30H31N3O7I, 672.1207; found, 672.1226.
Synthesis of N-Methyl Amide 25a
To a suspension of NaH (192 mg, 60% dispersion in mineral oil, 4.79 mmol) in THF (10.0 mL) was added a solution of amide 24a (2.92 g, 4.35 mmol) in THF (30.0 mL) at 0 °C. The mixture was stirred at 0 °C for 15 min and MeI (0.33 mL, 5.22 mmol) was added at this temperature. The reaction mixture was warmed to rt and stirred for 12 h before the addition of saturated aqueous NaHCO3. The aqueous mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 3/2) to provide N-methyl amide 25a (2.74 g, 92%). 1H NMR (300 MHz, CD3CN) δ 8.07 (dd, J = 1.1, 8.1 Hz, 1H), 7.76-7.73 (m, 1H), 7.62-7.56 (m, 1.7H), 7.47-7.44 (br m, 1H), 7.40-7.29 (m, 6H), 7.02 (br s, 0.3H), 6.40 (br s, 0.3H), 4.93 (s, 0.7H), 4.87 (s, 1.3H), 4.69 (s, 1H), 4.64 (s, 1.3H), 4.59 (s, 1.3H), 4.40 (br s, 1H), 4.20 (q, J = 7.1 Hz, 1.5H), 4.11-4.00 (m, 1H), 3.86-3.72 (m, 1.3H), 3.45 (br s, 0.3H), 3.11 (s, 2H), 2.99 (s, 0.3H), 2.92 (s, 0.8H), 2.87 (s, 0.3H), 2.58 (s, 1.3H), 2.26 (s, 0.6H), 1.30 (t, J = 6.9 Hz, 2H), 1.17 (t, J = 7.1 Hz, 1H); 13C NMR (75 MHz, CD3CN, 65 °C) δ 170.0, 156.6, 150.2, 147.0, 144.2, 139.7, 134.9, 134.2, 133.3, 132.0, 130.7, 130.6, 129.6, 129.3, 129.1, 128.8, 128.6, 126.1, 125.6, 103.1, 96.0, 75.3, 71.0, 62.5, 49.0, 48.1, 41.1, 39.0, 37.7, 27.2, 15.3; HRMS-ES (m/z): [M + H]+ calcd for C31H33N3O7I, 686.1363; found, 686.1371.
Synthesis of Tetracyclic Enamide 26a
To a solution of N-methyl amide 25a (2.21 g, 3.23 mmol) in DMA (56.0 mL) were added Pd(OAc)2 (145 mg, 0.65 mmol), PPh3 (339 mg, 1.29 mmol), n-Bu4NBr (2.08 g, 6.45 mmol) and K2CO3 (892 mg, 6.48 mmol). The mixture was stirred at 150 °C for 25 min, then cooled to rt and was diluted with H2O. The aqueous solution was extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/2) to provide spirocyclic enamide 26a (1.88 g, 90%). 1H NMR (300 MHz, CD3CN, 65 °C) δ 7.53-7.49 (m, 2H), 7.40-7.30 (m, 6H), 7.28-7.21 (m, 2H), 7.00 (d, J = 7.9 Hz, 1H), 6.80-6.84 (m, 2H), 4.85-4.80 (m, 2H), 4.36 (d, J = 11.9 Hz, 1H), 4.31, 4.26 (ABq, J = 7.1 Hz, 2H), 4.07-3.99 (m, 2H), 3.23 (s, 3H), 2.64-2.53 (m, 1H), 1.98-1.89 (m, 1H), 1.34 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CD3CN, 65 °C) δ 178.8, 154.5, 150.9, 145.2, 139.7, 136.3, 132.6, 132.4, 132.15, 132.11, 130.1, 129.6, 129.4, 129.0, 128.8, 125.2, 124.9, 110.4, 109.1, 95.8, 70.9, 66.7, 63.8, 50.5, 38.9, 31.5, 27.5, 15.0; HRMS-ES (m/z): [M + H]+ calcd for C31H32N3O7, 558.2240; found, 558.2238.
Synthesis of N-Boc Aniline 29a
To a beaker containing a solution of the Heck product 26a (1.71 g, 3.07 mmol) in toluene (30.0 mL) was added 5% platinum on carbon (514 mg). The beaker was then transferred into a high-pressure reaction vessel and flushed with H2. The hydrogen pressure was increased to 40 atm, and the reaction mixture was stirred at rt for 14 h. The pressure was released and the suspension was filtered through a pad of Celite. The solvent was removed to give the aniline which decomposed on standing and therefore was used immediately in crude form.
To a solution of the freshly prepared crude aniline in THF (90.0 mL) and H2O (30.0 mL) were added K2CO3 (8.49 g, 61.43 mmol) and (Boc)2O (10.05 g, 46.05 mmol). The mixture was stirred at 60 °C for 20 h. The aqueous solution was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/4) to provide N-Boc aniline 29a (1.68 g, 87% for 2 steps). 1H NMR (300 MHz, CD3CN, 65 °C) δ 7.72 (d, J = 8.9 Hz, 1H), 7.43-7.26 (m, 7H), 7.21 (s, 1H), 7.15-7.08 (m, 2H), 6.80-6.71 (m, 3H), 4.94, 4.91 (ABq, J = 6.7 Hz, 2H), 4.82, 4.73 (ABq, J = 11.6 Hz, 2H), 4.70 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 4.17-4.01 (m, 2H), 3.11 (s, 3H), 2.60-2.48 (m, 1H), 2.10-1.99 (m, 1H), 1.55 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CD3CN, 65 °C) δ 180.1, 154.7, 154.5, 145.1, 139.6, 138.6, 136.1, 131.8, 131.7, 129.99, 129.94, 129.6, 129.1, 129.0, 128.8, 125.1, 123.7, 122.9, 112.0, 109.2, 96.1, 80.9, 71.1, 67.0, 63.5, 51.8, 39.3, 32.1, 29.0, 27.4, 15.1; HRMS-ES (m/z): [M + H]+ calcd for C36H42N3O7, 628.3023; found, 628.3022.
Synthesis of Pentacyclic Aminal 30a
To a solution of N-Boc aniline 29a (302 mg, 0.48 mmol) in THF (15.0 mL) was added AlH3·Me2NEt (1.44 mL, 0.5 M in toluene, 0.72 mmol) dropwise at 0 °C. The reaction mixture was warmed to rt and stirred at rt for 4 h before saturated aqueous Na2SO4 was added. The aqueous mixture was extracted with EtOAc. The combined organic phases were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/9) to give the aminal 30a (250 mg, 74%). 1H NMR (300 MHz, CDCl3) δ 7.42-7.29 (m, 6H), 7.19 (dd, J = 1.4, 7.3 Hz, 2H), 7.05 (br m, 1H), 6.97 (t, J = 7.8 Hz, 2H), 6.56 (d, J = 7.7 Hz, 1H), 6.24 (d, J = 7.8 Hz, 1H), 5.82 (s, 1H), 4.84 (s, 2H), 4.74-4.65 (m, 3H), 4.50 (d, J = 11.6 Hz, 1H), 4.31-4.15 (m, 3H), 3.51 (br s, 1H), 3.00 (s, 3H), 2.50-2.35 (m, 2H), 1.53 (s, 9H), 7.1 (t, J = 1.35 Hz, 3H); 13C NMR (75 MHz, CD3CN, 65 °C) δ 154.3, 154.0, 151.8, 139.2,139.0, 134.4, 134.1, 130.4, 128.83, 128.76, 128.2, 127.9, 126.8, 125.7, 125.1, 124.5, 118.7, 116.8, 104.9, 94.9, 85.6, 81.5, 69.9, 65.5, 62.6, 52.0, 40.5, 34.6, 30.5, 28.0, 14.3; HRMS-ES (m/z): [M + H]+ calcd for C36H42N3O6, 612.3074; found, 612.3055.
Preparation of Cyanogen Azide
To a solution of cyanogen bromide (536 mg, 5.06 mmol) in CH3CN (10.0 mL) was added NaN3 (339 mg, 5.22 mmol) at 0 °C. The mixture was stirred at 0 °C for 4 h to give a solution of NCN3 in CH3CN (0.50 M) which can be stored at 0 °C for several weeks without noticeable decomposition.
Synthesis of N-Cyanoamidine 47
To a solution of enamide 30a (202 mg, 0.33 mmol) in EtOH (10.0 mL) was added 1 N aqueous KOH solution (10.0 mL, 10.0 mmol). The mixture was stirred at 94 °C for 3 h and then cooled to rt. After removal of EtOH in vacuo, the cloudy aqueous solution was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. Since the resulting enamine 45 was unstable and decomposed during chromatographic purification, the material was used directly in the next step.
To a solution of the crude enamine in MeCN (15.0 mL) was added NCN3 (1.0 mL, 0.5 M in MeCN, 0.50 mmol, freshly prepared). The solution was stirred at rt for 1 h and then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 2/3) to give the N-cyanoamidine 47 (178 mg, 93%). 1H NMR (300 MHz, CDCl3) δ 7.68 (br s, 1H), 7.44-7.29 (m, 5H), 7.20 (d, J = 3.0 Hz, 2H), 7.13 (d, J = 7.4 Hz, 1H), 7.06-7.00 (m, 1H), 6.96 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 7.6 Hz, 1H), 6.09 (d, J = 8.0 Hz, 1H), 6.07 (s, 1H), 4.92, 4.89 (ABq, J = 6.9 Hz, 2H), 4.81, 4.63 (ABq, J = 11.7 Hz, 2H), 4.74 (s, 2H), 4.34 (s, 1H), 3.87-3.84 (m, 1H), 3.55-3.51 (m, 1H), 2.78 (s, 3H), 2.75-2.64 (m, 1H), 2.12 (dd, J = 3.5, 14.3 Hz, 1H), 1.51 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 172.3, 150.1, 150.6, 137.5, 136.4, 132.8, 132.3, 129.8, 129.1, 128.5, 127.8, 127.6, 127.5, 125.9, 125.7, 119.5, 116.3, 104.5, 93.3, 81.6, 78.8, 69.8, 65.9, 53.9, 46.8, 39.1, 30.5, 29.9, 28.1; HRMS-ES (m/z): [M + H]+ calcd for C34H38N5O4, 580.2924; found, 580.2918.
Synthesis of Lactam 48
To a solution of N-cyanoamidine 47 (178 mg, 0.33 mmol) in EtOH (15.0 mL) was added 1 N aqueous KOH solution (15.0 mL, 15.0 mmol). The mixture was stirred at 94 °C for 12 h and then cooled to rt. After removal of EtOH in vacuo, the cloudy aqueous solution was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/1) to give the lactam 48 (102 mg, 60%). 1H NMR (300 MHz, CDCl3) δ 7.45-7.29 (m, 5H), 7.19-7.16 (m, 3H), 7.05-6.95 (m, 2H), 6.58 (d, J = 7.6 Hz, 1H), 6.44 (br s, 1H), 6.13 (s, 1H), 6.09 (d, J = 7.9 Hz, 1H), 4.92, 4.89 (ABq, J = 6.9 Hz, 2H), 4.83, 4.72 (ABq, J = 11.6 Hz, 2H), 4.78 (s, 2H), 4.17 (s, 1H), 3.86 (t, J = 11.7 Hz, 1H), 3.39-3.35 (m, 1H), 2.79 (s, 3H), 2.75-2.69 (m, 1H), 2.08-2.01 (m, 1H), 1.49 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 170.7, 153.4, 150.5, 137.6, 136.3, 132.8, 132.5, 129.9, 128.7, 128.4, 128.3, 127.7, 127.6, 127.2, 125.6, 119.1, 104.3, 93.4, 81.1, 79.1, 69.6, 65.7, 54.6, 49.1, 38.2, 31.5, 30.0, 28.1; HRMS-ES (m/z): [M + H]+ calcd for C33H38N3O5, 556.2811; found, 556.2820.
Synthesis of N-Boc Lactams 49
To a solution of lactam 48 (343 mg, 0.62 mmol) in THF (15.0 mL) was added LiHMDS (0.93 mL, 1.0 M in THF, 0.93 mmol). The mixture was stirred at rt for 10 min and (Boc)2O (141 mg, 0.65 mmol) was added. The reaction mixture was stirred at rt for another 10 min and quenched with aqueous saturated NaHCO3. The mixture was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 7/3) to give the N-Boc lactam (384 mg, 95%) as a 1:3 mixture of epimers 49a and 49b. For analytical purposes, the N-Boc lactam mixture was carefully purified by flash column chromatography on silica gel (hexanes/EtOAc, 9/1 then 7/3) to give pure samples of 49a and 49b.
49a
1H NMR (300 MHz, CDCl3) δ 8.31 (d, J = 5.7 Hz, 1H), 7.44-7.37 (m, 4H), 7.35-7.31 (m, 1H), 7.02-6.98 (m, 3H), 6.92 (t, J = 7.8 Hz, 1H), 6.46 (d, J = 7.7 Hz, 1H), 6.20 (d, J = 7.8 Hz, 1H), 5.90 (s, 1H), 4.84, 4.80 (ABq, J = 6.8 Hz, 2H), 4.69, 4.61 (ABq, J = 12.0 Hz, 2H), 4.50, 4.42 (ABq, J = 11.7 Hz, 2H), 4.36-4.33 (m, 1H), 3.87 (s, 1H), 3.83-3.77 (m, 1H), 2.97 (s, 3H), 2.52-2.37 (m, 2H), 1.60 (s, 9H), 1.49 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 169.4, 154.2, 152.1, 151.4, 138.1, 137.8, 133.0, 130.6, 128.6, 128.4, 128.3, 128.1, 127.8, 127.5, 126.1, 125.8, 125.2, 119.4, 105.2, 93.7, 84.9, 83.3, 81.2, 69.2, 66.8, 56.6, 45.2, 42.7, 34.5, 30.7, 28.1, 28.0; HRMS-ES (m/z): [M + H]+ calcd for C38H46N3O7, 656.3336; found, 656.3320.
49b
1H NMR (300 MHz, CDCl3) δ 7.42-7.38 (m, 4H), 7.35-7.32 (m, 1H), 7.28 (br s, 1H), 7.19-7.12 (m, 2H), 7.03-6.94 (m, 2H), 6.56 (d, J = 5.7 Hz, 1H), 6.10 (s, 1H), 6.09 (d, J = 5.9 Hz, 1H), 4.91, 4.88 (ABq, J = 5.1 Hz, 2H), 4.82, 4.69 (ABq, J = 8.8 Hz, 2H), 4.73 (d, J = 1.4 Hz, 2H), 4.35 (s, 1H), 4.10-4.04 (m, 1H), 3.96-3.93 (m, 1H), 2.84-2.76 (m, 1H), 2.78 (s, 3H), 2.17 (d, J = 10.6 Hz, 1H), 1.52 (s, 9H), 1.49 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 169.3, 152.8, 150.4, 137.5, 136.2, 132.6, 132.1, 129.7, 128.7, 128.3, 128.0, 127.63, 127.57, 127.3, 125.7, 125.5, 119.2, 104.4, 93.2, 82.8, 81.2, 79.7, 69.6, 65.7, 55.2, 51.6, 42.2, 32.3, 29.9, 28.0, 27.7; HRMS-ES (m/z): [M + H]+ calcd for C38H46N3O7, 656.3336; found, 656.3344.
Synthesis of C-Allyl Lactam 51
To a solution of N-Boc lactams 49 (207 mg, 0.32 mmol) in THF (20.0 mL) was added a solution of KOt-Bu (42 mg, 0.38 mmol) in THF (1.5 mL) dropwise at −78 °C, followed immediately by the addition of allyl iodide (0.48 mL, 1.0 M in THF, 0.48 mmol). The dry ice-acetone bath was removed and the mixture was warmed to rt and stirred for 40 min before aqueous saturated NaHCO3 was added. The mixture was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 9/1 then 7/3) to give the C-allyl lactam 51 (191 mg, 87%). 1H NMR (300 MHz, CD3CN, 65 °C) δ 8.31 (dd, J = 1.6, 7.7 Hz, 0.8H), 7.44-7.31 (m, 10H), 7.24(d, J = 9.0 Hz, 1H), 7.10-7.03 (m, 2H), 6.98-6.88 (m, 5H), 6.53 (d, J = 7.9 Hz, 1.8H), 6.36 (dd, J = 1.0, 7.8 Hz, 0.8H), 6.21 (d, J = 7.8 Hz, 1H), 5.75 (s, 1H), 5.70 (s, 1H), 5.64-5.55 (m, 1.8H), 5.13-5.03 (m, 2H), 4.93-4.83 (m, 4H), 4.80-4.75 (m, 2H), 4.67-4.52 (m, 5.8H), 4.35-4.17 (m, 4.8H), 3.84-3.77 (m, 0.8H), 3.73-3.62 (m, 0.8H), 3.13-3.02 (m, 1.7H), 3.00 (s, 2.8H), 2.93 (s, 3H), 2.91-2.84 (m, 1H), 2.70 (td, J = 5.7, 13.7 Hz, 1H), 2.26 (s, 2.3H), 2.25-2.12 (m, 2H), 1.93-1.88 (m, 1H), 1.58 (s, 9H), 1.55 (s, 9H), 1.51 (s, 15H); 13C NMR (75 MHz, CD3CN, 65 °C) δ 173.0, 155.3, 155.2, 155.1, 154.4, 152.6, 152.3, 146.4, 140.3, 139.9, 139.8, 139.6, 135.4, 135.3, 134.8, 134.4, 130.7, 130.4, 130.0, 129.7, 129.6, 129.5, 129.1, 128.8, 128.7, 128.6, 127.6, 127.4, 127.3, 125.9, 125.2, 120.3, 118.8, 118.7, 118.4, 107.4, 106.2, 105.3, 96.0, 95.8, 86.5, 84.6, 84.0, 82.8, 82.6, 82.1, 70.94, 70.88, 70.7, 68.1, 66.1, 60.8, 55.9, 52.4, 45.1, 40.7, 37.4, 32.1, 31.1, 30.9, 28.92, 28.89, 28.8, 28.7; HRMS-ES (m/z): [M + H]+ calcd for C41H50N3O7, 696.3649; found, 696.3651.
Synthesis of NH-Lactam 52
To a solution of the N-Boc lactam 51 (191 mg, 0.27 mmol) in EtOH (28.0 mL) was added 1 N aqueous KOH solution (2.8 mL, 2.8 mmol). The mixture was stirred at 80 °C for 13 h and then cooled to rt. After removal of EtOH in vacuo, the cloudy aqueous solution was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 7/3 then 1/1) to give the NH-lactam 52 (155 mg, 94%). 1H NMR (300 MHz, CDCl3) δ 8.27-8.22 (m, 1H), 7.40-7.24 (m, 5H), 6.97-6.82 (m, 4H), 6.53 (dd, J =0.8, 7.8 Hz, 1H), 6.26 (dd, J = 0.9, 7.8 Hz, 1H), 6.25 (s, 1H), 5.88-5.74 (m, 1H), 5.63 (s, 1H), 4.88-4.81 (m, 2H), 4.84, 4.77 (ABq, J = 6.7 Hz, 2H), 4.70, 4.61 (ABq, J = 11.9 Hz, 2H), 4.51 (d, J = 12.1 Hz, 1H), 4.24 (d, J = 12.1 Hz, 1H), 3.49 (td, J = 4.5, 12.8 Hz, 1H), 3.25-3.18 (m, 1H), 2.97 (s, 3H), 2.85 (d, J = 7.0 Hz, 2H), 2.54 (td, J = 6.0, 13.4 Hz, 1H), 1.95 (dd, J = 4.2, 13.5 Hz, 1H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 173.4, 154.1, 150.8, 138.5, 137.8, 134.4, 133.8, 132.7, 128.8, 128.4, 128.3, 127.8, 127.6, 126.8, 126.3, 125.9, 124.0, 119.4, 116.9, 106.1, 94.2, 83.1, 81.2, 69.6, 66.6, 58.6, 48.7, 40.6, 39.3, 31.4, 28.4, 28.1; HRMS-ES (m/z): [M + H]+ calcd for C36H42N3O5, 596.3124; found, 596.3136.
Synthesis of Mesylate 53
To a solution of allyl lactam 52 (155 mg, 0.26 mmol) in THF (9.0 mL) and H2O (3.0 mL) was added OsO4 (0.33 mL, 4 wt% solution in water, 0.052 mmol) and NMO (152 mg, 1.30 mmol). The mixture was stirred at rt for 12 h and then a solution of NaIO4 (278 mg) in H2O (3.0 mL) was added. The mixture was further stirred at rt for 2 h. The cloudy aqueous solution was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The resulting aldehyde was not purified and was used directly in the next step.
To a solution of the crude aldehyde in EtOH (20.0 mL) was added NaBH4 (25 mg, 0.66 mmol) at 0 °C. The mixture was stirred at 0 °C for 15 min and then quenched by the addition of saturated aqueous NH4Cl. After removal of EtOH in vacuo, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The alcohol was not purified and was used directly in the next step.
To a solution of the above crude alcohol in CH2Cl2 (10.0 mL) was added TEA (0.18 mL, 1.29 mmol) and MsCl (61 μL, 0.78 mmol) at 0 °C. The mixture was stirred at 0 °C for 15 min and then quenched by the addition of saturated aqueous NaHCO3. The mixture was diluted with H2O and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc/TEA, 2/3/0.1 then 3/7/0.1) to give the mesylate 53 (147 mg, 83% for 3 steps). 1H NMR (300 MHz, CDCl3) δ 8.18 (d, J = 7.4 Hz, 1H), 7.39-7.28 (m, 5H), 6.97-6.85 (m, 4H), 6.52 (d, J = 7.7 Hz, 1H), 6.25 (d, J = 7.3 Hz, 1H), 5.82 (s, 1H), 5.58 (s, 1H), 4.81, 4.76 (ABq, J = 6.7 Hz, 2H), 4.68, 4.61 (ABq, J = 11.8 Hz, 2H), 4.51-4.43 (m, 1H), 4.44 (d, J = 12.0 Hz, 1H), 4.14 (d, J = 12.1 Hz, 1H), 3.50 (td, J = 4.4, 12.5 Hz, 1H), 3.24-3.18 (m, 1H), 2.95 (s, 3H), 2.72 (s, 3H), 2.53-2.38 (m, 3H), 2.01 (dd, J = 3.8, 13.3 Hz, 1H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 173.2, 150.9, 138.9, 137.8, 133.8, 130.8, 128.6, 128.5, 127.8, 127.2, 126.8, 126.2, 124.4, 119.7, 106.4, 94.1, 83.2, 82.0, 69.7, 68.1, 66.6, 59.0, 46.5, 39.4, 36.7, 34.5, 31.4, 28.3, 28.1; HRMS-ES (m/z): [M + H]+ calcd for C36H44N3O8S, 678.2849; found, 678.2849.
Synthesis of Aldehyde 54
To a solution of mesylate 53 (49 mg, 0.072 mmol) in THF (5.0 mL) was added Pearlman’s catalyst (50 mg). After stirring under an atmosphere of hydrogen for 14 h, the reaction mixture was filtered through a pad of Celite and concentrated to give the corresponding alcohol which was not purified but used directly in the next step.
To a solution of the benzyl alcohol in CH2Cl2 (5.0 mL) was added Dess-Martin periodinane (35 mg, 0.083 mmol). The reaction was stirred at rt for 15 min and quenched by the addition of 10% aqueous Na2S2O3 solution. The aqueous mixture was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/3) to give the aldehyde 54 (30 mg, 75% for 2 steps). 1H NMR (300 MHz, CDCl3) δ 10.08 (s, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.06-6.88 (m, 4H), 6.78 (dd, J = 1.0, 7.8 Hz, 1H), 6.50 (dd, J = 1.0, 7.9 Hz, 1H), 6.25 (s, 1H), 5.70 (s, 1H), 4.55-4.46 (m, 1H), 4.03-3.92 (m, 1H), 3.41-3.32 (m, 1H), 3.17 (td, J = 4.5, 12.8 Hz, 1H), 3.01 (s, 3H), 2.74 (s, 3H), 2.60-2.49 (m, 3H), 1.96 (dd, J = 4.4, 13.8 Hz, 1H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 193.2, 172.6, 154.0, 151.1, 138.8, 134.6, 130.8, 130.0, 129.2, 127.6, 127.1, 126.2, 125.0, 117.4, 111.3, 82.6, 82.3, 67.9, 59.7, 46.8, 38.8, 36.7, 34.5, 31.2, 28.0, 27.9; HRMS-ES (m/z): [M + H]+ calcd for C28H34N3O7S, 556.2117; found, 556.2133.
Synthesis of Azide 55
To a solution of the mesylate 54 (25 mg, 0.045 mmol) in DMF (1.5 mL) was added NaN3 (50 mg, 0.77 mmol). The reaction mixture was stirred at 90 °C for 2 h and diluted with H2O. The aqueous mixture was extracted with Et2O. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/1) to give the azide 55 (14 mg, 61%). 1H NMR (300 MHz, CDCl3) δ 10.10 (s, 1H), 8.11 (dd, J = 2.5, 7.6 Hz, 1H), 7.05-6.94 (m, 4H), 6.77 (dd, J = 1.1, 7.8 Hz, 1H), 6.49 (dd, J = 1.0, 7.9 Hz, 1H), 6.14 (s, 1H), 5.70 (s, 1H), 3.72-3.63 (m, 1H), 3.38-3.32 (m, 1H), 3.17 (td, J = 4.7, 12.9 Hz, 1H), 3.00 (s, 3H), 2.92-2.82 (m, 1H), 2.55 (td, J = 6.1, 13.3 Hz, 1H), 2.33 (t, J = 8.8 Hz, 2H), 1.95 (dd, J = 4.5, 13.8 Hz, 1H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 193.4, 172.8, 154.0, 151.1, 138.6, 134.7, 131.2, 130.1, 129.1, 127.5, 126.9, 126.3, 125.1, 117.3, 111.1, 82.7, 82.0, 59.8, 48.7, 47.2, 38.8, 35.1, 31.1, 28.1, 28.0; HRMS-ES (m/z): [M + H]+ calcd for C27H31N6O4, 503.2407; found, 503.2418.
Synthesis of α,β-Unsaturated Ketone 56
To a solution of the aldehyde 55 (130 mg, 0.26 mmol) in acetone (20.0 mL) was added 10% aqueous NaOH solution (2.4 mL). The reaction mixture was stirred at 60 °C for 3 h and then cooled to rt. After removal of acetone in vacuo, the cloudy aqueous mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/1) to give the α,β-unsaturated ketone 56 (130 mg, 93%). 1H NMR (300 MHz, CDCl3) δ 8.35 (dd, J = 2.0, 6.6 Hz, 1H), 7.59 (d, J = 16.0 Hz, 1H), 6.98-6.91 (m, 4H), 6.35 (t, J = 7.9 Hz, 2H), 6.20 (d, J = 16.0 Hz, 1H), 5.74 (s, 1H), 5.63 (s, 1H), 3.66-3.59 (m, 1H), 3.29-3.26 (m, 1H), 3.16 (td, J = 4.4, 12.7 Hz, 1H), 3.00 (s, 3H), 2.89-2.82 (m, 1H), 2.55-2.44 (m, 1H), 2.49 (s, 3H), 2.31 (t, J = 8.6 Hz, 2H), 2.02-1.95 (m, 1H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 199.9, 173.2, 154.0, 151.1, 145.1, 138.8, 132.7, 131.5, 131.2, 129.1, 128.6, 127.4, 126.9, 125.9, 124.7, 118.6, 107.8, 82.6, 81.9, 58.9, 48.8, 46.5, 39.4, 35.2, 31.1, 28.8, 28.1, 26.5; HRMS-ES (m/z): [M + H]+ calcd for C30H35N6O4, 543.2720; found, 543.2720.
Synthesis of N-Boc Lactam 57
To a solution of lactam 56 (130 mg, 0.24 mmol) in THF (40.0 mL) was added LiHMDS (0.26 mL, 1.0 M in THF, 0.26 mmol) and (Boc)2O (63 mg, 0.29 mmol). The reaction mixture was stirred at rt for 10 min and quenched with aqueous saturated NaHCO3. The mixture was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 9/1, 3/1 then 1/1) to give N-Boc-lactam 57 (125 mg, 81%). 1H NMR (300 MHz, CDCl3) δ 8.34 (d, J = 7.2 Hz, 1H), 7.42 (d, J = 16.0 Hz, 1H), 6.97-6.88 (m, 4H), 6.32 (t, J = 7.2 Hz, 2H), 6.21 (d, J = 16.0 Hz, 1H), 5.61 (s, 1H), 3.63 (dd, J = 4.0, 13.3 Hz, 1H), 3.52-3.38 (m, 2H), 2.95 (s, 3H), 2.91-2.81 (m, 1H), 2.51 (td, J = 5.6, 13.6 Hz, 1H), 2.37 (s, 3H), 2.37-2.30 (m, 2H), 2.01-1.96 (m, 1H), 1.48 (s, 9H), 1.43 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 198.9, 171.5, 153.9, 152.8, 150.8, 143.2, 138.5, 133.1, 132.7, 131.1, 129.2, 128.4, 127.5, 127.0, 125.5, 124.9, 118.7, 107.7, 83.9, 82.4, 81.9, 59.0, 48.4, 48.2, 44.0, 34.2, 31.0, 29.4, 28.0, 27.6, 26.4; HRMS-ES (m/z): [M + H]+ calcd for C35H43N6O6, 643.3244; found, 643.3247.
Synthesis of Spiro-γ-Lactam 58
To a solution of the N-Boc lactam 57 (140 mg, 0.22 mmol) in THF (60.0 mL) and H2O (12.0 mL) was added PMe3 (2.0 mL, 1.0 M in THF, 2.0 mmol). The reaction mixture was stirred at 70 °C for 13 h and then cooled to rt. After removal of THF in vacuo, the cloudy aqueous mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 1/3) to give the spiro-γ-lactam 58 (118 mg, 88%). 1H NMR (300 MHz, CDCl3) δ 8.29 (br s, 0.5H), 7.80 (d, J = 16.0 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.20 (br s, 0.8H), 7.12 (t, J = 7.6 Hz, 1H), 6.98-6.93 (m, 1H), 6.88 (t, J = 7.8 Hz, 1H), 6.80 (d, J = 7.3 Hz, 1H), 6.49 (d, J = 16.0 Hz, 1H), 6.04 (s, 1H), 5.94 (d, J = 7.5 Hz, 1H), 4.66 (s, 0.8H), 3.44-3.38 (m, 1H), 3.21 (t, J = 9.7 Hz, 1H), 2.99-2.92 (m, 2H), 2.78 (br s, 2H), 2.37 (s, 3H), 2.42-2.37 (br m, 1H), 2.05-1.98 (m, 1H), 1.52 (s, 9H), 1.35 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 199.5, 176.0, 155.4, 153.8, 151.6, 143.5, 137.5, 136.9, 134.1, 129.3, 126.7, 125.9, 125.4, 125.3, 114.5, 105.6, 82.3, 81.6, 79.2, 62.1, 52.8, 40.1, 37.7, 35.5, 34.6, 30.0, 28.3, 28.2, 27.4; HRMS-ES (m/z): [M + H]+ calcd for C35H45N4O6, 617.3339; found, 617.3342.
Synthesis of Allylic Alcohol 59
To a solution of the spiro-γ-lactam 58 (113 mg, 0.18 mmol) in THF (30.0 mL) was added MeLi (0.57 mL, 1.6 M in Et2O, 0.91 mmol) dropwise at −78 °C. The reaction mixture was stirred at -78 °C for 15 min and then quenched with saturated aqueous NaHCO3. After removal of THF in vacuo, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (EtOAc) to give the allylic alcohol 59 (85 mg, 73%). 1H NMR (300 MHz, CDCl3) δ 7.45 (d, J = 7.5 Hz, 1H), 7.27 (br s, 0.8H), 7.21 (br s, 0.6H), 7.12 (td, J = 1.2, 7.4 Hz, 1H), 6.99-6.89 (m, 2H), 6.85 (d, J = 7.8 Hz, 1H), 6.69 (d, J = 7.8 Hz, 1H), 6.10 (d, J = 11.2 Hz, 1H), 5.90 (s, 0.8H), 5.87 (d, J = 7.8 Hz, 1H), 4.80 (br s, 0.8H), 4.27 (br s, 0.8H), 3.46-3.40 (m, 1H), 3.24 (t, J = 9.7 Hz, 1H), 3.09 (br s, 1H), 2.99-2.91 (m, 1H), 2.83-2.79 (m, 1H), 2.72-2.64 (br m, 1H), 2.47 (s, 3H), 2.18-2.06 (m, 1H), 1.92-1.84 (m, 1H), 1.51 (s, 9H), 1.41 (s, 3H), 1.37 (s, 9H), 1.31 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 175.5, 155.7, 153.9, 150.8, 138.9, 138.3, 137.6, 137.1, 129.0, 126.4, 126.1, 125.2, 124.9, 123.4, 114.7, 103.7, 82.8, 81.3, 79.6, 70.5, 60.4, 52.8, 40.1, 38.1, 35.4, 35.1, 30.7, 30.0, 28.8, 28.4, 28.3; HRMS-ES (m/z): [M + H]+ calcd for C36H49N4O6, 633.3652; found, 633.3647.
Synthesis of Hexacyclic γ-Lactam 61 and Diene 60
To a solution of the allylic alcohol 59 (83 mg, 0.13 mmol) in CHCl3 (20.0 mL) was added PPTS (3.3 mg, 0.013 mmol). The reaction mixture was stirred at rt for 1.5 h. After removal of CHCl3 in vacuo, the residue was purified by flash column chromatography on silica gel (hexanes/acetone, 5/1) to give the hexacycle 61 (51 mg, 62%) and diene 60 (20 mg, 24%).
Hexacycle 61
1H NMR (300 MHz, CDCl3) δ 7.36-7.32 (br m, 1H), 7.26-7.23 (br m, 1H), 7.13 (t, J = 7.7 Hz, 1H), 6.92 (td, J = 1.3, 7.7 Hz, 1H), 6.88 (t, J = 7.9 Hz, 1H), 6.28 (br d, J = 6.7 Hz, 1H), 5.98 (br s, 1H), 5.89 (d, J = 7.6 Hz, 1H), 5.84 (s, 1H), 5.55 (br s, 1H), 5.10 (br s, 1H), 3.91 (br s, 1H), 3.47 (br d, J = 9.0 Hz, 1H), 3.14-2.90 (m, 4H), 2.45 (s, 3H), 2.14-2.09 (br m, 1H), 1.98-1.92 (br m, 1H), 1.84 (s, 3H), 1.75 (s, 3H), 1.51 (s, 9H), 1.48 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 174.3, 156.2, 153.5, 150.6, 139.3, 137.3, 133.3, 128.8, 126.4, 125.9, 125.1, 125.0, 124.3, 116.9, 103.2, 89.5, 81.2, 79.6, 61.2, 59.7, 52.3, 41.0, 39.2, 36.1, 30.7, 28.6, 28.4, 25.6, 18.9; HRMS-ES (m/z): [M + H]+ calcd for C36H47N4O5, 615.3546; found, 615.3560.
Diene 60
1H NMR (300 MHz, CDCl3) δ 7.49 (d, J = 7.7 Hz, 1H), 7.45 (br s, 1H), 7.14 (t, J = 7.5 Hz, 1H), 6.97 (td, J = 1.4, 7.8 Hz, 1H), 6.88 (t, J = 7.8 Hz, 1H), 6.78 (d, J = 14.6 Hz, 1H), 6.74 (d, J = 7.3 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H), 5.97 (s, 1H), 5.88 (d, J = 7.6 Hz, 1H), 4.96 (s, 1H), 4.85 (s, 1H), 4.42 (br s, 1H), 3.48-3.42 (m, 1H), 3.26 (t, J = 9.8 Hz, 1H), 3.01-2.94 (m, 2H), 2.82 (br s, 1H), 2.52 (s, 3H), 2.46-2.37 (m, 1H), 2.03-1.86 (m, 1H), 1.91 (s, 3H), 1.53 (s, 9H), 1.38 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 175.8, 155.4, 151.3, 142.3, 138.2, 137.2, 131.5, 129.1, 128.2, 126.6, 126.1, 125.3, 125.1, 123.5, 116.7, 114.4, 103.9, 82.9, 81.4, 79.2, 61.5, 52.7, 40.1, 37.8, 35.2, 30.6, 28.4, 28.3, 19.2; HRMS-ES (m/z): [M + H]+ calcd for C36H47N4O5, 615.3546; found, 615.3554.
Synthesis of Methyl Imidate 62
To a solution of the hexacycle 61 (12.0 mg, 0.020 mmol) in CH2Cl2 (10.0 mL) was added DIPEA (34 μL, 0.20 mmol) and Me3OBF4 (29 mg, 0.20 mmol). The reaction mixture was stirred at rt for 30 min and quenched with saturated aqueous NaHCO3. The solution was diluted with H2O and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by preparative TLC (hexanes/acetone, 2/1) to give the methyl imidate 62 (10.5 mg, 86%). 1H NMR (300 MHz, CDCl3) δ 7.25 (d, J = 5.4 Hz, 1H), 7.13 (td, J = 1.4, 7.5 Hz, 1H), 6.95-6.89 (m, 2H), 6.77 (d, J = 7.8 Hz, 1H), 6.33 (br s, 1H), 5.96 (d, J = 6.8 Hz, 1H), 5.68 (s, 1H), 5.18 (br s, 1H), 3.92-3.65 (m, 4H), 3.78 (s, 3H), 3.48-3.36 (br m, 1H), 3.26-3.14 (br m, 1H), 2.98-2.73 (br m, 2H), 2.38 (s, 3H), 2.24-2.15 (m, 1H), 1.84 (s, 3H), 1.74 (s, 3H), 1.51 (s, 9H), 1.50 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 171.1, 155.8, 153.5, 150.7, 140.3, 136.8, 128.4, 126.3, 125.0, 124.9, 124.8, 124.5, 116.9, 103.5, 81.4, 80.2, 59.1, 57.9, 55.6, 51.1, 40.1, 38.4, 31.2, 29.7, 28.5, 28.4, 25.7, 18.8; HRMS-ES (m/z): [M + H]+ calcd for C37H49N4O5, 629.3703; found, 629.3723.
Synthesis of Amidine 63
To a solution of the imidate 62 (13.0 mg, 0.021 mmol) in CH2Cl2 (5.0 mL) was added TFA (0.25 mL). The reaction mixture was stirred at rt for 45 min and quenched with saturated aqueous NaHCO3. The solution was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by preparative TLC (hexanes/acetone, 2/1) to give the amidine 63 (9.0 mg, 88%). 1H NMR (300 MHz, CDCl3) δ 7.07-6.96 (m, 2H), 7.05 (dd, J = 1.4, 7.4 Hz, 1H), 6.86 (t, J = 8.5 Hz, 1H), 6.78 (t, J = 7.7 Hz, 1H), 6.05 (dd, J = 7.9, 12.9 Hz, 2H), 5.79 (br s, 0.6H), 5.61 (br s, 0.4H), 5.36-5.31 (m, 1H), 4.81 (d, J = 8.7 Hz, 1H), 3.83-3.73 (m, 2H), 3.36-3.29 (m, 1H), 3.26-3.18 (m, 1H), 2.90 (s, 3H), 2.87-2.77 (m, 1H), 2.29-2.20 (m, 1H), 2.07-2.00 (m, 1H), 1.85 (br s, 1H), 1.78 (s, 3H), 1.71 (s, 3H), 1.42 (br s, 5H), 1.23 (s, 4H); 13C NMR (75 MHz, CDCl3) δ 180.0, 154.0, 149.6, 137.7, 136.6, 134.3, 130.2, 129.7, 128.5, 127.5, 126.6, 124.6, 123.9, 121.9, 116.9, 104.3, 81.0, 79.1, 60.1, 58.6, 54.9, 54.8, 45.5, 38.3, 30.4, 28.3, 27.0, 25.7, 18.6; HRMS-ES (m/z): [M + H]+ calcd for C31H37N4O2, 497.2917; found, 497.2918.
Synthesis of (±)-Communesin F (8)
To a solution of the amidine 63 (14.0 mg, 0.028 mmol) in acetic acid (0.8 mL) and acetic anhydride (0.8 mL) was added NaBH4 (90 mg, 2.38 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and quenched with saturated aqueous Na2CO3. The aqueous solution was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The resulting N-acetyl-N-Boc-bis-aminal 64 was not purified and was used directly in the next step.
To a solution of the bis-aminal 64 in CH2Cl2 (5.0 mL) was added TFA (2.0 mL). The reaction mixture was stirred at rt for 12 h and quenched with saturated aqueous Na2CO3. The aqueous solution was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (hexanes/acetone, 5/1 then 2/1) to give (±)-communesin F (8) (8.2 mg, 66% for 2 steps).
As described by the Qin group6, (±)-communesin F exists as two amide rotamers in a ratio of 2.6: 1 in CDCl3 as shown by 1H NMR. Only the NMR data for the major rotamer is listed (see Supporting Information for copies of the spectra):
Major rotamer of 8
1H NMR (300 MHz, CDCl3) δ 6.98 (d, J = 1.7, 7.7 Hz, 1H), 6.80 (t, J = 7.7 Hz, 1H), 6.72-6.64 (m, 3H), 6.06 (d, J = 7.7 Hz, 1H), 5.84 (d, J = 7.6 Hz, 1H), 5.21 (br d, J = 8.9 Hz, 1H), 5.09 (s, 1H), 5.03 (d, J = 8.8 Hz, 1H), 4.64 (s, 1H), 3.83 (dd, J = 8.8, 11.4 Hz, 1H), 3.76 (br s, 0.5H), 3.34-3.27 (m, 1H), 3.25-3.17 (m, 1H), 3.04-2.96 (m, 1H), 2.80 (s, 3H), 2.75-2.70 (m, 1H), 2.38 (s, 3H), 2.30-2.18 (m, 2H), 1.97-1.91 (m, 1H), 1.83 (d, J = 1.2 Hz, 3H), 1.76 (d, J = 1.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.6, 150.1, 142.7, 140.6, 136.1, 132.7, 131.3, 128.3, 127.3, 124.6, 123.2, 120.6, 117.0, 114.7, 100.7, 82.6, 79.5, 64.4, 51.8, 51.2, 44.2, 37.8, 36.2, 30.8, 29.6, 26.0, 22.6, 18.5; HRMS-ES (m/z): [M + H]+ calcd for C28H33N4O, 441.2654; found, 441.2635.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (CA-034303) and the National Science Foundation (CHE-0806807) for financial support of this research.
Footnotes
Supporting Information Available: Copies of proton and carbon NMR spectra of new compounds and experimental procedures for some sequences. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355. [Google Scholar]
- 2.Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J Org Chem. 2001;66:8717. doi: 10.1021/jo010335e.For the retraction of the putative nomofungin structure see: Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J Org Chem. 2003;68:1640. doi: 10.1021/jo010335e.
- 3.(a) Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. J Nat Prod. 2004;67:78. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]; (b) Hayashi H, Matsumoto H, Akiyama K. Biosci Biotechnol Biochem. 2004;68:753. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]; (c) Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C. J Nat Prod. 2005;68:258. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]; (d) Kerzaon I, Pouchus YF, Monteau F, Le Bizec B, Nourrisson MR, Biard JF, Grovel O. Rapid Commun Mass Spectrom. 2009;23:3928. doi: 10.1002/rcm.4330. [DOI] [PubMed] [Google Scholar]
- 4.There is some confusion about nomenclature since some structurally different compounds were initially designated as the same communesins due to simultaneous publication by two groups (see refs 3a and 3b). The currently accepted communesin nomenclature can be found in ref 3c.
- 5.For a brief review of the communesins see: Siengalewicz P, Gaich T, Mulzer J. Angew Chem Int Ed. 2008;47:2. doi: 10.1002/anie.200801735.
- 6.Verbitski SM, Mayne CL, Davis RA, Concepcion GP, Ireland CM. J Org Chem. 2002;67:7124. doi: 10.1021/jo026012f. [DOI] [PubMed] [Google Scholar]
- 7.For labelling studies on the biosynthesis of the communesins see: Wigley LJ, Mantle PG, Perry DA. Phytochemistry. 2006;67:561. doi: 10.1016/j.phytochem.2005.10.011.
- 8.(a) Robinson R, Teuber HJ. Chem Ind. 1954:783. [Google Scholar]; (b) Woodward RB, Yand N, Katz TJ. Proc Chem Soc, London. 1960:76. [Google Scholar]; (c) Hendrickson JB, Rees R, Goschke R. Proc Chem Soc, London. 1962:383. [Google Scholar]
- 9.(a) May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203. [Google Scholar]; (b) May JA, Stoltz BM. Tetrahedron. 2006;62:5262. [Google Scholar]
- 10.Fuchs JR, Funk RL. J Am Chem Soc. 2004;126:5068. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 11.Sabhi A, Novikov A, Rainier JD. Angew Chem Int Ed. 2006;45:4317. doi: 10.1002/anie.200601278. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yang J, Song H, Xiao X, Wang J, Qin Y. Org Lett. 2006;8:2187. doi: 10.1021/ol0607138. [DOI] [PubMed] [Google Scholar]; (b) Yang J, Wu H, Shen L, Qin Y. J Am Chem Soc. 2007;129:13794. doi: 10.1021/ja075705g. [DOI] [PubMed] [Google Scholar]
- 13.Crawley SL, Funk RL. Org Lett. 2003;5:3169. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]
- 14.(a) Artman GD, III, Weinreb SM. Org Lett. 2003;5:1523. doi: 10.1021/ol034314d. [DOI] [PubMed] [Google Scholar]; (b) Artman GD., III . PhD Thesis. The Pennsylvania State University; University Park, PA: 2004. [Google Scholar]; (c) Seo JH, Artman GD, III, Weinreb SM. J Org Chem. 2006;71:8891. doi: 10.1021/jo061660a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Evans MA, Sacher JR, Weinreb SM. Tetrahedron. 2009;65:6712. [Google Scholar]
- 15.George JH, Adlington RM. Synlett. 2008:2093. [Google Scholar]
- 16.A preliminary account of a portion of this work has appeared: Liu P, Seo JH, Weinreb SM. Angew Chem Int Ed. 2010;49:2000. doi: 10.1002/anie.200906818.
- 17.Taken from the Ph.D. theses of: Seo JH. The Pennsylvania State University. University Park, PA: 2008. Liu P. The Pennsylvania State University; University Park, PA: 2010.
- 18.For a review of the intramolecular Heck reaction see: Link JT. Org React. 2002;60:157.
- 19.For examples of intramolecular Heck reactions with substrates bearing tetrasubstituted alkene moieties, see Grigg R, Sridharan V, Stevenson P, Worakun T. J Chem Soc Chem Commun. 1986:1697.Abelman MM, Oh T, Overman LE. J Org Chem. 1987;52:4133.Grigg R, Sridharan V, Stevenson P, Sukirthalingam S. Tetrahedron. 1989;45:3557.Cheng CY, Liou JP, Lee MJ. Tetrahedron Lett. 1997;38:4571.Liou JP, Cheng CY. Tetrahedron Lett. 2000;41:915.Hsin LW, Chang LT, Chen CW, Hsu CH, Chen HW. Tetrahedron. 2005;61:513.Frey DA, Duan C, Hudlicky T. Org Lett. 1999;1:2085.Fukuyama Y, Yuasa H, Tonoi Y, Harada K, Wada M, Asakawa Y, Hashimoto T. Tetrahedron. 2001;57:9299.Nicolaou KC, Roecker AJ, Follmann M, Baati R. Angew Chem Int Ed. 2002;41:2107.
- 20.For an intermolecular example, see: Dyker G, Korning J, Jones PG, Bubenitschek P. Angew Chem Int Ed. 1993;32:1733.
- 21.(a) Seno K, Hagishita S, Sato T, Kuriyama K. J Chem Soc Perkin Trans 1. 1984:2012. [Google Scholar]; (b) Ozlu Y, Cladingboel DE, Parsons PJ. Tetrahedron. 1994;50:2183. [Google Scholar]
- 22.Liedholm B. Acta Chem Scand. 1993;47:701. [Google Scholar]
- 23.Ferraris D, Ficco RP, Pahutski T, Lautar S, Huang S, Zhang J, Kalish V. Bioorg Med Chem Lett. 2003;13:2513. doi: 10.1016/s0960-894x(03)00465-7. [DOI] [PubMed] [Google Scholar]
- 24.For examples of Suzuki-Miyaura couplings with o-nitrobenzeneboronic acid, see: Ho TL, Hsieh SY. Helv Chim Acta. 2006;89:111.Gonzalez RR, Liguori L, Carrillo AM, Bjorsvik HR. J Org Chem. 2005;70:9591. doi: 10.1021/jo051589t.Myers AG, Herzon SB. J Am Chem Soc. 2003;125:12080. doi: 10.1021/ja0372006.Ghosez L, Franc C, Denonne F, Cuisinier C, Touillaux R. Can J Chem. 2001;79:1827.Murugesan N, Gu Z, Stein PD, Bisaha S, Spergel S, Girotra R, Lee VG, Lloyd J, Misra RN, Schmidt J, Mathur A, Stratton L, Kelly YF, Bird E, Waldron T, Liu ECK, Zhang R, Lee H, Serafino R, Abboa-Offei B, Mathers P, Giancarli M, Seymour AA, Webb ML, Moreland S, Barrish JC, Hunt JT. J Med Chem. 1998;41:5198. doi: 10.1021/jm970872k.Thompson WJ, Gaudino J. J Org Chem. 1984;49:5237.
- 25.For examples of replacement of a N-benzyl group with a carbamate moiety in a one-pot reaction, see: Ottenbrite RM, Alston PV. J Org Chem. 1974;39:1115.Ando K, Kankake M, Suzuki T, Kakayama H. Tetrahedron. 1995;51:129.Chenna A, Donnelly J, McCullough KJ, Proctor GR, Redpath J. J Chem Soc Perkin Trans 1. 1990:261.Winkler JD, Axten J, Hammach AH, Kwak YS, Lengweiler U, Lucero MJ, Houk KN. Tetrahedron. 1998;54:7045.Pichon N, Harrison-Marchand A, Mailliet P, Maddaluno J. J Org Chem. 2004;69:7220. doi: 10.1021/jo049037i.
-
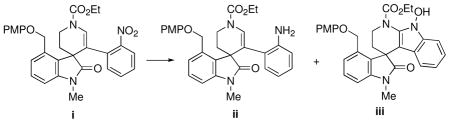
26.Attempts to reduce the nitro group of the related PMP-protected system i by chemical methods (e.g. Cu(acac)2/NaBH4/EtOH or SnCl2/NaBH4/THF/EtOH) led to the N-hydroxyindole iii as the major product along with some of the desired aniline ii.17a For related transformations, see: Stefanchi A, Leonetti F, Cappa A, Carotti A. Tetrahedron Lett. 2003;44:2121.Myers AG, Herzon SB. J Am Chem Soc. 2003;125:12080. doi: 10.1021/ja0372006.
- 27.For related lactam reductions leading to aminals, see for example: Govek SP, Overman LE. J Am Chem Soc. 2001;123:9468. doi: 10.1016/j.tet.2007.05.127.Miyamoto H, Okawa Y, Nakazaki A, Kobayashi S. Tetrahedron Lett. 2007;48:1805.Kawasaki T, Shinada M, Ohzono M, Ogawa A, Terashima R, Sakamoto M. J Org Chem. 2008;73:5959. doi: 10.1021/jo800984a.
- 28.For deacylation of an enamide with an alkyllithium reagent, see: Obika S, Nishiyama T, Tatematsu S, Nishimoto M, Miyashita K, Imanishi T. Heterocycles. 1997;44:537.Fowler FW. J Org Chem. 1972;37:1321.
- 29.For examples of cyclopropanation of an enamine or enamide with diazoacetate esters, see: Kaiser C, Tedeschi DH, Fowler PJ, Pavloff AM, Lester BM, Zirkle CL. J Med Chem. 1971;14:179. doi: 10.1021/jm00285a001.Wenkert E, Hudlicky T. J Org Chem. 1988;53:1953.Wenkert E, Hudlicky T, Showalter HDH. J Am Chem Soc. 1978;100:4894.Kaufman MD, Grieco PA. J Org Chem. 1994;59:7197.Grieco PA, Kaufman MD. J Org Chem. 1999;64:7586.Gnad F, Poleschak M, Reiser O. Tetrahedron Lett. 2004;45:4277.Bubert C, Cabrele C, Reiser O. Synlett. 1997:827.Beumer R, Bubert C, Cabrele C, Vielhauer O, Pietzsch M, Reiser O. J Org Chem. 2000;65:8960. doi: 10.1021/jo005541l.Beumer R, Reiser O. Tetrahedron. 2001;57:6497.Huang D, Yan M, Zhao WJ, Shen Q. Synth Commun. 2005;35:745.Wenkert E, McPherson A, Sanchez EL, Webb RL. Synth Commun. 1973;3:255.Paulini K, Reibig HU. Liebigs Ann Chem. 1991:455.Tanny SR, Grossman J, Fowler FW. J Am Soc Chem. 1972;94:6495.
- 30.For examples of dichlorocarbene additions to enamides, see: Castro JL, Castedo L, Riguera R. J Org Chem. 1987;52:3579.Perchonock CD, Lantos I, Finkelstein JA, Holden KG. J Org Chem. 1980;45:1950.Lantos I, Bhattacharjee D, Eggleston DS. J Org Chem. 1986;51:4147.Manikumar G, Shamma M. J Org Chem. 1981;46:386.
- 31.For examples of 1-aza-Cope rearrangements, see: Wu PL, Chu M, Fowler FW. J Org Chem. 1988;53:963.Wu PL, Fowler FW. J Org Chem. 1988;53:5998.Walters MA. Tetrahedron Lett. 1995;36:7055.Wu PL, Wang WS. J Org Chem. 1994;59:622.
- 32.Kamatani A, Overman LE. Org Lett. 2001;3:1229. doi: 10.1021/ol015696v. [DOI] [PubMed] [Google Scholar]
- 33.For examples of intramolecular hydride transfers, see: Bergmann R, Gericke R. J Med Chem. 1990;33:492. doi: 10.1021/jm00164a005.Wessig P, Henning HG. Liebigs Ann Chem. 1991:983.Vankar YD, Shah K, Bawa A, Singh SP. Tetrahedron Lett. 1991;47:8883.Marcuccio SM, Elix JA. Aust J Chem. 1985;38:1785.Mitscher LA, Gill H, Filppi JA, Wolgemuth RL. J Med Chem. 1986;29:1277. doi: 10.1021/jm00157a027.Wu A, Ghakraborty A, Witt D, Lagona J, Damkaci F, Ofori MA, Chiles JK, Fettinger JC, Isaacs L. J Org Chem. 2002;67:5817. doi: 10.1021/jo0258958.Bartels AB, Jones PG, Liebscher J. Synthesis. 2003:67.Padwa A, Wang Q. J Org Chem. 2006;71:3210. doi: 10.1021/jo060238r.Kocienski PJ, Kirkup M. J Org Chem. 1975;40:2998.Longridge JL, Nicholson S. J Chem Soc Perkin Trans 2. 1990:965.Ivanov IC, Karagiosov SK. Synthesis. 1995:633.Raucher S, Lawrence RF. Tetrahedron. 1983;39:3731.Craze GA, Watt I. Tetrahedron Lett. 1982;23:975.
- 34.(a) Marsh FD, Hermes ME. J Am Chem Soc. 1964;86:4506. [Google Scholar]; (b) Warren BK, Knaus EE. J Heterocycl Chem. 1982;19:1259. [Google Scholar]
- 35.For closely related cyclizations, see: Harrington PJ, Hegedus LS, McDaniel KF. J Am Chem Soc. 1987;109:4335.Yokoyama Y, Matsumoto T, Murakami Y. J Org Chem. 1995;60:1486.Yokoyama Y, Hikawa H, Mitsuhashi M, Uyama A, Hiroki Y, Murakami Y. Eur J Org Chem. 2004:1244.
- 36.Although Qin et al.12b have reported that it was necessary to stir this amine with silica gel at 50 °C in order to effect cyclization to the amidine, we observed that simple neutralization of the azepine TFA salt at room temperature was sufficient to produce 63.
- 37.For a review, see: Shibasaki M, Vogl EM, Ohshima T. Adv Synth Catal. 2004;346:1533.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.