

Abstract
Pancreatic cancer is a devastating disease with an extremely poor prognosis and thus there is a great need for better diagnostic and therapeutic tools. The 19q13 chromosomal locus is amplified in several cancer types, including pancreatic cancer, but the possible clinical significance of this aberration remains unclear. We used fluorescence in situ hybridization (FISH) on tissue microarrays (TMA) containing 357 primary pancreatic tumors, 151 metastases, and 24 local recurrences as well as 120 cancer cell lines from various tissues to establish the frequency of the 19q13 amplification and to find potential correlations to clinical parameters including patient survival. Copy number increases were found in 12.2% of the primary pancreatic tumors and 9.3% of the cell lines, including those derived from bladder, colorectal, ovarian, and thyroid carcinomas. Copy number changes were linked to high grade (P = 0.044) and stage (P = 0.025) tumors and the average survival time of patients with 19q13 amplification was shorter than of those without this aberration. Our findings revealed recurrent 19q13 amplification in pancreatic cancer and involvement of the same locus as in bladder, colorectal, ovarian, and thyroid carcinomas. More importantly, the 19q13 amplifications were associated with poor tumor phenotype and showed a trend towards shorter survival.
INTRODUCTION
Pancreatic cancer is a lethal disease which is typically diagnosed at an advanced stage when patients are no more eligible for curative surgery. Therefore, novel methods for early detection and better treatment of pancreatic cancer are urgently needed. Because the molecular mechanisms contributing to the development of pancreatic cancer are still not fully understood, additional information about the biology of pancreatic cancer is highly valuable and might eventually lead to the generation of novel tools for the clinical management of this disease.
Gene amplification, defined as a copy number increase of a restricted chromosomal region, is a typical mechanism for solid tumors to activate oncogenes and thereby amplified genomic regions are likely to harbor genes of importance for tumor development and progression. Excessive dosage of such genes may provide tumor cells a growth advantage or confer resistance to various therapies (Albertson, 2006). Identification of amplification target genes may offer better diagnostic and prognostic tools and even better treatment strategies. The ERBB2 oncogene is an excellent example of an amplification target gene with well established clinical significance as a prognostic and predictive marker and a therapeutic target in breast cancer (Carter et al., 1998). Other well-documented oncogenes that are activated via amplification and serve as prognostic markers or therapeutic targets include MYCN in neuroblastomas (Savelyeva and Schwab, 2001) and EGFR in gliomas (Etienne et al., 1998). Gene amplification has been studied widely also in pancreatic cancer and a number of presumptive oncogenes have been pinpointed, such as ARPC1A (Laurila et al., 2009), EMSY (van Hattem et al., 2008), GATA6 (Kwei et al., 2008), ERBB2, and MYC (Mahlamäki et al., 2002).
Genome-wide comparative genomic hybridization (CGH) surveys have identified multiple regions of copy number changes in several cancers, including pancreatic cancer. They provide a powerful approach for identifying novel cancer genes when combined with further genetic and functional studies. Our previous array CGH screen(Mahlamäki et al., 2004) revealed a 2.9 Mb region at 19q13 that is recurrently amplified in pancreatic cancer. This finding has been subsequently confirmed by several other microarray based copy number surveys (Aguirre et al., 2004; Holzmann et al., 2004; Bashyam et al., 2005; Gysin et al., 2005; Heidenblad et al., 2005). Recently, we characterized the structure and boundaries of the 19q13 amplicon in detail in pancreatic cancer cell lines and defined a 660 kb amplicon core region with exceptionally high level copy number increase (Kuuselo et al., 2007). This chromosomal region is extremely gene rich (Supplementary Figure 1). Only a subset of the genes within this region, such as MED29 (previously known as IXL), PAF1, DYRK1B, and PAK4, have been functionally validated and suggested to represent putative amplicon targets (Kuuselo et al., 2007; Moniaux et al., 2006; Deng et al., 2006; Chen et al., 2008).
In addition to pancreatic cancer, amplification of the 19q13 chromosomal region has also been reported in other tumor types, such as ovarian (Cheng et al., 1992; Bellacosa et al., 1995; Thompson et al., 1996; Tang et al., 2002), breast (Kallioniemi et al., 1994; Bellacosa et al., 1995), cervical (Rao et al., 2004), gastric (Staal et al., 1987), and lung cancer (Ried et al., 1994; Kim et al., 2005). However, these studies have been performed using a variety of technologies, ranging from chromosomal CGH to different array CGH platforms, and thus it is difficult to know whether they pinpoint a single common amplicon or correspond to multiple separate regions of copy number increase at 19q13. Here, we examined for the first time the presence of copy number aberrations at the 660 kb amplicon core (Kuuselo et al., 2007) in 120 cancer cell lines originating from various tissues.
In a previous study, we demonstrated using a small set of 31 tumors that the 19q13 amplification is present in about 10% of the primary pancreatic cancer cases (Kuuselo et al., 2007). Now, we evaluated the clinical significance of the 19q13 amplicon core in an extensive sample set containing more than 500 pancreatic tumors. We applied fluorescence in situ hybridization (FISH) to a tissue microarray (TMA) containing 357 primary tumors of the pancreas, 151 metastases, and 24 local recurrences to determine the 19q13 copy number levels and to reveal their possible association with clinicopathological parameters and patient survival.
MATERIALS AND METHODS
Pancreatic Cancer Patients
Primary pancreatic tumor samples and corresponding metastases were obtained from 356 patients who underwent pancreatic surgery at the Department of General- Visceral- and Thoracic Surgery, University Medical Center Hamburg-Eppendorf during the years 1993 to 2005. Formalin-fixed (buffered neutral aqueous 4% solution) paraffin-embedded material was utilized. All slides from all tumors were reviewed by two pathologists determining the histological type and grade of the samples (G1 = highly differentiated, G2 = moderately differentiated, G3 = poorly differentiated). The pathologic stage, nodal status and metastasis information were obtained from the primary reports of the Department of Pathology, University Medical Center Hamburg-Eppendorf. Follow-up and survival data were collected by the Department of General, Visceral and Thoracic Surgery. The following TNM classification was used: pT stage T1 = tumor size ≤ 2cm, T2 = tumor size > 2cm, T3 = tumor growth into surrounding tissues, T4 = tumor growth into the stomach, spleen, large bowel or nearby large blood vessels; pN stage N0 = no lymph node metastases, N1 = metastases in local lymph nodes; and pM stage M0 = no distant metastases, M1 = distant metastases. Median age of the patients was 62.8 years (range 21–88 years). The mean follow-up time for ductal adenocarcinomas was 18.52 months (range 1–74 month). Informed consent had been obtained from all patients upon admission to hospital.
Tissue Microarrays
The pancreatic cancer tissue microarray (TMA) contained a total of 600 samples. These included 357 primary tumors of the pancreas (213 ductal adenocarcinomas, 54 adenocarcinomas of the ampulla of Vater, 40 pancreatic endocrine tumors, 33 intraductal papillary mucinous neoplasms (IPMNs), 15 benign cystic tumors, 1 malignant cystic tumor, 1 acinar cell carcinoma), 129 corresponding lymph node metastases, 22 distant metastases, 24 local recurrences, and a standard control area containing 40 tumors from other organs, 10 healthy pancreatic tissues, and 18 healthy tissues from other sites. The cell line TMA had 120 cancer cell lines representing various tissue types and nine cell lines of non-neoplastic origin (Supplementary Table 1).
Fluorescence in situ hybridization (FISH)
To determine the 19q13 copy number levels, we used a contig of three partly overlapping locus-specific BAC probes (RP11-67A5, RP11-256O9, CTC-488F21) that were previously shown to correspond to the 660 kb core region of the amplicon (Supplementary Figure 1) and verified to give a single signal on normal lymphocytes (Kuuselo et al., 2007). The BAC clone DNA was isolated using standard alkaline lysis method and labeled with Spectrum Orange dUTP (Vysis, Downers Grove, IL) using random priming. A chromosome 19 pericentromere-specific reference probe (RP11-345J21) was labeled with fluorescein-12-dUTP (Life Sciences, US) and used as a control. FISH on TMA was carried out as described (Andersen et al., 1999) with some modifications. Briefly, the slides were deparaffinized in three changes of hexane for 10 min each, dipped twice in 100% EtOH, treated for 30 min with 0.3% NaBH4, and rinsed with PBS. Then the slides were treated for 40 min with Vysis Pretreatment Solution (Vysis, Downers Grove, IL) at 80°C, rinsed with H2O, and treated for 20 min with Vysis Protease at 37°C, followed by Proteinase K treatment for 10 min at 37°C. Finally the slides were washed in increasing series of EtOH (70%, 85%, 100%), dehydrated, denatured for 3 min at 70°C in 70% formamide/2X SSC, washed again in EtOH series, dehydrated, and hybridized with denatured probes in a humidified chamber at 37°C for 24h. The nuclei were counterstained with DAPI in Vectashield antifade solution.
Hybridization signals were evaluated using Olympus BX50 fluorescence microscope (Olympus, Tokyo, Japan) and relative copy numbers were calculated for each sample as ratios of mean absolute copy number of the locus-specific probe vs. the reference probe. Forty intact nuclei were scored per sample. Relative copy numbers over 1.5 but less than 2 were considered as gains whereas relative copy numbers greater than or equal to 2 were considered as amplifications. Polysomy was defined as samples with absolute copy number of the 19q13 locus over 5 but relative copy number less than two.
Statistical Analyses
Statistical analyses were done using JMP™ software (SAS Institute Inc. Cary, NC, USA). All P-values tested were two sided and P<0.05 was considered significant. The Pearson Chi-square test was used to assess the relationship between 19q13 copy number changes (categorized as normal, gain, amplification, and polysomy) and the clinicopathologic parameters T stage, N stage and tumor grade. The Kaplan-Meier method was used to visualize association of 19q13 copy number changes with cancer-specific survival and the log rank test was applied to test the significance between stratified groups.
RESULTS
The presence of copy number changes at the 19q13 amplicon core was first screened in a set of 120 cancer cell lines representing various tumor types (Supplementary Table 1). Ten (9.3%) of the 107 cell lines with successful hybridizations displayed increased copy number. Six of these were amplifications (relative copy number ≥ 2) and four were gains (relative copy number > 1.5 but < 2). Amplifications were detected in one (OVCAR-3) of four ovarian cancer cell lines, three (RT-112, KU-19-19, CRL-7930) of six bladder, one (SW-48) of twelve colorectal, and one (ONCO-DG-1) of four thyroid carcinoma cell lines.
We then determined the frequency of the 19q13 copy number changes in 357 primary pancreatic tumors, 151 metastatic lesions (including both lymph node and distant metastases), and 24 local recurrences. Among the primary tumors, copy number data were obtained in 303 cases and 12.2% of them had copy number increases (Fig. 1). Gains were detected in 7.3%, amplifications in 3.3%, and polysomy in 1.7% of the cases. Copy number changes were most frequently observed in ductal adenocarcinomas and pancreatic endocrine tumors (Table 1). In addition, copy number increase was also detected in one of 15 intraductal papillary mucinous neoplasms. Tumor samples with amplification typically showed a tight cluster of signals with an average of 3.4-fold copy number increase but a few cases with up to 10-fold amplification levels were observed. The polysomic samples had an average of five to ten copies of the 19q13 locus per cell, ranging up to twenty copies.
Figure 1.
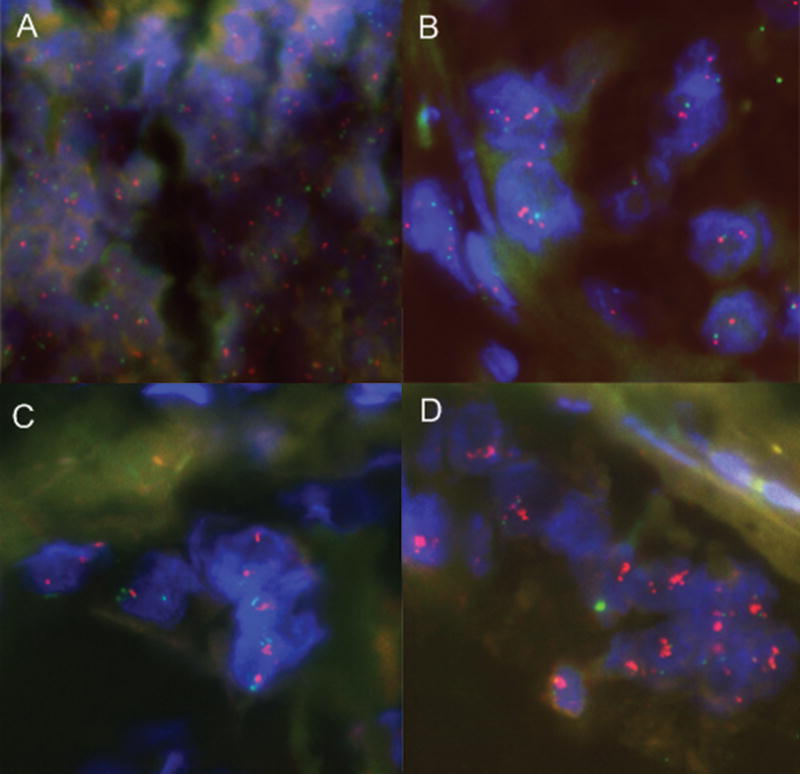
Analysis of 19q13 copy number levels in primary pancreatic tumors by FISH. Examples of tumors with (A) no copy number increase, (B) gain, (C) polysomy, and (D) amplification are shown. Red signals correspond to the 19q13 core region and green signals represent the chromosome 19 centromere probe. Nuclei were counterstained with DAPI (blue).
TABLE 1.
Primary Pancreatic Cancer Subtypes and Their Copy Number Status.
| 19q13 FISH result | ||||||
|---|---|---|---|---|---|---|
| n | analyzable (n) | normal (%) | gain (%) | amp (%) | poly (%) | |
| All samples | 357 | 303 | 87.8 | 7.3 | 3.3 | 1.7 |
| Ductal adenocarcinomas | 213 | 197 | 86.3 | 8.1 | 3.6 | 2.0 |
| Intraductal papillary mucinous neoplasms | 33 | 15 | 93.3 | 0.0 | 6.7 | 0.0 |
| Endocrine tumors | 40 | 35 | 88.6 | 8.6 | 0.0 | 2.9 |
| Cystic/benign tumors | 15 | 6 | 100 | 0.0 | 0.0 | 0.0 |
| Cystic/malignant tumors | 1 | 1 | 100 | 0.0 | 0.0 | 0.0 |
| Acinar cell cancers | 1 | 0 | - | - | - | - |
| Adenocarcinomas of the ampulla of Vater | 54 | 49 | 89.8 | 6.1 | 4.1 | 0.0 |
| Metastases | 151 | 121 | 94.2 | 3.3 | 2.5 | 0.0 |
| Lymph node metastases | 129 | 100 | 95.0 | 3.0 | 2.0 | 0.0 |
| Distant metastases | 22 | 21 | 90.4 | 4.8 | 4.8 | 0.0 |
| Local recurrences | 24 | 18 | 100 | 0.0 | 0.0 | 0.0 |
A total of seven (5.8%) of the 121 metastases with successful hybridizations showed copy number increase (Table 1). Of these, three were amplifications and four cases had gains. Copy number data were available on a subset of 91 cases where both the primary tumor and a matching lymph node metastasis were studied. Among this subset of cases, increased copy number was detected in four primary tumors and three corresponding metastases thus showing an overall concordance of 99% (90/91). Finally, none of the local recurrences (of a total of 18 analyzable cases) showed copy number increases (Table 1). However, 17 of the 18 corresponding primary tumors also had no copy number change while the one remaining case did show copy number gain.
To evaluate the possible clinical significance of the 19q13 amplification, the relationships between copy number data and clinicopathological characteristics were examined among the ductal adenocarcinomas, the most common histological subtype of pancreatic cancer (Table 2). Due to the small number of samples, tumors confined to pancreas (pT1 and pT2) were compared to those that had spread beyond the pancreas (pT3 and pT4). Similarly, moderately and well-differentiated tumors (G1 and G2) were combined and compared to poorly differentiated tumors (G3). Copy number increases (including gains, amplifications and polysomy) were linked to both tumor grade and stage (P = 0.044 and P = 0.025, respectively, Table 2). The frequency of gains and amplifications increased from low/moderate (G1-G2) to high grade (G3) tumors and from early (pT1-pT2) to late (pT3-pT4) stage tumors. Actually none of the low-grade (G1) tumors harbored copy number changes whereas 11% of grade 2 tumors and 16.8% of grade 3 tumors had increased copy number. All of the tumors with amplification were of grade 3.
TABLE 2.
Relationship Between the 19q13 Copy Number Changes and the Cancer Phenotype in Pancreatic Ductal Adenocarcinomas.
| n | normal (%) | gain (%) | amp (%) | poly (%) | P | ||
|---|---|---|---|---|---|---|---|
| pT stage | pT1 | 6 | 83.3 | 0.0 | 0.0 | 16.7 | 0.025a |
| pT2 | 53 | 92.5 | 1.9 | 1.9 | 3.8 | ||
| pT3 | 125 | 84.8 | 10.4 | 4.0 | 0.8 | ||
| pT4 | 9 | 66.7 | 22.2 | 11.1 | 0.0 | ||
| pN stage | pN0 | 68 | 83.8 | 10.3 | 4.4 | 1.5 | 0.826 |
| pN1 | 124 | 87.1 | 7.3 | 3.2 | 2.4 | ||
| Grade | G1 | 8 | 100 | 0.0 | 0.0 | 0.0 | 0.044b |
| G2 | 91 | 89.0 | 9.9 | 0.0 | 1.1 | ||
| G3 | 95 | 83.2 | 7.4 | 7.4 | 2.1 | ||
(T1+T2 vs. T3+T4)
(G1+G2 vs. G3)
Finally, the Kaplan-Meier analysis among the ductal adenocarcinomas showed that the survival of patients having high level copy number changes (amplifications and polysomy) was somewhat worse than those without 19q13 copy number changes, but this difference was not statistically significant (data not shown). Nonetheless, the average survival time for the normal copy number patient group was 26 months whereas it was only 16 and 17 months for the patients with amplification and polysomy, respectively.
DISCUSSION
Pancreatic cancer is a highly devastating disease with exceptionally poor prognosis. Consequently, there is a huge need for better understanding of the biology of this disease and for identification of novel diagnostic and prognostic markers as well as molecular targets for therapy. Every piece of knowledge about the mechanisms behind pancreatic cancer development may shift us towards novel strategies for improved clinical applications. To this end we evaluated the clinical significance of the 19q13 amplification that was initially discovered by us and others using genome-wide copy number screens of pancreatic cancer (Mahlamäki et al., 2004; Aguirre et al., 2004; Holzmann et al., 2004; Bashyam et al., 2005; Gysin et al., 2005; Heidenblad et al., 2005). Recently, we characterized this amplicon in detail in pancreatic cancer cell lines and in a small set of primary pancreatic tumors and defined a 660 kb amplicon core region (Kuuselo et al., 2007). Here we studied the clinical significance of the 19q13 amplification in a large collection of over 500 clinical pancreatic tumor samples using FISH to TMAs. We also examined whether this specific amplicon is present in other tumor types using more than 100 different cancer cell lines. The FISH technique is perhaps the most reliable and accurate method to detect different types of copy number changes from chromosomal rearrangements to amplifications and deletions (Fletcher et al., 1999). It now represents a standard diagnostic tool in the classification of hematological malignancies and in detecting clinically relevant gene amplification events, such as those involving the ERBB2 oncogene in breast cancer (Spiridon et al., 2002).
Copy number analysis in an extensive collection of over 500 pancreatic tumors revealed that 12.2% of the primary cancers had 19q13 copy number increases. This frequency is in good concordance with our preliminary data from a small set of 31 tumors (Kuuselo et al., 2007). The copy number aberrations were divided into three categories: gains were detectable in 7.3 % of the cases, amplification in 3.3%, and 1.7% of the tumors were polysomic. Moreover, a concordant copy number result between a primary tumor and a corresponding lymph node metastasis was obtained in 99% of the samples. Surprisingly, none of the local recurrences showed copy number changes. However, the number of samples analyzed was small and more importantly the corresponding primaries did not show copy number aberrations either. Thus these data do not allow us to draw conclusions on the possible genetic differences between the primaries and local recurrences.
Although amplification of the 19q13 chromosomal locus has been previously reported in a subset of other cancer types, here we characterized for the first time the presence of this specific amplicon core in a large number of cell lines representing various tumor types. In addition to pancreatic tumors, we found amplification of the 19q13 core region in ovarian, colorectal, urinary bladder, and thyroid cancer cell lines. These data now confirm previous studies in ovarian (Bellacosa et al., 1995; Cheng et al., 1992; Thompson et al., 1996; Tang et al., 2002), colorectal (Bardi et al., 1993), and urinary bladder (Richter et al., 2000) cancer. However, 19q13 amplification has not been previously reported in thyroid cancer. The cell line TMA data now allow rapid identification of numerous cell lines which are suitable for further functional analyses of putative target genes within the 19q13 amplicon.
Gene amplification has been shown to frequently associate with tumor progression, drug resistance, and poor clinical outcome in a variety of tumor types (Savelyeva and Schwab, 2001). For example, amplification of oncogenes, such as CCND1, EGFR, ERBB2, MDM2 and MYC, has been associated with high grade breast tumors (Al-Kuraya, 2004), MYCN amplification is a prognostic factor for patients with neuroblastoma (Savelyeva and Schwab, 2001), and amplification of genes such as DHFR and BCR-ABL1 has been reported to be associated with resistance to anti-cancer drugs (Albertson, 2006). Now we found that the 19q13 copy number increases were linked to both advanced tumor stage and grade in pancreatic cancer. Similarly, 19q13 amplification has been previously shown to associate with less differentiated and more aggressive tumors in ovarian carcinoma (Bellacosa et al., 1995).
Our results suggest a trend towards shorter survival time in patients with 19q13 amplification. The average survival time of patients with high level copy number increases (amplification or polysomy) was shorter than the survival time of patients with normal copy number status, although this difference was not statistically significant. The lack of statistical significance may be due to the rather small number of tumors with copy number changes but also on the overall poor prognosis of pancreatic cancer. In any case, our data are in concordance with the finding that high level copy number increases have a greater impact on tumor development than low level aberrations (Hogsdon et al., 2003). Previously, the 19q13 amplification has been associated with poor survival of patients with non–small cell lung cancers (Kim et al., 2005) but there are no other studies reporting associations of this specific amplification locus with patient survival. However, the connection of the 19q13 amplification to more aggressive and less differentiated tumors (Bellacosa et al., 1995; Tang et al., 2002) could support the hypothesis that this amplicon is indeed associated with poor prognosis.
Supplementary Material
Chromosomal location of the BAC clone contig and the genes within the 600kb amplicon core at 19q13. Arrowheads indicate the orientation of transcription of the genes but the size of the arrows does not reflect the size of the genes.
Composition of the cell line TMA and FISH results.
Acknowledgments
Supported by: Academy of Finland grant 122440, NIH grant PO1 CA109552, and the Sigrid Juselius Foundation.
We thank Kati Rouhento for her skillful technical assistance.
References
- Aguirre AJ, Brennan C, Bailey G, Sinha R, Feng B, Leo C, Zhang Y, Zhang J, Gans JD, Bardeesy N, Cauwels C, Cordon-Cardo C, Redston MS, DePinho RA, Chin L. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci USA. 2004;101:9067–9072. doi: 10.1073/pnas.0402932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson DG. Gene amplification in cancer. Trends Genet. 2006;22:447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M, Kochli O, Zuber M, Dieterich H, Mross F, Wilber K, Simon R, Sauter G. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004;64:8534–8540. doi: 10.1158/0008-5472.CAN-04-1945. [DOI] [PubMed] [Google Scholar]
- Andersen CL, Hostetter G, Grigoryan A, Sauter G, Kallioniemi A. Improved procedure for fluorescence in situ hybridization on tissue microarrays. Cytometry. 2001;45:83–86. doi: 10.1002/1097-0320(20011001)45:2<83::aid-cyto1149>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Bardi G, Johansson B, Pandis N, Mandahl N, Bak-Jensen E, Lindstrom C, Tornqvist A, Frederiksen H, Andren-Sandberg A, Mitelman F. Cytogenetic analysis of 52 colorectal carcinomas--non-random aberration pattern and correlation with pathologic parameters. Int J Cancer. 1993;55:422–428. doi: 10.1002/ijc.2910550317. [DOI] [PubMed] [Google Scholar]
- Bashyam MD, Bair R, Kim YH, Wang P, Hernandez-Boussard T, Karikari CA, Tibshirani R, Maitra A, Pollack JR. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia. 2005;7:556–562. doi: 10.1593/neo.04586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Auletta T, Dovirak O, Hutter C, Kuntz K, El-Ftesi S, Kendall J, Han H, Von Hoff DD, Ashfaq R, Maitra A, Iacobuzio-Donahue CA, Hruban RH, Lucito R. Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol Ther. 2008;7 doi: 10.4161/cbt.7.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Ewton DZ, Li S, Naqvi A, Mercer SE, Landas S, Friedman E. The kinase Mirk/Dyrk1B mediates cell survival in pancreatic ductal adenocarcinoma. Cancer Res. 2006;66:4149–4158. doi: 10.1158/0008-5472.CAN-05-3089. [DOI] [PubMed] [Google Scholar]
- Etienne MC, Formento JL, Lebrun-Frenay C, Gioanni J, Chatel M, Paquis P, Bernard C, Courdi A, Bensadoun RJ, Pignol JP, Francoual M, Grellier P, Frenay M, Milano G. Epidermal growth factor receptor and labeling index are independent prognostic factors in glial tumor outcome. Clin Cancer Res. 1998;4:2383–2390. [PubMed] [Google Scholar]
- Fletcher JA. DNA in situ hybridization as an adjunct in tumor diagnosis. Am J Clin Pathol. 1999;112:S11–18. [PubMed] [Google Scholar]
- Gysin S, Rickert P, Kastury K, McMahon M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer. 2005;44:37–51. doi: 10.1002/gcc.20216. [DOI] [PubMed] [Google Scholar]
- Hattinger CM, Stoico G, Michelacci F, Pasello M, Scionti I, Remondini D, Castellani GC, Fanelli M, Scotlandi K, Picci P, Serra M. Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosomes Cancer. 2009;48:289–309. doi: 10.1002/gcc.20640. [DOI] [PubMed] [Google Scholar]
- Heidenblad M, Lindgren D, Veltman JA, Jonson T, Mahlamaki EH, Gorunova L, van Kessel AG, Schoenmakers EF, Hoglund M. Microarray analyses reveal strong influence of DNA copy number alterations on the transcriptional patterns in pancreatic cancer: implications for the interpretation of genomic amplifications. Oncogene. 2005;24:1794–1801. doi: 10.1038/sj.onc.1208383. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Tr. 2003;78:337–345. doi: 10.1023/a:1023085825042. [DOI] [PubMed] [Google Scholar]
- Holzmann K, Kohlhammer H, Schwaenen C, Wessendorf S, Kestler HA, Schwoerer A, Rau B, Radlwimmer B, Dohner H, Lichter P, Gress T, Bentz M. Genomic DNA-chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res. 2004;64:4428–4433. doi: 10.1158/0008-5472.CAN-04-0431. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A, Kallioniemi OP, Piper J, Tanner M, Stokke T, Chen L, Smith HS, Pinkel D, Gray JW, Waldman FM. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci USA. 1994;91:2156–2160. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Yim SH, Lee JS, Kwon MS, Ryu JW, Kang HM, Fiegler H, Carter NP, Chung YJ. Genome-wide screening of genomic alterations and their clinicopathologic implications in non-small cell lung cancers. Clin Cancer Res. 2005;11:8235–8242. doi: 10.1158/1078-0432.CCR-05-1157. [DOI] [PubMed] [Google Scholar]
- Kuuselo R, Savinainen K, Azorsa DO, Basu GD, Karhu R, Tuzmen S, Mousses S, Kallioniemi A. Intersex-like (IXL) is a cell survival regulator in pancreatic cancer with 19q13 amplification. Cancer Res. 2007;67:1943–1949. doi: 10.1158/0008-5472.CAN-06-3387. [DOI] [PubMed] [Google Scholar]
- Kwei KA, Bashyam MD, Kao J, Ratheesh R, Reddy EC, Kim YH, Montgomery K, Giacomini CP, Choi YL, Chatterjee S, Karikari CA, Salari K, Wang P, Hernandez-Boussard T, Swarnalata G, van de Rijn M, Maitra A, Pollack JR. PLoS Genet. 2008;4:e1000081. doi: 10.1371/journal.pgen.1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurila E, Savinainen K, Kuuselo R, Karhu R, Kallioniemi A. Characterization of the 7q21-q22 amplicon identifies ARPC1A, a subunit of the Arp2/3 complex, as a regulator of cell migration and invasion in pancreatic cancer. Genes Chromosomes Cancer. 2009;48:330–339. doi: 10.1002/gcc.20643. [DOI] [PubMed] [Google Scholar]
- Mahlamäki EH, Bärlund M, Tanner M, Gorunova L, Höglund M, Karhu R, Kallioniemi A. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosomes Cancer. 2002;35:353–358. doi: 10.1002/gcc.10122. [DOI] [PubMed] [Google Scholar]
- Mahlamäki EH, Kauraniemi P, Monni O, Wolf M, Hautaniemi S, Kallioniemi A. High-resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia. 2004;6:432–439. doi: 10.1593/neo.04130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniaux N, Nemos C, Schmied BM, Chauhan SC, Deb S, Morikane K, Choudhury A, Vanlith M, Sutherlin M, Sikela JM, Hollingsworth MA, Batra SK. The human homologue of the RNA polymerase II-associated factor 1 (hPaf1), localized on the 19q13 amplicon, is associated with tumorigenesis. Oncogene. 2006;25:3247–3257. doi: 10.1038/sj.onc.1209353. [DOI] [PubMed] [Google Scholar]
- Rao PH, Arias-Pulido H, Lu XY, Harris CP, Vargas H, Zhang FF, Narayan G, Schneider A, Terry MB, Murty VV. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer. 2004;4:5. doi: 10.1186/1471-2407-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, Ackermann D, Maurer R, Alund G, Knonagel H, Rist M, Wilber K, Anabitarte M, Hering F, Hardmeier T, Schonenberger A, Flury R, Jager P, Fehr JL, Schraml P, Moch H, Mihatsch MJ, Gasser T, Kallioniemi OP, Sauter G. High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol. 2000;157:787–794. doi: 10.1016/s0002-9440(10)64592-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried T, Petersen I, Holtgreve-Grez H, Speicher MR, Schrock E, du Manoir S, Cremer T. Mapping of multiple DNA gains and losses in primary small cell lung carcinomas by comparative genomic hybridization. Cancer Res. 1994;54:1801–1806. [PubMed] [Google Scholar]
- Savelyeva L, Schwab M. Amplification of oncogenes revisited: from expression profiling to clinical application. Cancer Lett. 2001;167:115–123. doi: 10.1016/s0304-3835(01)00472-4. [DOI] [PubMed] [Google Scholar]
- Spiridon CI, Ghetie MA, Uhr J, Marches R, Li JL, Shen GL, Vitetta ES. Targeting multiple Her-2 epitopes with monoclonal antibodies results in improved antigrowth activity of a human breast cancer cell line in vitro and in vivo. Clin Cancer Res. 2002;8:1720–1730. [PubMed] [Google Scholar]
- Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TC, Sham JS, Xie D, Fang Y, Huo KK, Wu QL, Guan XY. Identification of a candidate oncogene SEI-1 within a minimal amplified region at 19q13.1 in ovarian cancer cell lines. Cancer Res. 2002;62:7157–7161. [PubMed] [Google Scholar]
- Thompson FH, Nelson MA, Trent JM, Guan XY, Liu Y, Yang JM, Emerson J, Adair L, Wymer J, Balfour C, Massey K, Weinstein R, Alberts DS, Taetle R. Amplification of 19q13.1-q13.2 sequences in ovarian cancer. G-band, FISH, and molecular studies. Cancer Genet Cytogen. 1996;87:55–62. doi: 10.1016/0165-4608(95)00248-0. [DOI] [PubMed] [Google Scholar]
- van Hattem WA, Carvalho R, Li A, Offerhaus GJ, Goggins M. Amplification of EMSY gene in a subset of sporadic pancreatic adenocarcinomas. Int J Exp Pathol. 2008;1:343–351. [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Tsuda H, Honda K, Onozato K, Takano M, Tamai S, Imoto I, Inazawa J, Yamada T, Matsubara O. Actinin-4 gene amplification in ovarian cancer: a candidate oncogene associated with poor patient prognosis and tumor chemoresistance. Modern Pathol. 2009;22:499–507. doi: 10.1038/modpathol.2008.234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chromosomal location of the BAC clone contig and the genes within the 600kb amplicon core at 19q13. Arrowheads indicate the orientation of transcription of the genes but the size of the arrows does not reflect the size of the genes.
Composition of the cell line TMA and FISH results.