

Abstract
N-Methyl-d-aspartate (NMDA) at a subtoxic concentration (100 μM) promotes neuronal survival against glutamate-mediated excitotoxicity via a brain-derived neurotrophic factor (BDNF) autocrine loop in cultured cerebellar granule cells. The signal transduction mechanism(s) underlying NMDA neuroprotection, however, remains elusive. The mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3-K) pathways alter gene expression and are involved in synaptic plasticity and neuronal survival. This study tested whether neuroprotective activation of NMDA receptors, together with TrkB receptors, coactivated the MAPK or PI3-K pathways to protect rat cerebellar neurons. NMDA receptor activation caused a concentration- and time-dependent activation of MAPK lasting 24 hr. This activation was blocked by the NMDA receptor antagonist MK-801 but was attenuated only partially by the tyrosine kinase inhibitor k252a, suggesting that activation of both NMDA and TrkB receptors are required for maximal neuroprotection. The MAPK kinase (MEK) inhibitor U0126 (10 μM) partially blocked NMDA neuroprotection, whereas LY294002, a selective inhibitor of the PI3-K pathway, did not affect the neuroprotective activity of NMDA. Glutamate excitotoxicity decreased bcl-2, bcl-XL, and bax mRNA levels,. NMDA increases Bcl-2 and Bcl-XL protein levels and decreases Bax protein levels. NMDA and TrkB receptor activation thus converge on the extracellular signal-regulated kinase (ERK) 1/2 signaling pathway to protect neurons against glutamate-mediated excitotoxicity. By increasing antiapoptotic proteins of the Bcl-2 family, NMDA receptor activation may also promote neuronal survival by preventing apoptosis. © 2005 Wiley-Liss, Inc.
Keywords: NMDA, cerebellar granule cells, TrkB receptors, MAPK, Akt, BDNF, excitotoxicity, glutamate, Bcl-2, Bcl-XL, Bax
Cerebral ischemic preconditioning is a phenomenon whereby a brief episode of sublethal ischemia, possibly due to the release of sublethal amounts of glutamate, provides tolerance against damage induced by a major ischemic insult. This phenomenon was first described in the heart (Murry et al., 1986) followed by the discovery of preconditioning in rat and young gerbil brain (Kitagawa et al.,1990; Kato et al.,1991; Liu et al.,1992) and in aged gerbils (Dowden and Corbett, 1999). Preconditioning in vivo requires N-methyl-d-aspartate (NMDA) receptors (Kato et al., 1992), extracellular signal-regulated kinase (ERK) activation (Shamloo et al.,1999) and nuclear factor-kB (NF-kB) (Blondeau et al., 2001; Ravati et al., 2001). Cycloheximide blocks preconditioning, suggesting that protein synthesis is required (Barone et al., 1998). These in vivo findings serve as criteria for in vitro preconditioning model systems.
Recently, NMDA was found to be neuroprotective against kainic acid excitotoxicity in the hippocampus in vivo (Ogita et al., 2003). One potential model to study neuroprotective mechanisms mediated by glutamate receptor activation is by employing cultured cerebellar granule cells. We have shown that subtoxic NMDA receptor activation protects vulnerable neurons against the excitotoxic actions of glutamate via a brain-derived neurotrophic factor (BDNF) autocrine loop (Marini et al., 1998). The protective effect was blocked by cycloheximide (Marini and Paul, 1992). NMDA activates NF-kB, and this is a critical element in NMDA neuroprotection (Lipsky et al., 2001). Cultured rat cerebellar granule cells thus represent the in vitro correlate of in vivo preconditioning or tolerance. Whether neuroprotective concentrations of NMDA activate ERK or PI3-K in cultured cerebellar granule cells is unknown.
Glutamate is the endogenous excitatory neurotransmitter required for normal physiologic responses in the mammalian central nervous system (CNS). Glutamate receptors mediate the pathophysiology of hypoxic-ischemic neuronal injury (Olney et al.,1971; Choi,1988; Novelli et al., 1988; Lee et al., 1999), as well as adaptive responses (Black, 1999; Ghosh and Greenberg, 1995; Katz and Shatz, 1996; Thompson, 2000; Steward and Worley, 2001). The molecular mechanisms by which such opposing effects are mediated, however, remain unclear.
The activation of a neurotrophin receptor via activation of an ionotropic glutamate receptor enhances survival in cerebellar neurons. Neuroprotective concentrations of NMDA and BDNF activate NF-kB, which in turn increases BDNF mRNA, suggesting that convergence on a common transcription factor provides a rapid and sustained response to increase BDNF synthesis and maintain neuronal survival (Lipsky et al., 2001). Different neuroprotective treatments may thus converge on common effectors, such as the mitogen-activated protein kinase (MAPK) signal transduction pathway or the Bcl-2 family of apoptosis-related genes, to protect neurons.
The focus of this work was to: (1) determine whether NMDA and TrkB receptors converge on the MAPK and the phosphatidylinositol-3 kinase (PI3-K)/Akt pathways to protect neurons against the excitotoxic effects of glutamate acting on NMDA receptors; and (2) determine the effect of neuroprotective concentrations of NMDA on the Bcl-2 family of genes and proteins against glutamate-mediated excitotoxicity.
MATERIALS AND METHODS
Materials
NMDA, dizocilpine (MK-801), and glutamic acid were obtained from Research Biochemicals (Natick, MA). K252a was purchased from Calbiochem (La Jolla, CA). Basal medium Eagle and fetal calf serum (FCS) were purchased from Life Technologies (Gaithersburg, MD). The anti-active (phosphorylated)-MAP kinase antibody (Thr202/Tyr204), the anti-active (phosphorylated)-Akt (Ser473) antibodies, and goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) were obtained from Cell Signaling (Beverly, MA). The Bcl-2 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The actin antibody was purchased from Oncogene Research Products (Boston, MA), and the Bcl-XL and Bax antibodies were purchased from Cell Signaling (Beverly, MA). The MAPK kinase (MEK) inhibitor U0126 was obtained from Calbiochem and the selective PI3-K inhibitor LY294002 as well as all other chemicals were obtained from Sigma (St. Louis, MO). Nupage 10% bis-tris gels were obtained from Invitrogen (Carlsbad, CA).
Cell Culture
Granule cells were prepared from postnatal Day 8 Sprague-Dawley rat pups. Briefly, meninges-free cerebella were minced and recovered by centrifugation. The pellets from 20 cerebella were subjected to trypsinization, followed by inactivation of the trypsin by the addition of soybean trypsin inhibitor. Cells were then dissociated by a series of triturations and recovered by centrifugation. The final pellet was reconstituted in basal Eagle medium containing glutamine (2 mM), FCS (10%), and potassium chloride (25 mM). No antibiotics were added, and the plating density was 1.8 106 cells/ml. The dishes were placed at 37°C in a 95% air/5% CO2 humidied incubator. Cytosine arabinoside (10 μM) was added 18-24 hr later to inhibit the proliferation of nonneuronal elements (Marini and Paul, 1992). On Day 7 in vitro, glucose (100 ml of a 100 mM solution) and sterile water (100 ml) were added to each 35 mm Nunc culture dish to maintain survival and to replace evaporative losses, respectively. The culture medium was not changed throughout the cultivation period. Granule cell neurons were used on Day 8 in vitro for all experiments unless otherwise indicated. Granule cell neurons represented about 95% of the total population of cells in the culture (Marini et al., 1989).
This research was conducted according to the principles set forth in the National Institutes of Health (NIH) Publication No. 85-23, Guide for the Care and Use of Laboratory Animals, and the Animal Welfare Act of 1986 as amended.
Drug Treatments
NMDA, glutamate, and MK-801 were prepared in sterile water. The MEK inhibitor U0126 and the PI3-K inhibitor LY294002 were prepared in dimethylsulfoxide (DMSO). U0126 or LY294002 were always added to the neurons 1 hr before the addition of either NMDA or glutamate.
Western Blot Analysis
For analysis of the levels of phosphorylated MAPK (ERK1/2 [p44/p42]), total MAPK, and phosphorylated (activated) levels of Akt and total Akt, cerebellar granule cells on Day 8 in vitro were treated with a maximum neuroprotective concentration of NMDA (100 μM) at various times (0-24 hr) or concentrations (0-100 μM) at 378C in a 95% air/5% CO2 humidied incubator. At the indicated time, the culture medium was removed and the cells washed twice with ice cold Locke’s buffer containing (in mM): 154 NaCl; 5.6 KCl; 1 MgCl2; 2.3 CaCl2;5.6 d-glucose; and 8.6 HEPES; pH 7.4. The cerebellar granule cells were disrupted in 200 ml of lysis buffer (1% Nonidet P-40, 20 mM Tris, pH 8.0, 137 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 0.15 units/ml aprotinin, 20 μM leupeptin, and 1 mM sodium vanadate) at 48C. After removal of cellular debris by centrifugation, protein levels in the lysates were measured by the Bradford Coomassie Blue colorimetric assay (Bio-Rad Laboratories, Hercules, CA) and equalized accordingly. The appropriate amount of cell lysate was mixed with sample buffer (2% SDS, 100 mM dithiothreitol, 10% glycerol, and 0.25% bromphenol blue) followed by fractionation through 10% bis-tris gels. Gel proteins were transferred to nitrocellulose membranes (0.22 μm; Schleicher and Schuell, Keene, NH). After blocking with 5% nonfat dry milk (for activated and total Akt and MAPK antibodies) in Tris-buffered saline containing 0.2% Tween 20 (TBST), the immobilized proteins were incubated overnight at 48C with specic antibodies (anti-phosphorylated MAPK 1:1,000, anti-MAPK 1:1,000, anti-phosphorylated Akt 1:1,000, and anti-Akt 1:1,000) and then incubated with goat anti-rabbit IgG conjugated to HRP for 1 hr. Blots were developed using enhanced chemiluminescence detection according to the manufacturer’s recommendations (Amersham Pharmacia Biotech, Piscataway, NJ). To determine total protein levels of either MAPK or Akt, the blots were stripped with Restore Western Blot Stripping Buffer (Pierce, Rockford, IL). The nitrocellulose membranes were washed with TBST followed by probing the blot with anti-MAPK or anti-Akt overnight at 48C. The blots were washed with TBST for 1 hr followed by incubation with the secondary antibody linked to HRP for 1 hr. Blots were developed as indicated above.
For the determination of Bcl-2, Bcl-XL, and Bax protein levels, cultured cerebellar granule cells on Day 8 in vitro were treated with NMDA (100 μM) for various times followed by the removal of culture medium. The nitrocellulose membranes were probed with either the Bcl-2 antibody (1:200), the Bcl-XL antibody (1:1,000), or the Bax antibody (1:1,000) overnight at 48C, washed three times in TBST for 30 min, and then incubated with HRP-conjugated goat anti-rabbit IgG (1:2,000; Cell Signaling). Immunoreactive bands were visualized using the chemiluminescence method described above. Only one immunoreactive band was detected when blots were probed for Bcl-2, Bcl-XL, and Bax. The blot was stripped and re-probed with the actin antibody (1:10,000) followed by goat anti-rabbit IgG antibody conjugated to HRP (Cell Signaling) and the bands detected using chemiluminescence to ensure that equivalent amounts of protein were present in each lane. Immunoreactive bands were quantitated by image analysis using a scanner (Hewlett-Packard) and Scanalytics image analysis software. When more than one band was detected by the antibodies (pERK1/2), each band was scanned separately and quantitated.
Determination of bcl-2, bcl-XL, and bax mRNA Levels
Cerebellar granule cells were treated with NMDA (100 μM) for 6 hr followed by the addition of an excitotoxic concentration of glutamate (100 μM) for 24 hr. The culture medium was aspirated and the granule cells were disrupted by 5 M guanidine thiocyanate (Fluka)/0.1M EDTA pH 8 (0.1mL) at room temperature. The culture dishes were scraped and the solubilized cells (∼107 cells/ml) were placed in 1.5 ml Eppendorf microcentrifuge tubes. The samples were then placed on dry ice and stored at -80°C. The RNase protection assays were carried out as described previously with the antisense cyclophilin RNA probe was used as the standard internal control (Strauss and Jacobowitz, 1993; Strauss et al., 2000). Briefly, an excess of 32P-labeled antisense riboprobes was hybridized to the target mRNA directly in the cell lysates at 378C overnight. Double-stranded RNA complexes were protected from RNase degradation, puried away from background contaminants by organic extractions and ethanol precipitation, and fractionated by native polyacrylamide gel electrophoresis. Gel pieces containing full-sized protected fragments were excised from the dried gel using the autoradiogram as a guide. The radioactivity detected by scintillation counting was converted to moles of mRNA. The internal control probe was included with each assay for normalization. Background counts were determined using specimens hybridized on dry ice, but otherwise treated the same as the experimental specimens were.
Antisense RNA probes were T7 RNA polymerase transcripts of probe templates labeled using [α-32P]UTP (10 mCi/ml, 800 Ci/mmol; NEN) to specic activities from 108-109 dpm/mg. Probe templates were generated from rat brain cDNA by reverse transcription (Superscript; Life Technologies) and PCR using high fidelity Taq/Pfu DNA polymerases (Boehringer-Mannheim, Indianapolis, IN). The rat bax template was synthesized using amplification primers corresponding to nucleotide (nt) 190-212 and nt 559-537 (GenBank accession number L22472), with respect to the initiating ATG codon, and subcloned into the NcoI and SalI sites of pGEM-5Zf (Promega, Madison, WI). The rat bcl-2 template was synthesized using primers at nt 13-35 and nt 651-629, with respect to the initiation codon (GenBank accession number L14680) and subcloned into the EcoRV site of pGEM-5Zf+. The rat bcl-xL template was synthesized using primers at nt 21-43 and nt 587-609, with respect to the initiation codon (GenBank accession number X82537) and subcloned into the EcoRV site of pGEM-5Zf+. The cyclophilin probe used the pGEM-4Z vector with the PstI/NcoI 309-base pair (bp) fragment of rat cyclophilin cDNA (-8 to 300) (Danielson et al.,1988).
Determination of Neuronal Survival
Cultured cerebellar granule cells were treated with the MEK inhibitor U0126 (10 μM) or the selective PI 3-kinase inhibitor LY294002 (10 μM), for 60 min before the addition of a maximum neuroprotective concentration of NMDA (100 μM) for 6 hr. Medium was then removed and replaced with sister culture medium. Either inhibitor and an excitotoxic concentration of glutamate (100 μM) was added, and neuronal viability was quantified 24 hr later using the 3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously (Lin and Long, 1996). Under the experimental condition where the culture medium was not removed, glutamate was added and neuronal viability quantified 24 hr later using the MTT assay. This colorimetric assay measured the reduction of MTT to an insoluble, colored formazan product by mitochondrial succinate dehydrogenase. This could occur only in metabolically active cells; thus, the colorimetric signal reflected cell viability. The final concentration of MTT was 0.3 mg/ml and the MTT was allowed to incubate with the neurons at 378C for 1.5 hr in the incubator. The cells were solubilized in 70% isopropanol acidified with 0.1 N HCl and the optical density at 595 nm was measured. Neuronal viability was determined from optical density readings of treated versus untreated neurons and expressed as a percent. Because most cells in the culture are neuronal, the formed product is strictly neuronal.
Statistical Analysis
All values are presented as mean ± standard deviation (SD) or percent of control (untreated). Analysis of variance (ANOVA) with post-hoc comparisons was used for analysis. P < 0.05 was considered “significant”.
RESULTS
NMDA Activates ERK1/2 Through NMDA and TrkB Receptors
To understand better how NMDA and TrkB receptors contribute to ERK1/2 activation, we determined the temporal kinetics of ERK1/2 after addition of a maximum neuroprotective concentration of NMDA (100 μM). We also determined the contribution of TrkB receptors to the activation of ERK1/2 by using k252a, a tyrosine kinase inhibitor shown previously to block NMDA neuroprotection (Marini et al., 1998) and to inhibit neurotrophin receptor activity (Lee et al., 2002). NMDA elicits a time-dependent increase in activated (phosphorylated) ERK1/2 beginning within 15 min and lasting at least 24 hr (1A). We have shown that a neuroprotective concentration of BDNF (100 ng/ml) (Marini et al., 1998) activates ERK1/2 with similar kinetics (Wu et al., 2004). The activation of ERK1/2 was also dependent upon the NMDA concentration (1B). ERK1/2 activation was blocked completely by MK-801 but only partially by k252a (2). NMDA and TrkB receptors are thus required to maximally activate ERK1/2.
MEK Inhibitor U0126 Blocks NMDA Receptor-Mediated Neuroprotection
We examined the effect of the MEK inhibitor U0126 to determine whether activated ERK1/2 is required for NMDA neuroprotection. The addition of U0126 (10 μM) completely blocked the activation of ERK1/2 by NMDA and reduced basal levels of activated ERK1/2 (3A). Reduced basal levels of activated ERK1/2 is expected because U0126 inhibits MEK, the kinase that activates MAPK (Subramaniam et al., 2004). Pretreatment of the cultured neurons with the MEK inhibitor partially blocked (˜30%) the neuroprotective activity of NMDA (3B). Interestingly, if NMDA was removed from the cultures before the addition of glutamate (100 μM), neuroprotection was preserved but the addition of U0126 (10 μM) completely abolished NMDA neuroprotection (3C). The MEK inhibitor did not alter the toxicity mediated by glutamate alone acting on NMDA receptors, consistent with previous findings (Skaper et al., 2001). None of the drugs affected basal neuronal survival (data not shown). These results suggest that preconditioning of neurons with NMDA requires activation of the ERK1/2 pathway to protect neurons against glutamate excitotoxicity.
Selective PI3-K Inhibitor LY294002 Fails to Block Neuroprotective Activity of NMDA
Akt, a downstream target of the PI3-K pathway, is activated by NMDA and the selective PI3-K inhibitor LY294002 (10 μM) blocks its activation (Zhu et al., 2002). Preincubation of cerebellar granule cells with LY294002 (10 μM) failed to block the protective properties of NMDA (3D). LY294002 did not affect basal neuronal survival (data not shown). MAPK and PI3-K are both involved in BDNF-mediated neuroprotection (Hetman et al., 1999); thus, the MEK inhibitor might also inhibit activation of Akt (inhibition of both cascades might be required to inhibit the neuroprotective activity of NMDA). Surprisingly, Akt phosphorylation increased after addition of NMDA in the presence of U0126 (4). These results demonstrate that inhibition of the MAPK pathway enhances Akt activation in cultured cerebellar granule cells; however, LY294002, an inhibitor known to block the PI3-K pathway and its downstream target Akt, does not affect the neuroprotective activity of NMDA. In contrast, our recent results show that AMPA-mediated neuroprotection requires activation of the PI3-K pathway; LY294002 blocks Akt phosphorylation, AMPA-mediated neuroprotection, and the AMPA-mediated increase in BDNF mRNA (Wu et al., 2004). The inotropic glutamate receptor activated thus dictates the signaling pathway required to mediate the survival-promoting effects of the agonist.
NMDA Neuroprotection Increases Bcl-2 and Bcl-XL Gene Expression but Reduces Bax after Excitotoxicity
Glutamate excitotoxicity induces apoptosis in cerebellar granule cells cultured in depolarizing concentrations of potassium chloride (Banaudha and Marini, 2000). We hypothesized that NMDA neuroprotection might be mediated, in part, by modulation of the Bcl-2 family of genes. To address this question, neurons were pretreated with NMDA (100 μM) in the presence or absence of glutamate or treated with glutamate alone (100 μM) for either 6 hr, when there is minimal cell death, or 24 hr, when about 50% of neurons die from the excitotoxic effects of glutamate acting on NMDA receptors. By 6 hr, neurons pretreated with NMDA (100 μM) alone increases antiapoptotic bcl-2 and bcl-XL mRNA levels. Addition of an excitotoxic concentration of glutamate (100 μM) markedly reduces levels of bcl-2 and bcl-XL mRNA. Pretreatment of the neurons with NMDA for 6 hr followed by the addition of a toxic concentration of glutamate for 6 hr attenuates this reduction (5A). These results suggest that NMDA may protect neurons against glutamate-mediated excitotoxicity by maintaining or increasing antiapoptotic gene expression levels. In contrast, pretreatment of the neurons with NMDA (100 μM) for 6 hr does not alter the mRNA levels of bax, a proapoptotic Bcl-2 family member. Addition of an excitotoxic concentration of glutamate (100 μM), however, results in a striking decrease in the mRNA levels, which are unaffected by pretreatment with a maximum neuroprotective concentration of NMDA (100 μM) (5A). These results suggest that the reduction in bax mRNA levels is mediated by glutamate. Alternatively, changes in bcl-2 and bcl-XL mRNA levels may be due coincidentally to enhanced neuronal survival induced by NMDA. This suggestion is not tenable because bax mRNA levels remain low. NMDA receptor activation thus differentially affects Bcl-2 family members in an antiapoptotic direction. Additional changes are observed after a 24-hr treatment with an excitotoxic concentration of glutamate (100 μM), a concentration that kills about 50% of neurons (Marini and Paul, 1992). NMDA (100 μM) induces a modest but significant increase in bcl-xL and bax mRNA levels (5B, hatched bars). Notably, the effect of NMDA on bax mRNA levels was delayed (i.e., no effect at 6 hr and a significant increase at 24 hr) in the absence of glutamate (5, stippled bar). As expected, addition of an excitotoxic concentration of glutamate (100 μM) resulted in a sustained reduction in bcl-2 and bcl-XL mRNA levels (5B, solid bars). Unexpectedly, however, pretreatment of the neurons with NMDA followed by the addition of glutamate for 24 hr attenuated the decrease in bax mRNA (5B, stippled bar) levels mediated by glutamate alone (5B, solid bar). Pretreatment with MK-801 (1 μM) had substantially the same effects as NMDA did (data not shown). Because of the unexpected increase in bax mRNA levels in neurons pretreated with NMDA, protein levels of the Bcl-2 family were determined in neurons pretreated with NMDA (100 μM). NMDA elicits an increase in Bcl-2 (6A) and Bcl-XL (6B) proteins whereas NMDA clearly decreased Bax protein levels under identical conditions (6C). MK-801 completely blocked the effect of NMDA on these proteins. The tyrosine kinase inhibitor, k252a, blocks NMDA neuroprotection and also blocks increases in Bcl-2 and Bcl-XL and the decrease in Bax levels suggesting that TrkB signaling mediates the observed effects on the Bcl-2 family of proteins by NMDA. In our hands, antibodies used to detect Bcl-2, Bcl-XL and Bax proteins produced only one immunoreactive band on Western blot analysis.
DISCUSSION
The diverse roles of NMDA receptors (Maren and Baudry, 1995) and neurotrophin receptors (Black, 1999) in neuronal plasticity are just beginning to be understood. One of the first examples of neuronal plasticity was the discovery that NMDA receptors were involved in induction (Bashir et al., 1991) and reversal (Villarreal et al., 2002) of long-term potentiation. NMDA receptor activation (Arvidsson et al., 2001) or inhibition (Nacher et al., 2003; Okuyama et al., 2004) increases neurogenesis in the adult rat dentate gyrus after hypoxic-ischemic neuronal injury, suggesting that formation of new neurons is enhanced by NMDA receptors. Understanding fundamental NMDA receptor plasticity may thus lead to protective strategies against acute and chronic neurodegenerative disorders in the highly vulnerable cerebral cortex.
BDNF has been shown to enhance long-term potentiation (Aoki et al., 2000; Gooney and Lynch, 2001) and activate NMDA receptors through phosphorylation of NMDA receptor subunit 1 (NR1) (Suen et al., 1997). Neurotrophin and NMDA receptors thus cooperate in neurophysiologic functions.
The intrinsic survival autocrine loop that cultured cerebellar granule cells use may exist in vivo (Lipsky et al., 2001; Marini et al., 2004). This may be one mechanism for the substantially reduced neuronal vulnerability that has been observed in the cerebellum in acute and chronic neurodegenerative disorders (Iwata et al., 2001; Marini et al., 2001). NMDA and TrkB receptors may act in a coordinated fashion to maintain these survival mechanisms.
NMDA exerts trophic activity against the excitotoxic actions of glutamate (Marini et al., 1998; Lipsky et al., 2001), which may be mediated through neurotrophin receptors. The signaling pathways employed by the neurotrophic factors have been studied extensively (Kaplan and Stephens, 1994). The first major pathway is the Ras-MAPK kinase (MEK)-MAPK pathway. This pathway serves diverse functions including survival, long-term potentiation, and synaptic plasticity (Grewal et al., 1999); however, the role of the MAPK pathway in protecting neurons is controversial. A role has been proposed for p44/42 MAPK (ERK1/2) in neuronal cell death mediated by various drugs, toxins, serum deprivation or neurotrophin withdrawal (Gunn-Moore et al., 1997; Maas et al., 1998; Murray et al., 1998; Runden et al.,1998; Dolcet et al.,1999; Encinas et al.,1999; Ryu et al., 1999; Ozawa et al., 1999; Linford et al., 2001; Subramaniam et al., 2004) and stroke (Namura et al., 2001). The role of MAPK in survival thus depends upon the downstream targets activated. The second major pathway is the PI3-K/Akt survival pathway; however, there is conflicting evidence for its role in neurotrophin-mediated survival (reviewed by Kaplan and Miller, 2000). The PI3-K/Akt pathway plays a predominant role in protecting neurons under conditions of serum deprivation (Hetman et al.,1999; Ryu et al.,1999).
NMDA receptors activate MAPK (Bading and Greenberg,1991), which may serve to protect neurons against glutamate-mediated excitotoxicity (Liu et al., 1999). We first confirmed that NMDA activates ERK1/2 (p44/p42) in cerebellar granule cells, consistent with previous findings (Bading and Greenberg, 1991). The temporal similarity between the rapid release of BDNF in the medium (2 min) and TrkB activation (10 min) shown previously (Marini et al., 1998) correlates well with the activation of ERK1/2 by NMDA (within 15 min). Activation of ERK1/2 was blocked completely by the NMDA receptor antagonist MK-801 and partially by k252a, a tyrosine kinase inhibitor that blocks neurotrophin activity. These results support the idea that cross talk between NMDA and TrkB receptors is necessary to maximally activate MAPK. Because ERK1/2 has been shown to be associated with either neuronal cell death or survival, we investigated the role of ERK1/2 in NMDA neuroprotection. We show that the MAPK inhibitor U0126 blocks NMDA-mediated ERK1/2 activation in cerebellar granule cells. Pretreatment with U0126 partially blocked NMDA neuroprotection during continuous exposure to NMDA, but completely blocked the neuroprotective activity when NMDA was removed from the culture medium after 6 hr pretreatment. The neuroprotective activity of NMDA therefore can circumvent inhibition of the MAPK pathway. One possible interpretation of this result is that MAPK is important for the survival effects of NMDA. This is similar to findings in which BDNF and MAPK protect neurons from death due to chemical toxicity (Skaper et al., 1998; Hetman et al.,1999). Because U0126 blocks about 75% of MAPK kinase activation (Davies et al., 2000), it is also possible that low-level activation of MAPK is sufficient to protect neurons but requires continuous activation achieved via NMDA receptor activation. Alternatively, as we have shown, a maximum neuroprotective concentration of NMDA activates Akt, a downstream target of the PI3-K pathway.
The PI3-K pathway is known to promote survival in many systems and may be another signal transduction pathway involved in the neuroprotective activity of NMDA. NMDA and TrkB receptors converge to activate Akt in cerebellar granule cells maintained in depolarizing concentrations of potassium chloride (Zhu et al., 2002). In addition, we show that U0126 increases phosphorylated (activated) Akt in the cultured neurons, suggesting that Akt may be important for NMDA to mediate its neuroprotective effect. Pretreatment with the selective PI3-K inhibitor LY294002 (10 μM), however, failed to block the neuroprotective activity of NMDA. This result suggests that unlike MAPK, the PI3-K pathway is not a requirement for the neuroprotective activity of NMDA. This is in contrast with previous studies where Akt plays an important role in preventing cerebellar granule cell death in an apoptotic cell death paradigm (Bhave et al., 1999).
We also present evidence that a maximum neuroprotective concentration of NMDA promotes survival through the antiapoptotic Bcl-2 family proteins. Bcl-2, Bcl-XL and other antiapoptotic family members reside in the outer mitochondrial membrane and suppress apoptosis by preventing cytochrome c release (Boise et al.,1993; Oltvai et al., 1993; Korsmeyer, 1996). TrkB receptor activation by in vivo administration of BDNF has been shown to increase antiapoptotic (Bcl-2 and Bcl-XL) and decrease proapoptotic (Bax) immunoreactivity after focal cerebral ischemia (Schäbitz et al., 2000). Consistent with the hypothesis that NMDA receptors activate multiple survival pathways, NMDA increases Bcl-2 and Bcl-XL protein levels in cerebellar granule cells through TrkB receptor activation. NMDA markedly reduces Bax protein levels, which is blocked by MK-801 or k252a; thus, it is likely that the NMDA-mediated release of BDNF activates TrkB receptors resulting in a reduction of Bax protein. Although the mechanism of NMDA receptor-mediated downregulation of Bax protein levels is unknown, it is possible that a neuroprotective concentration of NMDA reduces Bax protein levels through caspase activation as shown for Bcl-2 (Fujita and Tsuruo, 1998; Chen et al., 2000; Karran and Dyer, 2001; Subramanian and Chinnadurai, 2003), phosphorylation of Bax at sites other than those involved in apoptosis that ultimately enhance its degradation, i.e. phosphorylation of Bcl-2 reduces its prosurvival effects (Ishikawa et al., 2003; Linseman et al., 2004) or via calpain activation (Choi et al., 2001).
There seems to be a discrepancy with Bax expression in neurons treated with glutamate and NMDA. Treatment of cultured neurons with NMDA increases bcl-2 and bcl-XL mRNA levels at 6 hr, and NMDA pretreatment attenuates the glutamate-induced decline, whereas bax mRNA is not affected. Surprisingly, an excitotoxic concentration of glutamate markedly reduces bax mRNA levels at 6 hr; bax mRNA levels are increased at 24 hr. Although bcl-2 and bcl-XL mRNA levels are higher than bax mRNA levels are at 6 hr, it is likely that these antiapoptotic proteins are cleaved by caspases to promote apoptosis (Fujita and Tsuruo, 1998; Chen et al., 2000; Karran and Dyer, 2001; Subramanian and Chinnadurai, 2003). In glutamate-treated neurons, bax mRNA levels were attenuated markedly at 6 hr but did increase at 24 hr. The mechanism(s) of this effect requires further investigation. At 24 hr, NMDA elevates bax mRNA levels and normalizes bax gene expression in the presence of glutamate. Finally, there is a discrepancy between bax mRNA and protein levels in NMDA-treated neurons, i.e., mRNA levels are high and protein levels are low. NMDA may promote the degradation of Bax protein through caspase or calpain cleavage or through phosphorylation (e.g., as with IkB phosphorylation by IkB kinase; Lipsky et al., 2001) but these hypotheses require further investigation.
In summary, NMDA receptors protect neurons by activating TrkB receptors via a BDNF autocrine loop. The activations of NMDA and TrkB receptors converge to phosphorylate ERK1/2, a major protective signal cascade. Moreover, unique signaling cascades are activated via neurotrophin signaling that lead to the increase of antiapoptotic Bcl-2 and Bcl-XL proteins and the decrease in the proapoptotic protein Bax. The activation of common and unique signaling pathways via two different types of receptors cooperates to promote neuronal survival. NMDA receptor-mediated release of BDNF and TrkB receptor activation suggests a correlation between survival, protection, and activity-dependent synaptic remodeling. NMDA receptors, through the influx of calcium and BDNF release, may thus carry out multitask functions to enhance neuronal survival in cerebellar granule cells and may be viewed as a form of neuronal plasticity.
Fig. 1.
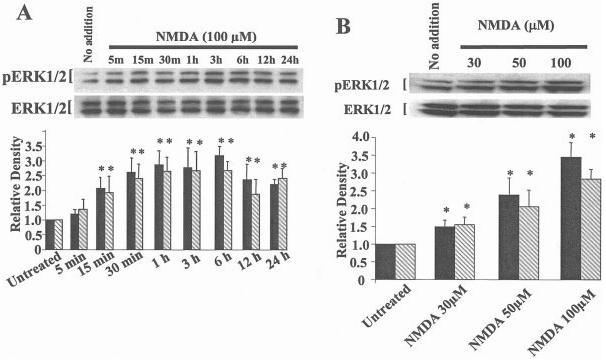
The temporal kinetics of NMDA and BDNF on phosphorylated (activated) MAPK. Cultured cerebellar granule cells were incubated for the indicated times with NMDA (100 μM; A), or with various concentrations of NMDA (B). Cerebellar lysates were prepared and proteins were separated by PAGE. Separated proteins were transferred to nitrocellulose membranes and subsequently probed for activated and total MAPK. Top:Western blot of pERK1/2. Bottom: Re-probing for total ERK1/2 protein to verify equal loading of protein across lanes. Results are expressed as the mean 6 SD of activated ERK1 compared to total ERK1 (solid bar) and activated ERK2 compared to total ERK2 (hatched bar) and normalized to untreated as shown in the lower panel of A and B. n = 9. *P < 0.01 vs. untreated (by ANOVA).
Fig. 2.
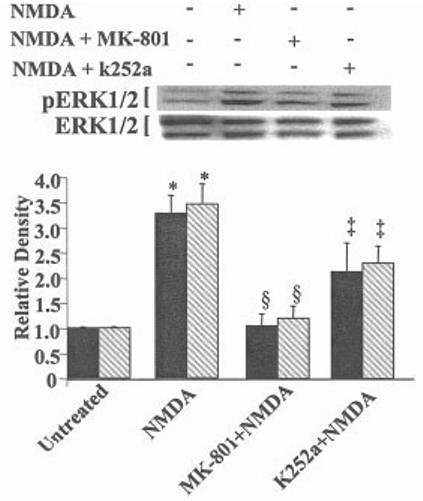
NMDA and TrkB receptors are required for maximal activation of ERK1/2. The contribution of NMDA and TrkB receptors to activate ERK1/2 (p44/p42) was determined using the specific NMDA receptor antagonist MK-801 and k252a, an inhibitor of neurotrophin activity. Cerebellar granule cells were incubated with NMDA (100 μM), MK-801 (1 μM) plus NMDA (100 μM), or k252a (20 nM) plus NMDA for 3 hr. n = 4. Scanning densitometry was carried out on activated ERK1 compared to total ERK1 (solid bar) or activated ERK2 compared to total ERK2 (hatched bar), quantitated and normalized to untreated (control) values on the same Western blots. Results are presented as in Fig. 1. §P < 0.01 vs. NMDA; *P < 0.01 vs. untreated alone;‡< 0.05 vs. NMDA alone (by ANOVA).
Fig. 3.

The MAPK but not the PI3-K pathway is involved in NMDA neuroprotection. Preincubation of cerebellar granule cells with U0126 (10 μM) for 1 hr followed by the addition of NMDA (100 μM) for 3 hr completely blocked the basal activity and activated pERK1/2, confirming its mechanism of action in blocking MAPK kinase (A). Cerebellar granule cells were exposed to medium alone (control) or medium containing glutamate, U0126, or NMDA for 24 hr. Neurons were also pretreated with U0126 (10 μM) or NMDA (100 μM; 6 hr) followed by the addition of glutamate or to a neuroprotective concentration of NMDA (100 μM). In those culture dishes exposed to glutamate (100 μM) for 24 hr, the culture medium was either not exchanged with sister culture media (B) or was exchanged (C). Cultured neurons were subjected to the identical experiment as outlined in B with the exception that LY294002 (10 μM) was used as the blocker of the PI3-K pathway instead of U0126. Neuronal viability was quantified using the MTT assay on Day 9 in vitro. Results are presented as percent control (mean ± SD) n = 9. *P < 0.001 vs. control; §P < 0.001 vs. glutamate; aP < 0.01 vs. NMDA; ‡P < 0.001 vs. NMDA plus glutamate (N + Glu) (by ANOVA).
Fig. 4.
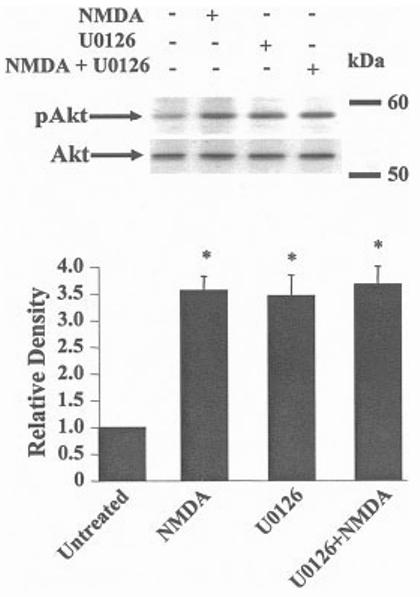
U0126 increases activated Akt (pAkt) in the presence or absence of NMDA. Cerebellar granule cells were exposed to medium alone, U0126 (10 μM) or to a neuroprotective concentration of NMDA (100 μM) for 3 hr. At the indicated time, lysates were prepared and the proteins were separated by gel electrophoresis followed by transfer to nitrocellulose. Levels of pAkt and total levels of Akt were determined. Top: NMDA or U0126 induced activation of Akt (pAkt). Bottom: The blot was stripped and re-probed with an antibody that recognizes phosphorylated and unphosphorylated forms of Akt to demonstrate that an equal amount of Akt proteins were present in each lane n= 6. *P < 0.001 vs. untreated.
Fig. 5.
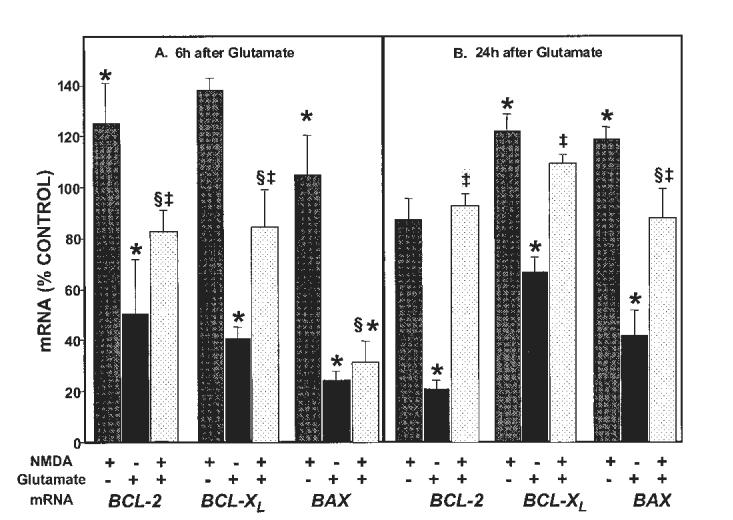
Neuroprotective NMDA treatment differentially regulates bcl-2, bcl-XL and bax mRNA. NMDA alone (100 μM, patterned bars) increased bcl-2 and bcl-XL but not bax mRNA at 6 hr (A). At 24 hr, NMDA treatment increased bcl-XL and bax mRNA (B). In contrast, an excitotoxic concentration of glutamate (100 μM, solid bars) decreased bcl-2, bcl-XL, and bax mRNA levels at 6 hr (A) and 24 hr (B). NMDA pretreatment (100 μM; 6 hr) with subsequent addition of 100 μM glutamate (stippled bars) attenuated the decreases in antiapoptotic bcl-2 and bcl-XL (but not proapoptotic bax) mRNA at 6 hr (A) whereas by 24 hr, NMDA attenuated the effect of glutamate on all three mRNA levels (B). n = 6. Results (mean ± standard error of the mean [SEM]) are percent of mRNA levels (mol target/mol cyclophilin) from control untreated cultures. *P < 0.05 vs. control; §P < 0.05 vs. glutamate; ‡P < 0.05 vs. NMDA alone (by ANOVA).
Fig. 6.
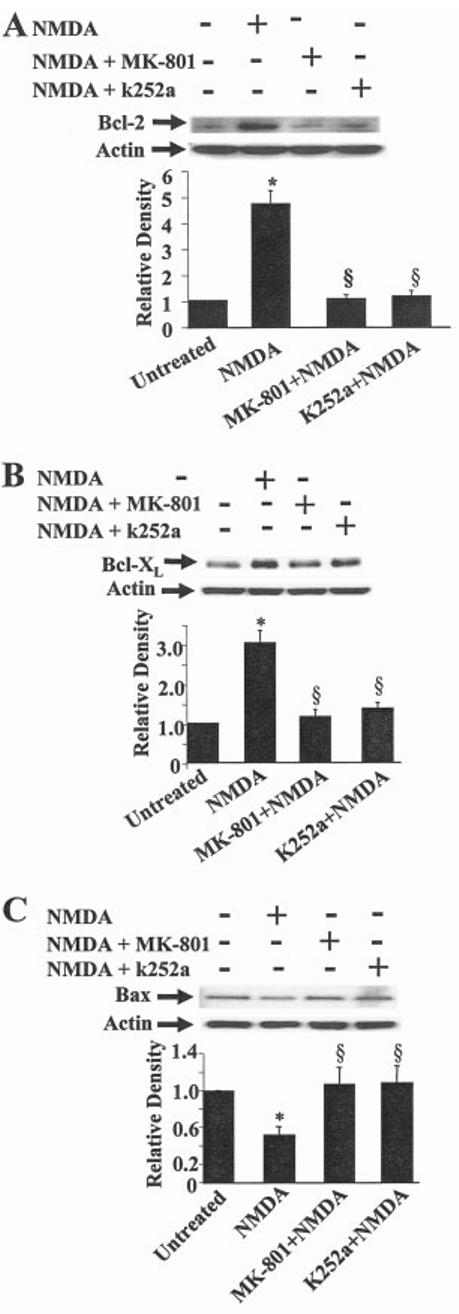
NMDA differentially modulates Bcl-2, Bcl-XL, and Bax protein expression. Cerebellar granule cells were exposed to medium alone or medium containing NMDA (100 μM) with or without MK-801 (1 μM) or k252a (10 nM) for 24 hr. Bcl-2 (A), Bcl-XL (B), and Bax (C) protein levels (top) were determined by Western blot analysis. Bottom: Re-probing of blots with anti-actin to verify equal loading of protein across lanes. Results are expressed as the average ± SEM n = 3. *P < 0.05 vs. untreated; §P < 0.001 vs. NMDA (by ANOVA).
REFERENCES
- Arvidsson A, Kokaia Z, Lindvall O. N-Methyl-d-aspartate receptor-mediated increase of neurogenesis in the adult rat dentate gyrus following stroke. Eur J Neurosci. 2001;14:10–18. doi: 10.1046/j.0953-816x.2001.01611.x. [DOI] [PubMed] [Google Scholar]
- Aoki C, Wu K, Elste A, Len GW, Lin SY, McAuliffe G, Black IB. Localization of brain-derived neurotrophic factor and TrkB receptors to postsynaptic densities of adult rat cerebral cortex. J Neurosci Res. 2000;59:454–463. doi: 10.1002/(SICI)1097-4547(20000201)59:3<454::AID-JNR21>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- Banaudha K, Marini AM. AMPA prevents glutamate-induced neurotoxicity and apoptosis in cultured cerebellar granule cell neurons. Neurotox Res. 2000;2:51–61. doi: 10.1007/BF03033327. [DOI] [PubMed] [Google Scholar]
- Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, Feuerstein GZ. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonists and early gene expression. Stroke. 1998;29:1937–1950. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bhave S, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci. 2001;21:4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Chen MC, Hsu TL, Luh TY, Hsieh SL. Overexpression of bcl-2 enhances LIGHT- and interferon-gamma-mediated apoptosis in Hep3BT2 cells. J Biol Chem. 2000;275:38794–38801. doi: 10.1074/jbc.M003292200. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and disease of the central nervous system. Neuron. 1988;1:623–628. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi WS, Lee EH, Chung CW, Jung YK, Jin BK, Kim SU, Oh TH, Saido TC, Oh YJ. Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J Neurochem. 2001;77:1531–1541. doi: 10.1046/j.1471-4159.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- Danielson PE, Forss-Petter S, Brow MA, Calavetta L, Douglass J, Milner RJ, Sutcliffe JG. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcet X, Egea J, Soler RM, Martin-Zanca D, Comella JX. Activation of phosphatidylinositol 3-kinase, but not extracellular-regulated kinases, is necessary to mediate brain-derived neurotrophic factor-induced moto-neuron survival. J Neurochem. 1999;73:521–531. doi: 10.1046/j.1471-4159.1999.0730521.x. [DOI] [PubMed] [Google Scholar]
- Dowden J, Corbett D. Ischemic preconditioning in 18- to 20-month old gerbils: long-term survival with functional outcome measures. Stroke. 1999;30:1240–1246. doi: 10.1161/01.str.30.6.1240. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuro-blastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- Fujita N, Tsuruo T. Involvement of Bcl-2 cleavage in the acceleration of VP-16-induced U937 cell apoptosis. Biochem Biophys Res Commun. 1998;246:484–488. doi: 10.1006/bbrc.1998.8587. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:249–252. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Gooney M, Lynch MA. Long-term potentiation in the dentate gyrus of the rat hippocampus is accompanied by brain-derived neurotrophic factor-induced activation of TrkB. J Neurochem. 2001;77:1198–1207. doi: 10.1046/j.1471-4159.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork JS. Extracellular-signal-regulated kinase signaling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Gunn-Moore FJ, Williams AG, Toms NJ, Tavare JM. Activation of mitogen-activated protein kinase and p70S6 kinase is not correlated with cerebellar granule cell survival. Biochem J. 1997;324:365–369. doi: 10.1042/bj3240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Kusaka E, Enokido Y, Ikeuchi T, Hatanaka H. Regulation of Bax translocation through phosphorylation at Ser-70 of Bcl-2 by MAP kinase in NO-induced neuronal apoptosis. Mol Cell Neurosci. 2003;24:451–459. doi: 10.1016/s1044-7431(03)00203-3. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- Karran L, Dyer MJ. Proteolytic cleavage of molecules involved in cell death or survival pathways: a role in the control of apoptosis? Crit Rev Eukaryot Gene Expr. 2001;11:269–277. [PubMed] [Google Scholar]
- Kato H, Liu Y, Araki T, Kogure K. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res. 1991;553:238–242. doi: 10.1016/0006-8993(91)90831-f. [DOI] [PubMed] [Google Scholar]
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz JC. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K. Ischemic tolerance phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ. Regulators of cell death. Trends Genet. 1996;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Kim AH, Chang PC, Chao MV. Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J Biol Chem. 2002;277:9096–9102. doi: 10.1074/jbc.M107421200. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(Suppl):7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Lin Y, Long JB. Acute or prolonged exposure to 1-aminocyclopropane-carboxylic acid protects spinal neurons against NMDA toxicity. Eur J Pharmacol. 1996;318:491–496. doi: 10.1016/s0014-2999(96)00811-4. [DOI] [PubMed] [Google Scholar]
- Linford NJ, Yang Y, Cook DG, Dorsa DM. Neuronal apoptosis resulting from high doses of the isoflavone genistein: role for calcium and p42/44 mitogen-activated protein kinase. J Pharmacol Exp Ther. 2001;299:67–75. [PubMed] [Google Scholar]
- Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3 phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky RH, Xu K, Zhu D, Kelly C, Terhakopian A, Novelli A, Marini AM. NF-kB is a critical determinant for NMDA receptor-mediated neuroprotection. J Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kato H, Nakata N, Kogure K. Protection of rat hippocampus against ischemic neuronal damage by pretreatment with sublethal ischemia. Brain Res. 1992;586:121–124. doi: 10.1016/0006-8993(92)91380-w. [DOI] [PubMed] [Google Scholar]
- Liu YZ, Boxer LM, Latchman DS. Activation of the Bcl-2 promoter by nerve growth factor is mediated by the p42/p44 MAPK cascade. Nucleic Acids Res. 1999;27:2086–2090. doi: 10.1093/nar/27.10.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas JW, Jr, Horstmann S, Borasio GD, Anneser JM, Shooter EM, Kahle PJ. Apoptosis of central and peripheral neurons can be prevented with cyclin-dependent/mitogen-activated protein kinase inhibitors. J Neurochem. 1998;70:1401–1410. doi: 10.1046/j.1471-4159.1998.70041401.x. [DOI] [PubMed] [Google Scholar]
- Maren S, Baudry M. Properties and mechanisms of long-term synaptic plasticity in the mammalian brain: relationships to learning and memory. Neurobiol Learn Mem. 1995;63:1–18. doi: 10.1006/nlme.1995.1001. [DOI] [PubMed] [Google Scholar]
- Marini AM, Choi J, Labutta R. Synaptic deprivation and age-related vulnerability to hypoxic-ischemic neuronal injury. Ann N Y Acad Sci. 2001;939:238–253. doi: 10.1111/j.1749-6632.2001.tb03631.x. [DOI] [PubMed] [Google Scholar]
- Marini AM, Jiang X, Wu X, Tian F, Zhu D, Okagaki P, Lipsky RH. Brain-derived neurotrophic factor and NF-kB: from genomics to molecular biology to phenotype. Restor Neurol Neurosci. 2004;22:121–130. [PubMed] [Google Scholar]
- Marini AM, Paul SM. N-Methyl-d-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini AM, Rabin SI, Lipsky RH, Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem. 1998;273:29394–29399. doi: 10.1074/jbc.273.45.29394. [DOI] [PubMed] [Google Scholar]
- Marini AM, Schwartz JP, Kopin IJ. The neurotoxicity of 1-methyl-4-phenylpyridinium in cultured cerebellar granule cells. J Neurosci. 1989;9:3665–3672. doi: 10.1523/JNEUROSCI.09-10-03665.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nacher J, Alonso-Llosa G, Rosell DR, McEwen BS. NMDA receptor antagonist treatment increases the production of new neurons in the aged rat hippocampus. Neurobiol Aging. 2003;24:273–284. doi: 10.1016/s0197-4580(02)00096-9. [DOI] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- Ogita K, Okuda H, Yamamoto Y, Nishiyama N, Yoneda Y. In vivo neuroprotective role of NMDA receptors against kainate-induced excitotoxicity in murine hippocampal pyramidal neurons. J Neurochem. 2003;85:1336–1346. doi: 10.1046/j.1471-4159.2003.01778.x. [DOI] [PubMed] [Google Scholar]
- Okuyama N, Takagi N, Kawai T, Miyake-Takagi K, Takeo S. Phosphorylation of extracellular-regulating kinase in NMDA receptor antagonist-induced newly generated neurons in the adult rat dentate gyrus. J Neurochem. 2004;88:717–725. doi: 10.1046/j.1471-4159.2003.02215.x. [DOI] [PubMed] [Google Scholar]
- Olney JW, Ho OL, Rhee V. Cytotoxic effects of acidic and sulphur-containing amino acids on the infant mouse central nervous system. Exp Brain Res. 1971;14:61–76. doi: 10.1007/BF00234911. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Ozawa H, Shioda S, Dohi K, Matsumoto H, Mizushima H, Zhou CJ, Funahashi H, Nakai Y, Nakajo S, Matsumoto K. Delayed neuronal cell death in the rat hippocampus is mediated by the mitogen-activated protein kinase signal transduction pathway. Neurosci Lett. 1999;262:57–60. doi: 10.1016/s0304-3940(99)00034-8. [DOI] [PubMed] [Google Scholar]
- Ravati A, Ahlemeyer B, Becker A, Klumpp S, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-kappaB. J Neurochem. 2001;78:909–919. doi: 10.1046/j.1471-4159.2001.00463.x. [DOI] [PubMed] [Google Scholar]
- Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu BF, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol. 1999;39:536–546. [PubMed] [Google Scholar]
- Schabitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–2217. doi: 10.1161/01.str.31.9.2212. [DOI] [PubMed] [Google Scholar]
- Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience. 1999;93:81–88. doi: 10.1016/s0306-4522(99)00137-2. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Strijbos PJ. Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann N Y Acad Sci. 2001;939:11–22. doi: 10.1111/j.1749-6632.2001.tb03606.x. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Floreani M, Negro A, Facci L, Giusti P. Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem. 1998;70:1859–1868. doi: 10.1046/j.1471-4159.1998.70051859.x. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley PF. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron. 2001;30:227–240. doi: 10.1016/s0896-6273(01)00275-6. [DOI] [PubMed] [Google Scholar]
- Strauss KI, Barbe MF, Marshall R, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KI, Jacobowitz DM. Quantitative measurement of calretinin and -actin mRNA in rat brain micropunches without prior isolation of RNA. Brain Res Mol Brain Res. 1993;20:229–239. doi: 10.1016/0169-328x(93)90045-q. [DOI] [PubMed] [Google Scholar]
- Subramanian T, Chinnadurai G. Pro-apoptotic activity of transiently expressed BCL-2 occurs independent of BAX and BAK. J Cell Biochem. 2003;89:1102–1114. doi: 10.1002/jcb.10573. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Zirrgiebel U, Von Bohlen Und Halbach O, Strelau J, Laliberte C, Kaplan DR, Unsicker K. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol. 2004;165:357–369. doi: 10.1083/jcb.200403028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM. Synaptic plasticity: building memories to last. Curr Biol. 2000;10:218–221. doi: 10.1016/s0960-9822(00)00370-5. [DOI] [PubMed] [Google Scholar]
- Villarreal DM, Do V, Haddad E, Derrick DE. NMDA receptor antagonists sustain LTP and spatial memory: active processes mediate LTP decay. Nat Neurosci. 2002;5:48–52. doi: 10.1038/nn776. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhu D, Jiang X, Okagaki P, Mearow K, Zhu G, McCall S, Banaudha K, Lipsky RH, Marini AM. AMPA protects cultured neurons against glutamate excitotoxicity through a phosphatidylinositol 3-kinase-dependent activation in extracellular signal-regulated kinase to upregulate BDNF gene expression. J Neurochem. 2004;90:807–818. doi: 10.1111/j.1471-4159.2004.02526.x. [DOI] [PubMed] [Google Scholar]
- Zhu D, Lipsky RH, Marini AM. Co-activation of the phosphatidylinositol-3-kinase/Akt signaling pathway by N-methyl-d-aspartate and TrkB receptors in cerebellar granule cell neurons. Amino Acids. 2002;23:11–17. doi: 10.1007/s00726-001-0103-9. [DOI] [PubMed] [Google Scholar]