

Abstract
The tumor specific, ligand-independent, constitutively active Epidermal Growth Factor Receptor (EGFR) variant, EGFRvIII, remains understudied in breast cancer. Here, we report that expression of EGFRvIII in the ErbB-2-overexpressing, estrogen-dependent MDA-MB-361 breast cancer cell line resulted in significant estrogen-independent tumor growth in ovariectomized, athymic nude mice in comparison to MDA-MB-361/wt cells. MDA-MB-361/vIII breast cancer cells maintained estrogen-induced tumor growth, but were tamoxifen-resistant in the presence of estrogen, while MDA-MB-361/wt cells had a significant reduction in tumor growth in the presence of estrogen and tamoxifen. Tamoxifen alone did not have a significant effect on EGFRvIII-mediated estrogen-independent tumor growth. Constitutive signaling from the EGFRvIII receptor resulted in increased activation of both the Akt and MAPK pathways. Compared to estrogen-dependent, tamoxifen-sensitive MCF-7/vIII breast cancer cells, which had unchanged levels of ERα, but an increase in progesterone receptor (PgR) in comparison to MCF-7/wt cells, MDA-MB-361/vIII cells had a reduction in ERα expression as well as a more pronounced reduction in progesterone receptor (PgR) compared to MDA-MB-361/wt cells. EGFRvIII expression was also significantly associated with an absence of PgR protein in invasive human breast cancer specimens. Alterations of pro-apoptotic proteins and anti-apoptotic proteins were observed in EGFRvIII transfectants. In conclusion, constitutive signaling through EGFRvIII and its down-stream effector proteins crosstalks with the ERα pathway, resulting in loss of PgR expression and alterations in the apoptotic pathway which may result in the estrogen-independent, tamoxifen-resistant phenotype conferred to EGFRvIII-expressing breast cancer cells.
Keywords: Breast Cancer, EGFRvIII, ErbB-2, ERα/PgR, Tamoxifen, Resistance
Introduction
The contribution of receptor tyrosine kinases, particularly those in the Epidermal Growth Factor Receptor (EGFR) family, to breast cancer progression has long been an interest due to the drug-target potential of these molecules. Just recently reports have attempted to evaluate the clinical significance of EGFRvIII expression in high-risk primary breast cancers as well as in circulating breast cancer cells in the peripheral blood of breast cancer patients.(1;2) However, the role of EGFR variants in breast cancer remains under-studied due to methodological difficulties in detecting the variant as well as a lack of over-expression of these variants in most cancers. The most common mutation of EGFR is the type-III variant (EGFRvIII) frequently observed in glioblastomas, but also present in various human cancers.(3-8) However, it is not found in normal adult tissue making it a potentially important area of study in the prognosis and treatment of breast cancer.(4;7)
EGFRvIII is constitutively active, therefore, leading to the activation of several down-stream signaling cascades such as MAPK, PI3K/Akt, and JNK.(9-11) Mouse fibroblasts expressing EGFRvIII are transformed and also show increased motility.(12;13)
Using laser capture microdissection (LCM)/RT-PCR and immunohistochemical analysis, we were able to detect a high incidence of EGFRvIII transcript expression in human primary invasive breast cancer cells along with full-length EGFR and EGFRvIII mRNA and protein in the same tumor specimens.(14) Further, constitutively active EGFRvIII does not undergo proper receptor internalization and degradation, and EGFRvIII can transform non-tumorigenic, IL-3-dependent murine hematopoietic cells and induce an IL-3-independent and ligand-independent malignant phenotype.(15;16) Moreover, co-expression of EGFRvIII with ErbB-2 has been detected in breast cancer.(17)
Although breast cancer is a heterogeneous disease, hormonal therapy for breast cancer has a profound impact on long-term survival. The anti-estrogen, tamoxifen, remains one of the most effective therapeutic agents for the treatment of ERα-positive breast cancer.(18-21) Therefore, estrogen receptor is one of the most prevailing predictive markers both in determining prognosis and in predicting response to hormone therapies.(18-21) The role of the progesterone receptor (PgR) in breast cancer is relatively less established than that of ERα, but epidemiological studies and clinical data suggest that PgR signaling plays a critical role in breast cancer development and progression.(22) PgR-negative tumors often have more aggressive features.(22) There is preclinical evidence that over-expression of EGFR and amplification of ErbB-2 correlates with endocrine therapy resistance.(23-29) Although the results are inconsistent, a vast amount of data suggests that increased cross-talk between ERα and ErbB-receptor signaling pathways has been implicated as one of the mechanisms involving tamoxifen resistant phenotype by which activation of one pathway leads to ligand-independent activation of the subsequent pathway.(30;31) ERα associated with G-proteins near the cell membrane can transactivate EGFR and the activation of mitogen-activated protein kinase (MAPK) and Akt pathways, which are downstream of ErbB-receptors, leads to phosphorylation of ERα, leading to increased ERα transcriptional activities in the absence of estrogen.(32-35) It is also believed that decreased antagonist activity of tamoxifen in high ErbB-2 expressing breast cancer cells results in cross-talk and transactivation of both the ERα and the EGFR/ErbB-2 receptors which leads to activation of Akt, MAPKs, and AIB1, along with aberrant co-repressor complexes which can be reversed with the tyrosine kinase inhibitor, gefinitib.(35) The role of constitutively active EGFR variants like, EGFRvIII, in endocrine therapy responsiveness has never been addressed and may be of value as this potent molecule may also be a driving force in endocrine therapy resistance in a subset of breast cancer patients.
Here we report that stable expression of EGFRvIII in the ErbB-2 over-expressing, ERα-positive MDA-MB-361 breast carcinoma cell line confers an estrogen-independent, tamoxifen-resistant phenotype in vivo. EGFRvIII-expressing MDA MB 361 cells exhibit increased Akt and MAPK activation and altered expression of pro- and anti-apoptosis proteins, as well as a marked decrease in ERα expression and aberrant PgR expression. Surprisingly, EGFRvIII expression was found to be significantly correlated to a lack of PgR, but not ERα expression in clinical invasive breast cancer specimens confirming that EGFRvIII expression is correlated to disease progression and an aggressive phenotype.
Material and methods
Reagents and cell lines
MCF-7 and MDA-MB-361 breast carcinoma cell lines and their derivatives were maintained in IMEM (MediaTech) supplemented with 10% FBS (Quality Biologicals) at 37°C, 5% CO2, and 90% humidity. Stable EGFRvIII-expressing MCF-7 and MDA-MB-361 cells were generated as previously described.(16) Anti-EGFR antibodies, Ab-1 and Ab-12, were purchased from Neomarkers (CA, USA) as previously described.(15) Anti-EGFRvIII antibody, Ab-18, was purchased from Neomarkers (CA, USA), and anti-EGFRvIII antibody, 4-5H, has been previously described.(15) Anti-Phosphotyrosine antibody was purchased Upstate (MA, USA). Antibodies for PgR and Akt (Akt1) were purchased from Santa Cruz BioTechology, Inc. (CA, USA). The ERα antibody was purchased from Vector Laboratories (CA, USA). Antibodies against p-Akt (Thr308), p-p44/42 MAPK (Thr202/Tyr204), p44/42 MAPK, Bcl-2, Bcl-xL, and BAD were purchased from Cell Signaling Technology (MA, USA). Antibodies for GAPDH and β-actin were purchased from Sigma-Aldrich (MO, USA).
Fluorescence-activated Cell Sorting (FACS) Analysis
Cells (0.5-1 × 106) cells were harvested and then stained for 1 h with anti-EGFRvIII antibody at 4°C. Stained cells were then washed with cold PBS. A secondary FITC-anti-mouse antibody was added for 30 minutes, and the EGFRvIII levels were quantified by flow cytometry.
Immunoprecipitation and Immunoblotting
Subconfluent cells were lysed in a lysis buffer containing 50 mM HEPES, 1% Triton X-100, 150 mM NaCl, 10% Glycerol, 1 mM EDTA, 1.5 mM MgCl2, 1 mM Na3VO4, 1 mM PMSF, 20 μg/mL leupeptin, 10 μg/mL aprotinin, 100 mM NaF, and 10 μg/mL of trypsin inhibitor. 500-1000 μg of the whole cell lysate was used for immunoprecipitation. After overnight precipitation at 4°C, protein A-agarose beads (Amersham Biosciences, NJ, USA) were added and left for a 2-hour incubation at 4°C. The immunocomplexes were then separated by SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences, NJ, USA) for western blot analysis. Immunoreactive bands were visualized by an enhanced chemiluminescence reagent (ECL, Amersham Biosciences, NJ, USA), and exposed to film (Amersham Biosciences, NJ, USA).
Equivalent amounts of whole cell lysates were subjected to SDS-PAGE and immunoblotted as mentioned above.
In vivo studies
Ovariectomized, athymic nude mice were inoculated subcutaneously with 5 × 106 MDA-MB-361/wt or MDA-MB-361/vIII cells in two fat pads of each mouse (n = 8 or n = 4). The estrogen source (0.72 mg) was a slow release pellet (60 day release) implanted subcutaneously into the cervical scapular space. The tamoxifen source, also a 60 day release implant, was placed subcutaneously. Tumor size was measured twice weekly and calculated by volume (length × width × thickness). Once tumors reached 2 cm in diameter or the estrogen or tamoxifen source was depleted, the mice were sacrificed.
Immunohistochemistry
Archival paraffin-embedded sections of primary invasive tumors or tissue micro-array with known ERα and PgR status were obtained from the Lombardi Comprehensive Cancer Center (LCCC) tissuebank. The sections were deparaffinized in xylene for 5 minutes. The specimens then were rinsed briefly in 1× PBS and stained using one of two different EGFRvIII-specific antibodies (4-5H or Ab-18). Microwave antigen retrieval using citrate-based solution is essential for detection of EGFRvIII in paraffin-embedded tissue specimens. Incubation times for staining tissues with 4-5H (1:30) and Ab-18 (1:5) was 2 hours and overnight, respectively. After washing with PBS, specimens were incubated in horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) secondary antibody (Kirkegard & Perry Lab, MD, USA) at a dilution of 1:250 for 30 minutes. Finally, diaminobenzidine (DAB) was used for colorimetric detection (BioGenex, CA, USA) and sections were then counterstained with hematoxylin (VWR). See our previous report on the quantification of the immunohistochemical staining.(17)
Statistics
Statistical analysis for the in vivo studies was performed using Repeated ANOVA. The relationship between ERα, PgR, and EGFRvIII in invasive human breast cancer was assessed using the Jonkheere Terpestra test. All tests of association were considered significant if the p-value was less than 0.05.
Results
EGFRvIII expression in breast carcinoma cells stably transfected with EGFRvIII cDNA
Enhanced growth factor signaling results in increased genomic and non-genomic activities of ERα and is believed to be one of the major mechanisms of acquired resistance to tamoxifen in breast cancer.(36) Although ERα-positive, low-ErbB-2-expressing MCF-7 cells stably expressing EGFRvIII exhibit enhanced tumorgenicity in comparison to their parental cells, MCF-7/EGFRvIII transfectants maintain an estrogen-dependent phenotype in vivo, but have a growth advantage in estrogen-stripped media, and are less sensitive to growth stimulation by estradiol and growth inhibition by the anti-estrogens, tamoxifen and ICI 182780, in vitro (data not shown).(16) To assess the impact of EGFRvIII on the estrogen-dependence and tamoxifen-responsiveness of an ERα-positive, ErbB-2-overexpressing breast cancer cell line, MDA-MB-361 cells were used for this study, with the assumption that this model could be relevant to progression of breast tumors from having a hormone-sensitive to a hormone-insensitive phenotype. Since cancer cell lines show loss of EGFRvIII expression under cell culture conditions, EGFRvIII expression was ectopically expressed in the MDA-MB-361 breast carcinoma cell line.(37) FACS and immunoprecipitation analysis revealed a moderate level of EGFRvIII (“vIII”) expression in MDA-MB-361 cells stably-expressing EGFRvIII and no expression in MDA-MB-361/wt cells (Figure 1, a and b), although a low level of endogenous (“wt”) EGFR was expressed in both cell lines (Figure 1b). Multiple EGFRvIII protein bands were present due to the different glycosylation states of the receptor (Figure 1b). The EGFRvIII expressed in the MDA-MB-361/vIII cells was shown to be highly phosphorylated, confirming the constitutive activation of the EGFRvIII receptor in the absence of ligand (Figure 1c).
Fig. 1.
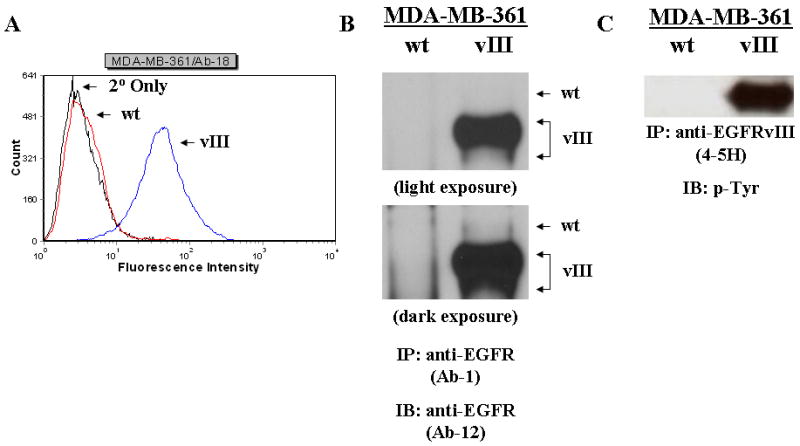
EGFRvIII expression in MDA-MB-361/vIII cells. a, the levels of EGFRvIII in the MDA-MB-361/vIII transfectants were quantitatively measured by flow cytometry using an anti-EGFRvIII antibody (Ab-18). The leftmost curve (black line) represents nonspecific staining (primary antibody omitted). The other curves represent the expression of EGFRvIII receptor in MDA-MB-361/wt (red line) and MDA-MB-361/vIII (blue line) cells. b and c, Immunoprecipitation and immunoblot analysis of EGFRvIII. Whole cell lysates (500-1000 μg) from MDA-MB-361/wt and MDA-361/vIII cells were immunoprecipitated with an anti-EGFR (Ab-1) or anti-EGFRvIII (4-5H) antibody, electrophoresed on SDS-PAGE, transferred onto nitrocellulose membranes, and immunoblotted with either an anti-EGFR (Ab-12) or anti-phosphotyrosine antibody. Protein bands were detected using a chemiluminescence detection system. b, MDA-MB-361/vIII cells expressed only full length EGFR (175-kDa), while MDA-MB-361/vIII cells expressed both full length EGFR and EGFRvIII (145-kDa). c, MDA-MB-361/vIII cells expressed a constitutively active EGFRvIII.
EGFRvIII induces an estrogen-independent, tamoxifen-resistant phenotype in breast carcinoma cells in vivo
We then conducted an in vivo study using ovariectomized, athymic nude mice to evaluate the EGFRvIII effects on breast cancer progression from a hormone-dependent to a hormone-independent phenotype. Figure 2 illustrates that in the absence of an estrogen source, the MDA-MB-361/vIII cells exhibited significant tumor growth, while the MDA-MB-361/wt cells were unable to establish xenografts or developed significantly smaller, dissipating tumors (216.9 ± 40.2 mm3 versus 1.3 ± 1.3 mm3, p<0.0001) indicating that the EGFRvIII was able to confer an estrogen-independent phenotype in estrogen-dependent breast cancer cells. Then, to determine the sensitivity of MDA-MB-361/vIII cells to estrogen-stimulation, xenografts were grown in the presence of an estrogen pellet. Both MDA-MB-361/wt and MDA-MB-361/EGFRvIII cells form tumors in the presence to estrogen, respectively (Figure 3, a and b). Although MDA-MB-361/wt cells produced estrogen-stimulated tumors, due to large variance in the size of the tumors, statistically significant results were not achieved (1709.0 ± 835.3 mm3 versus 5.1 ± 3.6 mm3, p=0.2012) (Figure 3a). As shown in Figure 3b, even though MDA-MB-361/vIII cells do not require estrogen for tumor formation, EGFRvIII-expressing cells maintained the ability to show estrogen-mediated tumor growth. MDA-MB-361/vIII cells were able to grow approximately 5-fold (1068.9 ± 113.5 mm3 versus 233.0 ± 48.1 mm3, p=0.0077) larger tumors at the end of the 60-day estrogen pellet release duration in comparison to tumors grown without an estrogen supplement. We then assessed whether an anti-estrogen could suppress tumor formation of MDA-MB-361/vIII cells. Tamoxifen suppression of estrogen-stimulated tumor growth was tested. As shown in Figure 3c, in the presence of estrogen and tamoxifen, MDA-MB-361/vIII cells formed significantly larger tumors, in contrast to MDA-MB-361/wt cells whereby tamoxifen sufficiently inhibited estrogen-induced tumors growth in the same treatment group (533.1 ± 39.0 mm3 versus 0 ± 0 mm3, p=0.0002), signifying tamoxifen sensitivity in these cells. These results suggest that MDA-MB-361/vIII cells were resistant to the antagonist activity of tamoxifen as tamoxifen was unable to suppress estrogen-mediated tumor growth. Furthermore, we determined whether tamoxifen had any agonist activity on EGFRvIII-expressing breast cancer cells. As shown in Figure 3d, MDA-MB-361/vIII cells were able to establish xenograft in the presence of tamoxifen and MDA-MB-361/vIII tumors were not stimulated by tamoxifen and there were no significant differences in the tumor growth with and without tamoxifen (139.9 ± 59.3 mm3 versus 283.1 ± 42.3 mm3, p=0.1034). In conclusion, EGFRvIII induces an estrogen-independent, tamoxifen-resistant phenotype in breast cancer cells although estrogen-responsiveness persists and tamoxifen does not act an as agonist for EGFRvIII-expressing breast cancer cells.
Fig. 2.
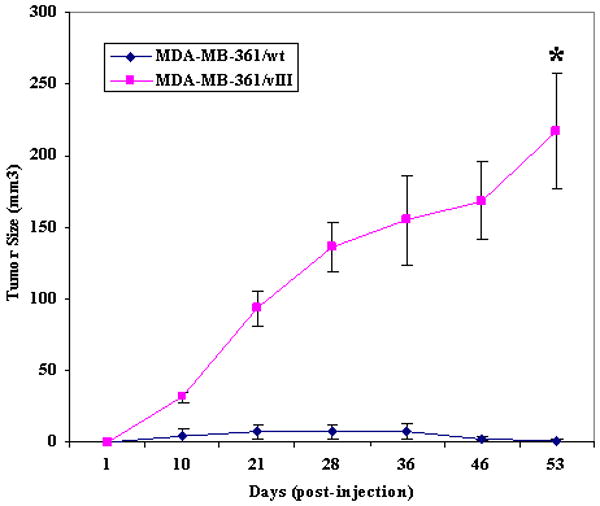
EGFRvIII induced estrogen-independent tumorgenicity in human breast cancer cells in ovariectomized, athymic nude mice. Five × 106 of MDA-MB-361/wt or MDA-MB-361/vIII cells were injected s.c. in ovariectomized athymic nude mice without an estrogen supplement. Eight mice for each cell line were used for this experiment, and each mouse received injections at both left and right mammary fat pads. All of the mice injected with MDA-MB-361/vIII cells produced tumors, while only 2 mice injected with MDA-MB-361/wt cells grew tumors, which eventually dissipated or decreased in size (216.9 ± 40.2 mm3 versus 1.3 ± 1.3 mm3, p<0.0001*). Bars, SE.
Fig. 3.
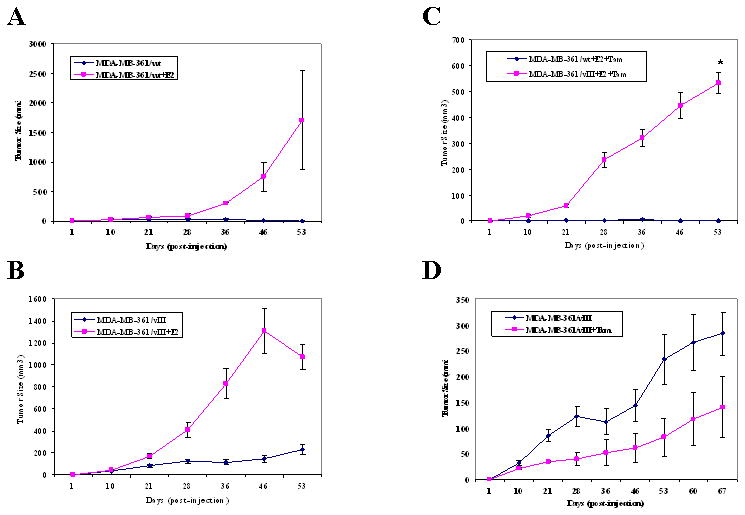
MDA-MB-361/wt cells grew tumors in nude mice with an estrogen pellet and sensitive to the growth inhibitory effects of tamoxifen, while EGFRvIII-expressing breast cancer cells were responsive to estrogen-stimulation, but were tamoxifen-resistant. Five × 106 MDA-MB-361/vIII cells were injected s.c. in ovariectomized, athymic nude mice with or without estrogen (E2) and/or tamoxifen (Tam). Four mice were used for each treatment group in this experiment, and each mouse received injections at both left and right mammary fat pads. a, MDA-MB-361/wt cells in the estrogen-treated group grew large tumors, whereas tumors that grew without an estrogen supplement were smaller and dissipating (1709.0 ± 835.3 mm3 versus 5.1 ± 3.6 mm3, p=0.2012). b, MDA-MB-361/vIII cells in the estrogen-treated group grew large tumors, whereas tumors that grew without an estrogen supplement were smaller (1068.9 ± 113.5 mm3 versus 233.0 ± 48.1 mm3, p=0.0077*). c, In the presence of estrogen and tamoxifen, MDA-MB-361/vIII cells continued to grow larger tumors than MDA-MB-361/wt cells, which had no tumor growth at the end of the treatment period (533.1 ± 39.0 mm3 versus 0 ± 0 mm3, p=0.0002*). d, EGFR/vIII cells did not have significantly reduced or enhanced tumor growth in the presence of tamoxifen alone in comparison to untreated cells (139.9 ± 59.3 mm3 versus 283.1 ± 42.3 mm3, p=0.1034). Bars, SE.
EGFRvIII increases the activation of Akt and MAPK in breast cancer cells
To explore the potential mechanisms of EGFRvIII-mediated tamoxifen resistance, we examined the levels of phosphorylated Akt and MAPK (p42/44) in MDA-MB-361/vIII cells, since these pathways have been shown to cross talk with the ERα pathway, and potentially leading to anti-estrogen resistance. Immunoblotting analysis was performed.(32-34;38;39) under normal growth conditions. Increased activation of both Akt and MAPK were observed in EGFRvIII-expressing breast cancer cells in comparison to their parental cells, although the increased activation is not as robust as often seen with growth factor stimulation of the signaling pathways (Figure 4, a and b). Since EGFRvIII mediates constitutively activated and sustained signaling pathways, whereas EGF-induces a robust transient effect on EGFR-mediated signaling pathways, these results implicate that constitutive activation of the EGFRvIII receptor in breast cancer cells leads to a persistent increased activation of downstream pathways, which is a probable explanation for the increased oncogenic signaling and biology found in cancer cells expressing EGFRvIII.(17)
Fig. 4.
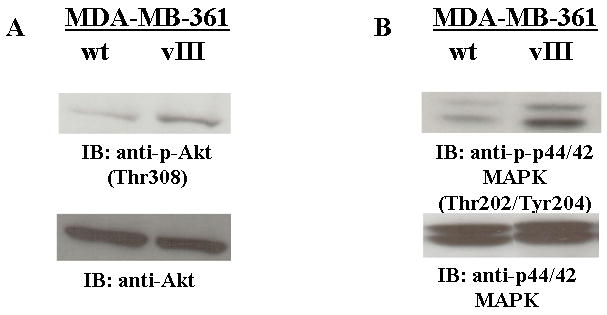
EGFRvIII enhanced the activation of Akt and MAPK in breast cancer cells. Immunoblot analysis of the Akt (a) and MAPK (b) activation. Whole cell lysates (25 μg) from MDA-MB-361/wt and MDA-361/vIII cells were electrophoresed on SDS-PAGE, transferred onto nitrocellulose membranes, and immunoblotted with phospho-specific antibodies for Akt or MAPK, stripped, and re-probed with total Akt or MAPK antibodies. Protein bands were detected using a chemiluminescence detection system. MDA-MB-361/vIII cells had increased activation of Akt (a) and MAPK (b) in comparison to MDA-MB-361/wt cells.
EGFRvIII down-regulates PgR expression, a potentially critical factor in promoting an estrogen-independent, tamoxifen-resistant phenotype
We further investigate the mechanism(s) involved in EGFRvIII-mediated estrogen-independent, tamoxifen-resistant phenotype. Cumulative studies have shown that increased growth factor signaling alters ERα signaling pathways, either by down-regulating ERα levels or it's transcriptional activity, therefore, we next determined the effects that EGFRvIII has on the expression of ERα, and it's down-stream target, PgR.(31-33;35;40-42) We compared the differences in the ERα pathway between MCF-7 and MDA-MB-361 cells, both with and without EGFRvIII expression. As shown in Figure 5, stable EGFRvIII expression in MCF-7 cells does not alter the ERα protein levels. However, it does appear that these cells may have hyper-activation of ERα transcriptional activity as these cells have a dramatic increase in PgR expression (Figure 5). In the MDA-MB-361 model system of EGFRvIII induced estrogen-independence, tamoxifen-resistance, ERα protein levels were reduced, although not substantially, and a more pronounced reduction in PgR protein levels (Figure 5). This observation leads us to believe that EGFRvIII cross-talks with the ERα pathway in breast cancer cells whether they express low or high levels of ErbB-2. However, it does appear that EGFRvIII enhances the cross-talk between ERα and ErbB-family receptors in cell lines expressing high levels of ErbB-2 to promote an estrogen-independent phenotype, while expressing EGFRvIII in low level of ErbB-2 cells may perhaps not be potent enough to confer an estrogen-independent, tamoxifen-resistance phenotype. Furthermore, reduction of PgR expression levels is likely to be one of the critical factors to promote estrogen-independent, tamoxifen-resistant phenotype through EGFRvIII/ErbB-2 network signaling.
Fig. 5.
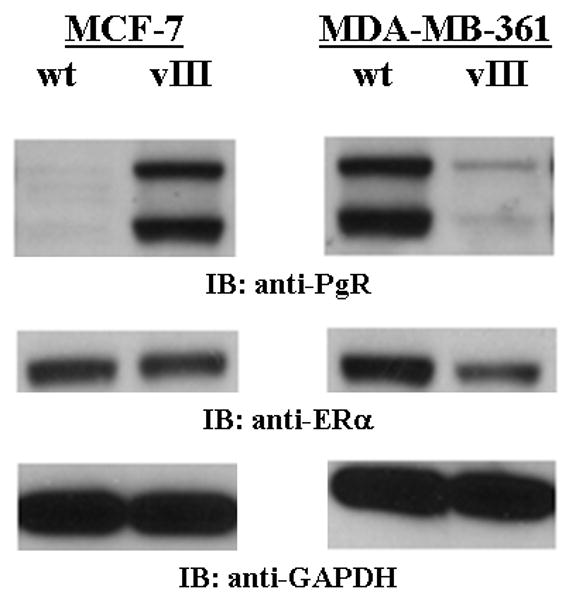
EGFRvIII decreased the expression of ER and PgR in estrogen-independent, tamoxifen-resistant breast cancer cells. Immunoblot analysis of the ERα and PgR expression of breast cancer cells expressing EGFRvIII in comparison to parental cells. Whole cell lysates (25 μg) from MCF-7/wt, MCF-7/vIII, MDA-MB-361/wt, and MDA-361/vIII cells were electrophoresed on SDS-PAGE, transferred onto nitrocellulose membranes, and immunoblotted with anti-ERα or anti-PgR antibodies. GAPDH was used as a loading control. Protein bands were detected using a chemiluminescence detection system. Estrogen-dependent, tamoxifen-sensitive MCF-7/vIII cells did not have altered ERα protein levels, but did not have increased PgR expression in comparison to MCF-7/wt cells. Conversely, estrogen-independent, tamoxifen-resistant MDA-MB-361/vIII cells had decreased levels of ERα and PgR protein levels in comparison to MDA-MB-361/wt cells.
To determine whether EGFRvIII-mediated reduction of ERα and PgR expression is also observed in clinical primary breast tumors, we investigated the correlation between EGFRvIII expression and ERα or PgR expression in primary invasive human breast cancer. Immunohistochemical analysis was performed on 137 paraffin-embedded breast cancer specimens for EGFRvIII expression to correlate with ER/PgR status of the tumors. Interestingly, a higher incidence of EGFRvIII expression was found in PgR-negative tumors in comparison to tumors which were found to express PgR (58% versus 39%, p=0.01) (Table I). However, the correlation between EGFRvIII and ERα expression was not able to achieve statistical significance, it is important to note that there seems to be higher incidence of EGFRvIII expression in invasive breast cancer specimens that were found to be ERα negative than those that expressed ERα (55% versus 43%, p=0.16). Nevertheless, in invasive human breast cancer, EGFRvIII correlates with the loss of ERα and PgR, both tumor characteristics which have been shown to be associated with a worse prognosis and decreased responsiveness to tamoxifen.(18-21;23-29;43) Most importantly, the clinical data is concomitant to our findings in our preclinical models.
Table I.
EGFRvIII, ERα, and PgR expression in invasive human breast cancer.
| Total N (%) | ER (+) N (%) | ER (-) N (%) | p-value | PgR (+) N (%) | PgR (-) N (%) | p-value | |
|---|---|---|---|---|---|---|---|
| 137 (100) | 77 (100) | 60 (100) | 72 (100) | 65 (100) | |||
| EGFRvIII (-) | 71 (52) | 44 (57) | 27 (45) | 44 (61) | 27 (42) | ||
| EGFRvIII (+) | 66 (48) | 33 (43) | 33 (55) | 0.16 | 28 (39) | 38 (58) | 0.01 * |
a significant association is detected at p<0.05 using the Jonkheere Terpstra test.
EGFRvIII in breast cancer cells alters expression levels of apoptotic proteins
The use of pro- and anti-apoptotic proteins as prognostic markers is well studied, particularly the relationship between the anti-apoptotic Bcl-2 expression and responsiveness to anti-tumor agents such as anti-estrogens.(44-49) However, as the results remain inconsistent between studies, we investigated the expression levels of Bcl-2, Bcl-xL, and BAD in EGFRvIII-expressing MCF-7 and MDA-MB-361 cells. Intriguingly, the expression of all three apoptotic proteins reflects alterations in PgR expression (Figure 5) in EGFRvIII expressing breast cancer cells regardless the expression levels of ErbB-2. In estrogen-dependent, tamoxifen-sensitive MCF-7/vIII cells, Bcl-2 was found to be up-regulated (Figure 6a) as was PgR levels, suggesting increased genomic activity of ER in these cells. Surprisingly, the anti-apoptotic Bcl-xL and anti-apoptotic BAD protein levels were also increased in MCF-7/vIII cells in comparison to MCF-7/wt cells (Figure 6, a and b). Interestingly, a similar correlation between the PgR and Bcl-2 modulation were also observed in estrogen-independent, tamoxifen-resistant MDA-MB-361/vIII cells. As shown in Figure 6, decreased Bcl-2 protein levels correlates to a loss of PgR expression and potentially decreased ERα signaling in these cells. Bcl-xL and BAD levels were also decreased in MDA-MB-361/vIII cells (Figure 6, a and b). These results suggest that EGFRvIII-mediated estrogen-independence and tamoxifen resistance may occur through the deregulation of the ERα signaling pathway as shown by altered expression of the down-stream target genes of ERα, PgR and Bcl-2, which in turn alters the expression of other proteins involved in apoptosis.
Fig. 6.
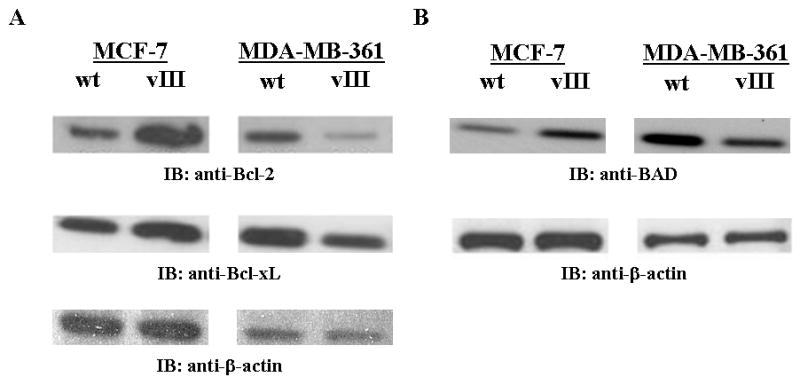
EGFRvIII alters the expression of apoptotic proteins in breast cancer cells. a and b, Immunoblot analysis of the Bcl-2, Bcl-xL, and BAD expression of breast cancer cells expressing EGFRvIII in comparison to parental cells. Whole cell lysates (25 μg) from MCF-7/wt, MCF-7/vIII, MDA-MB-361/wt, and MDA-361/vIII cells were electrophoresed on SDS-PAGE, transferred onto nitrocellulose membranes, and immunoblotted with anti-Bcl-2, anti-Bcl-xL, or anti-BAD antibodies. β-actin was used as a loading control. Protein bands were detected using a chemiluminescence detection system. Estrogen-dependent, tamoxifen-sensitive MCF-7/vIII cells have increased Bcl-2 (a), Bcl-xL (a), and BAD (b) protein expression in comparison to MCF-7/wt cells. Conversely, estrogen-independent, tamoxifen-resistant MDA-MB-361/vIII cells had decreased levels of Bcl-2 (a), Bcl-xL (a), and BAD (b) protein levels in comparison to MDA-MB-361/wt cells.
Discussion
Previous cell culture models on the impact of growth factor receptor signaling have shown both over-expression or gene amplification of ErbB-receptors in breast cancers can confer a tamoxifen-resistant phenotype, yet others have shown that acquired resistance to tamoxifen in breast cancer cells was not a result in changes in ErbB-receptors.(30;31;35;38;39;50-52) In order to address whether a naturally-occurring, constitutively active EGFR variant has any impact on the dependence of breast cancer cells on estrogen and their response to anti-estrogens, we used an isogenic MDA-MB-361 breast cancer cell line system. We clearly demonstrated that expressing EGFRvIII in MDA-MB-361 cells promote these estrogen-dependent, tamoxifen-sensitive cells to become estrogen-independent and tamoxifen-resistant in vivo (Figure 2 and 3). However, these cells still maintained their ability to respond to estrogen-induced tumor growth (Figure 3b). Various mechanisms can contribute to an estrogen-independent and tamoxifen-resistant phenotype, such as loss of PgR expression in breast cancer resulting from a loss or non-functioning ERα, low circulating levels of estrogen, hypermethylation of the PgR promoter, loss of the PgR gene locus, excessive growth factor signaling, increased ERα activity by SERMS or by growth factor simulation at the cell membrane, and altered ERα co-regulators.(53) Preclinical and clinical studies have recently suggested that PgR negativity in ER-positive breast cancer may be a marker of hyperactive growth factor signaling rather than a result of a nonfunctional ER signaling pathways, as previously suggested.(53;54) Our study provides direct evidence that suppressed/reduced PgR levels may derive from the hyperactivity in the signaling cascade generated by constitutively activated EGFRvIII. Our model system suggests that a constitutive activation of the EGFR receptor in high ErbB-2 expressing breast cancer cells leads to partial ERα loss and a significant reduction in PgR expression which may contribute to an estrogen-independent and tamoxifen-resistant phenotype (Figure 5). Conversely, a similar isogenic EGFRvIII breast cancer model using MCF-7 cells, which do not become estrogen-independent nor tamoxifen-resistant in vivo, showed an increase in PgR expression without any changes in ERα expression (Figure 5). In this context, PgR loss may be a marker of an active EGFRvIII/ErbB2 signaling network and a conferred estrogen-independent, tamoxifen- resistant phenotype. Furthermore, a similar observation was seen in our immunohistochemical analysis of invasive clinical breast cancer specimens. EGFRvIII expression was found to be significantly correlated to loss of PgR expression in invasive human breast cancer (Table I). Although our immunohistochemistry results only showed a significant association of EGFRvIII expression with loss of PgR expression (Table I), it is important to mention that the trend for ERα in the same set of breast cancer tissue samples is very similar to that of PgR expression. However, further investigation would be needed to confirm these results with a lager sample size.
Early studies have shown that increased MAPK activation may result in a decrease in ERα protein levels as was shown in MCF-7 cells which had hyperactivation of MAPK due to the over-expression of EGFR or ErbB-2 and inhibition of MAPK activity in ERα-negative breast cancer cells resulted in re-expression of ERα.(40;42) Growth factor stimulation has been shown to down-regulate PgR expression by inhibition of PgR gene transcription mediated by the PI3K/Akt/mTOR pathway and not by altered ERα levels or activity.(54) However, MDA-MB-361/vIII cells had enhanced activation of the pro-survival and proliferative, Akt and MAPK pathways (Figure 4). This synergistic effect by both a MAPK-induced down-regulation of ERα and a possible decrease in ERα activity combined with increased activation of the PI3K/Akt/mTOR may result in reduced PgR levels, an indicator for tumor aggressiveness and tamoxifen-resistance. Conversely, transactivation with EGFRvIII and other growth factor receptors, such as IGF receptors, has not been explored in breast cancer and may be critical in the potent oncogenic properties of the EGFRvIII receptor. In future studies, the use of Akt and MAPK inhibitors may help use dissect whether these two signaling pathways have any role in the changes in ERα and PgR protein levels as well as other genomic and non-genomic activities of ERα which lead to the estrogen-independent, tamoxifen-resistant phenotype of the MDA-MB-361/vIII cells.
Finally, cross-talk between EGFRvIII and ERα signaling pathways was further revealed through the altered expression of Bcl-2, which corresponded to PgR expression and potentially transcriptional activity of ERα in the isogenic MCF-7 and MDA-MB-361 cell lines (Figure 5 and 6). Bcl-2 confers a growth advantage to cells by inhibiting apoptosis and inducing chemotherapy resistance and has prognostic significance in certain types of lymphomas.(55-57) In this respect, Bcl-2 should correlate with more aggressive tumor and resistance to hormonal therapy and chemotherapy. However, the data has been conflicting in breast cancer. Clinical studies have revealed that high levels of Bcl-2 expression are associated with a number of favorable prognostic factors including ER positivity, PgR positivity, low histological grade and well-differentiated tumors.(45;46;49) Unexpectedly, numerous studies have shown that tumors with high Bcl-2 expression are more responsive to hormone therapy and have more favorable disease-free and overall survival.(58-60) Furthermore, low levels of BAD expression was associated with relapse in tamoxifen-treated breast cancer patients and Bcl-xL expression was often not associated with breast cancer patient outcomes.(44;49) We demonstrated that EGFRvIII-expressing breast cancer cells reveal a paradoxical relationship between proteins involved in apoptosis and tamoxifen-response. Tamoxifen-sensitive MCF-7/vIII cells express increased levels of Bcl-2, Bcl-xL, and BAD (Figure 6) as well as increased levels of PgR (Figure 5), whereas MDA-MB-361/vIII cells are tamoxifen-resistant, had decreased levels of all three apoptotic proteins and a reduction in PgR expression (Figure 5). The striking correlation between the apoptotic molecules and PgR expression observed in these EGFRvIII transfectants suggests a potentially important role for the apoptosis pathway in EGFRvIII expressing breast cancer cells and their response to estrogen and anti-estrogens. The predictive value of apoptotic proteins, ERα, PgR, and EGFRvIII in breast cancer requires further analysis and may potentially be used to identify breast cancer patients which may benefit from an aggressive chemotherapeutic regimen in place of endocrine therapy.
In conclusion, our study provides a direct evidence that expressing EGFRvIII in high ErbB-2-expressing breast cancer cells induces an estrogen-independent, tamoxifen-resistant phenotype. Unraveling the mechanisms of resistance to tamoxifen- mediated by EGFRvIII provides new insights and implicates that multiple mechanisms are involved in resistance. Acquired resistance to tamoxifen is likely through active EGFRvIII/ErbB-2 signaling networks, and PgR expression is negatively associated with tamoxifen resistance in breast cancer. Based on our MCF-7 and MDA-MB-361 model systems, reduced PgR or loss of PgR expression may be a predictive factor for the outcome of endocrine therapy in EGFRvIII/ErbB-2 expressing breast tumors. Furthermore, while evaluation of the apoptotic status of breast cancer, the regulators of apoptosis, such as Bcl-2, Bcl-xL, and BAD must be considered together with ERα and PgR. Since this EGFR variant is both naturally-occurring and tumor-specific, it may also prove to be a valuable drug-target and potentially a tumor-specific antigen that allows for targeting chemotherapeutic agents to breast cancer cells without causing systemic toxic to breast cancer patients. Although recently it was reported that EGFRvIII expression was not correlated to patient outcomes in breast cancer patients with locoregionally advanced breast cancer, a larger number of breast cancer specimens expressing EGFRvIII as well as a more stringent method of detecting EGFRvIII expression is required to make an accurate assessment of the clinical significance of EGFRvIII expression in breast cancer.(1) Our previous results have shown that EGFRvIII expression is not high in breast cancer specimens, however, this does not suggest that EGFRvIII is not involved in breast cancer tumorigenesis and progression since EGFRvIII expression can be readily transferred from expressing cells to non-expressing through “oncosomes”, which has been shown to enhance transformation of glioma cells.(17;61) Pre-clinical studies evaluating the use of other anti-estrogens alone and in combination with therapeutic agents targeting EGFRvIII and/or ErbB-2, such as antibodies or tyrosine kinase inhibitors, is also warranted.
Acknowledgments
This work was supported by the NIH Grant RO1 CA88871 (C.K. Tang). The authors thank the Flow Cytometry and Cell Sorting and the Biostatistics and Bioinformatics (Rebecca Slack, Ruihua Xu and Antai Wang) Core Facility Shared Resources of Lombardi Cancer Center, which are partially supported by National Institute of Health Grant 1P30-CA-51008 (Cancer Center Support Grant, to Lombardi Comprehensive Cancer Center).
Reference List
- 1.Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. 2007 Oct 1;25(28):4405–13. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- 2.Silva HA, Abraul E, Raimundo D, Dias MF, Marques C, Guerra C, de Oliveira CF, Regateiro FJ. Molecular detection of EGFRvIII-positive cells in the peripheral blood of breast cancer patients. Eur J Cancer. 2006 Oct;42(15):2617–22. doi: 10.1016/j.ejca.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Garcia dP I, Adams GP, Sundareshan P, Wong AJ, Testa JR, Bigner DD, Weiner LM. Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993 Jul 15;53(14):3217–20. [PubMed] [Google Scholar]
- 4.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995 Dec 1;55(23):5536–9. [PubMed] [Google Scholar]
- 5.Olapade-Olaopa EO, Moscatello DK, MacKay EH, Horsburgh T, Sandhu DP, Terry TR, Wong AJ, Habib FK. Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer. 2000 Jan;82(1):186–94. doi: 10.1054/bjoc.1999.0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990 Nov;87(21):8602–6. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, McLendon RE, Moscatello D, Pegram CN, Reist CJ. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995 Jul 15;55(14):3140–8. [PubMed] [Google Scholar]
- 8.Yamazaki H, Fukui Y, Ueyama Y, Tamaoki N, Kawamoto T, Taniguchi S, Shibuya M. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Mol Cell Biol. 1988 Apr;8(4):1816–20. doi: 10.1128/mcb.8.4.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonyak MA, Moscatello DK, Wong AJ. Constitutive activation of c-Jun N-terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem. 1998 Jan 30;273(5):2817–22. doi: 10.1074/jbc.273.5.2817. [DOI] [PubMed] [Google Scholar]
- 10.Montgomery RB, Moscatello DK, Wong AJ, Cooper JA, Stahl WL. Differential modulation of mitogen-activated protein (MAP) kinase/extracellular signal-related kinase kinase and MAP kinase activities by a mutant epidermal growth factor receptor. J Biol Chem. 1995 Dec 22;270(51):30562–6. doi: 10.1074/jbc.270.51.30562. [DOI] [PubMed] [Google Scholar]
- 11.Moscatello DK, Holgado-Madruga M, Emlet DR, Montgomery RB, Wong AJ. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem. 1998 Jan 2;273(1):200–6. doi: 10.1074/jbc.273.1.200. [DOI] [PubMed] [Google Scholar]
- 12.Batra SK, Castelino-Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS, Bigner DD. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995 Oct;6(10):1251–9. [PubMed] [Google Scholar]
- 13.Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004 Feb 20;108(5):643–53. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 14.Ge H, Gong X, Tang CK. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J Cancer. 2002 Mar 20;98(3):357–61. doi: 10.1002/ijc.10224. [DOI] [PubMed] [Google Scholar]
- 15.Han W, Zhang T, Yu H, Foulke JG, Tang CK. Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther. 2006 Oct;5(10):1361–8. doi: 10.4161/cbt.5.10.3226. [DOI] [PubMed] [Google Scholar]
- 16.Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000 Jun 1;60(11):3081–7. [PubMed] [Google Scholar]
- 17.Yu H, Gong X, Luo X, Han W, Hong G, Singh B, Tang CK. Co-expression of EGFRvIII with ErbB-2 enhances tumorigenesis: EGFRvIII mediated constitutively activated and sustained signaling pathways, whereas EGF-induced a transient effect on EGFR-mediated signaling pathways. Cancer Biol Ther. 2008 Nov 24;7(11) doi: 10.4161/cbt.7.11.6847. [DOI] [PubMed] [Google Scholar]
- 18.Balleine RL, Earl MJ, Greenberg ML, Clarke CL. Absence of progesterone receptor associated with secondary breast cancer in postmenopausal women. Br J Cancer. 1999 Mar;79(9-10):1564–71. doi: 10.1038/sj.bjc.6690249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuukasjarvi T, Kononen J, Helin H, Holli K, Isola J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J Clin Oncol. 1996 Sep;14(9):2584–9. doi: 10.1200/JCO.1996.14.9.2584. [DOI] [PubMed] [Google Scholar]
- 20.Osborne CK, Yochmowitz MG, Knight WA, III, McGuire WL. The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer. 1980 Dec 15;46(12 Suppl):2884–8. doi: 10.1002/1097-0142(19801215)46:12+<2884::aid-cncr2820461429>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 21.Ravdin PM, Green S, Dorr TM, McGuire WL, Fabian C, Pugh RP, Carter RD, Rivkin SE, Borst JR, Belt RJ. Prognostic significance of progesterone receptor levels in estrogen receptor-positive patients with metastatic breast cancer treated with tamoxifen: results of a prospective Southwest Oncology Group study. J Clin Oncol. 1992 Aug;10(8):1284–91. doi: 10.1200/JCO.1992.10.8.1284. [DOI] [PubMed] [Google Scholar]
- 22.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008 Apr;29(2):217–33. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arpino G, Weiss H, Lee AV, Schiff R, De PS, Osborne CK, Elledge RM. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005 Sep 7;97(17):1254–61. doi: 10.1093/jnci/dji249. [DOI] [PubMed] [Google Scholar]
- 24.Bamberger AM, Milde-Langosch K, Schulte HM, Loning T. Progesterone receptor isoforms, PR-B and PR-A, in breast cancer: correlations with clinicopathologic tumor parameters and expression of AP-1 factors. Horm Res. 2000;54(1):32–7. doi: 10.1159/000063434. [DOI] [PubMed] [Google Scholar]
- 25.De Laurentiis M, Arpino G, Massarelli E, Ruggiero A, Carlomagno C, Ciardiello F, Tortora G, D'Agostino D, Caputo F, Cancello G, Montagna E, Malorni L, et al. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin Cancer Res. 2005 Jul 1;11(13):4741–8. doi: 10.1158/1078-0432.CCR-04-2569. [DOI] [PubMed] [Google Scholar]
- 26.Dowsett M, Houghton J, Iden C, Salter J, Farndon J, A'Hern R, Sainsbury R, Baum M. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according oestrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann Oncol. 2006 May;17(5):818–26. doi: 10.1093/annonc/mdl016. [DOI] [PubMed] [Google Scholar]
- 27.Giltnane JM, Ryden L, Cregger M, Bendahl PO, Jirstrom K, Rimm DL. Quantitative measurement of epidermal growth factor receptor is a negative predictive factor for tamoxifen response in hormone receptor positive premenopausal breast cancer. J Clin Oncol. 2007 Jul 20;25(21):3007–14. doi: 10.1200/JCO.2006.08.9938. [DOI] [PubMed] [Google Scholar]
- 28.Kim HJ, Cui X, Hilsenbeck SG, Lee AV. Progesterone receptor loss correlates with human epidermal growth factor receptor 2 overexpression in estrogen receptor-positive breast cancer. Clin Cancer Res. 2006 Feb 1;12(3 Pt 2):1013s–8s. doi: 10.1158/1078-0432.CCR-05-2128. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson RI, McClelland RA, Gee JM, Manning DL, Cannon P, Robertson JF, Ellis IO, Blamey RW. Epidermal growth factor receptor expression in breast cancer: association with response to endocrine therapy. Breast Cancer Res Treat. 1994 Jan;29(1):117–25. doi: 10.1007/BF00666187. [DOI] [PubMed] [Google Scholar]
- 30.Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, Shepard HM, Osborne CK. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat. 1992;24(2):85–95. doi: 10.1007/BF01961241. [DOI] [PubMed] [Google Scholar]
- 31.Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995 Jun 15;10(12):2435–46. [PubMed] [Google Scholar]
- 32.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001 Mar 30;276(13):9817–24. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 33.Font de MJ, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol. 2000 Jul;20(14):5041–7. doi: 10.1128/mcb.20.14.5041-5047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003 Jan 24;278(4):2701–12. doi: 10.1074/jbc.M205692200. [DOI] [PubMed] [Google Scholar]
- 35.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004 Jun 16;96(12):926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 36.Normanno N, Di MM, De ME, De LA, de MA, Giordano A, Perrone F. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005 Dec;12(4):721–47. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 37.Lammering G, Valerie K, Lin PS, Hewit TH, Schmidt-Ullrich RK. Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother Oncol. 2004 Sep;72(3):267–73. doi: 10.1016/j.radonc.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, Gee JM, Nicholson RI. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006 Mar;96(2):131–46. doi: 10.1007/s10549-005-9070-2. [DOI] [PubMed] [Google Scholar]
- 39.Yang Z, Barnes CJ, Kumar R. Human epidermal growth factor receptor 2 status modulates subcellular localization of and interaction with estrogen receptor alpha in breast cancer cells. Clin Cancer Res. 2004 Jun 1;10(11):3621–8. doi: 10.1158/1078-0432.CCR-0740-3. [DOI] [PubMed] [Google Scholar]
- 40.Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clin Cancer Res. 2007 Dec 1;13(23):7029–36. doi: 10.1158/1078-0432.CCR-07-0587. [DOI] [PubMed] [Google Scholar]
- 41.Holloway JN, Murthy S, El-Ashry D. A cytoplasmic substrate of mitogen-activated protein kinase is responsible for estrogen receptor-alpha down-regulation in breast cancer cells: the role of nuclear factor-kappaB. Mol Endocrinol. 2004 Jun;18(6):1396–410. doi: 10.1210/me.2004-0048. [DOI] [PubMed] [Google Scholar]
- 42.Oh AS, Lorant LA, Holloway JN, Miller DL, Kern FG, El-Ashry D. Hyperactivation of MAPK induces loss of ERalpha expression in breast cancer cells. Mol Endocrinol. 2001 Aug;15(8):1344–59. doi: 10.1210/mend.15.8.0678. [DOI] [PubMed] [Google Scholar]
- 43.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004 Sep 1;10(17):5670–6. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 44.Cannings E, Kirkegaard T, Tovey SM, Dunne B, Cooke TG, Bartlett JM. Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res Treat. 2007 Apr;102(2):173–9. doi: 10.1007/s10549-006-9323-8. [DOI] [PubMed] [Google Scholar]
- 45.Hamilton A, Piccart M. The contribution of molecular markers to the prediction of response in the treatment of breast cancer: a review of the literature on HER-2, p53 and BCL-2. Ann Oncol. 2000 Jun;11(6):647–63. doi: 10.1023/a:1008390429428. [DOI] [PubMed] [Google Scholar]
- 46.Krajewski S, Krajewska M, Turner BC, Pratt C, Howard B, Zapata JM, Frenkel V, Robertson S, Ionov Y, Yamamoto H, Perucho M, Takayama S, et al. Prognostic significance of apoptosis regulators in breast cancer. Endocr Relat Cancer. 1999 Mar;6(1):29–40. doi: 10.1677/erc.0.0060029. [DOI] [PubMed] [Google Scholar]
- 47.Lee KH, Im SA, Oh DY, Lee SH, Chie EK, Han W, Kim DW, Kim TY, Park IA, Noh DY, Heo DS, Ha SW, et al. Prognostic significance of bcl-2 expression in stage III breast cancer patients who had received doxorubicin and cyclophosphamide followed by paclitaxel as adjuvant chemotherapy. BMC Cancer. 2007;7:63. doi: 10.1186/1471-2407-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Planas-Silva MD, Bruggeman RD, Grenko RT, Smith JS. Overexpression of c-Myc and Bcl-2 during progression and distant metastasis of hormone-treated breast cancer. Exp Mol Pathol. 2007 Feb;82(1):85–90. doi: 10.1016/j.yexmp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Sjostrom J, Blomqvist C, von BK, Bengtsson NO, Mjaaland I, Malmstrom P, Ostenstadt B, Wist E, Valvere V, Takayama S, Reed JC, Saksela E. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin Cancer Res. 2002 Mar;8(3):811–6. [PubMed] [Google Scholar]
- 50.Larsen SS, Egeblad M, Jaattela M, Lykkesfeldt AE. Acquired antiestrogen resistance in MCF-7 human breast cancer sublines is not accomplished by altered expression of receptors in the ErbB-family. Breast Cancer Res Treat. 1999 Nov;58(1):41–56. doi: 10.1023/a:1006232830161. [DOI] [PubMed] [Google Scholar]
- 51.Massarweh S, Osborne CK, Jiang S, Wakeling AE, Rimawi M, Mohsin SK, Hilsenbeck S, Schiff R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006 Aug 15;66(16):8266–73. doi: 10.1158/0008-5472.CAN-05-4045. [DOI] [PubMed] [Google Scholar]
- 52.Zhen LL, Zhu X, Zheng W, Wang XY, Wu ZY. Involvement of epidermal growth factor receptor signaling pathway in tamoxifen resistance of MCF-7 cells. Ai Zheng. 2006 Jul;25(7):839–43. [PubMed] [Google Scholar]
- 53.Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005 Oct 20;23(30):7721–35. doi: 10.1200/JCO.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Cui X, Zhang P, Deng W, Oesterreich S, Lu Y, Mills GB, Lee AV. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol Endocrinol. 2003 Apr;17(4):575–88. doi: 10.1210/me.2002-0318. [DOI] [PubMed] [Google Scholar]
- 55.Gascoyne RD, Adomat SA, Krajewski S, Krajewska M, Horsman DE, Tolcher AW, O'Reilly SE, Hoskins P, Coldman AJ, Reed JC, Connors JM. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin's lymphoma. Blood. 1997 Jul 1;90(1):244–51. [PubMed] [Google Scholar]
- 56.Miyashita T, Reed JC. bcl-2 gene transfer increases relative resistance of S49.1 and WEHI7.2 lymphoid cells to cell death and DNA fragmentation induced by glucocorticoids and multiple chemotherapeutic drugs. Cancer Res. 1992 Oct 1;52(19):5407–11. [PubMed] [Google Scholar]
- 57.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993 Jan 1;81(1):151–7. [PubMed] [Google Scholar]
- 58.Huang Y, Ray S, Reed JC, Ibrado AM, Tang C, Nawabi A, Bhalla K. Estrogen increases intracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis of human breast cancer MCF-7 cells. Breast Cancer Res Treat. 1997 Jan;42(1):73–81. doi: 10.1023/a:1005777219997. [DOI] [PubMed] [Google Scholar]
- 59.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996 Jul;2(7):1215–9. [PubMed] [Google Scholar]
- 60.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995 Sep 1;55(17):3902–7. [PubMed] [Google Scholar]
- 61.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008 May;10(5):619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]