

Summary
NeuroD, an insulin transactivator, is critical for development of the endocrine pancreas, and NeuroD mutations cause MODY6 in humans. To investigate the role of NeuroD in differentiated β cells, we generated mice in which neuroD is deleted in insulin-expressing cells. These mice exhibit severe glucose intolerance. Islets lacking NeuroD respond poorly to glucose and display a glucose metabolic profile similar to immature β cells, featuring increased expression of glycolytic genes and LDH-A, elevated basal insulin secretion and O2 consumption, and overexpression of NPY. Moreover, the mutant islets appear to have defective KATP channel-mediated insulin secretion. Unexpectedly, virtually all insulin in the mutant mice is derived from ins2, whereas ins1 expression is almost extinguished. Overall, these results indicate that NeuroD is required for β cell maturation and demonstrate the importance of NeuroD in the acquisition and maintenance of fully functional glucose responsive β cells.
Introduction
Diabetes is a metabolic disease that involves the death or dysfunction of the insulin-secreting β cells of the pancreas. Although diabetes can be managed with insulin and other drugs, physiological glucose homeostasis is difficult to achieve by these means, and hyperglycemia is largely responsible for the co-morbidities associated with diabetes. Consequently, much research on diabetes is aimed at understanding the molecular and cellular basis for pancreatic β cell development, survival, and regulated insulin secretion in order to discover ways to restore β cells or their functions in diabetic patients.
NeuroD is a basic helix-loop-helix (bHLH) transcription factor that is crucial for development of the pancreas (Chae et al., 2004; Chao et al., 2007; Huang et al., 2002; Malecki et al., 1999; Naya et al., 1997; Naya et al., 1995). NeuroD-null mice die of severe diabetes shortly after birth; their α and β cells are poorly differentiated, islets fail to form, and the majority of β cells are lost (Naya et al., 1997). Although the spatiotemporal expression pattern of NeuroD during pancreatic development has been characterized (Chae et al., 2004), its molecular, cellular, and physiological roles are still unknown. NeuroD has been shown to be critical for insulin gene expression in vitro (Naya et al., 1995; Qiu et al., 2002); however, neuroD-null pancreata contain 10–15% as much insulin as controls, an amount that has been shown to be sufficient to support viability in mice (Bonner-Weir et al., 1983). Therefore, the reduced amount of insulin in neuroD-null mice is unlikely to be the sole reason for their severe hyperglycemia and neonatal death.
In humans, mutations in neuroD can predispose individuals to develop maturity onset diabetes of the young (MODY6) (Malecki et al., 1999), suggesting a critical role for NeuroD in mature β cells. To separate out the β cell function of NeuroD, we generated mice in which neuroD is deleted in the cells that express insulin (NeuroDloxP/−; RIP:Cre mice, hereafter referred to as neuroD β-CKO mice). In parallel, we also generated mice in which neuroD is deleted in mature β cells in an inducible manner (tamoxifen-injected NeuroDloxP/−; Pdx1:CreER™ adult mice, hereafter referred to as neuroD PE-CKO mice). Unlike neuroD-null mice, neuroD β-CKO and neuroD PE-CKO mice have no impairment of pancreatic islet formation and survive to adulthood. However, they are mildly hyperglycemic and contain half the normal amount of insulin. Surprisingly, in each NeuroD mutant model tested, almost all insulin is derived from the expression of ins2, whereas little or no expression of ins1 is detected. Although differential regulation of the two rodent insulin genes has been described previously (Deltour et al., 1993; Giddings et al., 1991; Ling et al., 1998), no transcription factor has been linked to this phenomenon in vivo.
Although the amount of insulin found in neuroD β-CKO mice should be sufficient to maintain normoglycemia, these mice are severely glucose intolerant and display greatly reduced insulin secretion. Isolated islets from neuroD β-CKO mice respond poorly to high glucose and other fuel secretagogues, but are fully capable of insulin secretion following global membrane depolarization with exogenous KCl. Further physiological analysis of neuroD β-CKO islets suggests that they display many characteristics of neonatal islets, which respond poorly to glucose. For example, neuroD β-CKO islets have elevated levels of lactate dehydrogenase (LDH-A) and basal oxygen consumption, and overexpress Neuropeptide Y, all of which are features associated with fetal and neonatal beta cells (S. Bonner-Weir, personal comm.; (Asplund and Hellerstrom, 1972; Boschero et al., 1990; Freinkel et al., 1984; Rozzo et al., 2009)). Deletion of NeuroD in adult β cells (neuroD PE-CKO mice) similarly causes glucose intolerance and reversion to immature β cell characteristics. One of the key features of postnatal pancreatic maturation is an acquisition of glucose-stimulated insulin secretion (GSIS), which is critical for β cell function. The lack of this feature in neuroD β-CKO islets demonstrates for the first time that NeuroD is essential for the maintenance of β cell maturation and function, which could explain the role of NeuroD in MODY6 (Malecki et al., 1999).
Results
Impaired glucose tolerance and failure to secrete insulin in neuroD β-CKO mice
To study the role of NeuroD in mature β cells, we generated conditional knockout mice in which the RIP:Cre transgene (Herrera, 2000) was used to delete neuroD in approximately 90% of differentiated insulin-producing cells (neuroD β-CKO; Fig. S1A–C). Although NeuroD is known as a critical transcriptional activator of the insulin genes (Naya et al., 1995; Qiu et al., 2002), both male and female neuroD β-CKO mice survive and are indistinguishable from control littermates in their appearance and body weight (data not shown). In neonatal neuroD β-CKO mice (P1.5), the blood glucose concentration was higher and more variable than in the control mice. Periodic measurements of blood glucose during maturation (1–8 weeks) and adulthood (10–24 weeks) showed that the mutant mice fed ad libitum were mildly hyperglycemic (Fig. 1A) with greater variability in their blood glucose levels: 11% of readings were ≥ 250 mg/dL for mutant mice versus 0% for control mice (n=148–149 per genotype). These episodes of hyperglycemia occurred in mice that were normoglycemic at other times, indicating that the hyperglycemic episodes were not an inherent property of a few individual mice.
Figure 1. Physiological effects of β cell-specific ablation of neuroD.

(A) Blood glucose levels of neuroD β-CKO and control mice fed ad libitum: P1.5 (n=9–21), 1–8 weeks (n=114–115) and 10–24 weeks (n=34–35). (B) Blood glucose levels of neuroD β-CKO and control mice fasted for 16 h and fed mouse chow (n=9 per genotype). (C) Glucose tolerance test for neuroD β-CKO and control mice (n=10–11 per genotype). (D) Glucose tolerance test for neuroDloxP/−; PE-Cre and neuroDloxP/+; PE-Cre pre-tamoxifen injection and post-tamoxifen injection (n=8–9 each type). The neuroDloxP/−; PE-Cre mice with tamoxifen injection are considered as neuroD PE-CKO. (E) Plasma insulin levels after fasting (16 hours) or fed ad libitum (n=9–16 per genotype). (F) Plasma insulin levels after glucose injection (n=5–7 per genotype).. (G) Plasma glucagon levels fed ad libitum, fasted (5 hours) or fasted (16hours) (n=10–17 per genotype). (H) The expression of G6Pase in control and neuroD β-CKO after fasted for 16 hours (0) and 90 minutes after glucose injection (90′). Values were normalized to β2-microglobin mRNA and expressed as relative to control (n=5 per genotype). * P≤0.05, ** P≤0.01 and *** P≤0.001.
To determine whether neuroD β-CKO mice are glucose intolerant, young adult mice (1–3 months) were fasted during the day for 5 hours or overnight for 16 hours, followed by either feeding ad libitum, or intraperitoneal injection of glucose (2g/kg body weight). In both conditions, mutant mice had significantly higher fasting blood glucose levels. Following feeding or glucose injection, their blood glucose rose to levels twice as high as those in sibling control mice and took longer to return to homeostatic levels (Fig. 1B, C and Fig. S2A).
The glucose intolerance exhibited by the neuroD β-CKO mice is in sharp contrast to the phenotype of neuroD-null mice, which die shortly after birth with severe and sustained hyperglycemia (> 500 mg/dl). This difference indicates that NeuroD has a distinct function in committed β cells that is different from its earlier developmental function. To confirm that the phenotype of neuroD β-CKO mice is independent of a developmental defect, we generated tamoxifen-inducible mice by crossing neuroDloxP/loxP mice with Pdx:CreER™; neuroD +/− mice that express the inducible Cre recombinase (CreER™) under the control of the Pdx-1 promoter (Gu et al., 2002). Injection of tamoxifen in adult mice (neuroDloxP−; Pdx-1:CreER™) resulted in a 94% reduction in neuroD mRNA in fully developed β cells, and these mice (neuroD PE-CKO) were glucose intolerant by three weeks after treatment (Fig. 1D). These results indicate that glucose intolerance is a characteristic of mice that lack NeuroD in their β cells, regardless of their age at the time of deletion of neuroD.
Glucose intolerance can occur because of a lack of glucose-stimulated insulin secretion (GSIS), decreased action of insulin in the peripheral tissues, or both. To distinguish between these possibilities, we measured plasma insulin in neuroD β-CKO and control mice (Fig. 1E). In fasted animals, the plasma insulin level in control mice ranged between 0.29–0.63 ng/ml, while that of neuroD β-CKO was significantly lower at 0.18–0.32 ng/ml (p<0.001). Although mean plasma insulin was not significantly different in control and mutant mice fed ad libitum, it ranged from 0.47–2.64 ng/ml in controls, whereas it never exceeded 1.20 ng/ml in neuroD β-CKO mice. More dramatic differences were observed following intraperitoneal glucose injection, as the neuroD β-CKO mice were poor at mounting an insulin secretion response when challenged with glucose (Fig. 1F). Both the first and second phase responses were smaller and developed more slowly in the mutant mice. Thus, neuroD β-CKO mice are severely deficient in GSIS, resulting in glucose intolerance.
Mice were also tested for their ability to take up glucose in their peripheral tissues in response to exogenous insulin. Although the mutant mice had a higher fasting level of blood glucose, their ability to take up glucose was not significantly different from controls (Fig. S2B). This result indicates that neuroD β-CKO mice are not insulin-resistant, further supporting the hypothesis that they are glucose intolerant owing to defective insulin secretion.
Because glucose homeostasis partly depends on the balance of glucose storage and release, we investigated whether aberrant levels of glucagon or perturbation of hepatic gluconeogenesis contributed to the phenotype of neuroD β-CKO mice. Plasma glucagon levels were not significantly different in neuroD β-CKO mice versus control mice regardless of whether they were fed ad libitum, fasted for 5 hours, or fasted overnight for 16 hours (Fig. 1G). These results imply that aberrations in glucagon secretion do not account for the modest hyperglycemia of neuroD β-CKO mice. We also investigated whether the mice differed in their regulation of hepatic glucose-6-phosphatase (G6Pase), an indicator of gluconeogenesis. Mutant and control mice had a similar amount of hepatic G6Pase mRNA when fasted overnight, suggesting that gluconeogenesis was equally stimulated in both cases (Fig. 1H). However, at 90 minutes after glucose injection, G6Pase mRNA fails to decrease in the mutant mice. Since insulin is a powerful inhibitor of G6Pase expression (Onuma et al., 2009), the failure to downregulate G6Pase mRNA in neuroD β-CKO mice is likely due to their severe insulin secretion defect. Therefore, in neuroD β-CKO, sustained gluconeogenesis may exacerbate hyperglycemia during glucose challenge.
Islet morphology and β cell phenotype in the neuroD β-CKO pancreas
Because neuroD-null mice fail to form mature islets (Naya et al., 1997), we investigated whether neuroD β-CKO mice are glucose intolerant due to defective islet formation and/or maintenance. Pancreata from perinatal and 7 week-old mice were stained with antibodies to glucagon, insulin, somatostatin and pancreatic polypeptide, as markers for α, β, δ and PP cells, respectively. Although neuroD β-CKO mice formed islets similar in size to those of control mice, their α, δ and PP cells were intermingled in the β cell core instead of residing at the periphery (Fig. 2A–B, E–F; Fig. S3A–B; Fig. S5A–B; data not shown). Interestingly, the neuroD PE-CKO mice did not display disrupted islet architecture, suggesting this phenotype is associated with a developmental defect (Fig. 2C–D; Fig. S5C–D). The number of cells co-stained for insulin and somatostatin was increased in both the neuroD β-CKO and neuroD PE-CKO mice. The cells co-expressing insulin and somatostatin were elevated 60-fold in the islets of neuroD β-CKO mice: 0.77% of insulin-positive cells co-expressed somatostatin vs. 0.013% in controls (Fig. 2E–H). However, the total number of cells involved is small and thus may not contribute to the overall physiology of neuroD β-significantly different in neuroD β-CKO and control mice, indicating that other β cell characteristics are intact in the mutant β cells (Fig. S3C–D). To determine whether the failure to respond to glucose occurs at the level of glucose transport across the membrane, we examined the level of Glut-2 in neuroD β-CKO mice and found that the mutant β cells have only half as much Glut-2 protein as controls, as determined by quantification of immunostaining (Fig. 2I–J). However, neuroD PE-CKO mice, which are also glucose intolerant, have normal amounts of Glut2 in their islets at both 3 weeks and 2 months after tamoxifen treatment was initiated (Fig. 2K–L; Fig. S5E–F), indicating that reduction of Glut-2 is not responsible for glucose intolerance observed in both strains. Importantly, Pdx-1, Nkx6.1 and MafA, all of which are β cell transcription factors that are involved in the activation of insulin transcription and the regulation of insulin secretion (Ohlsson et al., 1993; Schisler et al., 2005; Wang et al., 2007), are unaffected by the absence of NeuroD (Fig. S3E–J, L). These results indicate that the defective GSIS observed in neuroD β-CKO mice is not a secondary consequence of altered regulation of these transcription factors.
Figure 2. Effect of β cell-specific ablation of neuroD on islet characteristics.

(A–D) Pancreatic sections from control, neuroD β-CKO and neuroD PE-CKO mice were immunostained for insulin (green) and for either glucagon (red, A–D) or somatostatin (red, E–H). Nuclei are stained with DAPI (blue). The white arrows in (F) and (H) indicate insulin and somatostatin co-stained cells. Original magnification was 200x. (I–L) The expression of Glut-2 (red) in the control, neuroD β-CKO and neuroD PE-CKO pancreas. Original magnification was 200x.
Because apoptosis is increased 10-fold in neuroD-null mice (Naya et al., 1997), we investigated whether apoptosis is also increased in neuroD β-CKO mice. We measured apoptosis by immunostaining for activated caspase 3; and also examined compensatory β cell proliferation by immunostaining for PCNA and Ki67. Overall, only a small fraction of the β cells were engaged in either proliferation (≤2%) or apoptosis (≤2%) in neuroD β-CKO and control islets, indicating that deletion of NeuroD in differentiated β cells does not cause increased apoptosis or proliferation, which is consistent with the observed normal islet area in these mice.
Expression of insulin 2, but not insulin 1, in neuroD β-CKO mice
Detection of a significant amount of insulin staining in the β cells of neuroD β-CKO islets was surprising because previous studies had shown that NeuroD is a critical factor for insulin gene transcription (Naya et al., 1995; Qiu et al., 2002). Morphometric quantification of >500 islets indicated that on average the intensity of insulin staining in neuroD β-CKO islets was half that of control islets. Moreover, although we observed no significant difference in either islet size or islet number between control and neuroD β-CKO mice, neuroD β-CKO pancreata contain 53% as much insulin as control pancreata (15.3 ± 4.2 ug/mg vs. 29.0 ± 7.8 ug/mg protein, n=7–13, p< 0.001). Taken together, these data indicate that neuroD β-CKO islets contain half the normal amount of insulin due to a corresponding reduction in insulin content per cell rather than a reduction in cell number.
Because rodents express two closely related insulin genes, ins1 and ins2 (Davies et al., 1994), we measured the level of expression of each insulin gene to determine the source of the remaining insulin in neuroD β-CKO mice. In pancreatic sections costained for insulin and C-peptide 1, >90% of the β cells in neuroD β-CKO pancreata lacked C-peptide 1 staining, suggesting that they fail to express ins1. In striking contrast, C-peptide 2 was present in all of the mutant β cells, implying that ins2 expression is relatively unaffected (Fig. 3A–D). Consistently, ins1 transcripts are also reduced by 95% in neuroD β-CKO islets, while ins2 transcripts are present at a level comparable to controls (Fig. 3I). These data indicate that neuroD is not required for ins2 expression in vivo, but is necessary for ins1 expression. Deletion of neuroD in adult β cells using the neuroD PE-CKO mice also resulted in the loss of ins1, but not ins2, expression (Fig. 3E–H, I). The differential regulation of ins1 and ins2 does not appear to be an artifact associated with the neuroD conditional allele because we also detect a dramatic decrease in immunostaining for C-peptide 1, but not C-peptide 2, in the few remaining β cells in e18.5 neuroD-null pancreata (Fig. S4A–D). Furthermore, in the neuroD-null embryos, there is a gradual decline in the ratio of ins1/ins2 mRNA between e14.5 and P0, (Fig. S4E). A similar phenomenon can be observed in the postnatal neuroD β-CKO mice, where the ratio of ins1/ins2 mRNA declines from 80% of control at e18.5 to <10% of control at 2 months (Fig. S4F).
Figure 3. Deletion of neuroD results in loss of insulin 1.
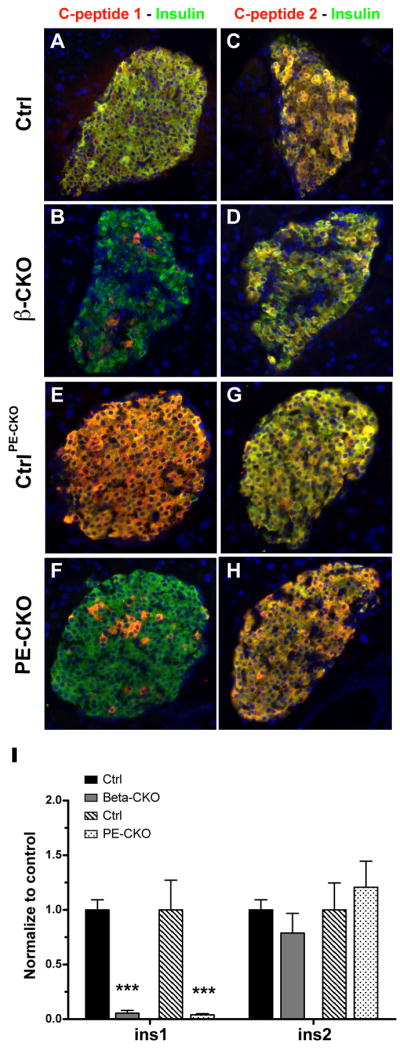
(A–D) Pancreatic sections from control, neuroD β-CKO and neuroD PE-CKO mice were co-stained with antibodies to insulin (green) and C-peptide 1 (red) (A–B, E–F) or C-peptide 2 (C–D, G–H). (K) Quantitative RT-PCR of ins1 and ins2 mRNA levels in control, neuroD β-CKO and neuroD PE-CKO islets. Values were normalized to β-actin mRNA and expressed as relative to the respective controls (n=4–6 per genotype). *** P≤0.001.
Previous studies have shown that mice can lack either one of the insulin genes and remain glucose tolerant (Leroux et al., 2001). We confirmed this result independently in ins1-null mice (data not shown). Moreover, studies of pancreatectomized animals have indicated that half the normal amount of insulin is more than sufficient to maintain glucose homeostasis (Bonner-Weir et al., 1983). Hence, neither the loss of insulin 1 per se, nor the reduction in total insulin, accounts for the defective insulin secretion and glucose intolerance of neuroD β-CKO mice.
Formation of Dense Core Granules in neuroD β-CKO mice
Insulin secretion is poor in some diabetic mouse models because of a paucity of insulin dense core granules (DCGs) in the β cells (Bruin et al., 2008; Like and Rossini, 1976; Pechhold et al., 2009; Piaggi et al., 2007). In addition, fewer DCGs are present in the underdeveloped β cells of neuroD-null mice (data not shown). We analyzed β cells of neuroD β-CKO mice using electron microscopy to determine whether they have normal DCG’s. The mutant β cells contain a large number of DCG’s, similar to those of control mice, implying that neither defective formation of DCG’s, nor depletion thereof, is the reason for defective insulin secretion in neuroD β-CKO mice (Fig. S4H–I).
Defects in stimulus-secretion coupling in isolated islets from neuroD β-CKO mice
To understand the physiological basis of defective GSIS observed in the NeuroD mutant mice, we stimulated islets isolated from control and neuroD β-CKO mice with a variety of insulin secretagogues (Fig. 4A). The amount of secreted insulin was normalized to total insulin content to take into account the reduction of insulin in the neuroD β-CKO islets. We found that the mutant islets secrete a larger percentage of their insulin (0.13 ± 0.01% vs. 0.05 ± 0.01% for controls) under basal conditions (2.8 mM glucose). However, during 1 hour of static incubation in 16.7mM glucose, the control islets secreted 1.0% of their insulin, whereas the neuroD β-CKO islets secreted only 0.26% of their insulin (Fig. 4A). The mutant islets also responded poorly to leucine (Fig. 4A). These secretion defects do not appear to be associated with a lack of readily releasable insulin granules because exposure of neuroD β-CKO islets to 30mM KCl induced robust insulin secretion that was not significantly different from control islets (Fig. 4A).
Figure 4. Insulin secretion profiles in neuroD β-CKO islets.

(A) Isolated islets from control and neuroD β-CKO mice treated with different insulin secretagogues (2.8mM glucose, 16.7mM glucose, 20mM leucine, 30mM KCl, 50uM glipizide and 10 mM methyl pyruvate). Secreted insulin was normalized to total insulin in the islets (n= 6–10 per genotype). (B) Quantitative RT-PCR of Kir6.2, Sur1, piccolo and Noc2 mRNA in control and neuroD β-CKO islets. The data were normalized to β-actin mRNA (n=3–8 per genotype). (C) Respiration rate of control and neuroD β-CKO mice islets incubated with glucose and oligomycin (OM) (n=6–17 per genotype). * P≤0.05, ** P≤0.01 and *** P≤0.001.
Extensive in vitro and in vivo studies of the gene products associated with MODY have suggested that they play a predominant role in glucose sensing-insulin secretion coupling (Giuffrida and Reis, 2005; Mitchell and Frayling, 2002). Because Sur1, a regulatory subunit of the pancreatic KATP channel, has been shown to be a transcriptional target of NeuroD in vitro (Keller et al., 2007; Kim et al., 2002), we assessed whether neuroD β-CKO mice display defective KATP channel function. Consistent with a defect in KATP channel function, neuroD β-CKO islets respond poorly to glipizide, a sulfonylurea drug, by secreting only 0.22% of their insulin content vs. 0.79% for control islets (Fig. 4A). Interestingly, there is no difference in the mRNA expression of the KATP channel gene (Kir6.2, kcnj11), or its regulatory subunit (Sur1, abcc8) between mutant and control islets (Fig. 4B), indicating that NeuroD does not regulate these genes at the level of transcription in vivo.
It remains possible that NeuroD affects other aspects of KATP channel activity. The neuroD β-CKO islets respond poorly to methyl pyruvate (Dufer et al., 2002)(Fig. 4A) and are deficient in molecules that link the activity of KATP channels to other parts of the machinery necessary for regulated insulin exocytosis. The expression of Piccolo (pclo) and Noc2 (rph3al) are both decreased in the β-CKO islets (Fig. 4B). Pclo encodes a scaffold protein that is necessary for assembly of insulin secretion complexes that link KATP channels, L-type calcium channels and insulin granules into functional units (Shibasaki et al., 2004), and Noc2 is a Rab effector that is required for GSIS through its interaction with small monomeric GTPases (Cheviet et al., 2004).
We also investigated whether there are defects in the steps that precede closure of the KATP channel in neuroD β-CKO islets. We measured O2 consumption in islets cultured in low and high glucose to measure the degree of oxidative metabolism that leads to ATP production. Compared to the controls, the mutant islets had a significantly greater rate of O2 consumption under the basal conditions (Fig. 4C). When challenged with high glucose, O2 consumption increased to a lesser extent in mutant than control islets, but achieved a similar rate overall. Virtually all of the O2 consumption in mutant and control islets was inhibited by treatment with the ATP synthase inhibitor Oligomycin A, implying that it is coupled to ATP production (Fig. 4C). Interestingly, these characteristics of respiration in neuroD β-CKO islets resemble those of GSIS-deficient neonatal islets (Boschero et al., 1990).
Defects in the amplification of GSIS in neuroD β-CKO islets
Although global membrane depolarization by KCl releases a normal amount of insulin from neuroD β-CKO islets, their reduced secretion of insulin in response to glucose, other fuel secretagogues, and glipizide implies that they are defective in the initial phase of GSIS upstream of Ca2+ influx. Therefore, to determine whether the mutant islets are capable of amplifying their insulin secretion following Ca2+ influx, we treated them with BayK8644, a drug that specifically opens L-type voltage sensitive calcium channels (VDCCs), together with 16.7mM glucose. The mutant islets secreted 2.6% of their total insulin, which is 10 times greater than their response to high glucose alone, but is still less than the 4.7% of insulin secreted by the control islets (Fig. 5A). Similar results were obtained using 20 mM L-arginine to enhance plasma membrane depolarization in the mutant and control islets. In addition, an agent (cAMP) that enhances the amplifying phase of GSIS (Doyle and Egan, 2003) also partially rescued insulin secretion in the mutant islets (Fig. 5A). Thus, neither type of treatment alone fully rescued insulin secretion in neuroD β-CKO islets, suggesting that the mutant islets have defects in both the initial and amplifying phases of GSIS.
Figure 5. Deletion of neuroD leads to increased NPY expression.
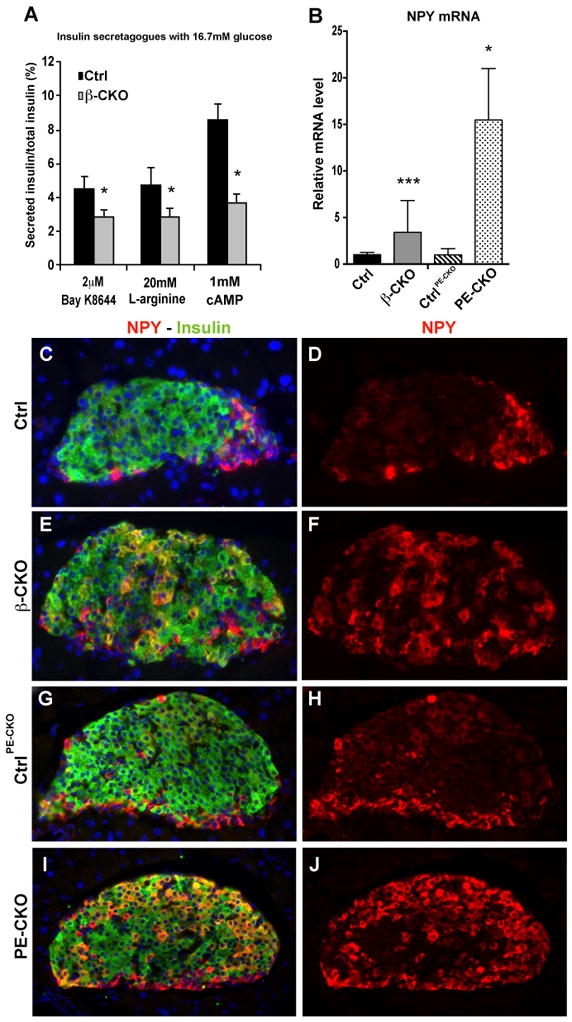
(A) Isolated islets from control and neuroD β-CKO mice treated with different insulin secretagogues (2uM BayK8644, 20mM L-arginine and 1mM dibutyryl cAMP) in the presence of 16.7mM glucose. Secreted insulin was normalized to total insulin in the islets (n= 6–9 per genotype). (B) Quantitative RT-PCR of NPY mRNA in control, neuroD β-CKO and neuroD PE-CKO islets. The data were normalized to β-actin mRNA (n=4–8 per genotype) and presented as relative to the respective controls. (C–J) Coimmunostaining of neuropeptide Y (red) and insulin (green) in the pancreatic sections of control for neuroD β-CKO (C–D), neuroD β-CKO (E–F), control for neuroD PE-CKO (G–H) and neuroD PE-CKO (I–J). Nuclei are stained with DAPI (blue). Original magnification was 200x. * P≤0.05, ** P≤0.01 and *** P≤0.001.
Adenyl cyclase activity, which is necessary for the conversion of ATP to cAMP, is inhibited by Neuropeptide Y (NPY), a hormone whose expression is normally decreased in islets after birth (Fig. S6A–J; (Motulsky and Michel, 1988)). We found that NPY mRNA is significantly increased in the neuroD β-CKO and neuroD PE-CKO islets, and that the immunostaining of NPY is clearly increased in >40% of the mutant β cells of each strain (Fig. 5B–J). The upregulation of NPY in the β cells of neuroD β-CKO and neuroD PE-CKO mice may contribute to their observed GSIS defects by decreasing the level of cAMP.
Increased glycolytic gene expression in the neuroD β-CKO islets
Impaired GSIS, as well as elevated rates of O2 consumption and insulin secretion in basal conditions, are properties shared by neuroD β-CKO islets and fetal beta cells (Freinkel et al., 1984; Hughes, 1994; Rozzo et al., 2009; Tu and Tuch, 1996). To investigate whether neuroD β-CKO islets display an expression profile similar to functionally immature β cells, we performed unbiased global gene expression analyses of adult islets isolated from the neuroD β-CKO pancreata and their littermate controls. Sixty-eight genes were significantly affected by the deletion of neuroD in β cells (Table S1–2). These results indicated that expression of lactate dehydrogenase A (LDHA) was significantly altered in the adult neuroD β-CKO islets. Normally, mature β cells are different from most mammalian cell types in that they have an unusually low amount of lactate dehydrogenase (LDH) (Schuit et al., 1997; Sekine et al., 1994). In contrast, fetal and neonatal β cells have elevated amounts of LDHA and an increased rate of glycolysis (Fig. S6L; S. Bonner-Weir, personal comm.; (Boschero et al., 1990)). Surprisingly, LDHA (ldha) mRNA and protein are increased dramatically in neuroD β-CKO islets in both low glucose and high glucose (Fig. 6A–E, J). Accordingly, the mutant islets exhibit a 3.5-fold increase in LDHA activity, a 2-fold increase in lactate production, and a >2-fold increase in LDHA immunostaining (Fig. 6K–L). Furthermore, neuroD β-CKO islets have elevated expression of several other glycolytic genes, including aldolase B, phosphofructokinase, liver form (PFKL), triose phosphate isomerase (TPI), enolase 1 (ENO1) and pyruvate kinase, liver and RBC form (PKLR) (Fig. 6A). These changes in gene expression suggest that glycolysis is enhanced in neuroD β-CKO islets. In contrast, there is no significant difference in the expression of key genes whose products participate in pyruvate metabolism and oxidative phosphorylation in mitochondria, such as pyruvate dehydrogenase A1 (Pdha-1) and its regulatory protein pyruvate dehydrogenase kinase 1 (PDK1), succinate dehydrogenase C (SDHC), and ATP synthase (ATP6) (Fig. 6A). Overall, the pattern of gene expression in neuroD β-CKO islets is consistent with the increase in glycolysis that is characteristic of neonatal β cells (Boschero et al., 1990). Comparable increases in LDHA mRNA and immunostaining, and glycolytic gene expression, also occur in neuroD PE-CKO islets, indicating that adult β cells require NeuroD to maintain their mature metabolic phenotype (Fig. 6A, F–I).
Figure 6. Increased expression of glycolytic genes.

(A) Quantitative RT-PCR of genes involved in glycolysis and mitochondrial function (n=4–8 per genotype). ¶: Error bar ±1.85; †: Error bar ±3.78. (B–I) Coimmunostaining of LDHA (red) and insulin (green) in the pancreatic sections of control for neuroD β-CKO (B–C), neuroD β-CKO (DE), control for neuroD PE-CKO (F–G) and neuroD PE-CKO (H–I). Nuclei are stained with DAPI (blue). Original magnification was 200x. (J) Increased LDHA protein in both low glucose (2.8mM) and high glucose (16.7mM) in the neuroD β-CKO islets. (K–L) Increased LDHA activity and increased production of lactate in neuroD β-CKO islets (n=8–11). * P≤0.05, ** P≤0.01 and *** P≤0.001.
Discussion
NeuroD is known to be important for β cell development and insulin transcription, however which aspects of β cell development and mature β cell function require NeuroD is not clearly understood. To determine the specific role of NeuroD in mature β cells, we generated neuroD β-CKO mice, in which NeuroD is deleted in the insulin-producing cell population at the onset of their formation. These mice survive and form islets that contain half the normal amount of insulin, and yet they are severely glucose intolerant. To determine whether continued function of NeuroD is required in mature adult β cells, we also deleted neuroD de novo in adult mice (neuroD PE-CKO) using inducible expression of Cre recombinase and found that these mice largely phenocopy the neuroD β-CKO mice. We performed extensive molecular, cellular and physiological analyses on both mouse models and found: 1) Despite the widely accepted belief that NeuroD is a critical transcription factor for insulin gene transcription, NeuroD is dispensable for ins2 gene expression in mice; 2) Although β cells lacking NeuroD produce sufficient insulin to support glucose homeostasis, the mutant mice are glucose intolerant, indicating that NeuroD regulates other aspects of β cell function that are unrelated to insulin transcription; 3) Continued activity of NeuroD is required for the proper function of β cells, providing the molecular and physiological basis for MODY6; and 4) β cells lacking NeuroD have a striking resemblance to immature β cells, indicating that NeuroD plays an important role in achieving and maintaining maturity of β cells.
NeuroD and activation of insulin transcription
NeuroD β-CKO mice retain half the normal amount of insulin because activation of the ins2 gene is unaffected in vivo both during development and in adult mice. Although differential expression of ins1 and ins2 has been detected under certain physiological conditions (Deltour et al., 1993; Giddings et al., 1991; Ling et al., 1998), this is the first time that a transcription factor has been associated with their differential regulation in vivo. This result was surprising because several prior in vitro studies have shown that NeuroD is capable of activating both ins1 and ins2 through conserved E-box elements (Clark and Docherty, 1993; German et al., 1991; Naya et al., 1995). Therefore, it was anticipated that expression of both genes would be affected in the absence of NeuroD. On the other hand, each of the E-box elements in the rat and human insulin genes has been shown to contribute variably to the regulation of ins expression (Crowe and Tsai, 1989; Karlsson et al., 1987). Our results demonstrate that such differential regulation of two rodent ins genes is mediated through NeuroD and that NeuroD is dispensable for ins2 gene expression in its native in vivo environment. To resolve the divergent mechanisms by which the insulin genes are regulated, it will be necessary to perform complementary in vitro and in vivo studies to understand fully the direct regulation of ins1 and ins2 by NeuroD and perhaps other E-box binding factors.
Mild hyperglycemia and severe glucose intolerance
Because neuroD β-CKO mice exhibit severe glucose intolerance, it seems paradoxical that they are only mildly hyperglycemic when fed ad libitum. It is unlikely that induced changes in hepatic glucose output can explain this phenotype since neuroD β-CKO mice fasted for 5 hours or 16 hours have similar amounts of plasma glucagon compared to control mice. It is also possible that insulin secretion is increased under ad libitum feeding conditions owing to mechanisms that do not depend on GSIS. The Sur1-knockout mice are also glucose intolerant and yet normoglycemic when fed ad libitum (Seghers et al., 2000), and it is believed that they secrete nearly normal amounts of insulin in response to feeding due to cholinergic stimulation of insulin secretion (Shiota et al., 2002). Similar compensatory mechanisms may regulate glucose homeostasis in neuroD β-CKO mice fed ad libitum.
NeuroD regulates β cell maturation
Isolated islets from neuroD β-CKO mice have a metabolic profile that resembles that of immature pancreatic β cells, which are found in late fetal or neonatal islets. Both the mutant β cells and immature β cells have insulin secretory granules, but lack GSIS. Compared to mature β cells, immature β cells have a higher rate of O2 consumption, produce more lactate, and secrete more insulin under basal conditions (Boschero et al., 1990; Rozzo et al., 2009), and these properties also characterize neuroD β-CKO islets. When challenged with high glucose, immature β cells fail to increase their oxidative metabolism as dramatically as do mature β cells, and consequently fail to secrete as much insulin (Hughes, 1994; Rozzo et al., 2009; Tu and Tuch, 1996). Glycolysis predominates in fetal and neonatal islets resulting in impaired GSIS, which relies on oxidative metabolism of glucose. Likewise, we found that expression of a number of glycolytic genes, including lactate dehydrogenase A (LDHA), was increased in neuroD β-CKO and neuroD PE-CKO islets. LDHA is of particular importance because it converts pyruvate to lactate in the cytosol, thereby preventing its oxidative metabolism in mitochondria. LDHA is expressed at high levels in the embryonic and neonatal pancreas, but becomes downregulated in the adult islets (Fig. S6L). Accordingly, the reduction of LDHA in mature β cells is believed to be critical for their ability to couple glucose metabolism to insulin secretion (Schuit et al., 1997; Sekine et al., 1994; Zehetner et al., 2008; Zhao and Rutter, 1998). Thus, it is likely that NeuroD plays a major role in β cell maturation by downregulating LDHA and other glycolytic genes because these changes are necessary for efficient oxidative metabolism of glucose and GSIS in mature β cells. Interestingly, mice with a β cell-specific knockout of the von Hippel-Lindau gene (vhlh) also displayed increased insulin secretion in low glucose and decreased GSIS owing to overexpression of LDHA and other glycolytic genes (Zehetner et al., 2008). The phenotype of the vhlh-deficient islets results from activation of Hif1α (Zehetner et al., 2008); however, it is unlikely that this pathway is operating in neuroD β-CKO islets because other genes that are strongly upregulated by Hif1α, including vegfa and pdk1 are unaffected in neuroD β-CKO islets (data not shown). Currently, we do not have evidence that LDHA or the glycolytic genes that we examined are direct targets for NeuroD (Keller et al., 2007). There is a canonical NeuroD E-box (CATCTG) at position -430 in the LDHA promoter of mice and rats, but this site is not conserved in the human gene.
NeuroD β-CKO mice have defects in the mechanism for GSIS beyond their immature oxidative metabolism of glucose. In particular, neuroD β-CKO islets secrete insulin poorly in response to glipizide-mediated closure of their KATP channels. Physiological induction of insulin secretion by closure of KATP channels involves the elevation of cytosolic [Ca2+] in a microenvironment around docked and primed insulin granules (Bokvist et al., 1995). This process is facilitated by the formation of an insulin secretion complex that links the ATP sensor (Kir6.1 and Sur1), cAMP sensor (cAMPGEFII), VDCCs and insulin granules into a functional unit (Shibasaki et al., 2004). Because NeuroD-deficient β cells have the basic components necessary for the first phase of GSIS, i.e., KATP channels, VDCCs, readily releasable insulin granules and ATP-coupled O2 consumption, we hypothesize that they are deficient in GSIS because they lack the structure necessary for those components to function together. In support of this hypothesis, we found that neuroD β-CKO islets have decreased expression of piccolo, a large scaffold protein and possible Ca2+ sensor that helps form this complex, and Noc2, a Rab effector that is associated with the insulin secretory granules and is required for efficient GSIS (Cheviet et al., 2004). NeuroD may also be important for the expression of key exocytotic proteins, such as SNAP25 and syntaxin1A (Ishizuka et al., 2007). Given the apparent immaturity of β cells that lack NeuroD, our hypothesis suggests further that formation of the insulin secretion complex may be a key step in β cell maturation.
Early precursor cells in the endocrine pancreas coexpress insulin and neuropeptide Y (NPY), but as development proceeds, NPY is decreased in β cells (Fig. S6A–J; (Teitelman et al., 1993)). NPY inhibits adenyl cyclase and cAMP production (Motulsky and Michel, 1988), which is required for efficient GSIS. The inhibitory role of NPY in GSIS has been demonstrated in several rodent models and in isolated islets (Imai et al., 2007; Myrsen-Axcrona et al., 1997; Myrsen et al., 1995; Wang et al., 1994). Consistent with the immature state and impaired GSIS of β cells that lack NeuroD, NPY mRNA and protein are greatly increased in both neuroD β-CKO and neuroD PE-CKO islets. Thus, it appears that NeuroD plays a global role in both activating β cell maturation-specific genes and down-regulating immature β cell-specific genes. A recent study has demonstrated that neonatal islets display a molecular profile that is distinct from the adult islets and identified a number of markers displaying transient expression in the perinatal period (Aye et al.). In support of our hypothesis, the gene encoding one of the markers of neonatal β cells, CK19, is highly upregulated in the neuroD β-CKO islets (Fig. S6M). NeuroD mRNA levels also gradually increase during this early postnatal maturation period (Fig. S6K); however, since NeuroD activity also depends on posttranscriptional and posttranslational regulation we need to further determine which of these gene expression changes are due to direct or indirect regulation by NeuroD.
Importance of NeuroD for creation of β cells suitable for therapy
The creation of functional β cells suitable for transplantation into patients with diabetes is a major goal of research on therapies for diabetes. Although many studies have attempted to produce β cells from many different cell sources, they have only been partially successful in inducing β cell differentiation (D’Amour et al., 2006; Jiang et al., 2007). Meanwhile, generation of mature β cells with tight control of insulin secretion in low glucose and a robust GSIS response in high glucose in vitro has not been successful. Our study of neuroD β-CKO and neuroD PE-CKO mice has shown that NeuroD is required for the transition of β cells from an immature to mature state during development and that NeuroD is essential for the maintenance of the mature β cell state in the adult. Thus, understanding the role of NeuroD in promoting β cell maturation could help point the way toward achieving β cell maturation in vitro.
Methods
Immunostaining and morphometric analysis of islets
Pancreata were fixed with 4% paraformaldehyde, cryosectioned, postfixed in methanol: acetone (1:1) for 5 min at −20°C and stained with the primary and secondary antibodies listed in Table S3. Antigen retrieval was used prior to staining for nuclear factors. Morphometric analysis was carried out using Slidebook 4.1 software (Intelligent Imaging Innovations) to quantify the area, intensity and overlap of staining for each antigen. The fraction of NPY-positive β cells was confirmed by counting immunostained cells in 3 islets of each genotype.
LDH activity and lactate assay
LDH activity was measured with an LDH assay kit (Cayman) and lactate production was analyzed with a lactate assay kit (Biovision). Additional details are provided in Supplemental Data.
Measurement of O2 consumption
O2 consumption was measured using a 96 well BD Oxygen Biosensor plate (BD). Equilibrated islets were added the BD Oxygen Biosensor plate at ~50 islets/well, and O2 consumption was measured at 1 min intervals in a fluorometric plate reader (Bio-Tek) at 37°C. DNA content of the islets was determined with Picogreen dye (Invitrogen).
Insulin secretion assay
Equilibrated islets were placed in wells of a 24-well plate at 10–15 islets/well with 400ul Kreb’s buffer containing 2.8mM glucose, incubated for 1 hour, and the supernatant was collected to measure basal insulin secretion. The islets were transferred to Kreb’s buffer containing different insulin secretagogues (see results) and incubated for 1 hour before the supernatant was collected. The islets were sonicated in 500 ul lysis buffer (10 mM Tris pH7.5, 200mM NaCl and 1mM EDTA), and insulin was extracted by acid ethanol. Insulin in supernatants and islet lysates was measured with a rat insulin RIA kit (Millipore). Methyl pyruvate, dibutyryl cAMP and Bay K8644 were purchased from Sigma.
Microarray
Total RNA was extracted from adult islets and islet purity was assessed and matched for 4 pairs of control and mutant RNA samples. 25 ng of each RNA sample was amplified using the Ovation™ RNA Amplification system V2 (Nugen, Inc.). The mouse PancChip 6.0 was used for microarray analysis (Kaestner et al., 2003). Genes that exhibited a fold change > 1.5, with a FDR ≤5% are shown in Tables S1 and S2. The full dataset is available at ArrayExpress under experiment accession number E-MTAB-152.
Statistics
Results are expressed as means ± S.E.M., and significance was determined by Student’s t-test for two-tailed unpaired groups. P≤0.05 was considered significant.
Supplementary Material
Acknowledgments
We are grateful to Dr. D. Melton for Pdx-1 ER™ Cre mice, Dr. J. Jami and Dr. A. Pugliese for Ins1−/− mice, Dr. C. Wright for Pdx-1 antibody, and the Beta Cell Biology Consortium for many other antibodies. We would especially like to thank Dr. S. Bonner-Weir for sharing data prior to publication and for providing critical insight into the neonatal β cell maturation process. We thank Julie Richheimer, Diana Ronai, Virginia Fonte, Michelle J. Doyle, Dina Balderes, Angela Minic, Sarah Stein, Teresa Mastracci, Nan Gao, Hans Hohmeier, Christine Andème Ondzighi and John Woo for tissue and/or technical support, Jonathon Schug for assistance with depositing microarray data, and Dr. Robert Poyton for helpful discussions. Support for the study came from NIH grant # UO1 DK072504 (LS and JL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asplund K, Hellerstrom C. Glucose metabolism of pancreatic islets isolated from neonatal rats. Horm Metab Res. 1972;4:159–163. doi: 10.1055/s-0028-1094091. [DOI] [PubMed] [Google Scholar]
- Aye T, Toschi E, Sharma A, Sgroi D, Bonner-Weir S. Identification of Markers for Newly Formed {beta} Cells in the Perinatal Period: A Time of Recognized {beta} Cell Immaturity. J Histochem Cytochem. doi: 10.1369/jhc.2009.954909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist K, Eliasson L, Ammala C, Renstrom E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. Embo J. 1995;14:50–57. doi: 10.1002/j.1460-2075.1995.tb06974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest. 1983;71:1544–1553. doi: 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschero AC, Bordin S, Sener A, Malaisse WJ. D-glucose and L-leucine metabolism in neonatal and adult cultured rat pancreatic islets. Mol Cell Endocrinol. 1990;73:63–71. doi: 10.1016/0303-7207(90)90045-a. [DOI] [PubMed] [Google Scholar]
- Bruin JE, Petre MA, Raha S, Morrison KM, Gerstein HC, Holloway AC. Fetal and neonatal nicotine exposure in Wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS ONE. 2008;3:e3371. doi: 10.1371/journal.pone.0003371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JH, Stein GH, Lee JE. NeuroD: the predicted and the surprising. Mol Cells. 2004;18:271–288. [PubMed] [Google Scholar]
- Chao CS, Loomis ZL, Lee JE, Sussel L. Genetic identification of a novel NeuroD1 function in the early differentiation of islet alpha, PP and epsilon cells. Dev Biol. 2007;312:523–532. doi: 10.1016/j.ydbio.2007.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheviet S, Coppola T, Haynes LP, Burgoyne RD, Regazzi R. The Rab-binding protein Noc2 is associated with insulin-containing secretory granules and is essential for pancreatic beta-cell exocytosis. Mol Endocrinol. 2004;18:117–126. doi: 10.1210/me.2003-0300. [DOI] [PubMed] [Google Scholar]
- Clark AR, Docherty K. How is the developmental timing and tissue-specificity of insulin gene expression controlled? J Endocrinol. 1993;136:187–190. doi: 10.1677/joe.0.1360187. [DOI] [PubMed] [Google Scholar]
- Crowe DT, Tsai MJ. Mutagenesis of the rat insulin II 5′-flanking region defines sequences important for expression in HIT cells. Mol Cell Biol. 1989;9:1784–1789. doi: 10.1128/mcb.9.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Davies PO, Poirier C, Deltour L, Montagutelli X. Genetic reassignment of the insulin-1 (Ins1) gene to distal mouse chromosome 19. Genomics. 1994;21:665–667. doi: 10.1006/geno.1994.1334. [DOI] [PubMed] [Google Scholar]
- Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, Bucchini D. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci U S A. 1993;90:527–531. doi: 10.1073/pnas.90.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev. 2003;55:105–131. doi: 10.1124/pr.55.1.7. [DOI] [PubMed] [Google Scholar]
- Dufer M, Krippeit-Drews P, Buntinas L, Siemen D, Drews G. Methyl pyruvate stimulates pancreatic beta-cells by a direct effect on KATP channels, and not as a mitochondrial substrate. Biochem J. 2002;368:817–825. doi: 10.1042/BJ20020657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freinkel N, Lewis NJ, Johnson R, Swenne I, Bone A, Hellerstrom C. Differential effects of age versus glycemic stimulation on the maturation of insulin stimulus-secretion coupling during culture of fetal rat islets. Diabetes. 1984;33:1028–1038. doi: 10.2337/diab.33.11.1028. [DOI] [PubMed] [Google Scholar]
- German MS, Blanar MA, Nelson C, Moss LG, Rutter WJ. Two related helix-loop-helix proteins participate in separate cell-specific complexes that bind the insulin enhancer. Mol Endocrinol. 1991;5:292–299. doi: 10.1210/mend-5-2-292. [DOI] [PubMed] [Google Scholar]
- Giddings SJ, Carnaghi LR, Fischer LJ, Miller CP. Differential regulation of rat insulin I and II messenger RNA synthesis: effects of fasting and cyproheptadine. Mol Endocrinol. 1991;5:549–554. doi: 10.1210/mend-5-4-549. [DOI] [PubMed] [Google Scholar]
- Giuffrida FM, Reis AF. Genetic and clinical characteristics of maturity-onset diabetes of the young. Diabetes Obes Metab. 2005;7:318–326. doi: 10.1111/j.1463-1326.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Huang HP, Chu K, Nemoz-Gaillard E, Elberg D, Tsai MJ. Neogenesis of beta-cells in adult BETA2/NeuroD-deficient mice. Mol Endocrinol. 2002;16:541–551. doi: 10.1210/mend.16.3.0784. [DOI] [PubMed] [Google Scholar]
- Hughes SJ. The role of reduced glucose transporter content and glucose metabolism in the immature secretory responses of fetal rat pancreatic islets. Diabetologia. 1994;37:134–140. doi: 10.1007/s001250050083. [DOI] [PubMed] [Google Scholar]
- Imai Y, Patel HR, Hawkins EJ, Doliba NM, Matschinsky FM, Ahima RS. Insulin secretion is increased in pancreatic islets of neuropeptide Y-deficient mice. Endocrinology. 2007;148:5716–5723. doi: 10.1210/en.2007-0404. [DOI] [PubMed] [Google Scholar]
- Ishizuka N, Minami K, Okumachi A, Okuno M, Seino S. Induction by NeuroD of the components required for regulated exocytosis. Biochem Biophys Res Commun. 2007;354:271–277. doi: 10.1016/j.bbrc.2006.12.197. [DOI] [PubMed] [Google Scholar]
- Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G, Majumdar AS. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25:1940–1953. doi: 10.1634/stemcells.2006-0761. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Lee CS, Scearce LM, Brestelli JE, Arsenlis A, Le PP, Lantz KA, Crabtree J, Pizarro A, Mazzarelli J, et al. Transcriptional program of the endocrine pancreas in mice and humans. Diabetes. 2003;52:1604–1610. doi: 10.2337/diabetes.52.7.1604. [DOI] [PubMed] [Google Scholar]
- Karlsson O, Edlund T, Moss JB, Rutter WJ, Walker MD. A mutational analysis of the insulin gene transcription control region: expression in beta cells is dependent on two related sequences within the enhancer. Proc Natl Acad Sci U S A. 1987;84:8819–8823. doi: 10.1073/pnas.84.24.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DM, McWeeney S, Arsenlis A, Drouin J, Wright CV, Wang H, Wollheim CB, White P, Kaestner KH, Goodman RH. Characterization of pancreatic transcription factor Pdx-1 binding sites using promoter microarray and serial analysis of chromatin occupancy. J Biol Chem. 2007;282:32084–32092. doi: 10.1074/jbc.M700899200. [DOI] [PubMed] [Google Scholar]
- Kim JW, Seghers V, Cho JH, Kang Y, Kim S, Ryu Y, Baek K, Aguilar-Bryan L, Lee YD, Bryan J, Suh-Kim H. Transactivation of the mouse sulfonylurea receptor I gene by BETA2/NeuroD. Mol Endocrinol. 2002;16:1097–1107. doi: 10.1210/mend.16.5.0934. [DOI] [PubMed] [Google Scholar]
- Leroux L, Desbois P, Lamotte L, Duvillie B, Cordonnier N, Jackerott M, Jami J, Bucchini D, Joshi RL. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes. 2001;50(Suppl 1):S150–153. doi: 10.2337/diabetes.50.2007.s150. [DOI] [PubMed] [Google Scholar]
- Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- Ling Z, Heimberg H, Foriers A, Schuit F, Pipeleers D. Differential expression of rat insulin I and II messenger ribonucleic acid after prolonged exposure of islet beta-cells to elevated glucose levels. Endocrinology. 1998;139:491–495. doi: 10.1210/endo.139.2.5749. [DOI] [PubMed] [Google Scholar]
- Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, Saad M, Warram JH, Montminy M, Krolewski AS. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–328. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- Mitchell SM, Frayling TM. The role of transcription factors in maturity-onset diabetes of the young. Mol Genet Metab. 2002;77:35–43. doi: 10.1016/s1096-7192(02)00150-6. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Michel MC. Neuropeptide Y mobilizes Ca2+ and inhibits adenylate cyclase in human erythroleukemia cells. Am J Physiol. 1988;255:E880–885. doi: 10.1152/ajpendo.1988.255.6.E880. [DOI] [PubMed] [Google Scholar]
- Myrsen-Axcrona U, Karlsson S, Sundler F, Ahren B. Dexamethasone induces neuropeptide Y (NPY) expression and impairs insulin release in the insulin-producing cell line RINm5F. Release of NPY and insulin through different pathways. J Biol Chem. 1997;272:10790–10796. doi: 10.1074/jbc.272.16.10790. [DOI] [PubMed] [Google Scholar]
- Myrsen U, Ahren B, Sundler F. Neuropeptide Y is expressed in subpopulations of insulin- and non-insulin-producing islet cells in the rat after dexamethasone treatment: a combined immunocytochemical and in situ hybridisation study. Regul Pept. 1995;60:19–31. doi: 10.1016/0167-0115(95)00114-5. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. Embo J. 1993;12:4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuma H, Oeser JK, Nelson BA, Wang Y, Flemming BP, Scheving LA, Russell WE, O’Brien RM. Insulin and epidermal growth factor suppress basal glucose-6-phosphatase catalytic subunit gene transcription through overlapping but distinct mechanisms. Biochem J. 2009;417:611–620. doi: 10.1042/BJ20080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechhold K, Zhu X, Harrison VS, Lee J, Chakrabarty S, Koczwara K, Gavrilova O, Harlan DM. Dynamic Changes in Pancreatic Endocrine Cell Abundance, Distribution, and Function in Antigen-Induced and Spontaneous Autoimmune Diabetes. Diabetes. 2009 doi: 10.2337/db08-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaggi S, Novelli M, Martino L, Masini M, Raggi C, Orciuolo E, Masiello P, Casini A, De Tata V. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the beta-cell line INS-1E. Toxicol Appl Pharmacol. 2007;220:333–340. doi: 10.1016/j.taap.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Guo M, Huang S, Stein R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol Cell Biol. 2002;22:412–420. doi: 10.1128/MCB.22.2.412-420.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, Newgard CB. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A. 2005;102:7297–7302. doi: 10.1073/pnas.0502168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, Prentki M. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem. 1997;272:18572–18579. doi: 10.1074/jbc.272.30.18572. [DOI] [PubMed] [Google Scholar]
- Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–9277. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J Biol Chem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- Shibasaki T, Sunaga Y, Fujimoto K, Kashima Y, Seino S. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J Biol Chem. 2004;279:7956–7961. doi: 10.1074/jbc.M309068200. [DOI] [PubMed] [Google Scholar]
- Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, et al. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277:37176–37183. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development. 1993;118:1031–1039. doi: 10.1242/dev.118.4.1031. [DOI] [PubMed] [Google Scholar]
- Tu J, Tuch BE. Glucose regulates the maximal velocities of glucokinase and glucose utilization in the immature fetal rat pancreatic islet. Diabetes. 1996;45:1068–1075. doi: 10.2337/diab.45.8.1068. [DOI] [PubMed] [Google Scholar]
- Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–358. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZL, Bennet WM, Wang RM, Ghatei MA, Bloom SR. Evidence of a paracrine role of neuropeptide-Y in the regulation of insulin release from pancreatic islets of normal and dexamethasone-treated rats. Endocrinology. 1994;135:200–206. doi: 10.1210/endo.135.1.8013354. [DOI] [PubMed] [Google Scholar]
- Zehetner J, Danzer C, Collins S, Eckhardt K, Gerber PA, Ballschmieter P, Galvanovskis J, Shimomura K, Ashcroft FM, Thorens B, et al. PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev. 2008;22:3135–3146. doi: 10.1101/gad.496908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Rutter GA. Overexpression of lactate dehydrogenase A attenuates glucose-induced insulin secretion in stable MIN-6 beta-cell lines. FEBS Lett. 1998;430:213–216. doi: 10.1016/s0014-5793(98)00600-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.