

Abstract
Over the past decade, molecular understanding of the congenital muscular dystrophies (CMDs) has greatly expanded. The diseases can be classified into 3 major groups based on the affected genes and the location of their expressed protein: abnormalities of extracellular matrix proteins (LAMA2, COL6A1, COL6A2, COL6A3), abnormalities of membrane receptors for the extracellular matrix (fukutin, POMGnT1, POMT1, POMT2, FKRP, LARGE, and ITGA7), and abnormal endoplasmic reticulum protein (SEPN1). The diseases begin in the perinatal period or shortly thereafter. A specific diagnosis can be challenging because the muscle pathology is usually not distinctive. Immunostaining of muscle using a battery of antibodies can help define a disorder that will need confirmation by gene testing. In muscle diseases with overlapping pathological features, such as CMD, careful attention to the clinical clues (e.g., family history, central nervous system features) can help guide the battery of immunostains necessary to target an unequivocal diagnosis.
Keywords: congenital muscular dystrophy, dystroglycans, glycosylation, muscle disease
INTRODUCTION
Mutations of 12 genes are known to cause congenital muscular dystrophies (CMDs). An orderly classification is essential to contend with the complexity of these disorders (Table 1). The most logical is to divide the CMDs into genes affecting extracellular matrix proteins (LAMA2 gene encoding laminin α2, COL6A1, COL6A2, and COL6A3) versus genes affecting membrane receptors for the extracellular matrix, including those that modify dystroglycan glycosylation, i.e., dystroglycanopathies [fukutin, fukutin-related protein (FKRP), protein O-linked mannose β1,2-N-acetylglucosaminyltransferase 1 (POMGnT1), protein-O-mannosyltransferase 1 (POMT1), protein-O-mannosyltransferase 2 (POMT2), and LARGE], and ITGA7, the gene encoding integrin α7. By convention, mutations of selenoprotein N (SEPN1), a constituent of the endoplasmic reticulum, are also included in the CMDs and remain part of this review despite overlap with the congenital myopathies (multi-minicore disease).
Table 1.
Congenital muscular dystrophies and associated gene defects
|
DEFECTS IN EXTRACELLULAR MATRIX PROTEINS
Laminin α2 or Merosin Deficiency (MDC1A)
Laminins are glycoproteins that, along with collagen IVs, form the scaffolding backbone of the basal lamina that surrounds individual myofibers [1] (Fig. 1). Each laminin is a heterotrimer composed of a heavy chain (α) and 2 light chains (β and γ). The major laminin of adult skeletal muscle is laminin-2 (or merosin), which is composed of the α2, β1, and γ1 chains. Only mutations of LAMA2 gene encoding laminin α2 (also referred to as merosin) cause muscular dystrophy. Laminins are secreted by myofibers and integrate into the basal lamina, where they bind to other extracellular matrix (e.g., collagen IV, agrin) and transmembrane proteins (e.g., dystroglycan, integrin α7β1), many of which are also related to CMD phenotypes.
Figure 1.

Illustration shows the major components of the dystrophin glycoprotein complex (DGC). Within the cytoplasm of the muscle fiber, the N-terminal of dystrophin links to the actin cytoskeleton. The cysteine-rich C-terminal domain of dystrophin links to the membrane via the dystroglycan complex. Dystroglycan consists of β-dystroglycan, a transmembrane protein, and α-dystroglycan, a highly glycosylated extracellular membrane protein. Several G domains of laminin-2 bind α-dystroglycan to link the complex from the extracellular matrix through the membrane to the actin cytoskeleton. Collagen VI is also a component of the muscle extracellular matrix. The sarcolemmal membrane is additionally anchored by integrin α7β1. Like dystroglycan, integrin α7β1 binds laminin-2 via its G domains, but it links the extracellular matrix to the cytoskeleton via integrin-associated proteins (examples shown are Pa = paxillin; T = talin; Vi = vinculin; FAK = focal adhesion protein).
Clinical features
MDC1A, the single most common form of CMD [2], is caused by laminin α2 or merosin deficiency. Typically patients are hypotonic at birth or shortly thereafter. A history of decreased fetal movements is not unusual. Facial, proximal, and distal limb muscles are affected. Contractures involve elbows, hips, knees, and ankles. Decreased suck and swallow may necessitate a feeding tube. Most patients achieve independent sitting, but fewer than 10% will learn to walk even a few steps [3]. Muscle strength tends to be static for long periods. Life-threatening problems relate to respiratory compromise. This may be improved with continuous positive airway pressure or bi-level positive airway pressure, but many patients require tracheostomy and assisted mechanical ventilation. Death may occur in the 1st decade or anytime thereafter after repeated episodes of pulmonary infection.
Clinically, most patients with complete laminin α2 deficiency are mentally normal, but learning disabilities and mental retardation have been reported [4]. Epilepsy has been estimated to occur in about 6% to 8% of cases, and the seizures are both partial and complex, with no consistent pattern [4–6]. Despite a minority with clinical central nervous system findings, a consistent finding common to all patients after 6 months of age is the presence of cerebral white matter abnormalities by magnetic resonance imaging (MRI) and computed tomography (Fig. 2). The changes are usually widespread, often most marked in the periventricular and frontal U fibers [7–9] and thought to be related to altered water distribution resulting from decreased laminin α2 in the extracellular matrix around cerebral blood vessels, which form the blood brain barrier [10]. Structural brain changes have been reported in occasional cases, and these include mild ventricular enlargement, focal cortical dysplasia, occipital polymicrogyria, and hypoplasia of pons and cerebellum [11,12].
Figure 2.
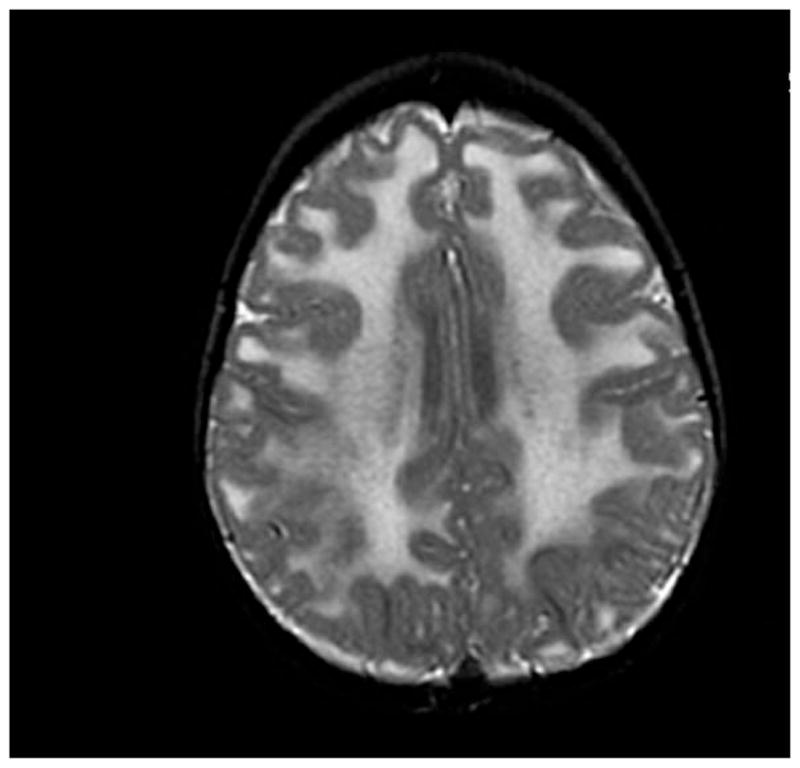
Axial T-2–weighted image of brain of a 2-year-old patient with laminin α2 deficiency shows high signal intensity in the white matter.
The peripheral nerve may be affected in laminin α2 deficiency [13–17]. Initial reports emphasized a motor neuropathy, but sensory fiber involvement is well documented [17]. The sural nerve shows reduction in the number of myelinated nerve fibers and short internodal segments in relation to fiber diameter, excessively wide nodes of Ranvier, and variability in myelin thickness with redundant folds and tomacula [17]. Laminin α2 is absent in the basement membrane surrounding Schwann cells and myelin sheath.
Clinically there has been no apparent cardiomyopathy in MDC1A, despite expression of laminin α2 in the heart. However, cardiac function by echocardiography demonstrates reduction in ejection fraction (43% ± 11%) compared with controls (53% ± 5%, P = 0.03) [18].
Late-onset disease with more favorable prognosis has been described in partial laminin α2 deficiency [19,20].
Genetics
Laminin α2 deficiency is inherited as an autosomal recessive disorder caused by mutations of the LAMA2 gene linked to chromosome 6q22-q23. Mutations of LAMA2 result in complete or partial laminin α2 deficiency; occur without identified hotspots; and include nonsense, missense, deletion, and splice-site mutations [8,9,21–23].
Molecular pathogenesis
The pathogenesis of MDC1A is not fully understood, but the structural organization of laminin-2 speaks to its critical interaction with proteins responsible for muscular dystrophies of varying types, including other CMDs (Fig. 1). Two important transmembrane proteins, α-dystroglycan and integrin α7β1, bind to laminin α2 through the G domains at its C-terminal. Disruption at these sites putatively contributes to loss of integrity of the sarcolemma. Loss of laminin α2 may also lead to an upregulation of laminin forms containing other laminin α chains in the muscle basal lamina (e.g., laminin α4) [24] that may ameliorate muscle pathology. Upregulation of laminin-binding proteins, for example, agrin, in skeletal muscles of the dy/dy mouse model for MDC1A significantly reduces the extent of muscle pathology [25].
Experimental models are available to further study laminin α2 deficiency in a variety of species, including dogs, cats, and mice [26–28]. Laminin α2–deficient mice share a dystrophic phenotype characteristic of the human disease and also demonstrate abnormalities in peripheral nerve myelination [29]. The majority of nerve fibers in the dorsal and ventral spinal roots at cervical, thoracic, lumbar, and sacral levels lack Schwann cells with resultant amyelination. Motor nerve conduction velocity is reduced by 25% to 30% [30], and there is widening of the nodes of Ranvier. The latter is also seen in the peripheral nerves of children with MDC1A, although amyelinated nerve roots are not encountered. Aberrant myelination in laminin α2 deficiency is related to the loss of basement membrane, a prerequisite for normal myelination [31].
Muscle Pathology
The muscle shows variability in fiber size with endomysial and perimysial connective tissue proliferation and fat infiltration in areas of muscle fiber loss (Fig. 3). Varying degrees of necrosis and regeneration are encountered. The changes can occur in an inflammatory milieu (especially in neonates), leading to a false diagnosis of infantile polymyositis [32]. Neurogenic changes may also be seen due to the concomitant abnormalities in nerves. Immune stains require a panel of antibodies to establish a specific diagnosis. With complete deficiency of laminin α2, both the C-terminal (80 kDa) and N-terminal (300 kDa) antibodies to this protein will fail to stain muscle fibers (Fig. 4). In contrast, the light chains of laminin-2 (β1 and γ1) will be preserved, and other laminin α chains (α4 and α5, in particular) are upregulated [24]. Western blots can be used to good advantage in problem cases or to reinforce findings by immune stains of tissue sections. Irrespective of the staining pattern, the tissue findings should be confirmed by mutation analysis.
Figure 3.
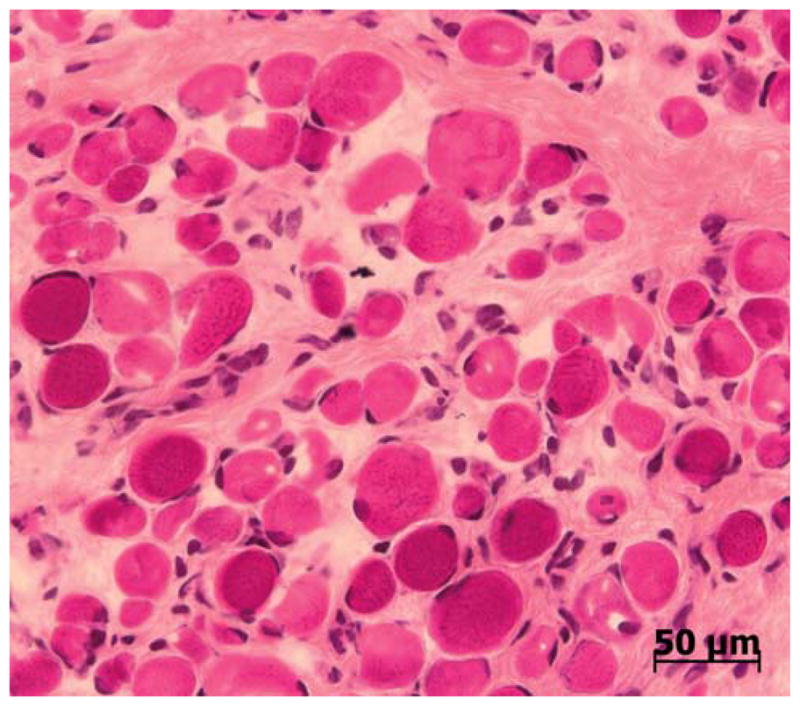
Muscle biopsy from 2-year-old patient with laminin α2 deficiency. The muscle shows marked variability in fiber size. Endomysial connective tissue proliferation surrounds virtually every muscle fiber in the field. Central nucleation is not prominent. H & E stain.
Figure 4.

Muscle fibers are not stained for laminin α2 (Vector Laboratories, Inc., Burlington, CA, USA) in a patient with laminin α2-deficient congenital muscular dystrophy (MDC1A) compared with normal control.
Skin biopsy provides a less invasive method of establishing a diagnosis in cases with complete laminin α2 deficiency [33,34] (Fig. 5). Laminin α2 will be absent from the basement membrane at the junction of the epidermis and dermis and from epithelial cells surrounding hair follicles. There is no known associated cutaneous pathology.
Figure 5.
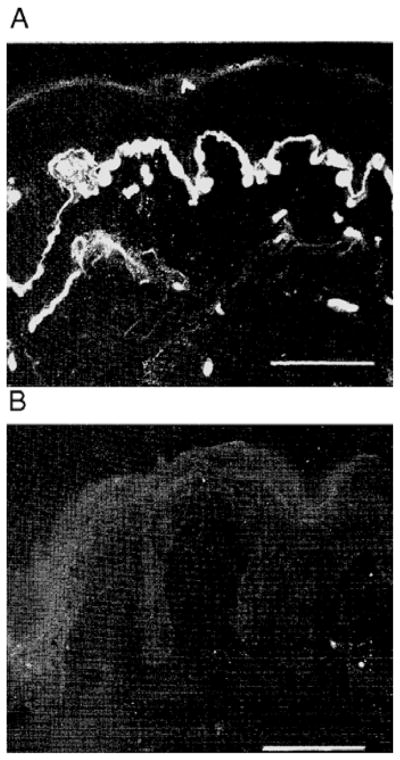
(A) Skin biopsy obtained from a normal control shows laminin α2 localized to the basement membrane at the junction of the epidermis and dermis. (B) Biopsy from a patient with laminin α2 deficiency lacks basement membrane staining for laminin α2. Previously published by Sewry et al. in The Lancet 1996;347:582–584. Reproduced with permission of Elsevier [33].
Ullrich Congenital Muscular Dystrophy (UCMD)
Ullrich’s insightful observations lead him to describe this disorder in 1930 [35]. He predicted an abnormality in connective tissue formation upon careful examination of a patient who died of pneumonia at age 14 months. Although his observations were astute, his suggested designation for the disease, “congenital atonic-sclerotic muscular dystrophy,” has fortunately been abandoned.
Clinical features
Typically patients with UCMD present in the neonatal period with hypotonia, muscle weakness, proximal joint contractures, and kyphosis of the spine. Congenital hip dislocation may be present. The distal joints present a contrasting feature with their hyperextensibility. The foot has a striking appearance because of a protruding calcaneous. Weakness can be severe, and children typically never achieve the ability to walk independently or do so for only short periods [36,37]. Mental capabilities are normal and brain MRI is normal. Disease progression results in contractures in fingers and heel cords that were previously hyperextensible. Spinal rigidity and scoliosis become more apparent [38]. Skin changes, particularly follicular hyperkeratosis (keratosis pilaris) over the extensor surfaces of the arms and legs, represent a consistent manifestation of UCMD. There is also a tendency toward keloid formation [39]. Respiratory failure related to poor expansion of chest wall and diaphragm muscle weakness leads to life-threatening infections in the 1st or 2nd decade [39].
Bethlem myopathy is a milder disorder, allelic to UCMD [40], inherited as an autosomal dominant condition. Although originally described as a benign disorder, additional experience reveals a slowly progressive condition leading to a need for ambulatory aids in more than two thirds of patients older than 50 years of age. It is not strictly a CMD, and space constraints preclude further discussion in this review [41].
Genetics
UCMD is generally considered to be an autosomal recessive disorder (see exceptions below) that involves the collagen VI genes (COL6A1, COL6A2, and COL6A3). As a ubiquitous extracellular matrix protein, collagen VI forms a microfibrillar network in close association with several other matrix constituents [42,43]. It is composed of 3 different peptide chains: α1(VI) and α2(VI), both 140 kDa in size, and a much larger α3(VI) at 260 to 300 kDa [44]. The α1(VI) and α2(VI) chains are encoded by COL6A1 and COL6A2, respectively, positioned head to tail on chromosome 21q22.3 [45]. COL6A3 locus is on chromosome 2q37. Mutations have been found in all 3 genes [46–48]. In some patients, the finding of only a single mutation suggests that some mutations may act in a dominant mode [48].
Molecular pathogenesis
As an extracellular matrix protein, collagen VI is uniquely positioned to have a profound effect on both the muscle fiber and the surrounding connective tissue (Fig. 1). Direct effects on the muscle have been reproduced in targeted gene disruption of COL6A1 in the mouse, exhibiting muscle fiber necrosis with regeneration and variation in muscle fiber size, and reduced contractile force [49].
Collagen VI mutations impair microfibrillar assembly by more than 1 mechanism. Some result in multiple aberrant transcripts that produce a truncated mRNA degraded through nonsense-mediated decay [50]. Others appear to have a dominant-negative effect [51].
Muscle pathology
The changes in the muscle range from mildly myopathic (limited to fiber size variability and scattered necrotic fibers) to overtly dystrophic with prominent endomysial and perimysial connective tissue proliferation with fat replacement. Although these findings are by no means specific, if accompanied by reduced or absent collagen VI staining, the diagnosis can be strongly implicated (Fig. 6) [52–55]. Rarely, collagen VI can be absent from the sarcolemmal basement membrane but not from the interstitium [56]. Expression of perlecan, collagen IV, and laminin α2 is normal in UCMD. If the biopsy demonstrates an abnormal pattern of collagen VI staining, molecular diagnostic confirmation should be pursued.
Figure 6.
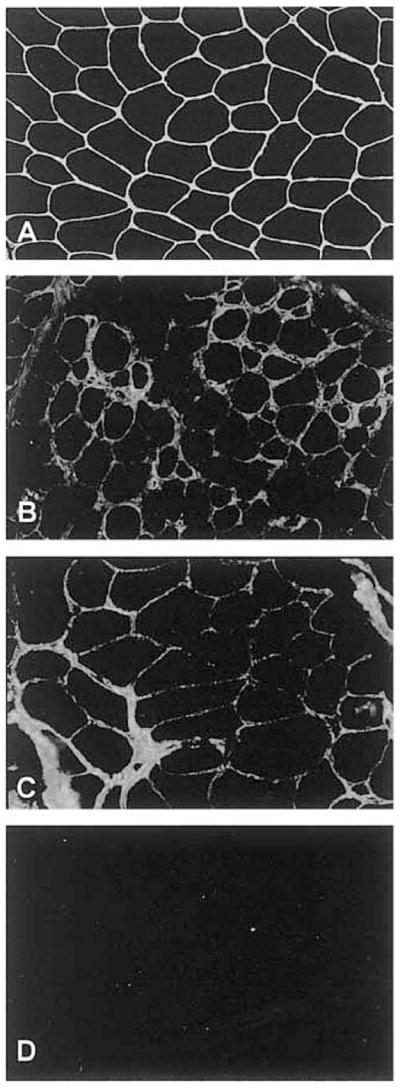
(A) Collagen VI is strongly expressed in the extracellular matrix of muscle fibers and around the blood vessel in a control. In collagen VI deficiency, the staining is reduced (B, C) or completely absent (D).
Previously published by Demir et al. in the Am J Hum Genet 2002;70:1446–1458. Reproduced with permission of University of Chicago Press.
DEFECTS IN EXTRACELLULAR MATRIX RECEPTORS
The dystroglycanopathies
The dystroglycanopathies are so named because all disorders center on genes that modify the glycosylation of α dystroglycan, a cell surface receptor for a number of extracellular matrix proteins, including laminin. Aberrant modification of α-dystroglycan by tissue-specific deletions in mouse muscle or brain resembles the underlying cellular pathology observed in clinical phenotypes.
Glycoproteins
Proteins with covalent links to carbohydrates (or sugars) are classified as glycoproteins. Between 0.5% and 1% of the genes in the human genome encode proteins that are involved in the synthesis, degradation, and function of glycoconjugates [57]. Dystroglycan, encoded by DAG1, is the focal point of the glycosylation defects leading to muscular dystrophy. Post-translational modification cleaves a single polypeptide into 2 proteins: α-dystroglycan and β-dystroglycan. α-dystroglycan is a secreted component that lies outside the muscle cell. It binds tightly but noncovalently to β-dystroglycan, which is a transmembrane protein. This complex of α- and β-dystroglycan chains serves as a vital component of the dystrophin glycoprotein complex that links the extracellular matrix to the actin cytoskeleton (Fig. 1). α-dystroglycan is heavily glycosylated and serves as a receptor for several proteins in the extracellular matrix that include laminin, neurexin (a family of neuronal-cell-surface proteins), agrin (a synaptic glycoprotein involved in the formation of neuromuscular junctions), biglycan (a small proteoglycan in the connective tissue), and perlecan (a ubiquitous heparan sulfate proteoglycan). β-dystroglycan, in turn, associates with a number of intracellular proteins, including dystrophin, that link the complex to filamentous actin. Increasingly, it is becoming apparent that dystroglycan not only serves a vital structural role in the membrane but also may subserve important roles in cell signaling. For example, dystroglycan can affect signaling via trimeric G proteins [58], Ras [59], Rac [60], Erk-MAP kinases [61], Akt/PI3 kinases [62], and Grb2 [63,64].
Dystroglycan and its sugar chains
There are 3 major groups of sugar-peptide linkages, N-linked glycans, O-linked glycans, and glycosaminoglycans (or GAGs, which are the sugar linkages found on proteoglycans). Both α- and β-dystroglycan have been shown to contain N-linked glycans, and α-dystroglycan is also highly glycosylated with several types of O-linked glycans. No evidence has been found that either α- or β-dystroglycan possess glycosaminoglycans [65]. In N-glycans, the reducing terminal N-acetylglucosamine (GlcNAc) is linked to the amide group of asparagine (Asn), via an aspartylglycosylamine linkage. In O-glycans, the reducing terminal sugar is usually N-acetylgalactosamine (GalNAc), which is attached to the hydroxyl group of serine (Ser) and threonine (Thr). In addition to O-linked GalNAc, other less common types of protein O-linked glycosylation also exist. O-linked mannose is one such linkage. In mammals, O-linked mannose has been found only on a limited number of glycoproteins, despite the fact that it is a more ubiquitous type of modification in lower organisms, such as yeast [66]. α-dystroglycan is the most extensively studied O-mannosyl–containing glycoprotein.
Figure 7 summarizes the biosynthetic pathway of O-mannosyl glycans on α-dystroglycan in mammals. This same pathway has been described for α-dystroglycan in brain, peripheral nerve, and skeletal muscle [67–70]. The 1st step requires the coexpression of protein-O-mannosyltransferase 1 (POMT1) and protein-O-mannosyltransferase 2 (POMT2) [71]. Mutations in either gene can cause Walker Warburg syndrome (WWS) [72]. In step 2, abnormalities in protein O-linked mannose β1,2-N-acetylglucosaminyltransferase 1 (POMGnT1) cause muscle-eye-brain (MEB) disease [73]. The 3rd and 4th steps, which synthesize β1,4-linked galactose and α2,3-linked sialic acid, are found on multiple types of N- and O-linked glycoproteins. Therefore, only steps 1 and 2 are unique to α-dystroglycan glycosylation, making defects in these steps the only ones likely to cause dystroglycanopathies. In addition, 3 other human dystroglycanopathy-related CMDs have mutations in genes encoding putative glycoslytransferases: fukutin (FCMD), FKRP (MDC1C), and LARGE (MDC1D). Although these genes perturb dystroglycan glycosylation, their function is not known.
Figure 7.
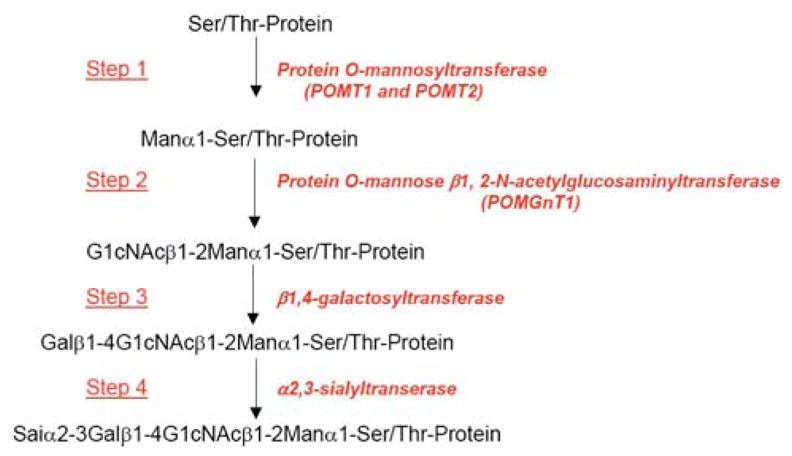
Summary of biosynthetic pathway for O-mannosyl glycans on α dystroglycan in mammals. Only steps 1 and 2 are unique to α–dystroglycan glycosylation. The 1st step requires the coexpression of protein-O-mannosyltransferase 1 (POMT1) and protein-O-mannosyltransferase 2 (POMT2). Step 2 requires protein O-linked mannose β1,2-N-acetylglucosaminyltransferase 1 (POMGnT1). The 3rd and 4th steps, which synthesize β1,4-linked galactose and α2,3-linked sialic acid, are found on multiple types of N- and O-linked glycoproteins.
Fukuyama Congenital Muscular Dystrophy (FCMD)
Clinical features
FCMD is characterized by severe CMD associated with mental retardation. Reduced fetal movements may be present in utero, and in the neonatal period patients are weak and floppy with poor suck and cry. Joints are hyperextensible. Facial weakness produces an open mouth appearance. Contractures appear by 1 year of age and include the hips, knees, and ankles. Most patients with FCMD never walk; if they do walk, though, it will be transient consisting of a few supported steps. Muscle hypertrophy of tongue and calf may be seen. Patients usually become bedridden before 10 years of age. Scoliosis accompanies loss of ambulation. Cardiomyopathy is common and may lead to congestive heart failure [74]. Most patients die by 20 years of age.
Severe mental retardation is characteristic, with IQ scores between 30 to 50. Seizures occur in 80% of patients, usually with onset at about 3 years of age [75]. Ocular abnormalities are present about 50% of patients, but in contrast to WWS, these patients are not blind. Abnormalities include high myopia, optic atrophy, and retinal changes (detachment, folding, fusion, or dysplasia) [76]. Cataracts are seen in some patients.
The most common and characteristic changes in the central nervous system are brain malformations, including polymicrogyria, pachygyria, and agyria of cerebrum and cerebellum (type II lissencephaly) with a highly disorganized cerebral cortex showing no recognizable lamination. In addition, neuronal overmigration into and within the leptomeninges, hydrocephalus, focal interhemispheric fusion, fusion of cerebellar folia, and hypoplasia of the corticospinal tracts are seen.
Genetics
FCMD is the most common autosomal recessive disorder in Japan (incidence is 0.7 to 1.2 per 10,000 births). It is caused by mutations of the FUKUTIN gene on chromosome 9q31 [77–79]. FUKUTIN encodes a protein of 461 amino acids with a predicted molecular weight of 56 kDa. A founder haplotype common to 87% of the FCMD alleles consists of 3-kb retrotransposal insertion of a tandemly repeated sequence located in the 3′ untranslated region of the gene (77). Other FCMD alleles include nonsense or missense mutations, insertions, and deletions [80,81]. Rarely, ethnic groups outside of Japan have been diagnosed with this disorder [82].
Molecular pathogenesis
FCMD, like other dystroglycanopathies (Table 1), shares an abnormally glycosylated α-dystroglycan protein. FUKUTIN encodes a putative glycosyl-transferase based on primary sequence analysis. The muscles of FCMD patients show reduced expression of glycosylated α-dystroglycan [83], although expression of α-dystroglycan polypeptide is present, along with β-dystroglycan, in the sarcolemmal membrane. Loss of glycosylation impairs binding of laminin α2 to α-dystroglycan. Consequently there may be secondary reduction in laminin α2 and basal lamina disruption [84–87]. This finding is a central theme common to all variants in this group of CMD. With regard to central nervous system findings, abnormal neuronal migration can be induced by brain-specific disruption of α-dystroglycan in mice, implicating aberrant glycosylation of dystroglycan in lissencephaly type II [86].
Muscle pathology
As a group, there are more similarities than differences in the muscle pathology of the dystroglycanopathies. Variations relate to causal mutations and related amount of residual glycosylated α-dystroglycan. In FCMD the muscle biopsy shows active muscle fiber degeneration (necrosis of individual fibers) accompanied by muscle regeneration. Fiber size variability is present with numerous hypertrophic fibers. There is increased endomysial and perimysial connective tissue and fat that replaces lost muscle tissue. By immunohistochemistry the sarcolemma shows normal expression of β-dystroglycan accompanied by absent or reduced glycosylated α-dystroglycan (Fig. 8) with preservation of core α-dystroglycan staining. In addition, the α-dystroglycan shows a shift in electrophoretic mobility [87].
Figure 8.
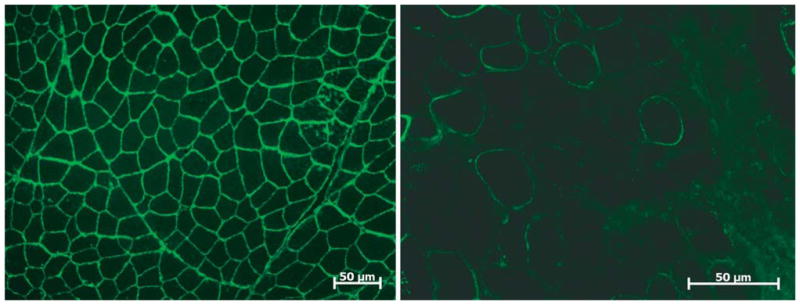
Stain for glycosylated α–dystroglycan (Upstate Cell Signaling Solutions, Lake Placid, NY, USA) in normal muscle compared with marked reduction in a patient with fukutin-related protein (FKRP) deficiency (MDC1C).
Muscle-eye-brain (MEB) disease
Clinical features
MEB disease is a disorder mainly seen in Finland [88–92]. The most profoundly affected patients exhibit decreased fetal movements in utero followed by marked hypotonia in the neonatal period. Impaired motor development results in a persistent bedridden state with profound facial and neck weakness and inability to turn over or even sit up. Many of the severely affected infants will die during the 1st year of life. In others, sitting may be achieved with minimal ambulation and speech development limited to a few spoken words [93]. In most cases life expectancy parallels that seen in FCMD, with death in late teenage years or early adulthood.
Seizures and mental retardation are common and represent the clinical manifestations of the neuronal migration disorder with lissencephaly type II. In MEB disease the brainstem is characteristically flattened. Visual impairment is caused by progressive myopia associated with retinal degeneration but does not reach the level of gravity seen in WWS. Optic colobomas, glaucoma, and cataracts are common [91,92].
Genetics
MEB disease is inherited as an autosomal recessive disorder with linkage to 1p34-p32 [94]. Loss-of-function mutations of POMGnT1 cause the disease. The spectrum of molecular defects includes nonsense, splice-site, exon skipping, and deletion mutations [93,95]. Recently a compound heterozygous missense mutation was associated with a disorder exclusively affecting the central nervous system without muscle involvement [96].
Most MEB disease patients have come from a small, isolated population in Finland, but it is now recognized that the disorder has a more widespread distribution, with patients reported from Italy, Belgium, Korea, Japan, and the United States [93,96]. FKRP gene mutations, usually associated with MDC1C, a disease without structural brain abnormalities, have also been reported as a cause of MEB disease [97].
Molecular pathogenesis
In MEB disease, the pathogenic events closely parallel those seen in FCMD. POMGnT1 is a glycosyltransferase responsible for the 2nd step in the biosynthesis of mammalian O-mannosyl glycans (Fig. 7). A loss of function mutation of this critical enzyme leads to a dramatic loss of glycosylation on muscle α-dystroglycan with reduced laminin α2 binding [87]. MEB disease can be diagnosed using an enzyme assay for POMGnT1 activity to show loss of function (or reduced function) in cultured fibroblasts or lymphoblasts [98]. The downstream manifestations include abnormal neuronal migration and skeletal muscle degeneration.
Muscle pathology
The muscle biopsy changes in MEB disease are not distinctly different from those described in FCMD. There is muscle necrosis, regeneration, and endomysial and perimysial fibrosis with fat replacing lost muscle fibers. Laminin α2 staining and glycosylated α-dystroglycan are reduced to absent with preservation of core α-dystroglycan.
Walker-Warburg Syndrome (WWS)
Clinical features
Walker originally reported the CNS manifestations of WWS in 1942 [99], but the muscular dystrophy component went unheralded for 40 years [100]. At that time diagnostic criteria for WWS were suggested to include type II lissencephaly, cerebellar abnormalities, retinal defects, and congenital muscular dystrophy [100].
Typically WWS is considered to be the most severe of the dystroglycanopathies. At birth patients lack spontaneous movements, with weak cry and suck, marked hypotonia, and inability to see. Microcephaly may be apparent and hydrocephalus, often related to aqueductal stenosis, represents a serious complication. Ocular abnormalities include microphthalmia, cataracts, iris malformations, and glaucoma. Retinal dysplasia with or without retinal detachment is typical with colobomas of the retina and hypoplastic optic nerves [101]. Cleft lip and palate and occipital encephalocele may distinguish WWS from other dystroglycanopathies [102].
The brain abnormalities include complete lissencephaly type II combined with pontocerebellar hypoplasia, with a Dandy-Walker malformation in 15% to 20%. The cerebellar cortex shows distortion of layering, malformation of dentate nucleus, and numerous cysts representing trapped arachnoid from aberrant neuronal migration [103]. Pyramidal tract hypoplasia is typical.
Recent reports expand the phenotype of WWS to include milder cases consisting of muscular dystrophy, microcephaly, and mental retardation in the absence of widespread structural brain abnormalities. These patients also exhibit fewer eye abnormalities, with myopia as the predominant clinical feature [104].
Genetics
WWS is inherited as an autosomal recessive disorder with both phenotypic and genetic heterogeneity. POMT1 mutations (chromosomal locus 9q34.1) represent 1 cohort [105,106] but by no means account for the majority of cases. POMT2 mutations (chromosomal locus 14q24.3) cause an indistinguishable clinical disorder [107], and fukutin mutations also account for some cases of WWS [108,109]. Adding further complexity, homozygous FKRP mutations have been reported with WWS [97].
Molecular pathogenesis
The consequences of hypoglycosylation of α-dystroglycan as described for the other dystroglycanopathies are responsible for the brain and muscle complications of WWS.
Muscle pathology
The changes in the muscle are indistinguishable from the other dystroglycanopathies. Antibody stains show markedly reduced to absent glycosylated α-dystroglycan, preservation of core α-dystroglycan, secondary decreased laminin α2, and normal β-dystroglycan.
Congenital Muscular Dystrophy Type 1C (MDC1C)
FKRP mutations provide a wide spectrum of phenotypic heterogeneity ranging in severity from the congenital muscular dystrophy (MDC1C) to a milder disease without central nervous system involvement classified with limb girdle muscular dystrophy, (LGMD2I) [110]. Due to space limitations, the milder phenotype will not be discussed in this review.
Clinical features
The hallmarks of MDC1C are severe muscle weakness and respiratory muscle compromise. In the neonatal period, hypotonia and feeding difficulties are apparent [111]. Motor milestones are markedly hampered by the dystrophic process. Children usually achieve independent sitting and may even take a few steps, but they never attain functional ambulation. Facial muscles are weak. Muscle hypertrophy may be present in the calf muscles or other lower limb muscles, and in some cases the tongue may be affected. Weak respiratory muscles result in pulmonary compromise [112], representing the most likely cause of death in the 1st decade or shortly thereafter. A dilated cardiomyopathy can add to their debilitated condition [113,114]. Cognitive development and vision are normal. Having said that, 1 variant was described with microcephaly, mild mental retardation, and cerebellar cysts, enlarging the spectrum of this congenital form of the disease [115].
Genetics
MDC1C is inherited as an autosomal recessive disorder with linkage to chromosome 19q13.3. Mutations of the FKRP gene include homozygous and heterozygous missense and nonsense mutations [115–117].
Molecular pathogenesis
FKRP encodes for a putative glycosyltransferase, the precise function of which is unknown. Overwhelming evidence, however, points to defects in glycosylation as the cause for the patient symptoms. The best evidence comes from side-by-side comparisons of muscle between the severe congenital muscular dystrophy, MDC1C, showing a marked reduction of glycosylated α-dystroglycan and the milder LGMD2I cases with subtle changes in this protein [118]. Recent work by Muntoni and colleagues [119] suggests that FKRP protein is localized to the Golgi in human skeletal muscle and that its localization is unchanged in MDC1C. Thus, mutations in FKRP giving rise to MDC1C are due to loss of function and not mislocalization to the endoplasmic reticulum, as previously reported [120].
Muscle pathology
The muscle pathology in MDC1C has no specific features by which to distinguish it from more severe CMD phenotypes with central nervous system involvement. Similar to these disorders, there is secondary deficiency of laminin α2 expression. In addition, there is a marked decrease in immunostaining using antibody to glycolsylated α-dystroglycan associated with a reduction in its molecular weight on western blots [117] (Fig. 8). β-Dystroglycan staining is normal.
Congenital Muscular Dystrophy Type 1D (MDC1D)
Clinical features
Only a single patient has so far been recognized with this form of dystroglycanopathy [121]. A 17-year-old girl had no recognized problems at birth but was found to be developmentally delayed at 5 months of age. She could not sit unsupported until she was 2.5 years of age and was not independently ambulatory until 4.5 years of age. Maximal motor function was achieved by 9 years of age when she was able to walk 200 yards, after which she gradually worsened. She had mild facial muscle weakness and muscle hypertrophy affecting the quadriceps and calf and arm muscles. Contractures were seen at ankles and elbows.
The patient was profoundly mentally retarded, with understanding limited to simple 1-step commands. Mirror movements were present in the upper limbs, and the fingers were held in a flexed position, with thumbs adducted. Gait was spastic, and muscle stretch reflexes were exaggerated with extensor plantar responses. Brain MRI showed minimal changes at 4 years of age but in teenage years showed extensive and symmetrical cerebral white matter changes sparing the internal capsule, corpus callosum, optic radiations, and infratentorial structures. In addition, neuronal migration defects consisting of mild pachygyria with moderately thickened cortex in the frontal lobes and mildly simplified gyri with shallow sulci in the posterior frontal, temporal, and parietal regions.
Genetics
A compound heterozygous mutation of the LARGE gene (chromosomal locus 22q12), missense at 1 allele and a 1-bp insertion at the other, was found in this patient. The human LARGE gene was named because it spans more than 660 kb of genomic DNA, although the mRNA is only about 4.4 kb [122]. LARGE also causes myodystrophy or myd, a mouse mutant with skeletal and cardiomyopathy [123].
Molecular pathogenesis
LARGE is considered to represent another putative glycosyltransferase, but further studies are required to better understand the pathogenesis of MDC1D. LARGE has 2 putative glycosyltransferase domains. The tandem nature of the primary sequence suggests that LARGE may be responsible for 2 glycosylation events, such as the synthesis of a disaccharide. Overexpression of LARGE in non-muscle cells and in cells from patients with FCMD, MEB disease, and WWS can stimulate glycosylation of α-dystroglycan and rescue laminin binding [124]. Thus, not only can LARGE stimulate the glycosylation of α-dystroglycan, but its overexpression can overcome the glycosylation defects caused by other dystroglycanopathy genes. How LARGE functions, therefore, will be important not only in defining the molecular basis for pathology in MDC1D but also for developing therapeutic strategies in related disorders.
Muscle pathology
Muscle biopsy showed reduced staining of α-dystroglycan. The extent of this reduction varied, with some fibers almost negative and others showing residual labeling, which was discontinuous through the basement membrane. Laminin α2 and β-dystroglycan expression were normal.
Integrin α7 deficiency
Clinical features
No clear phenotype has emerged in the cases reported with absent integrin α7 [125,126], making it a candidate disorder but not part of the official CMD classification [2]. Patients reported include (1) a 4-year-old boy with delayed motor milestones and mental retardation; (2) an 11-year-old girl with normal intelligence, congenital hip dislocation, and torticollis who did not walk until age 2; (3) a patient with hypotonia and torticollis at birth, delayed motor milestones, and ability to walk with support at age 5; and (4) a 4th patient with multiple joint contractures and respiratory insufficiency.
Genetics
Integrin a7 is linked to chromosome 12q13. The disease has been reported in 4 isolated patients without apparent gender preference [125,126]. The children were the products of nonconsanguineous parents. Mutations included deletions, splice-site mutations, and heterozygous missense mutations.
Molecular pathogenesis
Integrins are heterodimeric transmembrane glycoproteins consisting of an α and β chain. The α7 subunit is mainly expressed in skeletal and cardiac muscle, while the β1 chain is expressed throughout the body. Integrin α7β1 in skeletal and cardiac muscle binds via its extracellular domain to laminins, including laminin α2, and via its cytoplasmic domain to cytoskeletal-associated proteins [127] (Fig. 1). Like the dystrophin-glycoprotein complex, integrin α7β1 contributes to the overall integrity of the sarcolemma, each acting as an independently controlled laminin receptor. Mice that lack integrin α7 develop a mild but progressive form of muscle disease with similarities to the clinical condition [128]. Studies have raised the possibility that integrin α7β1 may functionally compensate for loss of the dystrophin-glycoprotein complex. Increased staining intensity of integrin α7β1 has been observed in Duchenne patients and mdx mice [129]. In addition, overexpression of integrin α7 improves mobility and increases life span in the dystrophin-utrophin double-mutant mice, supporting a compensatory role for integrin α7β1 in restoring muscle integrity.
Muscle pathology
The muscle biopsy features in patients with integrin α7 deficiency have not evolved to a level enabling a confident diagnostic pattern. Fiber size variability and type-1 predominance, both very nonspecific features, have been reported; fibrofatty replacement of muscle was described in the biopsy of the 11-year-old child [125]. Antibody staining for integrin α7 has been highly variable, especially in the 1st 2 years of life and may not be a reliable marker. In addition, laminin α2 appears to be preserved in these cases. Overall, more experience is needed to establish this disorder as a specific nosologic entity.
Rigid Spine with Muscular Dystrophy Type I (RSMD1): Deficiency of Selenoprotein N
The features of the “rigid spine syndrome” [130], as originally described by Dubowitz including spinal rigidity with varying degrees of limb contractures, are not unique to 1 single entity. They occur in X-linked and autosomal dominant Emery-Dreifuss muscular dystrophy, nemaline myopathy, multiminicore disease, Bethlem myopathy, and others (Fig. 9) [131,132]. In contrast, rigid spine muscular dystrophy type 1 (RSMD1) is a distinct disorder linked to chromosome 1p35 with a mutation in SEPN1 [133].
Figure 9.
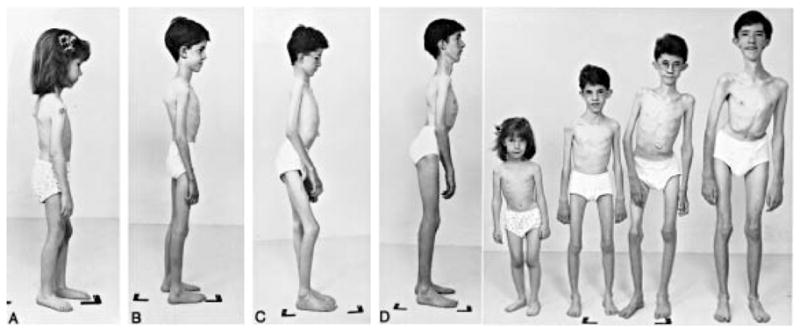
Four siblings affected by rigid spine muscular dystrophy type 1 (RSMD1) can be seen in side and frontal views. The loss of muscle bulk is striking, and the presence of scoliosis, varying degrees of lordosis, and joint contractures at elbows and knees can be seen. Previously published by Flanigan et al. in Ann Neurol 2000;47:152–161 with permission of John Wiley & Sons [132].
Clinical features
Hypotonia, neck weakness, early scoliosis, muscle weakness, and respiratory insufficiency dominate the clinical picture in RSMD1 [131,132].
At birth or in the neonatal period, hypotonia and poor head control are recognized. Motor milestones are usually not delayed. As the name implies, rigidity of the spine is a characteristic feature, and this evolves to scoliosis in most patients. Proximal weakness of the limbs can be significant, with resultant waddling gait and Gowers’ sign. Contractures of the extremities are mild, often including heel cord tightness. The temporomandibular joint may be affected, with limitation of mouth opening [134]. Respiratory failure can be significant in the 1st decade, related to stiffness of the rib cage and diaphragm muscle weakness, requiring nocturnal ventilatory assistance [132].
A late-onset variant has been described in a 26-year-old person with rapidly progressive respiratory and right heart failure with cough, orthopnea, interrupted sleep, morning headaches, and daytime somnolence [135].
Genetics
The disease is inherited as an autosomal recessive disorder. RSMD1 was the preferred name by the Human Gene Organization Nomenclature Committee (over RSS because of a previously assigned designation, Russell-Silver-Syndrome) [136]. Frameshift, nonsense, and missense mutations of the gene encoding selenoprotein N (SEPN1) were originally identified in 3 Turkish and 2 Iranian families [137,138]. More recently a novel mutation was found in the hairpin structure of a 3′ untranslated region of SEPN1 mRNA, resulting in reduced mRNA and protein in a patient with a mild phenotype [139].
Mutations in SEPN1 also cause multiminicore disease [140] and a desmin-related myopathy with Mallory body-like inclusions [141].
Molecular pathogenesis
As a cause for CMD, the protein involved in RSMD1 is quite distinct from the others so far discussed. Little is known about the pathogenic mechanism of the SEPN1-related myopathies. It has been established that the protein product of SEPN1 presides in the endoplasmic reticulum [142]. This localization suggests a role in membrane trafficking, protein processing, and regulation of calcium homeostasis. All of these functions are important in muscle function, but further studies are necessary to unravel the pathogenesis of this form of CMD.
Muscle pathology
Fiber size variability and type-1 fiber predominance are the usual features with variable findings, such as muscle fiber necrosis and regeneration, and endomysial connective tissue proliferation [142]. In one form of SEPN1-related myopathy, a congenital myopathy, multiminicore disease, with nondystrophic pathology is seen (multiple core-like areas of sarcoplasmic disorganization associated with mitochondrial depletion) [143].
Conclusions
Over the past decade, the understanding of the CMDs has rapidly expanded. The classification includes 3 major groups of disorders: abnormal extracellular matrix proteins, defects in glycoslyated dystroglycans, and an abnormal endoplasmic reticulum protein. The 1st 2 groups involve highly integrated proteins, all potentially linking the extracellular matrix to the muscle cytoskeleton. Furthermore, when defects in glycosylation are severe, neuronal migration is affected and collectively recognized as the type-II lissencephaly spectrum, encompassing cobblestone polymicrogyria-pachygyria on one end and complete agyria on the other. In group 3, the abnormal protein product of the SEPN1 gene is somewhat disconnected from the other CMDs. Even the muscle cannot be considered dystrophic (and with overlap to the spectrum of disease better labeled as a congenital myopathy, i.e., multiminicore disease).
In the big picture, looking at the muscle biopsy can help identify the dystrophic process in the 1st 2 groups of diseases, and a panel of antibodies directed at laminins and dystroglycans can be useful for providing direction toward making a molecular diagnosis needing confirmation by DNA tests. In the 3rd group, SEPN1 mutations, the pathology is very nonspecific and will require a strong clinical suspicion unless multiminicores are present to direct the work up.
References
- 1.Engvall E. Laminin variants: why, where, and when? Kidney Int. 1993;43:2–6. doi: 10.1038/ki.1993.2. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Voit T. The congenital dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635–649. doi: 10.1016/j.nmd.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001;38:649–657. doi: 10.1136/jmg.38.10.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Philpot J, Sewry C, Pennock J, Dubowitz Clnical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromusc Disord. 1995;5:301–305. doi: 10.1016/0960-8966(94)00069-l. [DOI] [PubMed] [Google Scholar]
- 5.Fardeau M, Tome FM, Helfling-Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency: clinical, histopathological, immunocytochemical and genetic analysis. Rev Neurol. 1996;152:11–19. [PubMed] [Google Scholar]
- 6.Pini A, Merlini L, Tome FM, Chevallay M, Gobbi G. Merosin-negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia: report of three Italian cases in two families. Brain Dev. 1996;18:316–322. doi: 10.1016/0387-7604(96)00028-9. [DOI] [PubMed] [Google Scholar]
- 7.Tezak Z, Prandini P, Boscaro M, et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. 2003;21:103–111. doi: 10.1002/humu.10157. [DOI] [PubMed] [Google Scholar]
- 8.Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology. 1998;51:101–110. doi: 10.1212/wnl.51.1.101. [DOI] [PubMed] [Google Scholar]
- 9.Allamand V, Sunada Y, Salih MA, et al. Mild congenital muscular dystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet. 1997;6:747–752. doi: 10.1093/hmg/6.5.747. [DOI] [PubMed] [Google Scholar]
- 10.Taratuto AL, Lubieniecki F, Diaz D, et al. Merosin-deficient congenital muscular dystrophy associated with abnormal cerebral cortical gyration: an autopsy study. Neuromuscul Disord. 1999;9:86–94. doi: 10.1016/s0960-8966(98)00112-6. [DOI] [PubMed] [Google Scholar]
- 11.Tsao CY, Mendell JR, Rusin J, Luquette M. Congenital muscular dystrophy with complete laminin-alpha2-deficiency, cortical dysplasia, and cerebral white-matter changes in children. J Child Neurol. 1998;13:253–256. doi: 10.1177/088307389801300602. [DOI] [PubMed] [Google Scholar]
- 12.Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology. 1995;45:2084–2089. doi: 10.1212/wnl.45.11.2084. [DOI] [PubMed] [Google Scholar]
- 13.Shorer Z, Philpot J, Muntoni F, Sewry C, Dubowitz V. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J Child Neurol. 1995;10:472–475. doi: 10.1177/088307389501000610. [DOI] [PubMed] [Google Scholar]
- 14.Mora M, Moroni I, Uziel G, et al. Mild clinical phenotype in a 12-year-old boy with partial merosin deficiency and central and peripheral nervous system abnormalities. Neuromuscul Disord. 1996;6:377–381. doi: 10.1016/0960-8966(96)00359-8. [DOI] [PubMed] [Google Scholar]
- 15.Prelle A, Comi GP, Rigoletto C, et al. An atypical case of partial merosin deficiency congenital muscular dystrophy. J Neurol. 1997;244:391–395. doi: 10.1007/s004150050110. [DOI] [PubMed] [Google Scholar]
- 16.Deodato F, Sabatelli M, Ricci E, et al. Hypermyelinating neuropathy, mental retardation and epilepsy in a case of merosin deficiency. Neuromuscul Disord. 2002;12:392–398. doi: 10.1016/s0960-8966(01)00312-1. [DOI] [PubMed] [Google Scholar]
- 17.Di Muzio A, De Angelis MV, Di Fulvio P, et al. Dysmyelinating sensory-motor neuropathy in merosin-deficient congenital muscular dystrophy. Muscle Nerve. 2003;27:500–506. doi: 10.1002/mus.10326. [DOI] [PubMed] [Google Scholar]
- 18.Spryrou N, Philpot J, Foale R, et al. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J. 1998;136:474–476. doi: 10.1016/s0002-8703(98)70222-4. [DOI] [PubMed] [Google Scholar]
- 19.Hermann R, Straub V, Meyer K, et al. Congenital muscular dystrophy with laminin alpha 2 chain deficiency: identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr. 1996;155:968–976. doi: 10.1007/BF02282889. [DOI] [PubMed] [Google Scholar]
- 20.Naom IS, D’Alessandro M, Tapaloglu H, et al. Refinement of the laminin alpha2 chain locus to human chromosome 6q2 in severe and mild merosin deficient congenital muscular dystrophy. J Med Genet. 1997;34:99–104. doi: 10.1136/jmg.34.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellbling-Ledlerc A, Zhang X, Topaloglue H, et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- 22.Di Blasi C, He Y, Morandi L, et al. Mild muscular dystrophy due to a nonsense mutation in the LAMA2 gene resulting in exon skipping. Brain. 2001;124:698–704. doi: 10.1093/brain/124.4.698. [DOI] [PubMed] [Google Scholar]
- 23.Coral-Vazquez RM, Rosas-Vargas H, Meza-Espinosa P, et al. Severe congenital muscular dystrophy in a Mexican family with a new nonsense mutation (R2578X) in the laminin alpha-2 gene. Hum Genet. 2003;48:91–95. doi: 10.1007/s100380300013. [DOI] [PubMed] [Google Scholar]
- 24.Patton BL, Connolly AM, Martin PT, et al. Distribution of ten laminin chains in dystrophic and regenerating muscles. Neuromuscul Disord. 1999;9:423–433. doi: 10.1016/s0960-8966(99)00033-4. [DOI] [PubMed] [Google Scholar]
- 25.Moll J, Barzaghi P, Lin S, et al. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 26.Shelton GD, Engvall E. Canine and feline models of human inherited muscle diseases. Neuromuscul Disord. 2005;15:127–138. doi: 10.1016/j.nmd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Wu X-R, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha-2 (LAMA2) gene. Nat Genet. 1994;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- 28.Guo LT, Zhang Xu, Kuang W. Laminin alpha2 deficiency and muscular dystrophy: genotype-phenotype correlation in mutant mice. Neuromuscul Disord. 2003;13:207–215. doi: 10.1016/s0960-8966(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 29.Miyagoe-Suzuki Y, Nakagawa M, Takeda S. Merosin and congenital muscular dystrophy. Microsc Res Tech. 2000;48:181–191. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<181::AID-JEMT6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 30.Rasminsky M, Kearney RE, Aguayo AJ, Bray GM. Conduction of nervous impulses in spinal root and peripheral nerves of dystrophic mice. Brain Res. 1978;143:71–85. doi: 10.1016/0006-8993(78)90753-9. [DOI] [PubMed] [Google Scholar]
- 31.Bunge MB, Williams AK, Wood PM. Neuron-Schwann cell interaction in basal lamina formation. Dev Biol. 1982;92:449–460. doi: 10.1016/0012-1606(82)90190-7. [DOI] [PubMed] [Google Scholar]
- 32.Pegoraro E, Mancias P, Swerdlow SH, et al. Congenital muscular dystrophy with primary laminin alpha2 (merosin) deficiency presenting as inflammatory myopathy. Ann Neurol. 1996;40:782–791. doi: 10.1002/ana.410400515. [DOI] [PubMed] [Google Scholar]
- 33.Sewry CA, Philpot J, Sorokin LM, et al. Diagnosis of merosin (laminin-2) deficient congenital muscular dystrophy by skin biopsy. Lancet. 1996;347:582–584. doi: 10.1016/s0140-6736(96)91274-x. [DOI] [PubMed] [Google Scholar]
- 34.Sewry CA, D’Alessandro M, Wilson LA, et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics. 1997;28:217–222. doi: 10.1055/s-2007-973703. [DOI] [PubMed] [Google Scholar]
- 35.Ulrich O. Kongenitale, atonisch-sklerotische Muskeldystrophie. Monatsschr Kinderheilkd. 1930;47:502–510. [Google Scholar]
- 36.Mercuri E, Yuva Y, Brown SC, et al. Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohistochemical study. Neurology. 2002:1354–1359. doi: 10.1212/wnl.58.9.1354. [DOI] [PubMed] [Google Scholar]
- 37.Voit T. Congenital muscular dystrophies: 1997 update. Brain Dev. 1998;20:65–74. doi: 10.1016/s0387-7604(97)00094-6. [DOI] [PubMed] [Google Scholar]
- 38.Muntoni F, Bertini E, Bonnemann C, et al. Neuromuscul Disord; 98th ENMC International Workshop on Congenital Muscular Dystrophy (CMD), 7th Workshop of the International Consortium on CMD, 2nd Workshop of the MYO CLUSTER Project GENRE; 26–28th October 2001; Naarden, The Netherlands. 2002. pp. 889–896. [DOI] [PubMed] [Google Scholar]
- 39.Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100th ENMC international workshop, 23–24 November 2001, Naarden, The Netherlands. Neuromuscul Disord. 2002;12:984–993. doi: 10.1016/s0960-8966(02)00139-6. [DOI] [PubMed] [Google Scholar]
- 40.Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance: a report on three pedigrees. Brain. 1976;99:91–100. doi: 10.1093/brain/99.1.91. [DOI] [PubMed] [Google Scholar]
- 41.Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claeysen S, Joubert L, Sebben M, et al. A single mutation in the 5-HT4 receptor (5-HT4-R D100(3.32)A) generates a Gs-coupled receptor activated exclusively by synthetic ligands (RASSL) J Biol Chem. 2003;278:37698–37704. doi: 10.1074/jbc.C200588200. [DOI] [PubMed] [Google Scholar]
- 43.Kuo HJ, Maslen CL, Keene DR, Glanville RW. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem. 1997;272:26522–26529. doi: 10.1074/jbc.272.42.26522. [DOI] [PubMed] [Google Scholar]
- 44.Engvall E, Hessle H, Klier G. Molecular assembly, secretion, and matrix deposition of type VI collagen. Cell Biol. 1986;102:703–710. doi: 10.1083/jcb.102.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heiskanen M, Saitta B, Palotie A, Chu ML. Head to tail organization of the human COL6A1 and COL6A2 genes by fiber-FISH. Genomics. 1995;29:801–803. doi: 10.1006/geno.1995.9008. [DOI] [PubMed] [Google Scholar]
- 46.Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA. 2001;98:7516–7521. doi: 10.1073/pnas.121027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pepe G, Lucarini L, Zhang RZ, et al. COL6A1 genomic deletions in Bethlem and Ullrich muscular dystrophy. Ann Neurol. 2006;59:190–195. doi: 10.1002/ana.20705. [DOI] [PubMed] [Google Scholar]
- 48.Lampe AK, Dunn DM, von Niederhausern AC, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet. 2005;42:108–120. doi: 10.1136/jmg.2004.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- 50.Lucarini L, Giusti B, Zhang RZ, Pan TC, Jimenez-Mallebrera C, Mercuri E, et al. A homozygous COL6A2 intron mutation causes in-frame triple-helical deletion and nonsense-mediated mRNA decay in a patient with Ullrich congenital muscular dystrophy. Hum Genet. 2005;117:460–466. doi: 10.1007/s00439-005-1318-8. [DOI] [PubMed] [Google Scholar]
- 51.Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355–369. doi: 10.1086/377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higuchi I, Shiraishi T, Hashiguchi T, et al. Frameshift mutation in the collagen VI gene causes Ullrich’s disease. Ann Neurol. 2001;50:261–265. doi: 10.1002/ana.1120. [DOI] [PubMed] [Google Scholar]
- 53.Stasia MJ, Bordigoni P, Martel C, Morel F. A novel and unusual case of chronic granulomatous disease in a child with a homozygous 36-bp deletion in the CYBA gene (A22(0)) leading to the activation of a cryptic splice site in intron 4. Hum Genet. 2002;70:446–458. doi: 10.1007/s00439-002-0720-8. [DOI] [PubMed] [Google Scholar]
- 54.Ishikawa H, Sugie K, Murayama K, et al. Ullrich disease: collagen VI deficiency: EM suggests a new basis for muscular weakness. Neurology. 2002;59:920–923. doi: 10.1212/wnl.59.6.920. [DOI] [PubMed] [Google Scholar]
- 55.Mercuri E, Yuva Y, Brown SC, et al. Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohistochemical study. Neurology. 2002;58:1354–1359. doi: 10.1212/wnl.58.9.1354. [DOI] [PubMed] [Google Scholar]
- 56.Ishikawa H, Sugie K, Murayama K, et al. Ullrich disease due to deficiency of collagen VI in the sarcolemma. Neurology. 2004;24:620–623. doi: 10.1212/01.wnl.0000113023.84421.00. [DOI] [PubMed] [Google Scholar]
- 57.Wopereis S, Lefeber DJ, Morava E, Wevers RA. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin Chem. 2006;52:574–600. doi: 10.1373/clinchem.2005.063040. [DOI] [PubMed] [Google Scholar]
- 58.Zhou YW, Oak SA, Senolges SE, et al. Laminin-alpha1 globular domains 3 and 4 induce heterotrimeric G protein binding to alpha-syntrophin’s PDZ domain and alter intracellular Ca2+ in muscle. Am J Physiol Cell Physiol. 2005;288:C377–C388. doi: 10.1152/ajpcell.00279.2004. [DOI] [PubMed] [Google Scholar]
- 59.Chockalingam PS, Cholera R, Oak SA, Zheng Y, Jarrett HW, Thomason DB. Dystrophin-glycoprotein complex and Ras and Rho GTPase signaling are altered in muscle atrophy. Am J Physiol Cell Physiol. 2002;283:C500–C511. doi: 10.1152/ajpcell.00529.2001. [DOI] [PubMed] [Google Scholar]
- 60.Spence HJ, Dhillon AS, James M, et al. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 2004;5:484–489. doi: 10.1038/sj.embor.7400140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langenbach KJ, Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- 62.Zhou YW, Thomason DB, Gullberg D, et al. Binding of laminin alpha1-chain LG4–5 domain to alpha-dystroglycan causes tyrosine phosphorylation of syntrophin to initiate Rac1 signaling. Biochemistry. 2006;45:2042–2052. doi: 10.1021/bi0519957. [DOI] [PubMed] [Google Scholar]
- 63.Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 64.Russo K, Di Stasio E, Macchia G, Rosa G, Brancaccio A, Petrucci TC. Characterization of the beta-dystroglycan-growth factor receptor 2 (Grb2) interaction. Biochem Biophys Res Commun. 2000;274:93–98. doi: 10.1006/bbrc.2000.3103. [DOI] [PubMed] [Google Scholar]
- 65.Martin PT. Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology. 2003;15:55R–66R. doi: 10.1093/glycob/cwg076. [DOI] [PubMed] [Google Scholar]
- 66.Endo T. Structure, function and pathology of O-mannosyl glycans. Glycoconj J. 2004;21:3–7. doi: 10.1023/B:GLYC.0000043740.26062.2c. [DOI] [PubMed] [Google Scholar]
- 67.Michele DE, Campbell KP. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 68.Smalheiser NR, Haslam SM, Sutton-Smith M, Morris HR, Dell A. Structural analysis of sequences O-linked to mannose reveals a novel Lewis X structure in cranin (dystroglycan) purified from sheep brain. J Biol Chem. 1999;273:23698–23703. doi: 10.1074/jbc.273.37.23698. [DOI] [PubMed] [Google Scholar]
- 69.Chiba A, Matsumura K, Yamada H, et al. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan: the role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J Biol Chem. 1997;272:156–162. doi: 10.1074/jbc.272.4.2156. [DOI] [PubMed] [Google Scholar]
- 70.Sasaki T, Yamada H, Matsumura K, Shimizu T, Kobata A, Endo T. Detection of O-mannosyl glycans in rabbit skeletal muscle alpha-dystroglycan. Biochim Biophys Acta. 1998;1425:599–606. doi: 10.1016/s0304-4165(98)00114-7. [DOI] [PubMed] [Google Scholar]
- 71.Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, Margolis RU, Endo T. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyl-transferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 74.Nakanishi T, Sakauchi M, Kaneda Y, et al. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics. 2006;117:e1187–e1192. doi: 10.1542/peds.2005-2469. [DOI] [PubMed] [Google Scholar]
- 75.Yoshioka M, Higuchi Y. Long-term prognosis of epilepsies and related seizure disorders in Fukuyama-type congenital muscular dystrophy. J child Neurol. 2005;20:385–391. doi: 10.1177/08830738050200041901. [DOI] [PubMed] [Google Scholar]
- 76.Yoshioka M, Kuroki S, Kondo T. Ocular manifestations in Fukuyama type congenital muscular dystrophy. Brain Dev. 1990;12:423–426. doi: 10.1016/s0387-7604(12)80076-3. [DOI] [PubMed] [Google Scholar]
- 77.Toda T, Segawa M, Nomura Y, et al. Localization of a gene for Fukuyama type congenital muscular dystrophy to chromosome 9q31–33. Nat Genet. 1993;5:283–286. doi: 10.1038/ng1193-283. [DOI] [PubMed] [Google Scholar]
- 78.Toda T, Miyake M, Kobayashi K, et al. Linkage-disequilibrium mapping narrows the Fukuyama-type congenital muscular dystrophy (FCMD) candidate region to <100 kb. Am J Hum Genet. 1996;59:1313–1320. [PMC free article] [PubMed] [Google Scholar]
- 79.Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;23:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 80.Saito K, Osawa M, Wang ZP, et al. Haplotype-phenotype correlation in Fukuyama congenital muscular dystrophy. Am J Med Genet. 2000;92:184–190. doi: 10.1002/(sici)1096-8628(20000529)92:3<184::aid-ajmg5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 81.Kobayashi K, Sasaki J, Kondo-Iida E, et al. Structural organization, complete genomic sequences and mutational analyses of the Fukuyama-type congenital muscular dystrophy gene, fukutin. FEBS Lett. 2001;489:192–196. doi: 10.1016/s0014-5793(01)02088-9. [DOI] [PubMed] [Google Scholar]
- 82.Silan F, Yoshioka M, Kobayashi K, et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol. 2003;53:392–396. doi: 10.1002/ana.10491. [DOI] [PubMed] [Google Scholar]
- 83.Hayashi YK, Ogawa M, Tagawa K, et al. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology. 2001;57:115–121. doi: 10.1212/wnl.57.1.115. [DOI] [PubMed] [Google Scholar]
- 84.Ishii H, Hayashi YK, Nonaka I, et al. Electron microscopic examination of basal lamina in Fukuyama congenital muscular dystrophy. Neuromuscul Disord. 1997;7:191–197. doi: 10.1016/s0960-8966(97)00462-8. [DOI] [PubMed] [Google Scholar]
- 85.Matsubara S, Mizuno Y, Kitaguchi T, Isozaki E, Miyamoto K, Hirai S. Fukuyama-type congenital muscular dystrophy: close relation between changes in the muscle basal lamina and plasma membrane. Neuromuscul Disord. 1999;9:388–939. doi: 10.1016/s0960-8966(99)00049-8. [DOI] [PubMed] [Google Scholar]
- 86.Moore SA, Saito F, Chen J, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- 87.Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 88.Raitta C, Lamminen M, Santavuori P, Leisti J. Ophthalmological findings in a new syndrome with muscle, eye and brain involvement. Acta Ophthalmol. 1978;56:465–472. doi: 10.1111/j.1755-3768.1978.tb05700.x. [DOI] [PubMed] [Google Scholar]
- 89.Santavuori P, Somer H, Sainio K, et al. Muscle-eye-brain disease (MEB) Brain Dev. 1989;11:147–153. doi: 10.1016/s0387-7604(89)80088-9. [DOI] [PubMed] [Google Scholar]
- 90.Santavuori P, Valanne L, Autti T, Haltia M, Pihko H, Sainio K. Muscle-eye-brain disease: clinical features, visual evoked potentials and brain imaging in 20 patients. EJPN. 1998;1:41–47. doi: 10.1016/1090-3798(98)01004-1. [DOI] [PubMed] [Google Scholar]
- 91.Haltia M, Leivo I, Somer H, et al. Muscle-eye-brain disease: a neuropathological study. Ann Neurol. 1997;41:173–180. doi: 10.1002/ana.410410208. [DOI] [PubMed] [Google Scholar]
- 92.Pihko H, Lappi M, Raitta C, Sainio K, Valanne L, Somer H, Santavuori P. Ocular findings in muscle-eye-brain (MEB) disease: a follow-up study. Brain Dev. 1995;17:57–61. doi: 10.1016/0387-7604(94)00101-3. [DOI] [PubMed] [Google Scholar]
- 93.Taniguchi K, Kobayashi K, Saito K, et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet. 2003;12:527–534. doi: 10.1093/hmg/ddg043. [DOI] [PubMed] [Google Scholar]
- 94.Cormand B, Avela K, Pihko H, et al. Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Gene. 1999;64:126–135. doi: 10.1086/302206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyl-transferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 96.Vervoort VS, Holden KR, Ukadike KC, Collins JS, Saul RA, Srivastava AK. POMGnT1 gene alterations in a family with neurological abnormalities. Ann Neurol. 2004;56:143–148. doi: 10.1002/ana.20172. [DOI] [PubMed] [Google Scholar]
- 97.de Bernabe D, Beltran-Valero, Voit T, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vajsar I, Zhang W, Dobyns WB, et al. Carriers and patients with muscle-eye-brain disease can be rapidly diagnosed by enzymatic analysis of fibroblasts and lymphoblasts. Neuromuscul Disord. 2006;16:132–136. doi: 10.1016/j.nmd.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 99.Walker AE. Lissencephaly Arch Neurol Psychol. 1942;48:13–29. [Google Scholar]
- 100.Dobyns WB, Pagon RA, Armstrong D, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32:195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 101.Gerding H, Gullotta F, Kuchemelster K, Busse H. Ocular findings in Walker-Warburg syndrome. Childs Nerv Syst. 1993;9:418–420. doi: 10.1007/BF00306196. [DOI] [PubMed] [Google Scholar]
- 102.Burton BK, Dillard RG, Weaver RG. Walker-Warburg syndrome with cleft lip and cleft palate in two sibs. Am J Med Genet. 1987;27:537–541. doi: 10.1002/ajmg.1320270306. [DOI] [PubMed] [Google Scholar]
- 103.Gelot A, Billette de Villemeur T, Bordarier C. Developmental aspects of type II lissencephaly: comparative study of dysplastic lesions in fetal and post-natal brains. Acta Neuropathol. 1995;89:72–84. doi: 10.1007/BF00294262. [DOI] [PubMed] [Google Scholar]
- 104.Van Reeuwijk J, Maugenre S, Van Den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat. 2006;27:453–459. doi: 10.1002/humu.20313. [DOI] [PubMed] [Google Scholar]
- 105.Beltran-Valero de Bernabe D, Currier S, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Currier SC, Lee CK, Chang BS, et al. Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am J Med Genet. 2005;133A:53–57. doi: 10.1002/ajmg.a.30487. [DOI] [PubMed] [Google Scholar]
- 107.Van Reeuwijk J, Janssen M, Van Den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.De Bernabe DB, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Gene. 2003;40:845–848. doi: 10.1136/jmg.40.11.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Silan F, Yoshioka M, Kobayashi K, et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol. 2003;53:392–396. doi: 10.1002/ana.10491. [DOI] [PubMed] [Google Scholar]
- 110.Boito CA, Melacini P, Vianello A, et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol. 2005;62:1894–1899. doi: 10.1001/archneur.62.12.1894. [DOI] [PubMed] [Google Scholar]
- 111.Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Quijano-Roy S, Galan L, Ferreiro A, et al. Severe progressive form of congenital muscular dystrophy with calf pseudohypertrophy, macroglossia and respiratory insufficiency. Neuromuscul Diord. 2002;12:466–475. doi: 10.1016/s0960-8966(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 113.Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- 114.Quijano-Roy S, Galan L, Ferreiro A, et al. Severe progressive form of congenital muscular dystrophy with calf pseudohypertrophy, macroglossia and respiratory insufficiency. Neuromuscul Disord. 2002;12:466–475. doi: 10.1016/s0960-8966(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 115.Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology. 2003;60:988–92. doi: 10.1212/01.wnl.0000052996.14099.dc. [DOI] [PubMed] [Google Scholar]
- 116.Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mercuri E, Brockington M, Quijano-Roy S, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- 118.Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol. 2004;164:727–737. doi: 10.1016/s0002-9440(10)63160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Torelli S, Brown SC, Brockington M, et al. Sub-cellular localisation of fukutin related protein in different cell lines and in the muscle of patients with MDC1C and LGMD2I. Neuromuscul Disord. 2005;15:836–843. doi: 10.1016/j.nmd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 120.Esapa CT, McIlinney RA, Blake DJ. Fukutin-related protein mutations that cause congenital muscular dystrophy result in ER-retention of the mutant protein in cultured cells. Hum Molec Genet. 2005;14:295–305. doi: 10.1093/hmg/ddi026. [DOI] [PubMed] [Google Scholar]
- 121.Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853–2861. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- 122.Peyrard M, Seroussi E, Sandberg-Nordquist AC, et al. The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc Natl Acad Sci USA. 1999;96:598–603. doi: 10.1073/pnas.96.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Holzfeind PJ, Grewal PK, Reitsamer HA, et al. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet. 2002;11:2673–2687. doi: 10.1093/hmg/11.21.2673. [DOI] [PubMed] [Google Scholar]
- 124.Barresi R, Michele DE, Kanagawa M, et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med. 2004;10:696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- 125.Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin alpha-7 gene cause congenital myopathy. Nature Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 126.Pegoraro E, Cepollaro F, Prandini P, et al. Integrin alpha 7 beta 1 in muscular dystrophy/myopathy of unknown etiology. Am J Pathol. 2002;160:2135–2143. doi: 10.1016/s0002-9440(10)61162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burkin DJ, Kaufman SJ. The α7β1 integrin in muscle development and disease. Cell Tissue Res. 1999;296:183–190. doi: 10.1007/s004410051279. [DOI] [PubMed] [Google Scholar]
- 128.Mayer U, Saher G, Fassler R, et al. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 129.Hodges BL, Hayashi YK, Nonaka I, Wang W, Arahata K, Kaufman SJ. Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J Cell Sci. 1997;110:2873–2881. doi: 10.1242/jcs.110.22.2873. [DOI] [PubMed] [Google Scholar]
- 130.Dubowitz V. Rigid spine syndrome: a muscle syndrome in search of a name. Proc R Soc Med. 1973;66:219–220. doi: 10.1177/003591577306600304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moghadaszadeh B, Topaloglu H, Merlini L, et al. Genetic heterogeneity of congenital muscular dystrophy with rigid spine syndrome. Neuromuscul Disord. 1999;9:376–382. doi: 10.1016/s0960-8966(99)00051-6. [DOI] [PubMed] [Google Scholar]
- 132.Flanigan KM, Kerr L, Bromberg MB, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol. 2000;47:152–161. [PubMed] [Google Scholar]
- 133.Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739–749. doi: 10.1086/342719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635–649. doi: 10.1016/j.nmd.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 135.Venance SL, Koopman WJ, Miskie BA, Hegele RA, Hahn AF. Rigid spine muscular dystrophy due to SEPN1 mutation presenting as cor pulmonale. Neurology. 2005;64:395–396. doi: 10.1212/01.WNL.0000149755.85666.DB. [DOI] [PubMed] [Google Scholar]
- 136.Moghadaszadeh B, Desqguerre I, Topaloglu H, et al. Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35–36. Am J Hum Genet. 1998;62:1439–1445. doi: 10.1086/301882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 2001;29:17–18. doi: 10.1038/ng713. [DOI] [PubMed] [Google Scholar]
- 138.Tajsharghi H, Darin N, Tulinius M, Oldfors A. Early onset myopathy with a novel mutation in the Selenoprotein N gene (SEPN1) Neuromuscul Disord. 2005;15:299–302. doi: 10.1016/j.nmd.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 139.Allamand V, Richard P, Lescure A, et al. A single homozygous point mutation in a 3′ untranslated region motif of selenoprotein N mRNA causes SEPN1-related myopathy. EMBO Rep. 2006;7:450–454. doi: 10.1038/sj.embor.7400648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;7:739–749. doi: 10.1086/342719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. 2004;55:676–686. doi: 10.1002/ana.20077. [DOI] [PubMed] [Google Scholar]
- 142.Petit N, Lescure A, Rederstorff M, et al. Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet. 2003;12:1045–1053. doi: 10.1093/hmg/ddg115. [DOI] [PubMed] [Google Scholar]
- 143.Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739–749. doi: 10.1086/342719. [DOI] [PMC free article] [PubMed] [Google Scholar]