

Abstract
Recognition of the importance of the endocannabinoid system in both homeostasis and pathologic responses raised interest recently in the development of therapeutic agents based on this system. The CB2 receptor, a component of the endocannabinoid system, has significant influence on immune function and inflammatory responses. Inflammatory responses are major contributors to central nervous system (CNS) injury in a variety of diseases. In this report, we present evidence that activation of CB2 receptors, by selective CB2 agonists, reduces inflammatory responses that contribute to CNS injury. The studies demonstrate neuroprotective effects in experimental autoimmune encephalomyelitis, a model of multiple sclerosis, and in a murine model of cerebral ischemia/reperfusion injury. In both cases, CB2 receptor activation results in reduced white cell rolling and adhesion to cerebral microvessels, a reduction in immune cell invasion, and improved neurologic function after insult. In addition, administration of the CB1 antagonist SR141716A reduces infarct size following ischemia/reperfusion injury. Administration of both a selective CB2 agonist and a CB1 antagonist has the unique property of increasing blood flow to the brain during the occlusion period, suggesting an effect on collateral blood flow. In summary, selective CB2 receptor agonists and CB1 receptor antagonists have significant potential for neuroprotection in animal models of two devastating diseases that currently lack effective treatment options.
Keywords: CB1, CB2, endocannabinoid system, neuroprotection, EAE, stroke
Introduction
Interest in the medicinal use of Cannabis sativa (marijuana) has a long historical record, extending back thousands of years. In comparison to the extensive history for medicinal applications of marijuana, the existence of an “endocannabinoid system”, with important homeostatic and pathologic functions, has only recently gained appreciation. The endocannabinoid system consists of endogenously produced cannabinoids, their receptors, and the enzymes responsible for their synthesis and degradation. The two most widely studied endogenous cannabinoids are N-arachidonyl ethanolamide and 2-arachidonoyl-glycerol (2-AG). Receptors for endogenous cannabinoids include the CB1 receptor, the CB2 receptor, vanilloid, GRP55, and other unidentified receptors. The enzymes responsible for the degradation of anandamide and 2-AG are the fatty acid amide hydrolase (FAAH) and monoacylglyceride (MGL), respectively. Although used in ancient Greece, Rome, and China for therapeutic purposes, concern about the use of cannabinoids as a drug of abuse has dampened interest in developing the potential therapeutic benefits of these compounds (Mechoulam et al. 1998). However, a better understanding of the biologic effects has led recently to an upsurge in interest for the development of therapeutic drugs through modification of the endocannabinoid system (Mackie 2006). An additional incentive was provided by the development of synthetic cannabinoid analogs and specific inhibitors of cannabinoid receptors. Several excellent reviews cover the therapeutic potential of cannabinoids, including their potential for immune suppression (Pertwee 1999; Klein 2005; Klein and Cabral 2006; Mackie 2006; Pacher et al. 2006). The present review is focused on the effects of CB2 receptor activation in models of multiple sclerosis (experimental autoimmune encephalomyelitis) and stroke (middle cerebral occlusion/reperfusion).
The central nervous system (CNS) is an obvious target for therapeutic manipulation of the endocannabinoid system. The most prevalent G-coupled protein receptors in the brain are CB1 receptors. Initial studies were focused on the activation of CB1 receptors to provide protection from excitotoxic injury, ischemia/reperfusion injury, and CNS autoimmune diseases (Nagayama et al. 1999; van der Stelt 2001a, b; Parmentier-Batteur et al. 2002; Muthian et al. 2004; Shouman et al. 2006). Subsequent recognition of the contribution of inflammation to CNS injury and of the immunomodulatory properties of CB2 receptors led to an increased focus on CB2 receptors as a therapeutic target. Unlike CB1 receptors, CB2 receptor activation is not associated with psychoactive effects. Therefore, targeting CB2 receptors with selective agonists has the added benefit of taking advantage of the immunomodulatory activity without psychoactive effects. In this manuscript, we will discuss the use of selective CB2 receptor agonists and CB1 receptor antagonists in two models of CNS injury, i.e., experimental autoimmune encephalomyelitis (EAE) and ischemia/reperfusion injury, as examples of diseases that might be treated through the manipulation of the endocannabinoid system.
Effects on EAE
Multiple sclerosis (MS), a chronic demyelinating disease of the central nervous system, is a major cause of neurological disability. MS is recognized as an autoimmune neurodegenerative disease triggered by inflammatory attacks in the CNS. Autoimmunity is considered the driving force behind the inflammatory processes leading to primary demyelination, whereas neurodegeneration becomes an essential player later on during disease progression. EAE is an animal model used to replicate many of the pathologic changes that occur in MS, and serves to characterize the sequential events leading to CNS autoimmune pathology (Steinman 1996). The characteristics of disease progression in EAE are dependent upon the method used to induce an autoimmune reaction against myelin. Immunization of C57BL/6 mice with myelin oligodendrocyte protein (MOG) results in the development of a chronic form of EAE. Immunization of SJL/J mice with proteolipid protein (PLP) results in the development of a remitting/relapsing form of EAE. EAE can also be induced by an adoptive transfer technique, in which myelin antigen-specific T cells are injected into naïve recipients.
Initially, the CB1 receptor was the primary focus of attention for studies using cannabinoid administration to alter EAE progression. It was hypothesized that, since the CB1 receptor inhibits synaptic transmission, its activation could be used to control clinical EAE including spasticity, tremor, and paralysis (Lyman et al. 1989; Wirguin et al. 1994; Baker et al. 2000). Later studies demonstrated that administration of competitive endocannabinoid uptake inhibitors and selective inhibitors of FAAH also attenuated spasticity in a chronic relapsing model of EAE (Baker et al. 2001). Further support for the potential protective effect of CB1 receptor activation was provided by an elegant study employing a conditional knockout EAE model in which neurons were depleted of the CB1 receptor, while the receptor remained intact in other cell types (Maresz et al. 2007).
The first evidence that selective activation of the CB2 receptor was protective in EAE emerged from an investigation of the effects of WIN55212–2 on the progression of EAE in C57BL/6 mice immunized with MOG35–55 (Ni et al. 2004). We were able to demonstrate that administration of WIN55212–2, which functions as an agonist for CB1 receptors and CB2 receptors, attenuated EAE progression. The fact that administration of the CB1 receptor antagonist SR141716A did not affect the protective effect, whereas the CB2 receptor antagonist SR144528blocked it, led to the conclusion that the protective effect of WIN55212–2 occurred through activation of CB2 receptors. However, although administration of the CB1 receptor antagonist did not attenuate the protective effects of WIN55212–2, CB1 receptor activation may play a neuroprotective role in later stages of the disease (Baker et al. 2000; Maresz et al. 2007).
The conclusion that the CB2 receptor played a key role in attenuating EAE progression through modulation of the immune response was supported by later studies utilizing the highly selective synthetic CB2 receptor agonist O-1966. The fact that O-1966 functions as a CB2 receptor agonist is based upon investigations using 3H-CP 55,940 binding to rat brain membranes and to Chinese hamster ovary cells (CHO) stably transfected with human CB1 and CB2 receptors. The affinity of O-1966 for CB1 and CB2 receptors was reported to be 5,055±984 and 23±2.1 nM, respectively. O-1966 stimulated 35S-GTPγS binding with an EC50 of 70±14 nM and an Emax of 74±5 (percent of maximal stimulation produced by the full agonist CP 55,940). The likelihood to produce psychoactive effects through activation of the CB1 receptor is tested using the “tetrad test” which consists of four individual tests: analgesia, sedation catalepsy, and hypothermia (Little et al. 1988). Intravenous administration of O-1966 to mice failed to produce effects in the tetrad test in doses up to 30 mg/kg, consistent with its very low CB1 receptor affinity. The ED50s of the CB1/CB2 receptor agonists used as control are approximately 1–2 mg/kg. Based on these characteristics, O-1966 was defined as a full CB2 receptor agonist (Wiley et al. 2002). Investigations of the effects of O-1966 on EAE progression demonstrated that administration of O-1966 resulted in significantly improved motor function in the chronic EAE model (C57BL/6/MOG), in the remitting–relapsing model (SJL/J/PLP), and in an adoptive transfer model (Fig. 1). In the C57BL/6/MOG model, we tested three concentrations of O-1966 (0.1, 1, and 10 mg/kg). All three concentrations were protective, with the 1 mg/kg dose being optimal. O-1966 was also found to effectively attenuate disease progression when administered after the onset of symptoms in the remitting–relapsing model. The protective effect of CB2 receptor activation on EAE has been confirmed recently in a report showing that encephalitogenic T cells from CB2-deficient mice were more aggressive in terms of CNS infiltration and caused more severe EAE (Maresz et al. 2007). Interestingly, although T cells from CB2-receptor-deficient mice were more aggressive in the CNS, their proliferation, apoptosis, and cytokine production in the spleen was not altered compared to wild-type T cells. It has been proposed that the difference between the activation of CB2-receptor-deficient T cells in spleen and CNS was the result of a lower level of endocannabinoid production in spleen (Dittel 2008).
Fig. 1.

Effects of O-1966 administration in models of EAE. a C57BL/6 mice were immunized with MOG35–55/CFA and pertussis toxin (days 0 and +2). One group received O-1966 (1 mg/kg) on day 7 and every fourth day thereafter (up to day 28). The control group received vehicle. *p<0.01. b One group of C57BL/6 mice was immunized with MOG35–55/CFA and a second group was immunized and treated with O-1966 on days 5 and 9. On day 11 post-immunization, the mice were sacrificed and splenocytes were restimulated ex vivo with MOG35–55 in the presence of IL-2 (2 U/ml), anti-IFNγ Ab (10 μg/ml), and rIL-12 (5 ng/ml). Four days later, splenic CD4+ T cells (1×107) were injected i.p. into naïve C57BL/6 recipients. *p<0.05. c SJL/J mice were immunized with PLP139–151/CFA and pertussis toxin (day 0). One group was treated with O-1966 (1 mg/kg) on day 7 and every fourth day thereafter (up to day 28). The control group received vehicle. *p< 0.01. d SJL/J mice were immunized with PLP139–151/CFA and pertussis toxin (day 0). One group was treated with O-1966 (1 mg/kg) on day 11 after development of clinical symptoms and every fourth day thereafter (up to day 27). The control group received vehicle. *p<0.05. Disease scores were assigned on the basis of motor function as follows: 0, no abnormality; 1, limp tail or hind limb weakness (waddling gait); 2, limp tail and hind limb weakness; 3, partial hind limb paralysis; 4, complete hind limb paralysis; and 5, moribund. Disease scores were compared using ANOVA with repeated measures
As illustrated in Fig. 2, there are numerous potential sites of action that could be affected by O-1966. Although the etiology of MS remains to be elucidated, it is widely accepted that cellular autoimmune responses, with heavy emphasis on T cells, play an essential role in the development and progression of MS immunopathology (Steinman 1996). EAE models can serve to define and characterize the sequential events leading to CNS autoimmune inflammation (Becher et al. 2006). The three major phases, the first occurring in the periphery and the next two in the CNS, consist of: priming/activation of T cells, recruitment of activated T cells into the CNS, followed by reactivation in the perivascular space, and the effector phase, characterized by inflammatory cell migration into parenchyma, microglial activation, and destruction of oligodendrocytes.
Fig. 2.

Proposed sites of CB2 agonist action in EAE include peripheral and perivascular dendritic cells, T cells, endothelial cells, and microglia
In EAE, the priming of encephalitogenic CD4+ T cells occurs in the periphery following immunization. Activation of naïve T cells requires cognate contact with dendritic cells (DC) which present the processed antigen in the context of MHC II and costimulatory molecules (CD40, CD80, CD86). At this stage, depending on the nature of DC, i.e., stimulatory or tolerogenic, CD4+ T cells could follow two alternative pathways, differentiating into either proinflammatory Th17/Th1 effectors or anti-inflammatory Th2/Treg cells. Following activation and maturation into effectors, T cells migrate to the CNS, cross the blood–brain barrier (BBB), and are reactivated by perivascular antigen-presenting cells. Recent studies proved that perivascular DC are the major players in encephalitogenic T cell reactivation (Hickey and Kimura 1988; Archambault et al. 2005; Greter et al. 2005; McMahon et al. 2005, 2006).
Reactivated T cells penetrate deeper into the CNS parenchyma where they activate resident microglia to secrete oxygen radicals, nitric oxide, proinflammatory cytokines and chemokines, and further promote local inflammation and attract additional inflammatory cells. CD40L (CD154)-expressing T cells upregulate IL-23 expression and release from microglia (Becher et al. 2006), and IL-23 maintains the function of the critical pathogenic cells in EAE/MS, the Th17 effectors. Although microglia do not function as APCs to allow T cell entry in the CNS, they play a critical role in the effector phase, and blocking microglial activation represses EAE (Heppner et al. 2005).
The possibility that the selective CB2 receptor agonist could modulate DC and T cell differentiation was investigated. Previously published reports indicate that delta(9)-tetrahydrocannabinol (THC), a CB1/CB2 receptor agonist, and the CB2 agonist JWH-133 shift the Th1/Th2 balance in favor of Th2, presumably through the inhibition of IL-12 production by antigen-presenting cells (Yuan et al. 2002; Correa et al. 2005; Lu et al. 2006). In mice infected with Legionella, THC was reported to inhibit IL-12 and IL-12 receptor expression through CB1 receptors and to promote IL-4 and GATA-3 expression through CB2 receptors (Klein et al. 2004). To more thoroughly investigate this possibility, dendritic cells from C57BL/6 mice treated with O-1966 (DCCB2) were washed extensively to remove all traces of O-1966 and added to allogeneic B10. A purified splenic CD4+ T cells. There was significant T cell proliferation in response to regular DC, whereas T cells exposed to DCCB2 did not proliferate (Fig. 3a). Culture supernatants were tested for secreted cytokines/chemokines by using Bioplex technology. Results for IFNγ, IL-4, and IL-10 were confirmed by regular ELISA. A number of proinflammatory cytokines and chemokines, including IFNγ, IL-6, MIP-1α, MCP-1, and KC (murine counterpart of human IL-8), were reduced in cultures containing DCCB2. In contrast, IL4, IL-13, and IL-10 (characteristic of Th2 and possibly Treg) were upregulated in the same cultures (Fig. 3b). In addition, DCCB2 strongly inhibited IL-17 release, presumably through the inhibition of Th17 differentiation (Fig. 3c). This suggests that DCCB2 induce a shift in T cell differentiation from Th1/Th17 to Th2/Treg. Since only DC were exposed to the CB2 agonist, the effects on T cell proliferation and cytokine profile were induced through the action of O-1966 on DC. While the results of these studies clearly showed an effect on DC function, we were unable to demonstrate any direct effect on T cells stimulated with anti-CD3 and anti-CD28 with respect to proliferation and cytokine profile.
Fig. 3.

Differentiation of DC in the presence of the CB2 receptor agonist O-1966 affects their capacity to stimulate T cell proliferation and the cytokine/chemokine profile. a Bone marrow-derived DC from C57BL/6 (H-2b) mice were differentiated in the presence or absence of 10−7 M O-1966 (DCCB2 or DC) for 7 days. Different numbers of purified CD11c+ DCCB2 or DC were co-cultured with purified CD4+ T cells from B10. A mice (H-2k) (1×106 cells/ml) for 4 days. Proliferation was determined by BrdU incorporation. Controls included T cells alone, DC or DCCB2 alone. b DCCB2 or DC (2.5×105 cells/ml) were co-cultured with CD4+ T cells (1×106 cells/ml) for 48 h. Culture supernatants were tested for cytokine/chemokine release by Bioplex technology. c DCCB2 were generated in the presence of various O-1966 concentrations (10−7, 10−8, and 10−9 M) and co-cultured with allogeneic T cells for 48 h. IFNγ and IL-17 secretion was determined by ELISA
As described previously, white blood cells must firmly adhere to endothelial cells before crossing the BBB. This involves interaction among adhesion molecules on endothelial cells and leukocytes. Expression of these molecules occurs sequentially in response to a variety of inflammatory stimuli. We have used implanted cranial windows to evaluate leukocyte/endothelial cell interactions in chronic EAE. Implantation of cranial windows allowed visualization of the pial microcirculation. White blood cells labeled with rhodamine 6G were clearly seen rolling and adhering to endothelial cells using epi-illumination fluorescence microscopy. The number of rolling leukocytes was considered to be the total number of leukocytes moving along the endothelial cells at a substantially slower velocity compared with the midstream blood cell velocity. They were counted as numbers of white blood cells passing an arbitrary line perpendicular to the longitudinal axis of the vessel over a period of 30 s. Sticking leukocytes were defined as the total number of leukocytes firmly attached to the microvascular endothelium not changing their location during the 30 s of observation. We have shown previously that treatment of EAE mice with WIN55212–2 and with WIN55212–2 in conjunction with a CB1 receptor antagonist significantly reduced the number of leukocytes rolling along the cerebral microvessels on day 15 post-immunization. Treatment with WIN55212–2 in conjunction with a CB2 receptor antagonist reversed the protective effect of WIN55212–2 (Ni et al. 2004). We obtained similar results upon treatment with O-1996. Increased leukocyte rolling and adhesion was evident on day 15 post-immunization, and i.p. administration of O-1966 significantly attenuated rolling and adhesion on cerebral microvessels (Fig. 4). These results support the hypothesis that adhesion molecules on T cells and/or endothelial cells may be directly affected by administration of CB2 receptor agonists.
Fig. 4.

In vivo administration of O-1966 reduces endogenous leukocyte rolling and adhesion. C57BL/6 mice were immunized with MOG35–55. One group was treated with O-1966 (1 mg/kg) on days 7, 11, and 15. The control group received vehicle. Sham—non-immunized mice. Leukocyte rolling and adhesion to pial microvessels was measured on day 15 by intravital microscopy (cranial window) as previously described. *p<0.05, ***p<0.001 (Ni et al. 2004)
In summary, the results described above provide evidence that treatment of EAE mice with a selective CB2 receptor agonist leads to a significant attenuation of white cell trafficking across the blood–brain barrier and that dendritic cells differentiated in the presence of a selective CB2 receptor agonist inhibit T cell proliferation and shift the cytokine response from IFNγ to IL-4/IL-13/IL-10. Activation of cannabinoid receptors on microglia was also reported to be protective (Stella 2004; Eljaschewitsch et al. 2006). All of these effects could contribute to an attenuation of EAE progression. Recently, it has been shown that high concentrations of IFNγ in the CNS of EAE mice may disrupt P2×7 purinergic receptor signaling and inhibit the neuroprotective effects of endogenous cannabinoids (Witting et al. 2006). In this context, inhibition of IFNγ by exogenous CB2 agonists may reduce or prevent the disruption of P2×7 signaling.
Although the studies described above emphasize the therapeutic potential of the CB2 receptor for treatment of multiple sclerosis through immunomodulation, there is evidence that CB1 receptor activation may have beneficial effects at later stages of the disease, since alleviation of spasticity appears to be mediated through direct effects on neuronal CB1 receptors (Baker et al. 2000; Maresz et al. 2007).
Effects on cerebral ischemia/reperfusion injury
While the mechanisms responsible for CNS damage following ischemia/reperfusion injury differ substantially from those responsible for damage in EAE, there are a number of processes common to both. One process contributing to damage in both cases is the capture of white blood cells from the flowing blood and invasion of the CNS parenchyma, where the cells become activated and release substances toxic to normal tissue. Numerous studies have provided evidence that inflammatory cell invasion and activation following ischemia contributes substantially to secondary injury (Hallenbeck et al. 1986, 1988; Dutka et al. 1987, 1988, 1989; Kochanek et al. 1987a, b, 1988; Kochanek and Hallenbeck 1992; Dawson et al. 1996; Weaver et al. 2002). Having demonstrated that administration of the selective CB2 receptor agonist O-1966 attenuated white cell rolling and adhesion to pial microvessels in EAE, we decided to determine whether this effect also occurred during reperfusion following ischemia. C57BL/6 mice were again used as the animal model in these studies. Ischemia was induced by an intravascular suture advanced through a branch of the external carotid artery to the origin of the middle cerebral artery. The suture was kept in place for 1 h and then removed to allow reperfusion. Brain and body temperature were maintained at 37°C. Maintaining body temperature is an important consideration, since CB1 receptor agonists can induce hypothermia, with a resulting neuroprotective effect. A LaserPro Blood Perfusion monitor was used to monitor regional cerebral blood flow before ischemia, during middle cerebral artery occlusion, and after reperfusion. In these studies, two different selective CB2 receptor agonists were injected either 1 h before middle cerebral artery occlusion or during reperfusion. White cell rolling and adhesion and infarct volume were monitored. As illustrated in Fig. 5, both CB2 agonists significantly attenuated leukocyte rolling and adhesion during reperfusion (Zhang et al. 2007). Immunohistochemical staining to evaluate the presence of activated macrophages/microglia (CD11b) and expression of an endothelial adhesion molecule (ICAM-1) 24 h following reperfusion indicate that CB2 receptor activation decreases both ICAM-1 and CD11b expression (Fig. 6). As would be predicted from the effect on white cell/endothelial cell interactions, administration of both selective CB2 agonists caused a significant reduction in infarct volume measured 24 h after reperfusion (Fig. 7).
Fig. 5.

In vivo administration of CB2 receptor agonists reduces leukocyte rolling and adhesion during cerebral ischemic/reperfusion injury. C57BL/6 mice were subjected to middle cerebral artery occlusion and reperfusion as described (Zhang et al. 2007). One group received O-1966, a second group received the CB2 receptor agonist O-3853, and the control group received vehicle. Leukocyte rolling and adhesion to cranial venules and arterioles before, 1 h, and 24 h after MCAO was determined by intravital microscopy as described previously. *p<0.05 (Zhang et al. 2007)
Fig. 6.

Administration of O-1966 reduces CD11b and ICAM-1 expression during cerebral ischemic/reperfusion injury. Expression of CD11b and ICAM-1 was determined by immunohistochemistry on brain slices obtained 24 h after ischemia. Four treatment groups were compared: vehicle treated, O-1966 administered prior to MCAO, O-1966 administered 1 h after occlusion, and O-1966 administered 3 h after occlusion
Fig. 7.
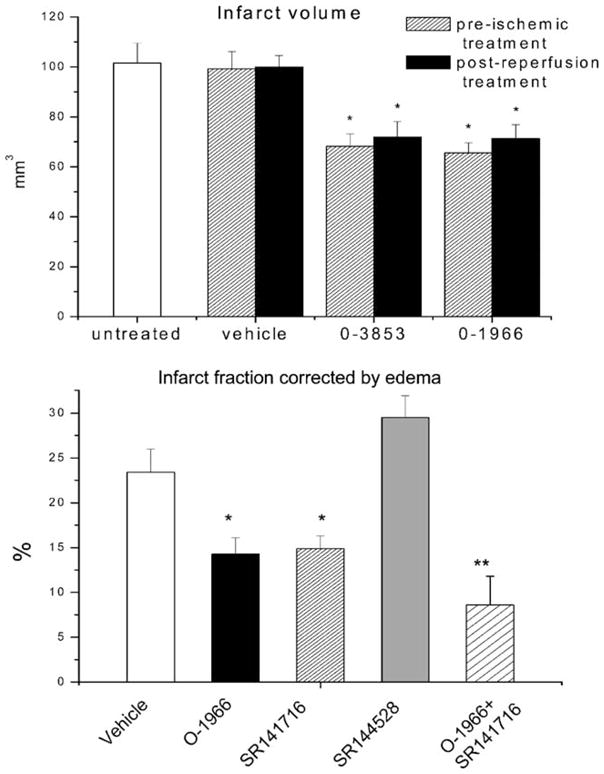
Effects of CB2 receptor agonists on infarct volume. a Administration of O-1966 or O-3853 1 h before (pre-ischemic) or 10 min after reperfusion reduced the infarct volume as compared to untreated or vehicle-treated animals. b The CB2 receptor agonist O-1966 (1 mg/kg), the CB1 receptor antagonist (SR141716A) (5 mg/kg), CB2 receptor antagonist (SR141528) (5 mg/kg), or combinations of O-1966 and the CB1 receptor antagonist were given prior to occlusion. Y axis represents % of infarcted hemisphere. *p<0.05, **p<0.01 (from Zhang et al. 2008)
CB2 receptor activation has been reported to protect other tissues from ischemia/reperfusion injury. There is a significant reduction in myocardial ischemia/reperfusion injury associated with CB2 receptor activation (Pacher et al. 2006; Pacher and Hasko 2008). It has also been proposed that CB2 receptor activation may be involved in ischemic reconditioning (Pacher and Hasko 2008). CB2 receptor activation has also been shown to significantly attenuate ischemia/reperfusion injury in the liver (Batkai et al. 2007). As reported for the CNS, this protection was associated with a reduction in inflammatory cell invasion, reduced inflammatory cytokine production, and reduction in the expression of adhesion molecules by endothelial cells. Ischemia/reperfusion in the liver was found to significantly enhance endocannabinoid production by hepatocytes, Kuppfer and endothelial cells, and correlated with the degree of hepatic damage and serum TNFα, MIP-1α, and MIP-2 levels. Exogenous CB2 receptor agonists were shown to be protective in hepatic ischemia/reperfusion, to reduce TNFα, MIP-1α, and MIP-2 release as well as caspase 3 activation and DNA fragmentation in hepatocytes (Rajesh et al. 2007).
Production of endogenous cannabinoids is upregulated following CNS ischemia as is the expression of cannabinoid receptors (Schmid et al. 1995; Jin et al. 2000; Muthian et al. 2004; Ashton et al. 2007; Zhang et al. 2008). To evaluate the contribution of endocannabinoids to ischemia/reperfusion injury, the CB1 receptor antagonist SR141716A and the CB2 receptor antagonist SR144528 were administered prior to ischemia. Consistent with the finding that activation of the CB2 receptor was protective, administration of the CB2 receptor antagonist increased infarct size following stroke (Zhang et al. 2008). In our experiments, the CB1 receptor antagonist was also protective, suggesting that CB1 receptor signaling played a detrimental role in ischemia/reperfusion (Fig. 7) This is in agreement with the protective effect of CB1 receptor antagonists reported in previous studies (Berger et al. 2004; Muthian et al. 2004). However, several other studies reported a protective effect for CB1 receptor signaling in ischemia/reperfusion. Nagayama et al. reported that the protective effect of WIN55212–2, a CB1/CB2 receptor agonist, was blocked by a CB1 receptor antagonist (Nagayama et al. 1999). The fact that CB1 receptor-deficient mice had larger infarct areas and lower blood flow in the ischemic penumbra also supported a protective role for CB1 receptors (Parmentier-Batteur et al. 2002). Several possible mechanisms could explain the different results. One explanation may be related to temperature. A number of investigators have reported that the protective effect of CB1 receptor activation was lost when animals were maintained at normal body temperature, suggesting that the protective effect of CB1 receptor signaling was related to hypothermia (Leker et al. 2003; Hayakawa et al. 2004). In our experiments, brain and body temperature were maintained at 37°C. Regarding the CB1 receptor-deficient mice, it must be recognized that embryonic deletion of the CB1 receptor may lead to abnormal CNS development, making these animals more susceptible to ischemia and therefore it may not reflect the actual contribution of the CB1 receptor in attenuating ischemic damage in the acute setting. As for the protective effect of the CB1 receptor antagonist SR141716A reported by us and others, this may be due to intrinsic neuroprotective properties of SR141716A, and not necessarily to CB1 receptor antagonism. Sommer et al. reported recently that, contrary to expectations, administration of SR141716A did not reduce glutamate receptor binding, indicating that the protective effect was not due to a reduction in excitotoxicity (Sommer et al. 2006).
When the CB2 agonist was given in combination with the CB1 receptor antagonist, changing the activity of both receptors simultaneously, the protective effects were additive indicating that these compounds might be working through different mechanisms (Fig. 7). An additional interesting result was the finding that this combination had significant influence on blood flow during occlusion. While neither the CB2 receptor agonist nor the CB1 receptor antagonist had an effect, when given together, they significantly increased blood flow during the occlusion (Fig. 8). This implies a potential for this combination to improve the collateral flow during cerebral ischemia. Determining the mechanism of action is however complicated by the fact that the CB1 receptor antagonist SR141716A has a number of effects in addition to its action as a CB1 receptor antagonist. SR141716A has inverse agonist properties and may function as an agonist at receptors other than CB1 or CB2 (Pacher et al. 2006). CB1 receptor-deficient mice exhibit vasodilation in response to cannabinoids and vasodilation is still inhibited by SR141716A (Jarai et al. 1999; O’Sullivan et al. 2004a, b). There is evidence that endogenous cannabinoids may induce vasodilation through vanilloid receptors present on endothelial cells (Louw et al. 2000; Golech et al. 2004; McCollum et al. 2007). In addition, CB1 receptors and vanilloid receptors are coexpressed on sensory nerves with vasoactive properties. There is also evidence that CB1 receptor activation may cause effects opposite to those produced by cannabinoids via vanilloid receptors (Ralevic et al. 2002). The mechanisms responsible for this unique effect on cerebral blood flow during middle cerebral artery occlusion may therefore involve complicated interactions among identified and unidentified cannabinoid receptors and remain to be elucidated.
Fig. 8.

Effects of drug treatment on blood flow during cerebral ischemic/reperfusion injury. The CB2 receptor agonist O-1966 (1 mg/kg), the CB1 receptor antagonist (SR141716A) (5 mg/kg), the CB2 receptor antagonist (SR141528) (5 mg/kg), or a combination of O-1966 and the CB1 receptor antagonist were given prior to occlusion. Blood flow was evaluated using laser Doppler measurements. N=6. *p<0.05 (from Zhang et al. 2008)
Summary
The CB2 receptor has significant influence on inflammatory responses during EAE and following ischemia/reperfusion injury. In both cases, CB2 receptor activation results in reduced white cell rolling and adhesion on cerebral micro-vessels, less immune cell invasion of the CNS, and improved neurological function after insult. In EAE, alterations in dendritic cell function may represent an important mechanism of action.
Administration of the commonly used CB1 receptor antagonist SR141716A was found to be protective against ischemia/reperfusion injury, and the combination of the CB2 receptor agonist O-1966 and the CB1 receptor antagonist SR141716A provided the highest level of protection in an animal model of stroke. The protection provided by SR141716A may be independent of the CB1 receptors and may involve activation of other, as yet unidentified, receptors.
These results provide evidence that alteration of the activation patterns of the various cannabinoid receptors warrant consideration for future therapeutic strategies.
Acknowledgments
This project is funded, in part, under a grant with the Pennsylvania Department of Health, a contract from BTG (London), and grants from DA P30 13429, DA 03672, and DA 05488 from the National Institute on Drug Abuse.
References
- Archambault AS, Sim J, et al. Defining antigen-dependent stages of T cell migration from the blood to the central nervous system parenchyma. Eur J Immunol. 2005;35(4):1076–1085. doi: 10.1002/eji.200425864. [DOI] [PubMed] [Google Scholar]
- Ashton JC, Rahman RM, et al. Cerebral hypoxia–ischemia and middle cerebral artery occlusion induce expression of the cannabinoid CB2 receptor in the brain. Neurosci Lett. 2007;412(2):114–117. doi: 10.1016/j.neulet.2006.10.053. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, et al. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404(6773):84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15(2):300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- Batkai S, Osei-Hyiaman D, et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J. 2007;21(8):1788–1800. doi: 10.1096/fj.06-7451com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Bechmann I, et al. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84(7):532–543. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- Berger C, Schmid PC, et al. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia. J Neurochem. 2004;88(5):1159–1167. doi: 10.1046/j.1471-4159.2003.02244.x. [DOI] [PubMed] [Google Scholar]
- Correa F, Mestre L, et al. Activation of cannabinoid CB2 receptor negatively regulates IL-12p40 production in murine macrophages: role of IL-10 and ERK1/2 kinase signaling. Br J Pharmacol. 2005;145(4):441–448. doi: 10.1038/sj.bjp.0706215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Ruetzler CA, et al. Polymorphonuclear leukocytes and microcirculatory perfusion in acute stroke in the SHR. Keio J Med. 1996;45(3):248–252. doi: 10.2302/kjm.45.248. discussion 252–253. [DOI] [PubMed] [Google Scholar]
- Dittel BN. Direct suppression of autoreactive lymphocytes in the central nervous system via the CB2 receptor. Br J Pharmacol. 2008;153(2):271–276. doi: 10.1038/sj.bjp.0707493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutka AJ, Hallenbeck JM, et al. A brief episode of severe arterial hypertension induces delayed deterioration of brain function and worsens blood flow after transient multifocal cerebral ischemia. Stroke. 1987;18(2):386–395. doi: 10.1161/01.str.18.2.386. [DOI] [PubMed] [Google Scholar]
- Dutka AJ, Kochanek P, et al. Air embolism may cause unrecognized ischemia of the gray–white junction. Undersea Biomed Res. 1988;15(2):99–106. [PubMed] [Google Scholar]
- Dutka AJ, Kochanek PM, et al. Influence of granulocytopenia on canine cerebral ischemia induced by air embolism. Stroke. 1989;20 (3):390–395. doi: 10.1161/01.str.20.3.390. [DOI] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49(1):67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Golech SA, McCarron RM, et al. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res Mol Brain Res. 2004;132(1):87–92. doi: 10.1016/j.molbrainres.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11(3):328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Hallenbeck JM, Dutka AJ, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17(2):246–253. doi: 10.1161/01.str.17.2.246. [DOI] [PubMed] [Google Scholar]
- Hallenbeck JM, Dutka AJ, et al. Stroke risk factors prepare rat brainstem tissues for modified local Shwartzman reaction. Stroke. 1988;19(7):863–869. doi: 10.1161/01.str.19.7.863. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, et al. Cannabidiol prevents infarction via the non-CB1 cannabinoid receptor mechanism. Neuroreport. 2004;15(15):2381–2385. doi: 10.1097/00001756-200410250-00016. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11(2):146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239(4837):290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Jarai Z, Wagner JA, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci U S A. 1999;96(24):14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, et al. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol. 2000;48(2):257–261. doi: 10.1002/1531-8249(200008)48:2<257::AID-ANA18>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5(5):400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- Klein TW, Cabral GA. Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol. 2006;1:50–64. doi: 10.1007/s11481-005-9007-x. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, et al. Cannabinoid receptors and T helper cells. J Neuroimmunol. 2004;147(1–2):91–94. doi: 10.1016/j.jneuroim.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23(9):1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Dutka AJ, et al. Indomethacin, prostacyclin, and heparin improve postischemic cerebral blood flow without affecting early postischemic granulocyte accumulation. Stroke. 1987a;18 (3):634–637. doi: 10.1161/01.str.18.3.634. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Dutka AJ, et al. Platelet activating factor receptor blockade enhances recovery after multifocal brain ischemia. Life Sci. 1987b;41(24):2639–2644. doi: 10.1016/0024-3205 (87)90278-5. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Dutka AJ, et al. Effects of prostacyclin, indomethacin, and heparin on cerebral blood flow and platelet adhesion after multifocal ischemia of canine brain. Stroke. 1988;19 (6):693–699. doi: 10.1161/01.str.19.6.693. [DOI] [PubMed] [Google Scholar]
- Leker RR, Gai N, et al. Drug-induced hypothermia reduces ischemic damage: effects of the cannabinoid HU-210. Stroke. 2003;34(8):2000–2006. doi: 10.1161/01.STR.0000079817.68944.1E. [DOI] [PubMed] [Google Scholar]
- Little PJ, Compton DR, et al. Pharmacology and stereo-selectivity of structurally novel cannabinoids in mice. J Pharmacol Exp Ther. 1988;247(3):1046–1051. [PubMed] [Google Scholar]
- Louw DF, Yang FW, et al. The effect of delta-9-tetrahydrocannabinol on forebrain ischemia in rat. Brain Res. 2000;857(1–2):183–187. doi: 10.1016/S0006-8993(99)02422-1. [DOI] [PubMed] [Google Scholar]
- Lu T, Newton C, et al. Cannabinoid treatment suppresses the T-helper cell-polarizing function of mouse dendritic cells stimulated with Legionella pneumophila infection. J Pharmacol Exp Ther. 2006;319(1):269–276. doi: 10.1124/jpet.106.108381. [DOI] [PubMed] [Google Scholar]
- Lyman WD, Sonett JR, et al. Delta 9-tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol. 1989;23(1):73–81. doi: 10.1016/0165-5728(89) 90075-1. [DOI] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- Maresz K, Pryce G, et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB(1) on neurons and CB(2) on autoreactive T cells. Nat Med. 2007;13(4):492–497. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- McCollum L, Howlett AC, et al. Anandamide-mediated CB1/CB2 cannabinoid receptor-independent nitric oxide production in rabbit aortic endothelial cells. J Pharmacol Exp Ther. 2007;321(3):930–937. doi: 10.1124/jpet.106.117549. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, et al. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11(3):335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, et al. CNS dendritic cells: critical participants in CNS inflammation. Neurochem Int. 2006;49(2):195–203. doi: 10.1016/j.neuint.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Fride E, et al. Endocannabinoids. Eur J Pharmacol. 1998;359(1):1–18. doi: 10.1016/S0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- Muthian S, Rademacher DJ, et al. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience. 2004;129(3):743–750. doi: 10.1016/j.neuroscience.2004.08.044. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, et al. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19(8):2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Geller EB, et al. Win 55212–2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler. 2004;10(2):158–164. doi: 10.1191/1352458504ms1009oa. [DOI] [PubMed] [Google Scholar]
- O’Sullivan SE, Kendall DA, et al. Characterisation of the vasorelaxant properties of the novel endocannabinoid N-arachidonoyl-dopamine (NADA) Br J Pharmacol. 2004a;141(5):803–812. doi: 10.1038/sj.bjp.0705643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE, Kendall DA, et al. Heterogeneity in the mechanisms of vasorelaxation to anandamide in resistance and conduit rat mesenteric arteries. Br J Pharmacol. 2004b;142(3):435–442. doi: 10.1038/sj.bjp.0705810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Hasko G. Endocannabinoids and cannabinoid receptors in ischaemia–reperfusion injury and preconditioning. Br J Pharmacol. 2008;153(2):252–262. doi: 10.1038/sj.bjp.0707582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, et al. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier-Batteur S, Jin K, et al. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22 (22):9771–9775. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Medical uses of cannabinoids: the way forward. Addiction. 1999;94(3):317–320. doi: 10.1080/09652149933801. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Pan H, et al. Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J Leukoc Biol. 2007;82(6):1382–1389. doi: 10.1189/jlb.0307180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevic V, Kendall DA, et al. Cannabinoid modulation of sensory neurotransmission via cannabinoid and vanilloid receptors: roles in regulation of cardiovascular function. Life Sci. 2002;71(22):2577–2594. doi: 10.1016/S0024-3205(02)02086-6. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Krebsbach RJ, et al. Occurrence and postmortem generation of anandamide and other long-chain N-acylethanolamines in mammalian brain. FEBS Lett. 1995;375(1–2):117–120. doi: 10.1016/0014-5793(95)01194-J. [DOI] [PubMed] [Google Scholar]
- Shouman B, Fontaine RH, et al. Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. Br J Pharmacol. 2006;148(4):442–451. doi: 10.1038/sj.bjp.0706755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Schomacher M, et al. Neuroprotective cannabinoid receptor antagonist SR141716A prevents downregulation of excitotoxic NMDA receptors in the ischemic penumbra. Acta Neuropathol. 2006;112(3):277–286. doi: 10.1007/s00401-006-0110-8. [DOI] [PubMed] [Google Scholar]
- Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85(3):299–302. doi: 10.1016/S0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- Stella N. Cannabinoid signaling in glial cells. Glia. 2004;48(4):267–277. doi: 10.1002/glia.20084. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Veldhuis WB, et al. Neuroprotection by Delta9-tetrahydrocannabinol, the main active compound in marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci. 2001a;21(17):6475–6479. doi: 10.1523/JNEUROSCI.21-17-06475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Veldhuis WB, et al. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci. 2001b;21(22):8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver M, Leshley K, et al. LEX032, a novel recombinant serpin, protects the brain after transient focal ischemia. Microvasc Res. 2002;63(3):327–334. doi: 10.1006/mvre.2002.2405. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Beletskaya ID, et al. Resorcinol derivatives: a novel template for the development of cannabinoid CB(1)/CB(2) and CB(2)-selective agonists. J Pharmacol Exp Ther. 2002;301(2):679–689. doi: 10.1124/jpet.301.2.679. [DOI] [PubMed] [Google Scholar]
- Wirguin I, Mechoulam R, et al. Suppression of experimental autoimmune encephalomyelitis by cannabinoids. Immunopharmacology. 1994;28(3):209–214. doi: 10.1016/0162-3109(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Witting A, Chen L, et al. Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proc Natl Acad Sci U S A. 2006;103(16):6362–6367. doi: 10.1073/pnas.0510418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Kiertscher SM, et al. Delta 9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J Neuroimmunol. 2002;133(1–2):124–131. doi: 10.1016/S0165-5728(02) 00370-3. [DOI] [PubMed] [Google Scholar]
- Zhang M, Martin BR, et al. Cannabinoid CB(2) receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27(7):1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Martin BR, et al. Modulation of the balance between cannabinoid CB(1) and CB(2) receptor activation during cerebral ischemic/reperfusion injury. Neuroscience. 2008;152(3):753–760. doi: 10.1016/j.neuroscience.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]