

Abstract
Shiga toxins comprise a family of structurally and functionally related protein toxins expressed by Shigella dysenteriae serotype 1 and multiple serotypes of Escherichia coli. While the capacity of Shiga toxins to inhibit protein synthesis by catalytic inactivation of eukaryotic ribosomes has been well described, it is also apparent that Shiga toxins trigger apoptosis in many cell types. This review presents evidence that Shiga toxins induce apoptosis of epithelial, endothelial, leukocytic, lymphoid and neuronal cells. Apoptotic signaling pathways activated by the toxins are reviewed with an emphasis on signaling mechanisms that are shared among different cell types. Data suggesting that Shiga toxins induce apoptosis through the endoplasmic reticulum stress response and clinical evidence demonstrating apoptosis in humans infected with Shiga toxin-producing bacteria are briefly discussed. The potential for use of Shiga toxins to induce apoptosis in cancer cells is briefly reviewed.
Keywords: apoptosis, caspase 8 activation, hemolytic uremic syndrome, microbial pathogenesis, Shiga toxin, Shiga toxin-producing Escherichia coli, Shigella dysenteriae serotype 1
Shiga toxins
Shiga toxins (Stxs) are cytotoxic proteins expressed by the enteric pathogens Shigella dysenteriae serotype 1 and certain serotypes of Escherichia coli designated Stx-producing E. coli (STEC). Stx is expressed by S. dysenteriae serotype 1. STEC may produce one or more genetic variants of Stxs, which are categorized based on their antigenic similarity to Stx. Stx type 1 (Stx1) is essentially identical to Stx, differing by a single amino acid residue in the A-subunit. Stx type 2 (Stx2) is approximately 56% homologous to Stx/Stx1 at the deduced amino acid sequence level. Antibodies raised against Stx/Stx1 fail to cross-react with Stx2 and vice versa. A number of genetic variants of Stx1 and Stx2 have been characterized (Table 1). In contrast to these genotypic differences, Stxs share many properties including molecular structure, enzymatic activity, receptor specificity and intracellular trafficking. All Stxs possess an AB5 structure with an enzymatically active A-subunit of approximately 32 kDa in noncovalent association with five identical B-subunits, with each B-subunit being approximately 7.7 kDa in size [1,2]. X-ray crystallographic analyses of Stxs have shown that the pentameric B-subunits form a ring with the carboxy terminus of the A-subunit interdigitated within the central pore (Figure 1) [3,4]. The A-subunits are highly specific N-glycosidases, which cleave a single adenine residue from the 28S rRNA component of eukaryotic ribosomes [5]. This adenine, located at position 4324 in the rat, is unpaired in a region of non-Watson–Crick base-pairing referred to as a GAGA tetraloop [6]. Toxin-mediated depurination of ribosomes leads to loss of elongation factor binding and protein synthesis inhibition. Stxs are potent protein synthesis inhibitors with 50% cytotoxic doses for many cell types in vitro in femtogram to picogram per milliliter amounts. Because of the extreme sensitivity of Vero cells (African green monkey renal epithelial cells) to the cytotoxic action of Stxs, the toxins are alternatively referred to as verotoxins or verocytotoxins. B-subunits mediate binding to the toxin receptor, a neutral glycolipid of the globo-series, globotriaosylceramide (Gb3) [7]. Gb3 may also be referred to as CD77 or the Pk blood antigen. Recent structure/function studies suggest that each toxin molecule may express 10–15 Gb3 binding sites per B-subunit pentamer [8,9], explaining the high affinity (dissociation constant [KD] ≈ 10-9 M) of toxin binding. All Stxs, with the exception of one Stx2 variant called Stx2e, bind Gb3; Stx2e shows preferential binding to the glycolipid globotetraosylceramide (Gb4).
Table 1. Shiga toxin variants and disease progression.
| Toxin | Amino acid homology† (%) | Disease progression‡ |
|---|---|---|
| Stx1 | – | D → HC → HUS |
| Stx1c | (A) 97; (B) 95 | D → HC → HUS |
| Stx1d | (A) 93; (B) 92 | D |
| Stx2 | – | D → HC → HUS |
| Stx2c | (A) 100; (B) 97 | D → HC → HUS |
| Stx2c2 | (A) 100; (B) 97 | D |
| Stx2d | (A) 99; (B) 97 | D |
| Stx2dactivatable | (A) 99; (B) 97 | D → HC → HUS |
| Stx2e | (A) 93; (B) 84 | D |
| Stx2f | (A) 63; (B) 57 | D |
Figure 1. Ribbon diagram of Shiga toxin.
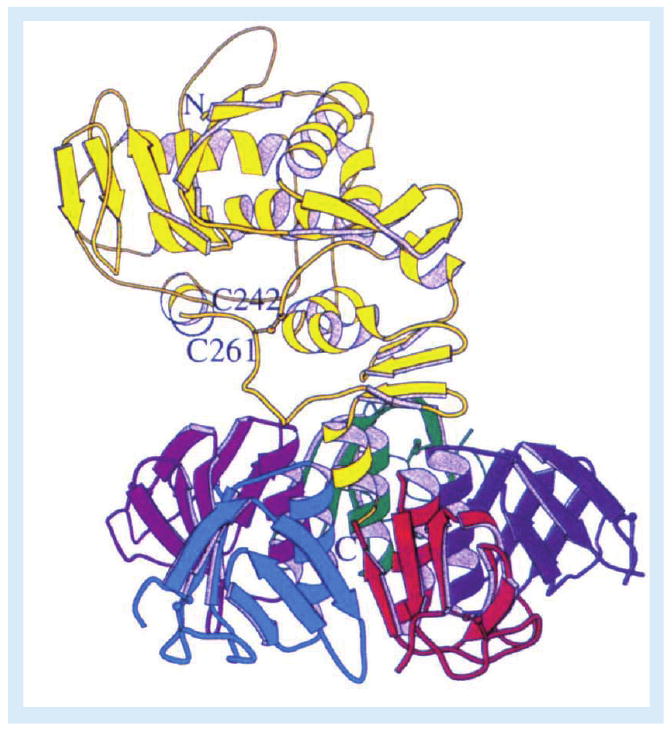
The enzymatic A-subunit (yellow) noncovalently associates with a pentameric ring of receptor-binding B-subunits. Following toxin internalization and proteolysis of the A-subunit by furin or calpain, a disulphide bond between C242 and C261 links the A1- and A2-fragments. Reproduced with permission from [3].
Shiga toxin genes are encoded by temperate lambdoid bacteriophages. STEC express multiple Stx variants because they harbor multiple Stx-encoding bacteriophages (Stx-phages). Stx-phages display extensive genetic mosaicism; however, genes encoding the Stx A- and B-subunits are generally located downstream of the antiterminator Q and the P′R promoter. As a consequence of this orientation, the toxin genes are late genes optimally expressed upon induction of the lytic cycle. STEC may possess cryptic lambdoid prophages that serve as sources for recombination events, yielding novel toxin-converting phages, and Stx-phages expressing new tail assemblies may expand the host range of toxin-producing organisms [10]. Lysogenic conversion to the toxigenic phenotype may occur if recipient bacteria display phage receptors and possess integration sites within the genome. Thus, Stx-phages are responsible for the dissemination of stx genes in E. coli and other enteric bacteria. Stx genes in S. dysenteriae serotype 1 are associated with prophage sequences containing multiple insertion sequences that disrupt phage excision; S. dysenteriae serotype 1 may not be an effective donor of Stx genes. What are the selective advantages of maintaining the toxin genes in the phage genome? Free Stx-phages have been found to persist in aquatic and terrestrial environments after the death of their bacterial hosts (reviewed in [10]). Whether Stxs contribute to increased phage survival ex vivo will require additional studies. Readers are referred to recent reviews on Stx-phages for additional information on genome organization, regulation of toxin gene expression and dissemination of stx genes by transduction [10–12].
Interaction of Stxs with host cells
To be effective protein synthesis inhibitors, Stxs must reach the cytoplasm to access ribosomes. Stxs utilize a highly orchestrated transport pathway to reach the endoplasmic reticulum (ER), an intracellular compartment rich in membrane-associated ribosomes and containing the cellular machinery necessary for protein translocation into the cytoplasm. Following binding and cross-linking of Gb3, Stxs are internalized by clathrin-dependent or clathrin-independent mechanisms [13–16]. Membrane Gb3 expression is a critical determinant of toxin sensitivity. Cells expressing low Gb3 levels are sensitized to toxicity by increased membrane expression of toxin receptors, while cells selected for loss of Gb3 expression are resistant to Stxs [17,18]. The innate immune response to Stxs and other bacterial components, such as flagellin or lipopolysaccharides (LPS), may be important in sensitizing cells to Stxs, as the proinflammatory cytokines TNF-α and IL-1β upregulate genes involved in Gb3 biosynthesis in some cell types [19]. Structural differences in the toxin receptor also contribute to toxin susceptibility. Gb3 is heterogeneous, displaying variability in fatty acid chain length, degree of bond saturation and hydroxylation. Expression of Gb3 isoforms with long-chain, unsaturated fatty acids was associated with increased toxin sensitivity [7,18]. Römer et al. demonstrated that long-chain, unsaturated Gb3 isoforms were more likely to induce negative membrane curvature leading to Gb3 clustering and formation of tubular invaginations [16]. Finally, Gb3 association with lipid rafts was necessary for efficient intracellular transport of Stx1 B-subunits [20]. Polyunsaturated fatty acid incorporation into cell membranes, known to disrupt lipid rafts, protected cells from Stx intoxication [21]. Readers are directed to a recent review of Gb3 heterogeneity and its role in Stx binding and internalization [22].
Stxs are not inert cargo molecules. Stx B-subunits induced membrane invaginations in energy-depleted HeLa cells and cytosol-free model membranes, while cross-linking Gb3 with anti-Gb3 antibodies failed to induce tubular invaginations [16]. These data suggest that initiation of toxin internalization may be a property inherent to the B-subunit pentamer. Stxs also activate signaling cascades that facilitate toxin uptake and intracellular routing. Signaling molecules reported to be activated by toxin binding and uptake include Src family kinase members Yes and Lyn, the tyrosine kinase Syk, the serine/threonine kinase PKCδ, and p38 MAPK [23–27]. Activation is rapid and transient; for example, Stx1 treatment of a human renal epithelial cell line activated Yes, resulting in increased tyrosine phosphorylation of lipid raft-associated proteins 10 min after toxin exposure, which returned to normal levels 1 h after toxin treatment [23]. Stx-containing endosomes are routed to different intracellular compartments in a cell-specific manner. In primary human monocytes, Stx-containing endosomes were routed to lysosomes [20]. Peptides derived from chimeric antigens containing Stx1 B-subunits were processed and presented in association with MHC class I molecules on dendritic cells [28]. In many cell types, Stxs in early endosomes traffic to the trans-Golgi network (TGN) and through the Golgi stacks to deliver Stxs to the ER. This intracellular routing process is called retrograde transport (Figure 2). Stxs may utilize direct endosome-to-TGN sorting pathways used in recycling the host-cell proteins mannose-6-phosphate receptor and TGN38. Immunogold-electron microscopy was used to show toxin trafficking to the Golgi. Mechanisms of TGN-to-Golgi transfer and intra-Golgi cisternal transport are poorly understood. Recent studies using inhibitors of Ras superfamily (Rab) GTPases identified at least six Rabs that may be involved in toxin transport from the cell surface to the Golgi apparatus [29]. Drugs that blocked toxin transit through the Golgi protected cells from cytotoxicity [30]. Stxs may be directed from the Golgi to the ER using a Rab6 GTPase-dependent protein retrieval pathway. Clearly, retrograde transport represents an excellent target for development of pharmacologic inhibitors, and libraries of small-molecule transport inhibitors are being evaluated for their ability to protect cells from Stxs [31]. Pavelka et al. contains a more detailed discussion of retrograde transport processes [32].
Figure 2. Retrograde transport of Shiga toxins.

Following toxin internalization via clathrin-dependent or clathrin-independent mechanisms, holotoxin within an early endosome is routed to the TGN, through the cis-, medial- and trans-cisternae of the Golgi apparatus, to the ER. In the process, the A-subunit is nicked at a single site but the A1- and A2-fragments remain associated via a disulphide bond. Translocation of the enzymatically active A1-fragment into the cytoplasm occurs in the ER.
ER: Endoplasmic reticulum; TGN: trans-Golgi network.
Reproduced with permission from [1].
Stx1 holotoxins containing fluorescein-labeled A-subunits and rhodamine-labeled B-subunits were used to show that dual-labeled holotoxins were transported to the ER [33]. However, during retrograde transport, Stx A-subunits undergo limited proteolysis by furin or calpain [34,35]. The resulting A1- and A2-fragments are linked by a disulfide bond (between Cys242 and Cys261 in Stx; Figure 1); the latter fragment maintains association with the B-pentamer. In the reductive environment of the ER, the disulfide bond is broken, and the 27-kDa A1-fragment translocates across the ER membrane, a process termed retrotranslocation. N-glycosidase activity is associated with the A1-fragment. How does the A1-fragment cross the ER membrane? Proteins that improperly fold or are improperly glycosylated may cross from the ER lumen into the cytoplasm where they are routed to the proteasome for degradation. The translocation machinery involved in the transport of nascent proteins into the ER lumen is also involved in the retrieval and degradation of misfolded proteins. Several bacterial and plant toxins utilize components of the host ER-associated degradation (ERAD) pathway to unfold and enter the cytosol [36]. Stx retrotranslocation is thought to involve the Sec61p translocon, although attempts to co-immunoprecipitate Sec61α with Stx1 A1-fragments were unsuccessful [33]. While the A2-fragment and B-subunits do not appear to be translocated into the cytoplasm, experiments using inhibitors of glycolipid biosynthesis demonstrated that the holotoxin must be delivered to the ER associated with Gb3 in lipid rafts in order for efficient A1-fragment retrotranslocation to occur [37]. A-subunit processing exposes a hydrophobic domain on the A1-fragment that interacts with ER chaperones [38–40]. Stxs also activate sensors of misfolded proteins localized within the ER membrane [41]. Thus, mammalian cells may recognize Stxs delivered to the ER as misfolded proteins, activate the ER stress response, and the A1-fragment ‘co-opts’ the ERAD pathway to reach ribosomes. How do A1-fragments escape routing to the proteasome? A1-fragments contain few (Stx/Stx1) or no (Stx2) lysine residues [42,43], which may minimize ubiquitination, and A1-fragments associate with ribosomal proteins, which may inhibit proteasomal transport [44]. However, treatment of Vero cells with a proteasome inhibitor increased the amount of 125I-Stx1 A1-fragments entering the cytoplasm and increased cytotoxicity, suggesting that some fraction of translocated A1-fragments may be degraded by target cells [33]. Finally, activation of the ribotoxic and ER stress responses may, in some cell types, initiate apoptotic signaling pathways (see below).
Diseases caused by Stx-producing bacteria
Shigella spp. are the causative agents of bacillary dysentery, a disease characterized by abdominal cramping, fever and diarrhea with blood and mucus in the stool. Shigella spp. are human-adapted pathogens transmitted by the fecal–oral route. The infectious dose may be as low as 10–100 organisms. The incidence of fatal bacillary dysentery is estimated to be over 1 million cases per year [45]. All species of Shigella are invasive. Following internalization by M-cells, the bacteria mediate pathogen-directed endocytosis at the basolateral surfaces of intestinal epithelial cells. Once internalized, the bacteria rapidly escape the endosome to replicate within the cytoplasm. Pathogen-directed unipolar polymerization of host cell cytoskeletal elements propels the bacteria into adjacent uninfected epithelial cells. Following replication within infected cells, the host cells lyse, releasing bacteria. Thus, bacillary dysentery is characterized by colonic ulceration and a brisk inflammatory response. Stx appears to be an ancillary virulence determinant in bacillary dysentery. Of the four Shigella species, only S. dysenteriae serotype 1 expresses Stx. Feeding studies in macaque monkeys using toxigenic and atoxigenic S. dysenteriae isogenic strains revealed that both strains caused fulminant dysentery, but only the toxigenic strain damaged colonic capillaries, suggesting that Stx targets vascular endothelial cells for destruction [46]. Additional information on the pathogenesis of bacillary dysentery can be found in Schroeder and Hilbi [47].
Shiga toxin-producing E. coli are the causative agents of bloody diarrhea or hemorrhagic colitis. STEC are not invasive, but many strains are capable of tightly adhering to the intestinal epithelium, causing substantial alterations in gut histology. Ruminant animals used in food production appear to be the major natural reservoirs for STEC, although outbreaks of bloody diarrhea have been linked to ingestion of improperly washed vegetables, unpasteurized fruit juices, contaminated well water, under-chlorinated swimming pool water and to animal contact during petting zoo visits [2,48]. Person-to-person transmission is also responsible for disease spread. Almost 500 different STEC serotypes have been isolated from humans with bloody diarrhea [2]; however, E. coli O157:H7 is the predominant STEC serotype associated with outbreaks of bloody diarrhea in many countries, and is most often associated with severe disease [49]. In the USA, the annual estimated incidence of foodborne disease caused by E. coli O157:H7 is approximately 73,000 cases per year while non-O157:H7 strains cause approximately 37,000 cases per year [50]. Like the Shigellae, the infectious dose of STEC for humans may be as low as 50 bacteria. At 3–8 days following the ingestion of STEC, patients develop watery diarrhea, which may progress to bloody diarrhea with abdominal cramping, nausea and vomiting. Fever is not a common sign following STEC ingestion. The use of antibiotics or antimotility agents is contraindicated; some antibiotics have been shown to increase toxin expression by STEC and antimotility agents may increase toxin retention in the GI tract [51].
Patients infected with Stx-producing bacteria are at an increased risk of developing a life-threatening extraintestinal disease termed the hemolytic uremic syndrome (HUS). HUS is primarily due to the production and translocation of Stxs across the intestinal epithelial barrier. HUS may manifest after STEC are no longer detectable in the stool. Following toxin-mediated damage to colonic blood vessels, Stxs may enter the bloodstream although free toxins have not been detected in the circulation of HUS patients. The risk of progression to extraintestinal complications is increased in patients infected with STEC expressing Stx2, either alone or in combination with Stx1 or Stx2c [52]. The kidneys and CNS are most frequently damaged by Stxs. HUS is a constellation of hematological, renal and neurological complications that develop in 10–15% of patients with hemorrhagic colitis. Complications include thrombocytopenia and hemolytic anemia with schistocytes (fragmented erythrocytes) present in blood smears. The characteristics of acute renal failure, which may follow STEC infection, include oliguria or anuria, swollen glomerular endothelial cells detached from the basement membrane, intraglomerular fibrin deposition and thrombotic microangiopathy. Mesangiolysis or mesangial hyperplasia have been described in some HUS cases. Renal tubular injury may be present but is not a consistent finding late in the course of HUS [53,54]. Approximately 66% of HUS patients require dialysis. CNS involvement may present as lethargy, irritability, seizures, paresis and coma. Long-term sequelae include renal insufficiency, hypertension, hyperactivity and distractability, and insulin-dependent diabetes mellitus. HUS mortality is 3–5%. There is variability in signs and symptoms following ingestion of STEC. For example, patients may present with acute renal failure in the absence of bloody diarrhea [54].
Apoptosis
Apoptosis is a form of cell death resulting from the activation of intracellular signaling pathways, or ‘programmed cell death.’ It is critical for normal development and tissue homeostasis. Many pathogenic microorganisms have evolved mechanisms of altering apoptosis to favor pathogen survival or dissemination. Apoptotic cells display characteristic morphological changes such as cell shrinkage, cytoplasmic vacuolation, chromatin condensation (pyknosis), nuclear fragmentation (karyorhexis) and cell blebbing to produce apoptotic bodies. Many of the morphological changes seen in apoptotic cells are mediated by the sequential activation of a series of proteases called caspases. Caspases are aspartyl-specific cysteine proteases, synthesized as zymogens called procaspases. Caspases are activated by limited proteolysis at sequence-specific sites to generate N-terminal prodomains and C-terminal protease domains. Once activated, caspases form heterotetramers (two prodomains plus two protease domains) that possess protease activity. Caspases with large prodomains (e.g., caspase 8) also associate with adaptor molecules to form death-inducing signaling complexes (DISCs). Caspase-dependent cleavage of downstream substrates usually results in substrate activation although in some cases negative regulatory proteins are cleaved, thereby activating substrate function [55]. Caspases 2, 8, 9 and 10 are characterized as initiator caspases, those that initiate signaling cascades leading to cell death, while caspases 3, 6 and 7 are executioner caspases, those that activate proximal mediators of cell death. In humans, caspases 1, 4 and 5 are involved in the processing of proinflammatory cytokines.
Apoptosis induction is categorized into two major pathways: the extrinsic or death receptor pathway, and the intrinsic or mitochondrial pathway. The extrinsic pathway is activated when death-inducing ligands, such as FasL, TNF-α or TRAIL interact with their receptors, Fas, TNFR1 or death receptor (DR)4/DR5, respectively. After engagement of death receptors, adaptor proteins Fas-associated death domain protein (FADD) or TNFRI-associated death-domain protein (TRADD) associate with the receptors which, in turn, recruit procaspase 8 or procaspase 10 to form the DISC. The resultant proximity of procaspase 8 molecules induces autoproteolysis, and the caspase 8 fragments dimerize to form the active protease. Caspase 8 may directly cleave and activate the executioner caspase, caspase 3, or may cleave B-cell lymphoma (Bcl)-2 inhibitory Bcl-2 homology (BH)3-domain-containing protein (Bid), which in its truncated form (tBid) is sufficient to translocate to mitochondria and facilitate Bax/Bak pore formation and increased mitochondrial outer membrane permeability. The intrinsic or mitochondrial pathway is activated by diverse signals, which may damage DNA (UV light or cytotoxic chemotherapeutic drugs) or induce oxidative stress. These stressors activate pro-apoptotic proteins to form pores in mitochondrial membranes. Pore formation results in the release of cytochrome c which, with procaspase 9, apoptosis protease activating factor (APAF)-1 and dATP, triggers the formation of a macromolecular complex called the apoptosome. Apoptosome formation leads to caspase 9 activation and cleavage of procaspase 3. Caspase 3 cleaves many downstream substrates that contribute to apoptosis. For example, caspase 3 cleaves a negative regulatory domain from caspase-activated DNase, activating its ability to cut internucleosomal genomic DNA into fragments that are multiples of 180–200 bp. Caspase-3 also inactivates the DNA repair protein poly(ADP-ribose) polymerase (PARP). Loss of PARP function facilitates DNA fragmentation and apoptosis. DNA fragmentation and PARP cleavage are frequently used as indicators of apoptosis.
Members of the Bcl-2 protein family are critical regulators of apoptosis. Bcl-2 proteins may be pro- or anti-apoptotic. Anti-apoptotic proteins Bcl-2 and Bcl-XL possess four conserved BH domains and hydrophobic C-termini, which allow them to intercalate into mitochondrial or ER membranes. Pro-apoptotic Bcl-2 proteins (e.g., Bax, Bak and Bid) possess similar structures but lack one or more BH domains. Bax and Bak may translocate to mitochondrial outer membranes where they oligomerize, or associate with the voltage-dependent anion channel, to form pores in the membrane. Bcl-2 and Bcl-XL translocation to mitochondrial membranes is associated with inhibition of pore formation. tBid facilitates the oligomerization of Bax and Bak to form membrane pores that disrupt mitochondrial membranes and release cytochrome c. Thus, caspase 8-mediated cleavage of Bid links extrinsic and intrinsic pathways of apoptosis induction. In addition to cytochrome c, additional apoptosis inducing factors may be released from the mitochondrial intermembrane space. Finally, apoptosis may also ensue following perturbation of lysosomal membrane permeability. Additional details on apoptosis initiators derived from mitochondria, lysosomes and mechanisms of apoptosis induction may be found in recent reviews [55–58].
The precise mechanism(s) by which Stxs activate apoptosis remain to be clarified, although recent evidence indicates that signaling through MAPK pathways may contribute to the induction of cell death. MAPKs are serine/threonine-specific protein kinases first shown to be activated by extracellular growth factors or mitogens. In addition to mitogens, other mediators of cell stress, such as UV light, cytotoxic drugs, heat shock and reactive oxygen species may activate MAPK signaling. MAPKs are part of a signaling cascade in which upstream MAPK kinase kinases (MAP3Ks) activate MAPK kinases (MAP2Ks) which, in turn, activate MAPKs. The MAPKs can be categorized into three major groups, each group containing multiple isoforms: c-Jun N-terminal kinases (JNK), p38 MAPKs and extracellular signal-regulated kinases (ERKs). JNK, p38 and ERK regulate (via phosphorylation) an extensive number of substrates, many of which serve as transcription factors to control the expression of genes involved in cell proliferation, differentiation, inflammation and apoptosis [59–61]. In 1997, Iordanov et al. showed that protein-synthesis-inhibiting toxins that act on the peptidyltransferase reaction center of eukaryotic ribosomes (α-sarcin, ricin and anisomycin) share the capacity of activating the JNK cascade, a phenomenon referred to as the ribotoxic stress response [62]. As would be expected, given the mode of action of Stxs, they were also shown to activate not only JNK, but also the p38 and ERK MAPK cascades in epithelial and myeloid cells [63–65]. Stx-mediated activation of the ribotoxic stress response requires toxin enzymatic activity, suggesting that the capacity of the A1-fragment to retrotranslocate and act on the peptidyltransferase center of ribosomes is necessary for MAPK activation. MAPK-mediated regulation of apoptosis induction may be quite complex, with minor differences in patterns of MAPK activation manifesting significant changes in cell death or survival. For example, ERK-1 and -2 may directly phosphorylate caspase 9, thereby inhibiting apoptosis [66]. Signaling through ERK and JNK pathways appears to phosphorylate Bcl-2 on serine 70, a modification that may be necessary for anti-apoptotic function [67,68]. By contrast, signaling through p38 MAPKs appears to phosphorylate Bcl-2 at alternative serine and threonine residues, which reduces anti-apoptotic function [69]. A more complete understanding of the mechanisms by which Stxs induce apoptosis will require clarification of the patterns of MAPKs activated by the toxins in different cell types.
Many assays have been used to detect apoptosis in Stx-treated cells including detection of DNA fragmentation by electrophoresis in agarose gels, terminal dUTP nick-end labeling (TUNEL) staining to detect nicked DNA, FACS analysis with annexin V staining to detect altered membrane phosphatidylserine in apoptotic cells, electron microscopy, western blotting using antibodies specific for cleaved caspases, assessment of caspase activity using fluorophore-labeled substrates, fluorescence microscopy to detect the translocation of Bcl-2 family members to mitochondria and changes in mitochondrial membrane potential detected by the loss of fluorescent compounds that intercalate into polarized membranes. Readers are referred to Jones [70] for a more complete listing of laboratory methods and protocols used to detect Stx-mediated apoptosis.
Stx-induced apoptosis
Epithelial cells
Given the exquisite sensitivity of Vero cells to Stxs, it is not surprising that these cells were among the first used to study Stx-induced apoptosis. Inward et al. used light microscopy and detection of DNA laddering to show that Stx1 induced apoptosis in Vero cells [71]. Early studies designed to detect Stx-induced apoptosis in primary cells and cell lines have been reviewed previously [72]. More recent studies have characterized signaling pathways activated by Stxs that lead to apoptosis. Using human intestinal explants maintained ex vivo, it was shown that intestinal epithelial cells express very low levels of Gb3. Toxin binding was limited to Gb3-expressing crypt cells and Paneth cells [73,74]. A comparative study of toxin uptake and apoptosis in polarized Gb3-expressing (Gb3+) and Gb3-deficient (Gb3-) human intestinal epithelial cell lines demonstrated that Caco-2 cells (Gb3+ colon carcinoma cells) internalized Stxs, routed the toxins to the ER and nuclear membrane, and protein synthesis inhibition and apoptosis followed. In contrast, T84 cells (Gb3- colon carcinoma cells) also internalized Stxs and delivered Stx1 A-subunits to the ER, yet protein synthesis inhibition and apoptosis did not occur [75]. In addition to toxin routing to the ER, it was shown that Stx1 and Stx2 interacted with polarized intestinal epithelial cell monolayers, including Gb3- T84 cells, in such a way that the toxins were transported intact across monolayers by paracellular or transcytotic mechanisms without disrupting transmembrane electrical resistance [76]. Toxin transmural transport showed an apical to basal membrane directional preference, and toxins delivered across epithelial barriers were active. Thus, Stxs delivered across Gb3- intestinal epithelium may damage Gb3+ submucosal microvascular endothelial cells without affecting epithelial cell viability. Toxin receptor(s) utilized by T84 cells and differential mechanisms of toxin retrograde versus transcytotic transport remain to be clarified, although it was recently shown that purified Stx1 B-subunits were taken-up by polarized T84 cells via clathrin-independent, actin-mediated macropinocytosis [77].
In contrast to intestinal epithelium, human renal tubule cells express Gb3 and may be damaged early in the course of HUS [78]. Given the critical role of Bcl-2 proteins in regulating homeostatic programmed cell death, Jones et al. examined the capacity of Stxs to alter expression of Bcl-2 family members in epithelial cell lines [79]. Treatment of the Gb3+ laryngeal epithelial cell line HEp-2 with Stx1 or Stx2 increased the expression of the pro-apoptotic protein Bax, while pro-apoptotic Bak and anti-apoptotic Bcl-2 expression were not altered. However, Wilson et al. showed that human renal epithelial cells were protected from Stx2-induced apoptosis by siRNA silencing of Bak expression, implicating both Bak and Bax in Stx-induced epithelial cell apoptosis [80]. The pan-caspase inhibitor ZVAD-fmk, or the ectopic expression of Bcl-2, protected HEp-2 cells from toxin-induced apoptosis. Toxin treatment activated caspases 6 and 9 and triggered cleavage of Bid and PARP [81]. Purified toxin B-subunits did not effectively initiate HEp-2 cell apoptosis. Collectively, these data suggest that Stxs trigger apoptosis by altering the balance of pro- and anti-apoptotic Bcl-2 proteins. Furthermore, while caspase 9, 6 and 3 activation may have directly contributed to Stx-mediated cell death, alterations in mitochondria through tBid generation, leading to caspase 9 activation, were also involved. Additional mechanisms may be employed by Stxs to alter Bcl-2 proteins so as to favor induction of apoptosis. Suzuki et al. noted that the Stx2 A1-fragment contains a pentameric amino acid sequence homologous with the BH1 domain of Bcl-2. Stx2, but not Stx1, co-immunoprecipitated with Bcl-2 and translocated to mitochondria in a recombinant human hepatoma cell line overexpressing Bcl-2 [82]. The authors speculated that Bcl-2, normally a pro-survival signaling protein, may direct Stx2 A1-fragments to mitochondrial membranes to trigger apoptosis. However, it should be noted that Stx1 and Stx2 activate apoptosis at roughly equivalent doses for many cell types in vitro. Additional studies to examine differential intracellular routing of Stx1 and Stx2 A1-fragments to mitochondria may clarify these results.
HeLa cells (Gb3+ cervical adenocarcinoma) express keratins and possess an epithelial morphology. Fujii et al. carried out an extensive characterization of the HeLa cell response to Stxs [83]. The toxins rapidly induced apoptosis with more than 60% cell death detected by TUNEL staining and DNA fragmentation 4 h after toxin treatment. HeLa cell apoptosis required toxin enzymatic activity and was characterized by rapid activation of caspases 3, 6, 8 and 9. Caspase-specific peptide inhibitors for caspases 3, 6 and 8 blocked Stx1-induced apoptosis while a caspase 9-specific inhibitor was ineffective. Caspase 3 activation was blocked by a caspase 8-specific inhibitor, placing caspase 8 activation upstream of executioner caspase activation. Stxs were shown to alter HeLa cell mitochondria. Stx1 treatment triggered Bid cleavage, decreased mitochondrial transmembrane potential and mediated the release of cytochrome c into the cytoplasm. Cytochrome c release is important for apoptosome formation and caspase 9 activation, yet data from experiments using a caspase 9-specific inhibitor suggested that this pathway played a minor role in Stx-induced HeLa cell apoptosis. This seeming paradox was resolved when it was shown that the expression of the anti-apoptotic factor x-linked inhibitor of apoptosis protein (XIAP) was increased in Stx-treated cells. XIAP binds caspase 9 to inhibit its activity. Stxs failed to induce Fas, TNF-α and IL-1β expression in HeLa cells. In summary, Stx-induced apoptosis in HeLa cells is rapid and involves elements of extrinsic and intrinsic pathways. The early activation of caspase 8 may directly activate caspase 3, and subsequent caspase 6 activation may contribute to a strictly caspase-dependent cell death pathway by cleaving lamin A, thereby facilitating chromatin condensation. However, the rapid cleavage of procaspase 8 also leads to Bid cleavage and the permeabilization of mitochondrial membranes. However, the mitochondrial pathway may not effectively induce apoptosis as caspase 9 activity may be neutralized by increased XIAP expression. Finally, the activation of caspase 8 did not involve the formation of DISC associated with death receptors Fas or TNFR1.
To examine the correlation between apoptotic signaling and toxin enzymatic activity, HeLa cells were transfected with plasmids expressing the genes encoding the Stx1 A- or B-subunits under control of a tetracycline-inducible promoter [84]. Induction of stx1B expression induced apoptosis as assessed by changes in cell morphology, release of histone proteins and DNA laddering. Induced expression of the stx1A gene resulted in necrotic cell death as assessed by release of the cytoplasmic protein LDH in the absence of DNA fragmentation. Apoptosis induced by Stx1 B-subunits involved activation of caspases 1 and 3. In contrast to experiments showing that treatment of HeLa cells with Stx1 failed to elicit cytokine expression [83], induction of stx1B expression in transfected HeLa cells triggered the release of soluble TNF-α and IL-1β. The authors speculated that apoptosis mediated by B-subunits alone may be induced through cytokine autocrine signaling pathways. A number of caveats should be considered in interpreting these data. First, plasmids used in the study contained a strong cytomegalovirus promoter, and the inducer doxycycline could activate high levels (0.1–1.0 μg) of Stx1 B-subunit expression. Thus, toxin subunits may have been overexpressed in this system relative to amounts of toxin internalized via retrograde transport. Second, toxin B-subunits localize to the ER, but are not thought to cross the ER membrane to enter the cytoplasm. Finally, apoptotic signaling pathways activated during the course of toxin binding and retrograde transport would not be reproduced in this protocol.
Signaling pathways that may link toxin enzymatic activity with apoptosis have been explored using epithelial cells. Treatment of the intestinal epithelial cell line HCT8 with Stx1, but not with Stx1 holotoxin containing a point mutation in the active site of the A-subunit, activated JNK and p38 MAPK signaling cascades, and induced DNA fragmentation and procaspase 3 cleavage. A p38 MAPK inhibitor blocked Stx-induced cell death and caspase 3 activation [63]. DHP-2, a pharmacological inhibitor of the upstream kinase MAP3K, zipper sterile-α-motif kinase (ZAK), blocked Stx2-mediated activation of JNK and p38 MAPKs, partially protected HCT8 cells from apoptosis and partially blocked caspase 3 activation [85]. However, DHP-2 was reported not to affect DNA laddering, suggesting that multiple signaling pathways contribute to apoptosis induction in HCT8 cells. Interestingly, the ZAK inhibitor did not alter protein synthesis inhibition caused by Stx2. Thus, ZAK appears to specifically link signals generated by Stxs with stress-activated protein kinases and apoptosis. Treatment of a human proximal tubule epithelial cell line with curcumin, an anti-inflammatory agent that blocks NF-κB activation, protected cells from Stx1- and Stx2-induced apoptosis [86]. Taken together, these data suggest that signaling pathways responsible for Stx-induced epithelial cell apoptosis include stress-activated protein kinases, the upstream MAP3K ZAK, and the NF-κB pathway.
The ability of Stxs to alter cell functions other than protein synthesis may contribute to apoptosis induction. In this regard, Stxs have been shown to alter the cell cycle progression of epithelial cells and to induce the expression of inflammatory and apoptosis-related genes. Treatment of the human colon cancer cell line HCT116 with Stx1 induced apoptosis, which was preceded by the arrest of cells in S phase, and was followed by an increase in a sub-G0/G1 cell population. Extensive washing of HCT116 cells treated with thymidine for 6 h released cells from S phase arrest, while extensive washing of cells treated with Stx1 for 6 h failed to restore normal cell cycling, suggesting that the action of Stxs on the cells was irreversible. Despite the fact that Stxs may be potent protein synthesis inhibitors, Stx1 treatment of HCT116 cells selectively increased expression of growth arrest and DNA damage proteins GADD34, GADD45α and GADD45β [87]. DNA damage is a major inducer of GADD genes. The enzymatic activity of Stxs does not appear to be limited to rRNA as the toxins have been shown to be capable of removing adenine from DNA [88]. Evidence that Stxs are routed to nuclear membranes [89] and mediate DNA depurination suggest that DNA damage may serve as an alternative mechanism of apoptosis operating concurrently with signaling initiated following rRNA damage. Additional experiments will be necessary to mechanistically link toxin-induced DNA damage with increased gene expression, cell cycle arrest and induction of apoptosis. These studies also highlight that gene expression may be maintained or induced following intoxication in many cell types. For example, numerous studies have documented the capacity of Stxs to induce chemokine expression, and these chemoattractants may serve as indirect mediators of apoptosis by eliciting migration of inflammatory cells expressing FasL, perforin and granzymes. Stxs induce the expression of TNF-α from human monocytes and macrophages and this cytokine may act as an apoptosis inducer for some cell types.
Endothelial cells
Fujii et al. reported similar apoptotic pathways activated in human brain microvascular endothelial cells (HBMECs) and HeLa cells treated with Stx2 [90]. Thus, epithelial and endothelial cell responses to Stxs shared the rapid activation of caspase 8 leading to Bid cleavage, tBid translocation to the mitochondria, mitochondrial outer membrane disruption, release of cytochrome c, formation of the apoptosome, procaspase 9 and 3 cleavage, DNA fragmentation, and apoptosis. The transcriptional regulator CCAAT-enhancer-binding protein homologous protein (CHOP) was activated by Stx2 treatment of HBMECs. Microarray analysis of expression of 51 apoptosis-related genes in Stx2-treated HBMECs revealed that a subset of genes was consistently (> one time point) upregulated by toxin treatment: GADD34, GADD45A, GADD45B, protein kinase-like ER kinase (PERK), CREB5, CHOP and activating transcription factor 4 (ATF4). Interestingly, PERK, CHOP and ATF4 are critical signaling components of the ER stress response. Microarray analysis of gene expression induced by exposure of human umbilical vein endothelial cells to Stxs showed 25 and 24 genes upregulated by Stx1 and Stx2, respectively. Among the genes upregulated were TRAF-1 and TNFαIP-3, known to encode apoptosis regulatory proteins [91].
Studies using endothelial cells have provided information on the regulation of caspase 8 activation in Stx-treated cells. Expression of the anti-apoptotic protein cellular FADD-like IL-1β-converting enzyme (FLICE)-like inhibitory protein-long form (c-FLIPL) was rapidly downregulated in endothelial cells following Stx exposure [92]. Decreased c-FLIPL expression sensitized endothelial cells to other cytotoxic mediators, but maintenance of c-FLIPL expression did not protect cells from Stx-induced apoptosis, suggesting that the role of reduced c-FLIPL expression in the vascular damage characteristic of HUS may be indirect, sensitizing target cells to other bacterial products such as LPS. c-FLIPL may inhibit caspase 8 activity and the de novo synthesis of c-FLIPL may be necessary for maintaining an anti-apoptotic state, leading to speculation that Stx-mediated protein synthesis inhibition was required for early caspase 8 activation. However, c-FLIPL levels declined within 2 h of toxin exposure, suggesting that c-FLIPL expression may not be regulated through transcriptional or translational repression, but rather through the activation of proteases that degrade c-FLIPL. Finally, the role of c-FLIPL in apoptosis induction may be more complex than originally appreciated. Under some circumstances, c-FLIPL translocates to the DISC and promotes caspase activation [93].
The anti-apoptotic Bcl-2 family member myeloid cell leukemia sequence-1 (Mcl-1) also appears to be important in control of apoptosis in Stx-treated endothelial cells. Mcl-1 was rapidly degraded in human microvascular and arterial endothelial cells following toxin treatment. Mcl-1 degradation preceded apoptosis induction as measured by caspase 3 activation. Treatment of cells with the proteasome inhibitor lactacystin prevented Stx1- and Stx2-induced Mcl-1 degradation and protected cells from apoptosis. Expression of other Bcl-2 family members, Bax, Bcl-2 and Bcl-XL did not change following toxin treatment [94]. Treatment of human endothelial cells with TNF-α or IFN-γ prior to toxin exposure upregulated Gb3 expression and increased the percentage of cells undergoing apoptosis. Treatment of the cells with both TNF-α and IFN-γ produced a synergistic effect on toxin receptor expression and apoptosis induction [95].
Leukocytes
Despite expressing membrane Gb3, primary human monocyte-derived macrophages are relatively insensitive to Stxs compared with epithelial and endothelial cells. This reduced sensitivity is associated with the failure of Gb3 to associate with lipid rafts, and to subsequently direct Stxs to the ER. Rather, Stxs appear to be routed into degradative pathways in macrophages and dendritic cells [20,28]. However, treatment of human monocyte-derived macrophages with LPS increased Gb3 expression and Stx B-subunit retrograde transport [20], suggesting that macrophage activation by other bacterial products may increase sensitivity of the cells to Stxs. Interestingly, primary bovine intestinal crypt cells express Gb3 and internalize Stxs but are resistant to toxin-mediated killing. Like primary human monocytes, Gb3 in bovine intestinal crypt cells appears to be distributed throughout the membrane rather than localized into lipid rafts, and toxins are translocated to lysosomes for destruction [96].
In contrast to primary cells, human myeloid leukemia cells frequently express abundant membrane Gb3 [97]. The human Gb3+ myeloid leukemia cell line THP-1 is susceptible to apoptosis induction by Stxs [98]. Caspases 2, 6, 8 and 9 were reported to be activated by Stxs and THP-1 cells were protected from Stx-induced apoptosis by treatment with a caspase 3-specific inhibitor. DNA fragmentation, chromatin condensation and caspase 3 activation were blocked by brefeldin A, suggesting that Stxs must be transported through the Golgi apparatus for apoptosis to occur. The sensitivity of THP-1 cells for Stx-mediated apoptosis is cell maturation-dependent. In the undifferentiated monocyte-like state, the cells grew in suspension with a characteristic doubling time. Monocyte-like THP-1 cells underwent rapid apoptosis when treated with Stx1 with greater than 80% cell death noted 12 h after toxin treatment [99]. Treatment of THP-1 cells with differentiation agents, such as phorbol esters, IFN-γ or vitamin D3, stopped cell replication, and the cells became plastic-adherent and actively phagocytic. THP-1 cell differentiation was associated with increased expression of CD14 and decreased expression of membrane Gb3 [100]. Differentiated, macrophage-like THP-1 cells underwent apoptosis when treated with Stx1, but the kinetics of cell death were significantly delayed compared with undifferentiated, monocyte-like cells, with only 11% cell death noted 12 h after toxin treatment [99].
Caspase 8- and 3-specific inhibitors blocked, and caspase 6- and 9-specific inhibitors partially blocked, the rapid onset of apoptosis induced by Stx1 in monocyte-like THP-1 cells [101]. Caspase 3, 8 and 9 activities were elevated at 2 h, and peaked 8 h, after toxin treatment. Stx1 treatment signaled Bid cleavage, loss of mitochondrial transmembrane potential and release of cytochrome c into the cytoplasm. Caspase 8 activation occurred upstream of mitochondrial membrane disruption as a caspase 8-specific inhibitor reduced mitochondrial membrane depolarization. However, a caspase 6-specific inhibitor was shown to reduce caspase 8 and 9 activation, suggesting that in addition to activating the mitochondrial pathway, Stx1 may activate caspase 6 to directly activate caspase 8 (Figure 3). Stx1-mediated apoptosis of monocytic THP-1 cells required toxin enzymatic activity and occurred independently of TNF-α/TNFR1 signaling. In many ways, signaling for cell death in monocytic THP-1 cells was similar to that characterized for HBMECs and HeLa cells except that XIAP production in HeLa cells may inhibit caspase 9 function, while a caspase 9-specific inhibitor partially reduced apoptosis in THP-1 cells, suggesting that the intrinsic or mitochondrial pathway of apoptosis induction is functional in monocytes [101]. However, apoptosis in monocytic THP-1 cells involved two mechanisms that were not shown to be activated in other cell types. First, Stx1 treatment of THP-1 cells triggered the release of Ca2+ from intracellular stores leading to activation of calpain. Calpain may directly cleave procaspase 8 and a calpain inhibitor partially protected cells from Stx1-induced apoptosis. Second, Stx1 treatment of monocytic cells upregulated the expression of TRAIL and DR5, suggesting that autocrine signaling through this ligand/death receptor pair may contribute to apoptosis [41]. The capacity of Stxs to kill Gb3+ myeloid leukemia cells holds promise for the development of Stxs or engineered toxin chimeric proteins as chemotherapeutic agents to treat cancer. The sensitivity of monocytic THP-1 cells to apoptosis induction by Stxs has also been used to devise a high-throughput, flow cytometry-based assay for the detection of functional toxins in contaminated foods at varying pH and temperature conditions [102].
Figure 3. Shiga toxin-induced apoptotic signaling pathways in monocytic THP-1 cells.
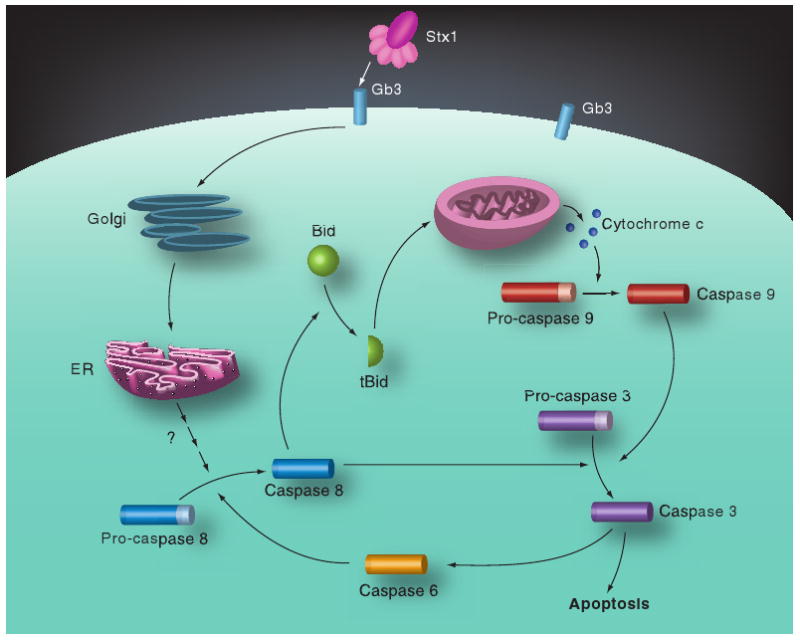
Following toxin internalization, caspase 8 is rapidly activated, which in turn triggers caspase-dependent and mitochondrial-dependent apoptotic signaling cascades. Caspase 8 may directly activate caspase 3, which in turn may activate caspase 6 to create an amplication loop for activation of the executioner caspase. Caspase 8 also cleaves Bid. tBid molecules translocate to mitochondria and facilitate the release of cytochrome c. With dATP and Apaf-1, cytochrome c forms the apoptosome, leading to sequential activation of caspases 9 and 3. Apoptotic signaling requires the retrograde transport of holotoxin to the ER.
bid: BH3-domain-containing protein; ER: Endoplasmic reticulum; Gb3: Globotriaosylceramide; Stx: Shiga toxin.
Adapted with permission from [101]; © American Society for Microbiology.
Many of the apoptotic signaling molecules activated in monocytic THP-1 cells were also activated in macrophage-like THP-1 cells, yet the differentiated cells survived initial challenge with Stxs. Unlike monocytic cells, Stxs appeared to simultaneously activate pro- and anti-apoptotic signaling cascades in macrophage-like cells [103]. Pro- and anti-apoptotic signaling required toxin enzymatic activity. Unlike monocytic THP-1 cells, macrophage-like cells acquired the ability to express soluble TNF-α and membrane TNFRs when stimulated with Stxs. However, the cells were refractory to apoptosis mediated by TNF-α, and treatment with antibodies directed against TNFRs or TNF-α prior to Stx1 treatment did not alter the percentages of apoptotic cells. These data suggest that autocrine signaling through the TNF-α/TNFR1 pathway does not contribute to Stx1-induced macrophage apoptosis. In monocytic cells, the apoptosis inhibitors cellular inhibitor of apoptosis (cIAP)-1 and cIAP-2 were degraded beginning 12 h after Stx1 treatment, while apoptosis inhibitor levels were slightly elevated or maintained in Stx1-treated macrophage-like cells. Differential signaling that may be involved in the initial survival response of macrophage-like cells may include the prolonged activation of stress-activated protein kinases JNK and p38 [65]. The capacity to simultaneously trigger apoptotic and survival pathways is not unique to Stxs; ribotoxic trichothecene mycotoxins expressed by Fusarium spp. also elicit coincident apoptosis and survival signaling in macrophages [104].
Protein synthesis inhibition mediated by Stxs may lead to the increased accumulation of improperly folded proteins within the ER lumen. Three sensors of misfolded proteins reside within the ER membrane: the kinase PERK, the dual kinase/endoribonuclease inositol-requiring enzyme-1 (IRE1) and the transcription factor ATF6. Following detection of misfolded proteins, these sensors initiate intracellular signaling cascades, which may result in decreased protein translation, although expression of select proteins, such as chaperones, is maintained. Prolonged signaling through PERK, IRE1 and ATF6, due to a failure to clear the ER lumen of misfolded proteins, may lead to apoptosis [105]. Stx enzymatic activity may activate the ER stress response leading to apoptosis. Alternatively, holotoxin molecules per se may be sensed as misfolded proteins. Lee et al. showed that Stx1 activates all three ER stress sensors in toxin-sensitive monocytic THP-1 cells [41]. Downstream signals of ER stress, phosphorylation of the translation initiation factor eIF2α, increased expression of the transcription factor CHOP and alternative splicing of X-box protein-1 mRNA, were activated by Stx1 treatment. Protein and mRNA expression of the anti-apoptotic factor Bcl-2 was downregulated in monocytic THP-1 cells. By contrast, Stx1 treatment of differentiated, macrophage-like THP-1 cells, which initially survive toxin challenge, activated the ER stress sensors PERK and IRE1, but failed to activate ATF6, and Bcl-2 expression remained elevated in these cells [106]. Furthermore, Bcl-2 is phosphorylated and fails to translocate to mitochondria following Stx1 treatment of macrophage-like THP-1 cells. The ability of Bcl-2 to neutralize the pro-apoptotic Bcl-2 family members is known to be reduced by phosphorylation and failure to translocate to mitochondria [57]. Thus, Bcl-2 is a key regulator controlling the onset of apoptosis in response to toxin treatment, and cell maturation appears to be correlated with a transient dissociation of ER stress signaling from the downregulation of Bcl-2 expression. Stxs are not the only AB5 toxins shown to activate the ER stress response. Subtilase cytotoxin, a newly described protein toxin that may be expressed by STEC and reportedly cleaves the ER chaperone binding immunoglobin protein, also activates ER stress sensors [107].
In marked contrast to studies exploring apoptosis, studies using Stxs and human polymorphonuclear neutrophils (PMNs) have focused on cell survival. PMNs normally have short lifespans, on the order of 12–24 h. Liu et al. reported that Stx2 inhibits spontaneous PMN apoptosis in a dose-dependent manner, increasing the time necessary to reach 50% apoptosis from approximately 12 to 48 h [108]. A subsequent study did not support this finding [109]; however, differences in the methods used to isolate human PMNs may affect toxin binding to the cells [110,111]. The mechanism of prolonged survival of intoxicated PMNs remains to be fully characterized, but appears to involve signaling through protein kinase C [108] and the delayed activation of caspase-3 [111]. In contrast to murine, bovine and porcine PMNs, human PMNs do not express Gb3, yet Scatchard analysis estimated 2.1 × 105 toxin binding sites per human PMN, with binding sites possessing a lower toxin binding affinity than that characterized for Gb3 [112]. Stxs appear to selectively bind mature PMNs and retinoic acid-differentiated, granulocytic HL-60 cells [111]. Studies using purified Stx1 A- and B-subunits showed that toxin binding to PMNs involved both subunits, and the binding of Stx2 to human serum amyloid P appeared to facilitate Gb3-independent binding to human PMNs [110].
Free Stxs have not been detected in plasma of patients with bloody diarrhea or HUS. Flow cytometric analyses of human blood incubated with fluoresceinated Stxs ex vivo showed that Stxs associated almost exclusively with PMNs and monocytes, and fluorescent Stxs could be transferred from PMNs to cytokine-activated endothelial cells expressing Gb3 in vitro [111,112]. Clinical studies detected PMN-associated Stxs in the blood of HUS patients and showed that PMN-associated Stxs persisted for an average of 5 days after the toxins were no longer detectable in stool [113,114], a remarkable finding given the short lifespans of PMNs. The influx of PMNs into the lamina propria during the course of STEC infection may provide a cell type for the hematogenous spread of the toxins. However, to successfully employ the PMN as a ‘toxin carrier’, apoptosis would have to be delayed. Additional mechanisms may be contributing to the prolonged presence of Stxs associated with PMNs. For example, Brigotti et al. have recently described interleukocytic transfer of Stxs from senescent to nascent PMNs, suggesting that toxin persistence in the circulation may exceed the lifespan of a single neutrophil [111]. Circulation of Stx-laden PMNs to tissues with Gb3-rich microvascular beds would result in the transfer of the toxins from PMNs to target endothelial cells. Experiments to characterize Gb3-independent binding of Stxs to PMNs and toxin-mediated alterations in PMN cell signaling responsible for delayed spontaneous apoptosis, remain to be performed. However, the data suggest, that inhibiting the apoptosis-delaying action of Stxs for human PMNs may represent an approach to limit toxin dissemination following bloody diarrhea.
The curious case of Burkitt's lymphoma cells
Germinal centers found in secondary lymphoid organs are sites of apoptosis. Germinal centers are largely comprised of centroblasts, Gb3+ B-cells undergoing rapid proliferation in response to antigen stimulation. Centroblasts are subjected to a mutational program called somatic hypermutation that introduces point mutations in rearranged immunoglobulin gene segments encoding the antigen-binding sites of membrane-bound immunoglobulins. The result of many of the point mutations will be B-cell clones with reduced affinity for antigen, and these cells are destined to undergo apoptosis. Burkitt's lymphoma (BL) cells are thought to represent transformed counterparts of normal centroblasts, sharing the properties of high membrane Gb3 expression and low expression of the anti-apoptotic factor Bcl-2. The observation that Stx binding to Gb3 recruits Src family kinases and Ig μ-heavy chains to lipid rafts in BL cells [24] suggests that Gb3 may be a necessary signaling molecule for apoptosis induction of centroblasts undergoing negative selection for loss of antigen reactivity. Thus, one could consider BL cells as ‘primed’ for apoptotic cell death. As would be expected of Gb3+ cells, BL cells are killed when incubated with Stxs. Apoptosis induction was rapid; treatment of BL Ramos cells with Stx1 resulted in an approximate 50% reduction in cell viability in 9 h [115]. Apoptosis involved the early activation of caspase 8, followed by caspase 7 and 3 activation. Caspases 1, 2, 4, 5, 6 and 9 were reported not to be involved in apoptotic signaling. Use of a caspase 3 inhibitor resulted in only partial protection of the BL Ramos cell line from Stx1-induced apoptosis, suggesting that caspase-independent mechanisms may contribute to cell death. Stx1-mediated apoptosis did not appear to involve signaling through Fas or TNFRs.
What makes BL cells most interesting is that multiple signaling mechanisms appear to contribute to Stx-induced apoptosis. BL cells are killed when incubated with holotoxin, but are also killed when exposed to purified Stx B-subunits or immobilized anti-Gb3 antibodies, thereby dissociating cell death by toxin enzymatic activity from cell death induced by Gb3 cross-linking. Tétaud et al. examined apoptosis induction in several BL cell lines [116]. Gb3+ Ramos and BL2 cells were sensitive to apoptosis induction by Stx1 or anti-Gb3 antibody treatment, while the Gb3+ cell line P3HR1 failed to undergo apoptosis when treated with Stx1 but was still sensitive to killing by anti-Gb3 treatment. Treatment of toxin-sensitive Ramos cells with Stx1 resulted in the rapid (within 2 h) cleavage of procaspase 8 and PARP, and loss of mitochondrial membrane potential. Caspase 8 activation occurred upstream of effects on mitochondrial membranes, as treatment with a caspase 8-specific inhibitor protected Ramos cells from mitochondrial membrane depolarization. Stx1 treatment of toxin-resistant P3HR1 cells failed to cleave procaspase 8 or PARP, and mitochondrial membrane potential was not disrupted. The induction of apoptosis by anti-Gb3 antibody was caspase 8- and 3-independent in both toxin-sensitive Ramos cells and toxin-resistant P3HR1 cells. Interestingly, anti-Gb3 antibody treatment induced modest changes in mitochondrial membrane potential in P3HR1 cells but failed to alter mitochondrial membranes in Ramos cells, even though both cell lines were sensitive to killing by toxin receptor cross-linking. The generation of reactive oxygen species was critical in apoptosis induced by antibody-mediated Gb3 cross-linking, as antioxidants protected BL cells from cell death. In contrast, antioxidants did not protect toxin-sensitive BL cells from Stx1. A recombinant Stx1 B-subunit engineered to express an ER-specific glycosylation site was transported to the ER in both toxin-sensitive and toxin-resistant BL cell lines, suggesting that retrograde transport of B-subunits to the ER was not sufficient to induce apoptosis.
An important caveat in interpreting experiments using BL cells in apoptosis experiments is the characterization of the cells. BL cell lines are categorized based on the maintenance of centroblast-like properties during cell passage. Group 1 BL cells, which are Gb3+, Bcl-2low, are readily induced to undergo apoptosis by signals such as Gb3 cross-linking by Stx1 B-subunits or anti-Gb3 antibodies, or by antigen receptor cross-linking by anti-mIgM antibodies. At high passage numbers, some BL cells may undergo ‘phenotypic drift’ associated with changes in the expression of Epstein–Barr virus-encoded genes, so that membrane Gb3 expression is reduced and Bcl-2 expression is upregulated. At high passage numbers, these cells are less susceptible to apoptosis induction by Stx1 B-subunits and anti-mIgM. Gordon et al. found that low passage number group 1 BL cells transfected with plasmids encoding Bcl-2 or Bcl-XL, or cultured in the presence of soluble CD40L, were protected from killing by Stx1 B-subunits or anti-IgM antibodies, but not from killing by Stx1 holotoxin [117]. Therefore, delivery of functional toxins appears to overcome the pro-survival signaling of Bcl-2 family members and CD40L. In summary, the data suggest that Stxs signal apoptosis in Gb3+ BL cells through caspase 8 and 3 activation, and changes in mitochondrial membrane potential, similar to apoptosis induction pathways defined in other cell types. However, simply cross-linking Gb3 induces apoptosis in BL cells via a caspase 8/3-independent, oxidative stress-dependent pathway that may or may not involve changes in mitochondrial membrane potential. Signaling for apoptosis triggered by receptor cross-linking is circumvented by Bcl-2 or Bcl-XL expression, while the holotoxin overcomes the protective effects of anti-apoptotic Bcl-2 proteins.
Neurons
Animals have been employed to model Stx-mediated neuropathogenesis. Similar to some humans infected with STEC, rabbits develop CNS microvascular thromboses with focal hemorrhages and edema as well as convulsions and paralysis following toxin administration [118]. Mice primarily develop hindlimb paralysis and lethargy as a consequence of intoxication [119]. Electron microscopy of brain lesions in rabbits given intravenous Stx2 showed damage to ependymal cells of the third ventricles, and the use of intravenously injected tracers showed that Stx2 administration disrupted the blood–brain and CSF–brain barriers. Immunoelectron microscopy studies of brain tissues demonstrated the presence of Stx2 in edematous capillary endothelial cells, ependymal cells and in myelin sheaths [118]. However, in addition to mediating vascular damage, the administration of Stx2 to rabbits via intravenous or intrathecal routes resulted in neuronal damage in the CNS characterized by atrophy, chromatin condensation and nuclear pyknosis [120]. Immunohistochemical techniques were used to verify the ability of Stx2 to cause neuronal apoptosis in rabbits [121]. Early in the course of disease, TUNEL+ neurons in the dentate gyrus of the hippocampus and granular layers of the cerebellum were noted. Later in the disease, occasional apoptotic neurons were noted in the basal ganglia, thalamus and cerebral cortex. Apoptosis of glial cells within the pons and endothelial cells of cerebral capillaries was also detected. Interestingly, apoptosis in the rabbit CNS did not correlate with the localization of Gb3. Toxin receptor was primarily limited to endothelial cells comprising small blood vessels in the spinal cord gray matter and, to a lesser extent, to blood vessels in spinal cord white matter, root ganglia and brain parenchyma. Gb3 was not detected on microglial cells or neurons. Prior to the onset of neurological symptoms, microglial cell activation was noted in ischemic neural tissues, and proinflammatory cytokine expression was upregulated in all regions of the brain and spinal cord examined. These results led to speculation that Stxs may indirectly trigger neuronal apoptosis via a mechanism involving the focal induction of cytokine expression [121]. Stxs may directly activate Gb3- microglia via an unknown mechanism, but activation may be indirect as endogenously generated thrombin has been shown to induce TNF-α, IL-1β, IL-6, inductible nitric oxide synthase and cyclooxygenase-2 expression by microglial cells [122]. Thus, Stx-induced thrombotic microangiopathy may activate microglial cells, which in turn produce proinflammatory mediators. The localized production of TNF-α and IL-1β may not only activate genes involved in Gb3 biosynthesis leading to increased vascular damage, but may also activate programmed cell death in neuronal cells.
The mouse has been extensively used as an animal model for Stx-mediated renal damage. However, the location of Gb3 in the mouse CNS is controversial. Ren et al. used immunohistochemical techniques in a comparative analysis of toxin receptor expression in dorsal root ganglia of humans, rabbits, mice and rats [123]. Human and rabbit sensory neurons expressed Gb3 while galactosyl-globotriaosylceramide was detected on rodent sensory neurons. Only human and rabbit CNS capillary endothelial cells expressed Gb3. In contrast to these studies, Okuda et al. detected the presence of “low, but definite” Gb3 expression by microvascular endothelial cells of the mouse brain cortex and pia mater [124]. Using immunofluorescent microscopy, Obata et al. showed that CD31-reactive endothelial cells in murine nervous tissues were Gb3- while neurons in the CNS and peripheral nervous tissues were Gb3+ [125]. Gross histopathology revealed no major changes in murine CNS tissues following toxin administration. However, electron microscopy of lumbar spinal cord samples prepared from moribund Stx2-treated mice exhibited increased association of microglial cells with neurons and the formation of microglial processes, which appeared to intrude on synapses. As is the case for experiments employing rabbits to study neuronal apoptosis, studies to examine the mechanistic aspects of Stx signaling leading to direct or indirect apoptosis of rodent neurons are needed.
Role of Stx-induced apoptosis in disease
Apoptosis was noted in tubular epithelial cells in mice fed an Stx2-producing E. coli O157:H7 strain, although scant glomerular fluorescence (one–two TUNEL+ nuclei per glomerulus) was detected. Mice fed a nontoxigenic E. coli O157:H7 strain showed TUNEL+ cells in renal specimens, but the apoptotic cells appeared to be infiltrating inflammatory cells rather than renal cells [126]. Clinical studies of human HUS cases revealed extensive damage to glomeruli and evidence of damage to renal tubular epithelial cells, including pyknotic nuclei and sloughing of cells into tubule lumina. TUNEL staining performed on renal cortical tissues from HUS cases revealed apoptotic nuclei localized to tubular epithelial cells as well as cells within glomeruli [126]. Renal biopsies from seven HUS cases were subjected to a dual staining protocol combining the TUNEL assay with measurement of the loss of an RNA splicing factor [127]. This study confirmed a role for apoptosis in HUS in glomeruli and renal tubules. Despite the fact that neurological complications are a major prognostic indicator of poor outcome [51], the relevance of animal studies demonstrating a role for Stx-induced neuronal apoptosis in human disease remains to be explored. Clinical studies measuring delayed apoptosis of PMNs isolated from HUS patients have confirmed the results of in vitro experiments using freshly isolated PMNs. HUS disease severity appears to be correlated with markers of PMN deactivation, that is, reduced expression of CD11b, CD66 and myeloperoxidase, and reduced respiratory burst in response to PMA treatment [128]. Thus, Stxs may not only delay apoptosis, but also affect PMN function in humans.
Role for Stx-induced apoptosis in the treatment of cancer
Tumor cells frequently possess alterations in programmed cell death pathways that render the cells resistant to apoptosis induction. The evidence that Stxs induce apoptosis in transformed cells has led to the examination of the toxins as potential anticancer chemotherapeutic agents. Johansson et al. demonstrated extensive variability in breast cancer cell lines for apoptosis induction by Stx1 in vitro [129]. Sensitivity was directly correlated with Gb3 expression, and use of the drug PPMP, which downregulates glucosylceramide production, inhibited Stx1-mediated apoptosis as assessed by TUNEL staining. In the Gb3+ breast cancer cell line T47D, apoptosis induction by Stx1 was associated with activation of caspases 8, 9 and 3, and activation (phosphorylation) of the stress-activated protein kinases JNK and MAPK kinase (MKK)3/6. Immunohistochemical detection of Gb3 in cryostat sections of 25 breast cancer tissues revealed Gb3 expression primarily in arterioles and small arteries; Gb3 was detected in tumor cells in 17 of the 25 specimens. These data suggest that Stxs may not only be directly cytotoxic for tumor cells, but may also disrupt the extensive vascular network established in some tumors. Many Gb3+ tumors have been shown to be highly aggressive and refractive to antineoplastic therapy. The single intratumor injection of Stx1 into athymic nude mice harboring a Gb3+ human malignant meningioma dramatically improved survival and increased apoptosis from a constitutive level of 4.3% TUNEL+ cells to 51% TUNEL+ cells within the tumor xenograft [130].
The possibility that Stxs may mediate protein synthesis inhibition and apoptosis in normal cells constitutes a concern for the use of the holotoxin as an anticancer agent. Since toxin binding and retrograde transport are primarily functions of the Stx B-subunits, the toxic A-subunit could be dissociated from the holotoxin, and chimeric proteins comprising antineoplastic agents linked to the B-subunits created to deliver the drugs to cancer cells. Viels et al. used whole-body imaging to show that Cy5-labeled Stx B-subunits specifically home to Gb3+ human colorectal carcinoma cells xenografted in nude mice, with fluorescent signal being detected up to 7 days after intravenous injection [131]. The investigators used confocal microscopy with an HT29 human colon cancer cell line engineered to express the green fluorescent protein to show that Cy5-labeled Stx B-subunit expression co-localized to green fluorescent protein-positive xenograft cells. Studies were also carried out with Cy5-Stx B-subunits and antibodies directed against CD11b (expressed on granulocytes and monocytes) and CD34 (expressed on vascular endothelial cells) to show that the B-subunit co-localized with inflammatory cells and blood vessels within the xenograft. Thus, Stx B-subunits coupled to chemotherapeutic agents may be effective in reducing inflammation and angiogenesis associated with tumors.
Conclusion
Are conserved mechanisms of apoptosis induction by Stxs beginning to emerge? Consistent features of apoptosis induction appear to be the necessity of transporting active holotoxin to the ER and the subsequent disruption in the balanced expression of pro- and anti-apoptotic Bcl-2 proteins. Not only is the ER the site of toxin retrotranslocation, it is also the screening site for proper folding and glycosylation of host proteins destined to be secreted or translocated to membranes. Truncated host proteins produced by intoxicated cells and/or Stx A1-fragments unfolding during retrotranslocation may activate the ER stress response. The prolonged activation of the transcriptional regulator CHOP appears necessary for the diminished expression of the anti-apoptotic protein Bcl-2. The cleavage of Bid would further shift the balance toward the pro-apoptotic state. Other members of the Bcl-2 family of proteins, such as Mcl-1, may be inactivated by proteolysis.
Additional conserved features of apoptosis induction by Stxs are the rapid activation of caspase 8 and activation of the mitochondrial pathway. The link between caspase 8 activation and mitochondrial membrane disruption appears to be caspase 8-mediated cleavage of Bid. However, cytochrome c release and apoptosome formation do not appear to be essential in killing as this pathway was neutralized in HeLa cells. How is caspase 8 rapidly activated by Stxs so as to initiate apoptosis? The data suggest that Stxs activate caspase 8 through multiple mechanisms, signaling through TRAIL–DR5 ligation in myeloid cells, the degradation of the caspase 8 inhibitor c-FLIPL in endothelial cells. However, while Stx1 treatment may trigger increased expression of TRAIL, DR5 and putative c-FLIPL inhibitors, the time necessary for transcriptional/translational activation would appear to preclude a role for these pathways in rapid caspase 8 activation. The rapid response suggests that Stxs may initiate apoptotic signaling events directly from the ER. B-cell receptor-associated protein 31 (BAP31) is an ER membrane protein that tethers procaspase 8 to the cytoplasmic face of the ER [132]. Following caspase 8 activation, BAP31 is cleaved and induces mitochondrial fission and cytochrome c release [133]. It would be interesting to determine if the rapid induction of apoptosis by Stxs involves BAP31 cleavage and activation of caspase 8 associated with the ER membrane. Src tyrosine kinases phosphorylate caspase 8 [134]. Given the capacity of Stxs to signal through Src family members, one might speculate that caspase 8 is phosphorylated following Stx treatment, thereby altering its apoptosis inducing functions.
Other signaling pathways contributing to apoptosis induction remain to be explored. Stxs activate caspase 6 in epithelial and monocytic cells. caspase 6 may then directly cleave procaspase 8, thereby establishing an amplification loop for caspase 8 activation. Caspase 8 may associate with the death effector domain-containing (DEDD) proteins DEDD and DEDD2 to translocate into the nucleolus, and nucleolar translocation of the complex signals caspase 6 activation and apoptosis [135]. It is not known if Stxs trigger DEDD/DEDD2 translocation, but toxin B-subunits bind the nucleolar protein B23 and accumulate in nucleoli in human macrophages [40]. Increased TRAIL and DR5 expression has been implicated in Stx-induced apoptosis of THP-1 cells. Signaling through death receptors leading to DISC formation and caspase 8 activation requires that death receptors be internalized in a lipid raft- and clathrin-dependent manner (reviewed in [136,137]). Thus, as was the case for toxin internalization and retrograde transport, Stx-induced signaling for apoptosis may be dependent upon the nature of the target cell membrane. Interpretation of data on the role of caspase 8 in activating apoptosis is complicated by the fact that an alternative splicing event introduces a 136-bp insertion between exons 8 and 9 of caspase 8 mRNA. The alternatively spliced mRNA encodes a stop codon, producing a truncated protein called caspase 8L. Caspase 8L binds to DISC formed at death receptors but lacks enzymatic activity. Thus, caspase 8L negatively regulates apoptotic signaling [134]. It would be interesting to determine if resistance to Stx-induced apoptosis, for example in P3HR1 BL cells, or delayed onset of apoptosis, for example in macrophage-like THP-1 cells, is associated with alternative caspase 8 mRNA splicing.
Finally, it appears that a conserved feature of direct Stx-induced apoptosis is the lack of signaling through Fas or TNFR1. Genotoxic stress, such as Stx-induced DNA damage, may trigger procaspase 8 cleavage in a death receptor-independent process. For example, the cytotoxic drug paclitaxel induced apoptosis of transformed B-lymphoid cells, which was initiated by the mitochondrial pathway leading to caspase 8 activation in the absence of death receptor ligation. Release of cytochrome c led to sequential activation of caspases 9, 3 and 8, and to Bid cleavage, establishing an autoamplification loop and resulting in apoptosis. Paclitaxel-induced apoptosis and caspase activation were inhibited by overexpression of Bcl-XL showing that mitochondrial perturbation preceded caspase 8 activation. Paclitaxel triggered caspase 8 activation in cells stably transfected with a dominant negative FADD mutant lacking the N-terminal death effector domain showing that apoptotic signaling was CD95/Fas independent [138]. Other proteases, such as granzyme B, have been reported to activate caspase 8, although it is not known if Stxs induce granzyme B expression or release. In addition to proteolysis, evidence has been presented that the simple dimerization of procaspase 8 may be sufficient to trigger activation. Dimerization of a protease-resistant procaspase 8 mutant produced a protease with enzymatic activity identical to the wild-type caspase 8 heterotetramer [139]. Whether Stxs are capable of activating protease-independent mechanisms of caspase 8 activation remain to be explored.
Future perspective
Shigella dysenteriae serotype 1 and STEC have evolved mechanisms to modulate programmed cell death. Much work remains to be done to understand the role of Stxs in this process. Apoptosis caused by Stxs does not appear to be a critical factor in the initiation of disease. The toxins translocate across Gb3- intestinal epithelial barriers without cell destruction. Experiments to clarify the mechanisms of toxin transport are needed. Bovine Gb3+ crypt cells bind, internalize and degrade toxins; whether a similar response is operative in human crypt cells remains to be characterized. The role, if any, of Paneth cells in the host response to Stxs is unknown. The inflammatory response elicited by pathogen invasion or colonization may provide a cellular toxin transporter, and apoptosis of the carrier cell may be suppressed. The mechanism(s) by which Stxs shift the balance of pro- and anti-apoptotic signaling to favor PMN survival are not well understood.
In contrast to the intestinal epithelial barrier, vascular endothelial cells and renal tubular epithelial cells express Gb3 and undergo apoptosis following intoxication. A limited number of clinical studies of HUS cases verify glomerular and renal tubular apoptosis in situ. The development of therapeutic agents that neutralize Stx signaling for apoptosis may spare the kidney and improve the outcome of renal disease caused by the toxins. Studies are needed to clarify the role of the innate immune response in apoptosis. Apoptosis induction for some cell types may be indirect, that is, the capacity of Stxs to elicit the expression of proinflammatory cytokines may sensitize cells to apoptosis mediated by host response factors or other microbial products. Stx-induced chemokine production may also indirectly contribute to apoptosis by signaling an inflammatory cell infiltrate in damaged tissues. Rabbit neurons appear to be susceptible to Stx-induced apoptosis; however, clinical studies linking apoptosis with CNS complications in humans are lacking. Stx signaling for apoptosis frequently involves the early activation of caspase 8, the activation of the mitochondrial pathway and an alteration in the balance of expression of Bcl-2 family members. However, many details on apoptotic signaling pathways initiated by Stxs remain to be clarified.
Executive summary.
Shiga toxins
Shiga toxins (Stxs) are a family of genetically and functionally related cytotoxins expressed by enteric pathogens.
Stxs inhibit protein synthesis by altering ribosomes.
Stxs are encoded by bacteriophages, which disseminate the genes in Escherichia coli.
Interaction of Stxs with host cells
Expression of the membrane glycolipid globotriaosylceramide (Gb3) is essential for toxin binding and internalization. Toxin binding to Gb3 triggers membrane invagination and activates protein kinase pathways.
Following internalization, toxins are routed to the endoplasmic reticulum (ER) in a process called retrograde transport. The expression of Gb3 isoforms with long-chain, unsaturated fatty acids in association with lipid rafts facilitates retrograde transport.
Toxin A1-fragments possessing enzymatic activity may use the ER-associated degradation pathway to enter the cytosol, and in the process, activate the ribotoxic and ER stress responses.
Diseases caused by Stx-producing bacteria
Stx-producing bacteria cause bloody diarrheal diseases, which may progress to life-threatening extraintestinal complications. The kidneys and CNS are the organs most frequently involved.
Hematogenous spread of Stxs is responsible for renal and CNS damage.
Apoptosis
Apoptosis is a form of cell death that ensues following activation of intracellular signaling pathways.
The two major mechanisms of apoptosis induction are extrinsic or death receptor-mediated and intrinsic or mitochondrial-mediated pathways.
Stx-induced apoptosis
Stxs induce apoptosis in epithelial, endothelial, myeloid and lymphoid cells in vitro and appear to induce apoptosis in rabbit neurons in vivo.
Human intestinal epithelial cells are Gb3 deficient, and Stxs are transported across intestinal epithelial barriers using transcytotic or paracellular mechanisms. Mechanisms of toxin transmural transport are poorly defined.
Many human epithelial and endothelial cells express Gb3 and are sensitive to Stx-induced apoptosis. Apoptosis induction involves activation of both extrinsic (caspase 8, 6 and 3 activation) and intrinsic (truncated BH3-domain-containing protein [tBid] generation, cytochrome c release, caspase 9 activation) pathways. Alterations in the balanced expression of pro- and anti-apoptotic Bcl-2 family members also contribute to apoptosis induction.
Stxs may indirectly induce apoptosis by inhibition of normal cell cycling, induction of proinflammatory and apoptosis-related gene expression, and attracting and activating cells capable of expressing apoptosis-inducing factors.
Primary human monocytes express low levels of membrane Gb3 and route Stxs to lysosomal compartments. Human myeloid leukemia cells express Gb3 and extrinsic and intrinsic pathways of apoptosis induction are activated. Additional mechanisms of apoptosis induction operative in myeloid cells include the release of Ca2+ stores to activate calpain, and increased expression of TRAIL and DR5.
Human polymorphonuclear neutrophils (PMNs) are Gb deficient, yet bind to Stxs via uncharacterized receptor(s). Toxin binding to PMNs is associated with delayed spontaneous apoptosis. PMNs are proposed to act as toxin ‘carriers’ to Gb3-rich microvascular beds, although further studies to support this model are needed.
Two separate apoptosis signaling pathways have been described in Burkitt's lymphoma cell lines: one requires holotoxin and the other is mediated by Gb3 cross-linking.
Studies on Stx-induced neuron apoptosis are limited and mechanisms of apoptosis induction largely uncharacterized. Microglial cell activation and the localized production of cytokines may contribute to apoptosis in the CNS.
Role of Stx-induced apoptosis in disease
A limited number of clinical studies have shown apoptotic renal tubule and glomerular cells in hemolytic uremic syndrome kidney specimens. The role of apoptosis in CNS disease is unknown.
Role for Stx-induced apoptosis in the treatment of cancer
Stxs may be developed as cytotoxic agents to induce apoptosis of tumor cells, targeting Gb3+ tumor cells or inflammatory and vascular cells associated with tumors.
Conclusion
Conserved features of Stx-induced apoptosis include: routing active toxin to the ER where prolonged signaling through the ribotoxic and ER stress responses may activate apoptosis; alterations in the balanced expression of pro- and anti-apoptotic Bcl-2 proteins; rapid activation of caspase 8, which, in turn, activates both caspase-dependent and mitochondrial-dependent apoptotic signaling pathways; and lack of signaling through Fas and TNFRs to trigger apoptosis.
Disruption of Stx-induced apoptosis may represent a therapeutic target to ameliorate tissue damage caused by Stxs.
Acknowledgments
The editorial assistance of Sean Cleghorn is gratefully acknowledged.
Work in the author's laboratory was supported by US Public Health Services grant RO1 AI34530–12 from the National Institute of Allergy and Infectious Diseases (MD, USA).
Footnotes
Financial & competing interests disclosure: The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from that disclosed.
No writing assistance was utilized in the production of the manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Sandvig K. Shiga toxins. Toxicon. 2001;39:1629–1635. doi: 10.1016/s0041-0101(01)00150-7. [DOI] [PubMed] [Google Scholar]; ■ Overview of the early work characterizing the Shiga toxin family.
- 2.Gyles CL. Shiga toxin-producing Escherichia coli: an overview. J Anim Sci. 2007;85:E45–62. doi: 10.2527/jas.2006-508. [DOI] [PubMed] [Google Scholar]; ■■ Thorough review of Shiga toxins, other Shiga toxin-producing Escherichia coli (STEC) virulence determinants, and diseases caused by STEC.
- 3.Fraser ME, Chernaia MM, Kozlov YV, James MNG. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat Struct Biol. 1994;1:59–64. doi: 10.1038/nsb0194-59. [DOI] [PubMed] [Google Scholar]
- 4.Fraser ME, Fujinaga M, Cherney MM, et al. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem. 2004;279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 5.Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes: RNA N-glycosidase activity of the toxins. Eur J Biochem. 1988;171:45–50. doi: 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]
- 6.Correll CC, Munishkin A, Chan YL, Ren Z, Wool IG, Steitz TA. Crystal structure of the ribosomal RNA domain necessary for binding elongation factors. Proc Natl Acad Sci USA. 1998;95:13436–13441. doi: 10.1073/pnas.95.23.13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lingwood CA. Shiga toxin receptor glycolipid binding: pathology and utility. In: Philpott D, Ebel F, editors. Methods in Molecular Medicine Volume 73: E coli Shiga Toxin Methods and Protocols. Humana Press Inc.; Totowa, NJ, USA: 2003. pp. 165–186. [DOI] [PubMed] [Google Scholar]; ■ Reviews the interaction of Shiga toxins with Gb3 at molecular and cellular levels.
- 8.Nyholm PG, Magnusson G, Zheng Z, Norel R, Binnington-Boyd B, Lingwood CA. Two distinct binding sites for globotriaosyl ceramide on verotoxins: identification by molecular modelling and confirmation using deoxy analogues and a new glycolipid receptor for all verotoxins. Chem Biol. 1996;3:263–275. doi: 10.1016/s1074-5521(96)90106-4. [DOI] [PubMed] [Google Scholar]
- 9.Bast DJ, Banerjee L, Clark C, Reed RJ, Brunton JL. The identification of three biologically relevant globotriaosylceramide receptor binding sites on the verotoxin 1 B subunit. Mol Microbiol. 1999;32:953–960. doi: 10.1046/j.1365-2958.1999.01405.x. [DOI] [PubMed] [Google Scholar]
- 10.Allison HE. Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2007;2:155–174. doi: 10.2217/17460913.2.2.165. [DOI] [PubMed] [Google Scholar]; ■■ Reviews Shiga toxin phages and the mechanisms by which the toxin genes disseminate within the microbial gene pool.
- 11.Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence. Infect Immun. 2002;70:3985–3993. doi: 10.1128/IAI.70.8.3985-3993.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herold S, Karch H, Schmidt H. Shiga toxin-encoding bacteriophages – genomes in motion. Int J Med Microbiol. 2004;294:115–121. doi: 10.1016/j.ijmm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Sandvig K, Garred Ø, Prydz K, Kozlov JV, Hansen SH, van Deurs B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature. 1992;358:510–511. doi: 10.1038/358510a0. [DOI] [PubMed] [Google Scholar]
- 14.Khine AA, Tam P, Nutikka A, Lingwood CA. Brefeldin A and filipin distinguish two globotriaosyl ceramide/verotoxin-1 intracellular trafficking pathways involved in Vero cell cytotoxicity. Glycobiology. 2004;14:701–712. doi: 10.1093/glycob/cwh085. [DOI] [PubMed] [Google Scholar]
- 15.Torgersen ML, Lauvrak SU, Sandvig K. The A-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005;272:4103–4113. doi: 10.1111/j.1742-4658.2005.04835.x. [DOI] [PubMed] [Google Scholar]
- 16.Römer W, Berland L, Chambon V, et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature. 2007;450:670–675. doi: 10.1038/nature05996. [DOI] [PubMed] [Google Scholar]
- 17.Pudymaitis A, Armstrong G, Lingwood CA. Verotoxin-resistant cell clones are deficient in the glycolipid globotriosylceramide: differential basis of phenotype. Archiv Biochem Biophys. 1991;286:448–452. doi: 10.1016/0003-9861(91)90064-p. [DOI] [PubMed] [Google Scholar]
- 18.Schweppe CH, Bielaszewska M, Pohlentz G, et al. Glycosphingolipids in vascular endothelial cells: relationship of heterogeneity in Gb3Cer/CD77 receptor expression with differential Shiga toxin 1 cytotoxicity. Glycoconj J. 2008;25:291–304. doi: 10.1007/s10719-007-9091-7. [DOI] [PubMed] [Google Scholar]
- 19.Stricklett PK, Hughes AK, Ergonul Z, Kohan DE. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxicity and globotriaosylceramide expression. J Infect Dis. 2002;186:976–982. doi: 10.1086/344053. [DOI] [PubMed] [Google Scholar]
- 20.Falguiéres T, Mallard F, Baron C, et al. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol Biol Cell. 2001;12:2453–2468. doi: 10.1091/mbc.12.8.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spilsberg B, Llorente A, Sandvig K. Polyunsaturated fatty acids regulate Shiga toxin transport. Biochem Biophys Res Commun. 2007;364:283–288. doi: 10.1016/j.bbrc.2007.09.126. [DOI] [PubMed] [Google Scholar]
- 22.Müthing J, Schweppe CH, Karch H, Friedrich AW. Shiga toxins, glycosphingolipid diversity and endothelial cell injury. Thromb Haemost. 2009;101:252–264. [PubMed] [Google Scholar]; ■■ Review focusing on the biophysical features of Gb3 that influence Shiga toxin binding and intracellular trafficking.
- 23.Katagiri YU, Mori T, Nakajima H, et al. Activation of Src family kinase Yes induced by Shiga toxin binding to globotriaosylceramide (Gb3/CD77) in low density, detergent-insoluble microdomains. J Biol Chem. 1999;274:35278–35282. doi: 10.1074/jbc.274.49.35278. [DOI] [PubMed] [Google Scholar]
- 24.Mori T, Kiyokawa N, Katagiri YU, et al. Globotriaosyl ceramide (CD77/Gb3) in the glycolipid-enriched membrane domain participates in B-cell receptor-mediated apoptosis by regulating Lyn kinase activity in human B-cells. Exp Hematol. 2000;28:1260–1268. doi: 10.1016/s0301-472x(00)00538-5. [DOI] [PubMed] [Google Scholar]
- 25.Lauvrak SU, Wälchli S, Iversen TG, et al. Shiga toxin regulates its entry in a Syk-dependent manner. Mol Biol Cell. 2006;17:1096–1109. doi: 10.1091/mbc.E05-08-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torgersen ML, Wälchli S, Grimmer S, Skanland SS, Sandvig K. Protein kinase Cδ is activated by Shiga toxin and regulates its transport. J Biol Chem. 2007;282:16317–16328. doi: 10.1074/jbc.M610886200. [DOI] [PubMed] [Google Scholar]
- 27.Wälchli S, Skanland SS, Gregers TF, et al. The MAP kinase p38 links Shiga toxin dependent signaling and trafficking. Mol Biol Cell. 2008;19:95–104. doi: 10.1091/mbc.E07-06-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haicheur N, Bismuth E, Bosset S, et al. The B-subunit of Shiga toxin fused to a tumor antigen elicits CTL and targets dendritic cells to allow MHC class I restricted presentation of peptides derived from exogenous antigens. J Immunol. 2000;165:3301–3308. doi: 10.4049/jimmunol.165.6.3301. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs E, Haas AK, Spooner RA, Yoshimura S, Lord JM, Barr FA. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J Cell Biol. 2007;177:1133–1143. doi: 10.1083/jcb.200612068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenzie J, Johannes L, Taguchi T, Sheff D. Passage through the Golgi is necessary for Shiga toxin B subunit to reach the endoplasmic reticulum. FEBS J. 2009;276:1581–1595. doi: 10.1111/j.1742-4658.2009.06890.x. [DOI] [PubMed] [Google Scholar]
- 31.Saenz JB, Doggett TA, Haslam DB. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect Immun. 2007;75:4552–4561. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavelka M, Neumüller J, Ellinger A. Retrograde traffic in the biosynthetic-secretory route. Histochem Cell Biol. 2008;129:277–288. doi: 10.1007/s00418-008-0383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Excellent review of retrograde (and anterior grade) intracellular trafficking.
- 33.Tam PJ, Lingwood CA. Membrane-cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology. 2007;153:2700–2710. doi: 10.1099/mic.0.2007/006858-0. [DOI] [PubMed] [Google Scholar]
- 34.Garred Ø, van Deurs B, Sandvig K. Furin-induced cleavage and activation of Shiga toxin. J Biol Chem. 1995;270:10817–10821. doi: 10.1074/jbc.270.18.10817. [DOI] [PubMed] [Google Scholar]
- 35.Garred Ø, Dubinina E, Holm PK, et al. Role of processing and intracellular transport for optimal toxicity of Shiga toxin and toxin mutants. Exp Cell Res. 1995;218:39–49. doi: 10.1006/excr.1995.1128. [DOI] [PubMed] [Google Scholar]
- 36.Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr Topics Microbiol Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]; ■■ Reviews the evidence that protein toxins that translocate to the endoplasmic reticulum are capable of ‘co-opting’ the endoplasmic reticulum-associated degradation pathway to enter the cytoplasm.
- 37.Smith DC, Sillence DJ, Falguières T, et al. The association of Shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol Biol Cell. 2006;17:1375–1387. doi: 10.1091/mbc.E05-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaPointe P, Wei X, Gariepy J. A role for the protease sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J Biol Chem. 2005;280:23310–23318. doi: 10.1074/jbc.M414193200. [DOI] [PubMed] [Google Scholar]
- 39.Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falguiéres T, Johannes L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol Cell. 2006;98:125–134. doi: 10.1042/BC20050001. [DOI] [PubMed] [Google Scholar]
- 41.Lee SY, Lee MS, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770–780. doi: 10.1111/j.1462-5822.2007.01083.x. [DOI] [PubMed] [Google Scholar]; ■ First study to show that Shiga toxins activate the endoplasmic reticulum stress response.
- 42.Strockbine NA, Jackson MP, Sung LM, Holmes RK, O'Brien AD. Cloning and sequencing of the genes for Shiga toxin from Shigella dysenteriae type 1. J Bacteriol. 1988;170:1116–1122. doi: 10.1128/jb.170.3.1116-1122.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson MP, Neill RJ, O'Brien AD, Holmes RK, Newland JW. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol Lett. 1987;44:109–114. doi: 10.1016/0882-4010(87)90106-9. [DOI] [PubMed] [Google Scholar]
- 44.McCluskey AJ, Poon GMK, Bolewska-Pedyczak E, et al. The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J Mol Biol. 2008;378:375–386. doi: 10.1016/j.jmb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 45.Kotloff KL, Winickoff JP, Ivanoff B, et al. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull WHO. 1999;77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 46.Fontaine A, Arondel J, Sansonetti PJ. Role of Shiga toxin in the pathogenesis of bacillary dysentery studied by using a Tox- mutant of Shigella dysenteriae 1. Infect Immun. 1988;56:3099–3109. doi: 10.1128/iai.56.12.3099-3109.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tesh VL. Foodborne enterohemorrhagic Escherichia coli infections. In: Beier RC, Pillai SD, Phillips TD, Ziprin RL, editors. Preharvest and Postharvest Food Safety: Contemporary Issues and Future Directions. Blackwell Publishing; Ames, IA, USA: 2004. pp. 27–42. [Google Scholar]
- 49.Karmali MA, Mascarenhas M, Shen S, et al. Association of genomic O island 122 of Escherichia coli EDL 933 with verocytotoxin-producing Escherichia coli seropathotypes that are linked to epidemic and/or serious disease. J Clin Microbiol. 2003;41:4930–4940. doi: 10.1128/JCM.41.11.4930-4940.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mead PS, Slutsker L, Dietz V, et al. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarr PI, Gordon CA, Chandler WI. Shiga toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 52.Orth D, Grif K, Khan AB, Naim A, Dierich MP, Würzner R. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn Microbiol Infect Dis. 2007;59:235–242. doi: 10.1016/j.diagmicrobio.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 53.Proulx F, Tesh VL. Renal diseases in the pediatric intensive care unit: thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. In: Wheeler DS, Wong HR, Shanley TP, editors. Pediatric Critical Care Medicine: Basic Science and Clinical Evidence. Springer Verlag; London, UK: 2007. pp. 1189–1204. [Google Scholar]
- 54.Scheiring J, Andreoli SP, Zimmerhackl LB. Treatment and outcome of Shiga toxin-associated hemolytic uremic syndrome (HUS) Pediatr Nephrol. 2008;23:1749–1760. doi: 10.1007/s00467-008-0935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Recent review of the pathophysiology of Shiga toxin-associated hemolytic uremic syndrome.
- 55.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Broad overview of intrinsic and extrinsic pathways of apoptosis induction.
- 56.Kroemer G, Galuzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 57.Szegezdi E, MacDonald DC, Chonghaile TN, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 58.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 59.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 60.Pearson G, Robinson F, Beers-Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 61.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 62.Iordanov MS, Pribnow D, Magun JL, et al. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith WE, Kane AV, Campbell ST, Acheson DWK, Cochran BH, Thorpe CM. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect Immun. 2003;71:1497–1504. doi: 10.1128/IAI.71.3.1497-1504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foster GH, Tesh VL. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-α. J Leukoc Biol. 2002;71:107–114. [PubMed] [Google Scholar]
- 65.Cherla RP, Lee SY, Mees PL, Tesh VL. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J Leukoc Biol. 2006;79:397–407. doi: 10.1189/jlb.0605313. [DOI] [PubMed] [Google Scholar]
- 66.Allan LA, Clarke PR. Apoptosis and autophagy: regulation of caspase-9 by phosphorylation. FEBS J. 2009;276:6063–6073. doi: 10.1111/j.1742-4658.2009.07330.x. [DOI] [PubMed] [Google Scholar]
- 67.Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS. Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 68.Mathura VS, Soman KV, Varma TK, Braun W. A multimeric model for murine anti-apoptotic protein Bcl-2 and structural insights for its regulation by post-translational modification. J Mol Model. 2003;9:298–303. doi: 10.1007/s00894-003-0152-y. [DOI] [PubMed] [Google Scholar]
- 69.DeChiara G, Marcocci ME, Torcia M, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biological consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 70.Jones NL. Detection of Shiga toxin-mediated programmed cell death and delineation of death signaling pathways. In: Philpott D, Ebel F, editors. Methods in Molecular Medicine Volume 73: E coli Shiga Toxin Methods and Protocols. Humana Press Inc.; Totowa, NJ, USA: 2003. pp. 229–241. [DOI] [PubMed] [Google Scholar]; ■ Resource for detailed protocols and materials necessary to measure Shiga toxin-induced apoptosis.
- 71.Inward CD, Williams J, Chant I, et al. Verocytotoxin-1 induces apoptosis in Vero cells. J Infect. 1995;30:213–218. doi: 10.1016/s0163-4453(95)90693-2. [DOI] [PubMed] [Google Scholar]
- 72.Cherla RP, Lee SY, Tesh VL. Shiga toxins and apoptosis. FEMS Microbiol Lett. 2003;228:159–166. doi: 10.1016/S0378-1097(03)00761-4. [DOI] [PubMed] [Google Scholar]; ■ Reviews early studies on Shiga toxin-induced apoptosis.
- 73.Schüller S, Frankel G, Phillips AD. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004;6:289–301. doi: 10.1046/j.1462-5822.2004.00370.x. [DOI] [PubMed] [Google Scholar]
- 74.Schüller S, Heuschkel R, Torrente F, Kaper JB, Phillips AD. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microbes Infect. 2007;9:35–39. doi: 10.1016/j.micinf.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 75.Philpott DJ, Ackerley CA, Kiliaan AJ, Karmali MA, Perdue MH, Sherman PM. Translocation of verotoxin-1 across T84 monolayers: mechanism of bacterial toxin penetration of epithelium. Am J Physiol Gastrointest Liver Physiol. 1997;273:G1349–G1358. doi: 10.1152/ajpgi.1997.273.6.G1349. [DOI] [PubMed] [Google Scholar]
- 76.Hurley BP, Jacewicz M, Thorpe CM, et al. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect Immun. 1999;67:6670–6677. doi: 10.1128/iai.67.12.6670-6677.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malyukova I, Murray KF, Zhu C, et al. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G78–G92. doi: 10.1152/ajpgi.90347.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takeda T, Dohi S, Igarashi T, Yamanaka T, Yoshiya K, Kobayashi N. Impairment by verotoxin of tubular function contributes to the renal damage seen in haemolytic uraemic syndrome. J Infect. 1993;27:339–341. doi: 10.1016/0163-4453(93)92474-b. [DOI] [PubMed] [Google Scholar]
- 79.Jones NL, Islur A, Haq R, et al. Escherichia coli Shiga toxins induce apoptosis in epithelial cells that is regulated by the Bcl-2 family. Am J Physiol Gastrointest Liver Physiol. 2000;278:G811–G819. doi: 10.1152/ajpgi.2000.278.5.G811. [DOI] [PubMed] [Google Scholar]
- 80.Wilson C, Foster GH, Bitzan MM. Silencing of Bak ameliorates apoptosis of human proximal tubular epithelial cells by Escherichia coli-derived Shiga toxin 2. Infection. 2005;33:362–367. doi: 10.1007/s15010-005-5073-5. [DOI] [PubMed] [Google Scholar]
- 81.Ching JCY, Jones NL, Ceponis PJM, Karmali MA, Sherman PM. Escherichia coli Shiga-like toxins induce apoptosis and cleavage of poly(ADP-ribose) polymerase via in vitro activation of caspases. Infect Immun. 2002;70:4669–4677. doi: 10.1128/IAI.70.8.4669-4677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki A, Doi H, Matsuzawa F, et al. Bcl-2 antiapoptotic protein mediates verotoxin II-induced cell death: possible association between Bcl-2 and tissue failure by E. coli O157:H7. Genes Dev. 2000;14:1734–1740. [PMC free article] [PubMed] [Google Scholar]
- 83.Fujii J, Matsui T, Heatherly DP, et al. Rapid apoptosis induced by Shiga toxin in HeLa cells. Infect Immun. 2003;71:2724–2735. doi: 10.1128/IAI.71.5.2724-2735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ Thorough evaluation of Shiga toxin-induced apoptosis induction in an epithelial cell line.
- 84.Nakagawa I, Nakata M, Kawabata S, Hamada S. Regulated expression of the Shiga toxin B gene induces apoptosis in mammalian fibroblastic cells. Mol Microbiol. 1999;33:1190–1199. doi: 10.1046/j.1365-2958.1999.01564.x. [DOI] [PubMed] [Google Scholar]
- 85.Jandhyala DM, Ahluwalla A, Obrig T, Thorpe CM. ZAK: a MAP3kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol. 2008;10:1468–1477. doi: 10.1111/j.1462-5822.2008.01139.x. [DOI] [PubMed] [Google Scholar]
- 86.Sood A, Mathew R, Trachtman H. Cytoprotective effect of curcumin in human proximal tubule epithelial cells exposed to Shiga toxin. Biochem Biophys Res Commun. 2001;283:36–41. doi: 10.1006/bbrc.2001.4749. [DOI] [PubMed] [Google Scholar]
- 87.Bhattacharjee RN, Park KS, Uematsu S, et al. Escherichia coli verotoxin 1 mediates apoptosis in human HCT116 colon cancer cells by inducing overexpression of the GADD family of genes and S phase arrest. FEBS Lett. 2005;579:6604–6610. doi: 10.1016/j.febslet.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 88.Brigotti M, Alfieri R, Sestili P, et al. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002;16:365–372. doi: 10.1096/fj.01-0521com. [DOI] [PubMed] [Google Scholar]
- 89.Arab S, Lingwood CA. Intracellular targeting of the endoplasmic reticulum/nuclear envelope by retrograde transport may determine cell hypersensitivity to verotoxin via globotriaosyl ceramide fatty acid isoform traffic. J Cell Physiol. 1998;177:646–660. doi: 10.1002/(SICI)1097-4652(199812)177:4<646::AID-JCP15>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 90.Fujii J, Wood K, Matsuda F, et al. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect Immun. 2008;76:3679–3689. doi: 10.1128/IAI.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matussek A, Lauber J, Bergau A, et al. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood. 2003;102:1323–1332. doi: 10.1182/blood-2002-10-3301. [DOI] [PubMed] [Google Scholar]
- 92.Erwert RD, Winn RK, Harlan JM, Bannerman DD. Shiga-like toxin inhibition of FLICE-like inhibitory protein expression sensitizes endothelial cells to bacterial lipopolysaccharide-induced apoptosis. J Biol Chem. 2002;277:40567–40574. doi: 10.1074/jbc.M206351200. [DOI] [PubMed] [Google Scholar]
- 93.Chang DW, Xing Z, Pan Y, et al. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Erwert RD, Eiting KT, Tupper JC, Winn RK, Harlan JM, Bannerman DD. Shiga toxin induces decreased expression of the anti-apoptotic protein Mcl-1 concomitant with the onset of endothelial apoptosis. Microb Pathog. 2003;35:87–93. doi: 10.1016/s0882-4010(03)00100-1. [DOI] [PubMed] [Google Scholar]
- 95.Molostvov G, Morris A, Rose P, Basu S. Interaction of cytokines and growth factor in the regulation of verotoxin-induced apoptosis in cultured human endothelial cells. Br J Haematol. 2001;113:891–897. doi: 10.1046/j.1365-2141.2001.02835.x. [DOI] [PubMed] [Google Scholar]
- 96.Hoey DEE, Sharp L, Currie C, Lingwood CA, Gally DL, Smith DGE. Verotoxin 1 binding to intestinal crypt epithelial cells results in localization to lysosomes and abrogation of toxicity. Cell Microbiol. 2003;5:85–97. doi: 10.1046/j.1462-5822.2003.00254.x. [DOI] [PubMed] [Google Scholar]
- 97.Kniep B, Monner DA, Schwuléra U, Mühlradt PF. Glycosphingolipids of the globo-series are associated with the monocytic lineage of human myeloid cells. Eur J Biochem. 1985;149:187–191. doi: 10.1111/j.1432-1033.1985.tb08910.x. [DOI] [PubMed] [Google Scholar]
- 98.Kojio S, Zhang HM, Ohmura M, Gondaira F, Kobayashi N, Yamamoto T. Caspase-3 activation and apoptosis induction coupled with the retrograde transport of Shiga toxin: inhibition by brefeldin A. FEMS Immunol Med Microbiol. 2000;29:275–281. doi: 10.1111/j.1574-695X.2000.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 99.Harrison LM, Cherla RP, van den Hoogen C, van Haaften WCE, Lee SY, Tesh VL. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb Pathog. 2005;38:63–76. doi: 10.1016/j.micpath.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 100.Ramegowda B, Tesh VL. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect Immun. 1996;64:1173–1180. doi: 10.1128/iai.64.4.1173-1180.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee SY, Cherla RP, Caliskan I, Tesh VL. Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line THP-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect Immun. 2005;73:5115–5126. doi: 10.1128/IAI.73.8.5115-5126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Babu US, Gaines DM, Wu Y, Westphal CD, Pereira M, Raybourne RB. Use of flow cytometry in an apoptosis assay to determine pH and temperature stability of Shiga-like toxin 1. J Microbiol Meth. 2008;75:167–171. doi: 10.1016/j.mimet.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 103.Lee SY, Cherla RP, Tesh VL. Simultaneous induction of apoptotic and survival signaling pathways in macrophage-like THP-1 cells by Shiga toxin 1. Infect Immun. 2007;75:1291–1302. doi: 10.1128/IAI.01700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou HR, Islam Z, Pestka JJ. Induction of competing apoptotic and survival signaling pathways in the macrophage by the ribotoxic trichothecene deoxynivalenol. Toxicol Sci. 2005;87:113–122. doi: 10.1093/toxsci/kfi234. [DOI] [PubMed] [Google Scholar]
- 105.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee MS, Cherla RP, Leyva-Illades D, Tesh VL. Bcl-2 regulates the onset of Shiga toxin 1-induced apoptosis in THP-1 cells. Infect Immun. 2009;77:5233–5244. doi: 10.1128/IAI.00665-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wolfson JJ, May KL, Thorpe CM, Jandhyala DM, Paton JC, Paton AW. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress signaling pathways. Cell Microbiol. 2008;10:1775–1766. doi: 10.1111/j.1462-5822.2008.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu J, Akahoshi T, Sasahana T, et al. Inhibition of neutrophil apoptosis by verotoxin 2 derived from Escherichia coli O157:H7. Infect Immun. 1999;67:6203–6205. doi: 10.1128/iai.67.11.6203-6205.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Flagler MJ, Strasser JE, Chalk CL, Weiss AA. Comparative analysis of the abilities of Shiga toxins 1 and 2 to bind to and influence neutrophil apoptosis. Infect Immun. 2007;75:760–765. doi: 10.1128/IAI.01594-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Greiner TP, Mulvey GL, Marcato P, Armstrong GD. Differential binding of Shiga toxin 2 to human and murine neutrophils. J Med Microbiol. 2007;56:1423–1430. doi: 10.1099/jmm.0.47282-0. [DOI] [PubMed] [Google Scholar]
- 111.Brigotti M, Carnicelli D, Ravanelli E, et al. Interactions between Shiga toxins and human polymorphonuclear leukocytes. J Leukoc Biol. 2008;84:1019–1027. doi: 10.1189/jlb.0308157. [DOI] [PubMed] [Google Scholar]
- 112.te Loo DMWM, Monnens LAH, van der Velden TJAM, et al. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood. 2000;95:3396–3402. [PubMed] [Google Scholar]
- 113.te Loo DMWM, van Hinsbergh VWM, van den Heuvel LPWJ, Monnens LAH. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12:800–806. doi: 10.1681/ASN.V124800. [DOI] [PubMed] [Google Scholar]
- 114.Brigotti M, Caprioli A, Tozzi AE, et al. Shiga toxins present in the gut and in the polymorphonuclear leukocytes circulating in the blood of children with hemolytic-uremic syndrome. J Clin Microbiol. 2006;44:313–317. doi: 10.1128/JCM.44.2.313-317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kiyokawa N, Mori T, Taguchi T, et al. Activation of the caspase cascade during Stx1-induced apoptosis in Burkitt's lymphoma cells. J Cell Biochem. 2001;81:128–142. doi: 10.1002/1097-4644(20010401)81:1<128::aid-jcb1029>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 116.Tétaud C, Falguières T, Carlier K, et al. Two distinct Gb3/CD77 signaling pathways leading to apoptosis are triggered by anti-Gb3/CD77 mAb and verotoxin-1. J Biol Chem. 2003;278:45200–45208. doi: 10.1074/jbc.M303868200. [DOI] [PubMed] [Google Scholar]
- 117.Gordon J, Challa A, Levens JM, et al. CD40 ligand, Bcl-2, and Bcl-xL spare group I Burkitt lymphoma cells from CD77-directed killing via verotoxin-1 B chain but fail to protect against the holotoxin. Cell Death Diff. 2000;7:785–794. doi: 10.1038/sj.cdd.4400710. [DOI] [PubMed] [Google Scholar]
- 118.Fujii J, Kinoshita Y, Kita T, et al. Magnetic resonance imaging and histopathological study of brain lesions in rabbits given intravenous verotoxin 2. Infect Immun. 1996;64:5053–5060. doi: 10.1128/iai.64.12.5053-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fujii J, Kita T, Yoshida S, et al. Direct evidence of neuron impairment by oral infection with verotoxin-producing Escherichia coli O157:H- in mitomycin-treated mice. Infect Immun. 1994;62:3447–3453. doi: 10.1128/iai.62.8.3447-3453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mizuguchi M, Tanaka S, Fujii I, et al. Neuronal and vascular pathology produced by verocytotoxin-2 in the rabbit central nervous system. Acta Neuropathol. 1996;91:254–262. doi: 10.1007/s004010050423. [DOI] [PubMed] [Google Scholar]
- 121.Takahashi K, Funata N, Ikuta F, Sato S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. BMC J Neuroinflamm. 2008;5:11. doi: 10.1186/1742-2094-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee DY, Park KW, Jin BK. Thrombin induces neurodegeneration and microglial activation in the cortex in vivo and in vitro: proteolytic and non-proteolytic actions. Biochem Biophys Res Commun. 2006;346:727–738. doi: 10.1016/j.bbrc.2006.05.174. [DOI] [PubMed] [Google Scholar]
- 123.Ren J, Utsunomiya I, Taguchi K, et al. Localization of verotoxin receptors in nervous system. Brain Res. 1999;825:183–188. doi: 10.1016/s0006-8993(99)01196-8. [DOI] [PubMed] [Google Scholar]
- 124.Okuda T, Tokuda N, Numata S, et al. Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J Biol Chem. 2006;281:10230–10235. doi: 10.1074/jbc.M600057200. [DOI] [PubMed] [Google Scholar]
- 125.Obata F, Tohyama K, Bonev AD, et al. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J Infect Dis. 2008;198:1398–1406. doi: 10.1086/591911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Karpman D, Håkansson A, Perez MTR, et al. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: in vivo and in vitro studies. Infect Immun. 1998;66:636–644. doi: 10.1128/iai.66.2.636-644.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.te Loo DMWM, Monnens LAH, van den Heuvel LPWJ, Gubler MC, Kockx MM. Detection of apoptosis in kidney biopsies of patients with D+ hemolytic uremic syndrome. Pediatr Res. 2001;49:413–416. doi: 10.1203/00006450-200103000-00017. [DOI] [PubMed] [Google Scholar]
- 128.Fernandez GC, Gomez SA, Ramos MV, et al. The functional state of neutrophils correlates with the severity of renal dysfunction in children with hemolytic uremic syndrome. Pediatr Res. 2007;61:123–127. doi: 10.1203/01.pdr.0000250037.47169.55. [DOI] [PubMed] [Google Scholar]
- 129.Johansson D, Kosovac E, Moharer J, et al. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer. 2009;9:67. doi: 10.1186/1471-2407-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Salhia B, Rutka JT, Lingwood C, Nutikka A, van Furth WR. The treatment of malignant meningioma with verotoxin. Neoplasia. 2002;4:304–311. doi: 10.1038/sj.neo.7900243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Viels T, Dransart E, Nemati F, et al. In vivo tumor targeting by the B-subunit of Shiga toxin. Mol Imaging. 2008;7:239–247. [PubMed] [Google Scholar]
- 132.Chandra D, Choy G, Deng X, Bhatia B, Daniel P, Tang DG. Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol Cell Biol. 2004;24:6592–6607. doi: 10.1128/MCB.24.15.6592-6607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Breckinridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fulda S. Caspase-8 in cancer biology and therapy. Cancer Lett. 2009;281:128–133. doi: 10.1016/j.canlet.2008.11.023. [DOI] [PubMed] [Google Scholar]; ■■ Reviews caspase 8, the initiator caspase, which has emerged as a key activator of Shiga toxin-induced apoptosis.
- 135.Alcivar A, Hu S, Tang J, Yang X. DEDD and DEDD2 associate with caspase-8/10 and signal cell death. Oncogene. 2003;22:291–297. doi: 10.1038/sj.onc.1206099. [DOI] [PubMed] [Google Scholar]
- 136.Schulze S, Tchikov V, Schneider-Brachert W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat Rev Mol Cell Biol. 2008;9:655–662. doi: 10.1038/nrm2430. [DOI] [PubMed] [Google Scholar]
- 137.Valmiki MG, Ramos JW. Death effector domain-containing proteins. Cell Mol Life Sci. 2009;66:814–830. doi: 10.1007/s00018-008-8489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■■ Comprehensive review of apoptosis and cell survival signaling pathways activated by the death effector domain-containing proteins FADD, c-FLIPL and caspase-8.
- 138.von Haefen C, Wieder T, Essmann F, Schulze-Osthoff K, Dörken B, Daniel PT. Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8 driven mitochondrial amplification loop. Oncogene. 2003;22:2236–2247. doi: 10.1038/sj.onc.1206280. [DOI] [PubMed] [Google Scholar]
- 139.Donepudi M, MacSweeney A, Briand C, Grütter MG. Insights into the regulatory mechanism for caspase 8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 140.Melton-Celsa AR, O'Brien AD. Structure, biology and relative toxicity of Shiga toxin family members for cells and animals. In: Kaper JB, O'Brien AD, editors. Escherichia coli O157:H7 and Other Shiga Toxin-Producing E coli Strains. ASM Press; Washington, DC, USA: 1998. pp. 121–128. [Google Scholar]
- 141.Zhang W, Bielaszewska M, Kuczius T, Karch H. Identification, characterization and distribution of a Shiga toxin 1 gene variant (stx1c) in Escherichia coli strains isolated from humans. J Clin Microbiol. 2002;40:1441–1446. doi: 10.1128/JCM.40.4.1441-1446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bürk C, Dietrich R, Açar G, Moravek M, Bülte M, Märtlbauer E. Identification and characterization of a new variant of Shiga toxin 1 in Escherichia coli ONT:H19 of bovine origin. J Clin Microbiol. 2003;41:2106–2112. doi: 10.1128/JCM.41.5.2106-2112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jelacic JK, Damrow T, Chen GS, et al. Shiga toxin-producing Escherichia coli in Montana: bacterial genotypes and clinical profiles. J Infect Dis. 2003;188:719–729. doi: 10.1086/376999. [DOI] [PubMed] [Google Scholar]
- 144.Melton-Celsa AR, Kokai-Kun JF, O'Brien AD. Activation of Shiga toxin type 2d (Stx2d) by elastase involves cleavage of the C-terminal two amino acids of the A2 peptide in the context of the appropriate B pentamer. Mol Microbiol. 2002;43:207–215. doi: 10.1046/j.1365-2958.2002.02733.x. [DOI] [PubMed] [Google Scholar]
- 145.Schmidt H, Scheef J, Morabito S, Caprioli A, Wieler LH, Karch H. A new Shiga toxin 2 variant (Stx2f) from Escherichia coli isolated from pigeons. Appl Environ Microbiol. 2000;66:1205–1208. doi: 10.1128/aem.66.3.1205-1208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]