

Abstract
Pyruvate decarboxylase (PDC) uses thiamine diphosphate as an essential cofactor to catalyze the formation of acetaldehyde on the pathway of ethanol synthesis. Here we report the crystallographic image of a prereaction intermediate of a bacterial pyruvate decarboxylase prepared by cocrystallizing the enzyme with pyruvate and a stable analogue of the cofactor’s activated ylid form. A second crystal structure of PDC in complex with fluoride shows that the ion organizes a water molecule that occludes the pyruvate binding site, accounting for the inhibitory effect of the halide. Also reported is a structure of the cofactor-free apo form, which when compared to the structure of the holo form indicates how thiamine diphosphate organizes the active site pocket of pyruvate decarboxylase to support catalysis. Guided by the structural and enzymatic data, we propose roles for several key residues in the catalytic mechanism.
Thiamine diphosphate (ThDP,11 in Figure 1a) is a required cofactor for several enzymes from the central metabolism in species from all domains of life. ThDP-dependent enzymes include the α-keto acid decarboxylases and dehydrogenases, transketolase, acetohydroxyacid and acetolactate synthases, 1-deoxyxylulose 5-phosphate synthase, and benzaldehyde lyase (1,2). Collectively, they catalyze a diversity of reactions that involve the cleavage and formation of bonds adjacent to the carbon of a carbonyl group. Pyruvate decarboxylase (PDC, EC 4.1.1.1), the subject of this study, catalyzes the conversion of pyruvate to acetaldehyde with release of carbon dioxide as part of the fermentation process that generates ethanol.
Figure 1.

(a) Catalytic cycle of pyruvate decarboxylation by PDC. Protonation at N-1′ and deprotonation at 4′-NH2 of ThDP cofactor (1) give the imino tautomer (2) which promotes the deprotonation of C2 on the thiazolium ring to form the active ylid (3). The ylid attacks C2 of pyruvate to give the lactyl adduct (4). Decarboxylation then gives the enamine intermediate (5); protonation gives hydroxyethyl-ThDP (6), and then release of the product acetaldehyde regenerates the ylid (3). (b) Structure of the intermediate analogue, triazole-ThDP 7. The C2 and S atoms of ThDP are replaced with nitrogen atoms in the triazole derivative.
In common with other known ThDP-dependent enzymes, PDC is thought to stabilize the imino tautomer of ThDP (2 in Figure 1a), which in turn promotes deprotonation of C2 of the thiazolium ring to generate the reactive ylid (3) (3−5). In PDC, the ylid attacks pyruvate to generate a lactyl adduct (4). Decarboxylation of the lactyl adduct yields the enamine intermediate (5), which is then protonated to give hydroxyethyl ThDP (6). Finally, release of the acetaldehyde product regenerates the ylid. The reaction cycle is very similar in the E1p subunit of pyruvate dehydrogenase, except that the C2 group is transferred from the enamine intermediate onto the dithiolane ring of a lipoyl group, giving an acetyl dihydrolipoyl moiety. The E1p enzyme active site has evolved to prevent the proton-activated release of free acetaldehyde, which would represent an off-pathway misreaction for pyruvate dehydrogenase.
Much of the detailed understanding of how ThDP is activated and progresses through the reaction cycle has come from crystal structures that are available for several ThDP-dependent enzymes, including, among others, transketolase (6,7) (e.g., PDB entry 1TRP), pyruvate decarboxylase (8−12) (PDB entries 1ZPD and 1PVD), the E1 subunits of pyruvate and other 2-oxo acid dehydrogenase complexes (13,14) (PDHc, PDB entry 1QS0), and pyruvate oxidase (PDB entries 1POX and 1POW) (15,16) . These structures reveal common features for ThDP binding in the active site pocket (10,17). The binding of ThDP requires Mg2+ (or another bivalent metal ion), which is bound to the pyrophosphate and a number of conserved residues in the active site (10,16). The thiamine-dependent enzymes are all oligomeric and are organized as homodimers, homotetramers, or heterotetramers, with the ThDP cofactor bound at the interface between subunits.
Studies of PDCs from fungi and plants reveal that the substrate itself can be an allosteric activator of those enzymes. The artificial activator pyruvamide accelerates the catalytic reaction by at least 3 orders of magnitude (3,11). It has been demonstrated that these enzymes undergo a large conformational change upon binding of the substrate (11) and potential allosteric sites have been identified in the yeast enzyme (12) and the close prokaryotic homologue, phenylpyruvate decarboxylase (18). In contrast to its eukaryotic homologues, the PDC from the α-protobacterial species, Zymomonas mobilis (ZmPDC), is not allosterically activated by the substrate (10,19). It is striking that the bacterial and eukaryotic enzymes are structurally homologous, yet they differ markedly in cooperative behavior. The crystal structure of ZmPDC has been determined (PDB entry 1ZPD), but in this structure, it was apparent that the cofactor had been degraded, with the loss of C2 by hydrolysis, and the enzyme was inactive (10).
We have previously reported (20,21) the synthesis of a triazole analogue of ThDP (Figure 1b), which has both the C2 and S atoms replaced with nitrogen. The triazole ring is neutral and thus resembles the reactive zwitterionic ylide 3. The triazole derivative is an extremely potent inhibitor of ZmPDC with a KI value of ca. 20 pM, compared to a KD value of 0.35 μM for ThDP (20,21). One reason why the triazole derivative binds so much more tightly than ThDP itself is probably due to the influence of the protein to provide greater stabilization of the neutral thiazole-like ring relative to the positively charged thiazolium ring (22,23). In this way, the enzyme promotes both the formation of the ylid and the decarboxylation reaction.
We report here three crystal structures of ZmPDC: (i) the apoenzyme, (ii) the complex with triazole-ThDP and fluoride, and (iii) the complex with triazole-ThDP and pyruvate. This last structure represents a prereaction complex of substrate in the active site. Comparison of the holo and apo forms of the enzyme reveals a significant conformational change resulting in a clamping of a loop that interacts with the ThDP analogue in the active site. We discuss the implications of these structural data for the enzyme reaction mechanism.
Materials and Methods
Protein Expression and Purification
ZmPDC was overexpressed and purified to homogeneity as described previously (24). Apo-PDC was prepared by dissolving the holo-PDC in HEPES-KOH buffer (50 mM, pH 8.2) containing dipicolinic acid (1 mM). After incubation at 5 °C for 30 min, the apo-PDC was separated from cofactors by repeated ultrafiltration (100 kDa cutoff). The buffer was finally replaced with MES-KOH buffer (10 mM, pH 6.5). The preparation of the apoenzyme was confirmed by the complete lack of enzymic activity without added ThDP. Addition of ThDP (4 μM) and MgCl2 (10 mM) to the apoenzyme restored the fully active enzyme (>95% of its original activity).
Enzyme Activity Assay
Activity was quantified using a coupled enzyme assay, monitoring the NADH-dependent reduction of acetaldehyde by alcohol dehydrogenase, as described previously (21,22).
Crystallizations
ZmPDC was crystallized using the hanging drop vapor diffusion method. Apo-PDC (7.6 mg/mL) was incubated with a triazole analogue (Figure 1b) (3 mM) and MgCl2 (5 mM) in MES-KOH buffer (10 mM, pH 6.5) at 4 °C overnight. The binding of the triazole analogue was confirmed by an assay for PDC activity (20). Crystallization droplets were set up at 18 °C by mixing 1 μL of enzyme solution with 1 μL of reservoir solution containing potassium fluoride (0.2 M) and PEG 3350 (20%, w/v). Thin platelike crystals were obtained within 7 days, but the plates grew as misaligned layers. To improve crystal size and morphology, crystallization droplets were set up as described above with xylitol (3%, w/v), pre-equilibrated for 24 h, and then seeded with serially diluted suspensions of crushed microcrystals. This procedure gave larger single crystals within 1−14 days (maximum size of ca. 0.4 mm × 0.2 mm × 0.1 mm). The procedure was repeated to give apo crystals using an enzyme solution without the triazole analogue and with potassium chloride (0.2 M) in place of potassium fluoride, and seeding was done with seed stock solutions prepared from apo-PDC crystals (obtained without xylitol).
Cocrystallization of PDC with pyruvate was conducted using a reservoir solution containing potassium chloride (0.2 M), PEG 3350 (17%, w/v), and xylitol (3 or 1.5%, w/v), which was mixed in equal volumes (1 μL each) with a preincubated (overnight) enzyme solution including the triazole analogue (3 mM), MgCl2 (5 mM), and pyruvate (5 mM). Droplets were pre-equilibrated for 24 h and then seeded as described above (using seed stock solutions obtained from holo crystals). Crystals grew from this condition within 7 days. All crystals were transferred to a cryoprotectant solution containing potassium chloride (or potassium fluoride) (0.2 M), PEG 3350 (20%, w/v), and xylitol (15%, w/v) and immediately frozen in liquid nitrogen.
Structure Determination, Refinement, and Modeling
X-ray diffraction data of apo and holo form crystals were collected at 100 K at station ID23-1 (ESRF, Grenoble, France), and data for the complex with pyruvate were collected at station I03 (Diamond Light Source, Didcot, U.K.). Data were processed, scaled, and truncated in MOSFLM, SCALA, and TRUNCATE (CCP4 suite). Data processing statistics are summarized in Table 1.
Table 1. Data Collection and Refinement Statistics.
| apo form | holo form | pyruvate form | |
|---|---|---|---|
| no. of cofactors in the asymmetric unit | none | four triazole-ThDP molecules | eight triazole-ThDP molecules |
| substrate | − | − | three pyruvate molecules |
| Data Collection Statistics | |||
| space group | P1 | P1 | P1 |
| unit cell [a, b, c (Å); α, β, γ (deg)] | 70.90, 111.18, 167.12; 89.89, 90.87, 101.27 | 70.28, 92.09, 98.63; 73.44, 85.76, 67.82 | 70.62, 111.58, 167.43; 89.82, 90.11, 78.94 |
| resolution (Å) | 50−2.3 | 34.25−1.7 | 50−2.2 |
| wavelength (Å) (synchrotron station) | 0.976 (ID23.1, ESRF) | 0.92 (I03, Diamond) | 0.951 (I02, Diamond) |
| no. of homotetramers per asymmetric unit | 2 | 1 | 2 |
| no. of observations (unique) | 798597 (218210) | 811429 (223238) | 812084 (214702) |
| multiplicitya | 3.7 (3.7) | 3.6 (2.6) | 3.8 (3.8) |
| Rmergea | 0.130 (0.413) | 0.125 (0.473) | 0.147 (0.877) |
| ⟨I/sd⟩a | 11.5 (4.0) | 10.7 (2.3) | 11.5 (2.0) |
| I/σa | 4.0 (1.4) | 3.4 (1.2) | 2.9 (0.6) |
| completenessa (%) | 98.1 (97.3) | 92.5 (87.5) | 95.0 (91.0) |
| Wilson B factor (Å2) | 26.9 | 19.0 | 40.7 |
| Refinement Statistics | |||
| resolution (Å) | 50−2.3 | 34−1.75 | 50−2.2 |
| Rfree | 0.194 | 0.187 | 0.213 |
| Rwork | 0.171 | 0.157 | 0.194 |
| Rfree+work | 0.170 | 0.158 | 0.195 |
| no. of residues | 2256 | 1128 | 2256 |
| no. of waters | 2797 | 2065 | 1807 |
| mean B factor (Å2) | |||
| main chain | 17.2 | 22.3 | 44.9 |
| side chain | 20.7 | 27.3 | 45.6 |
| waters | 24.4 | 35.6 | 43.4 |
| rmsd [lengths (Å)/bond angles (deg)/chiral restraints (Å3)] | 0.005/0.87/0.071 | 0.006/0.98/0.099 | 0.005/0.89/0.111 |
| Ramachandran plot (preferred/allowed/outlier) (%) | 97.9/2.0/0.1 | 98.2/1.6/0.2 | 97.8/2.0/0.2 |
| PDB entry | 2WVH | 2WVG | 2WVA |
Data shown in parentheses correspond to the high-resolution shell: in the apo form, 2.3−2.42 Å; in the holo form without pyruvate, 1.7−1.79 Å; in the holo form with pyruvate bound, 2.2−2.32 Å.
The three structures reported here were determined by molecular replacement using AMORE (CCP4 suite). The holo form of ZmPDC (PDB entry 1ZPD) was used as the search model with cofactors, ligands, and solvent molecules removed. The cofactor, Mg2+, and waters were added at later stages of refinement. The models were refined by simulated annealing with CNS version 1.1 (25) followed by REFMAC with noncrystallographic symmetry restraints (NCS) and translation-libration-screw disorder parameters (26). The model was manually rebuilt using COOT (27). Refinement parameters for the triazole ThDP analogue were generated by PRODRG2 (28). The triazole derivative and pyruvate were modeled into the structure model at a late stage of the refinement using difference maps as a guide, and their locations were confirmed by omit maps calculated after simulated annealing by slow cooling from 3000 K with CNS version 1.1 (25). The crystallographic refinement parameters are summarized in Table 1. For the apo, triazole-ThDP, and pyruvate-bound triazole-ThDP structures, 97.9, 98.2, and 97.8% of the residues have the most favorable values in the Ramachandran plot, respectively. There is no residue with disallowed geometry in any of the three structures.
Results
The Triazole Derivative of ThDP Binds at the Active Site
The triazole derivative of ThDP is an unreactive analogue, resembling the zwitterionic ylid except that the S and C2 atoms are both replaced by N. In the crystal of ZmPDC prepared in the presence of the triazole derivative, a homotetramer occupies the crystal asymmetric unit, and there is well-defined electron density corresponding unambiguously to the triazole molecule in all four active sites. Dominating positive peaks (>3σ) were observed around the active site in the difference map calculated from an initial model in which the cofactor was absent. These peaks correspond to the electron density for the cofactor and solvent molecules. To confirm that the density represents the triazole derivative, the native cofactor ThDP was modeled into the structure at the beginning of model refinement. After refinement with ThDP, a negative peak appeared at the position of the S atom in the thiazolium ring consistent with the presence at that position of a nitrogen atom in the triazole molecule.
The quaternary structure of PDC (Figure 2) is organized as a dimer of dimers in which the surfaces differ for the principal dimer and dimer−dimer interfaces. The overall fold, secondary structural elements, tertiary structure, and subunit assembly are identical to those of the previously determined structure of ZmPDC bound to a degraded form of ThDP (PDB entry 1ZPD). The root-mean-square (rms) fit between the two structures for main chain Cα positions is 0.29 Å. The active sites are deeply buried in the protein (∼15 Å from the surface) and are located at the interface of two protomers. The interface generating the active site is composed of the PYR domain of one protomer and the PP domain of another protomer (domains are labeled in Figure 2c). The triazole analogue binds to PDC in this cleft and is held by extensive interactions with the surrounding residues (Figure 3). There are hydrophobic interactions with the triazole ring contributed from the Tyrb470 aromatic ring and the Ileb415 and Ileb472 side chains. Ileb415 is the hydrophobic residue supporting the V shape of the cofactor that is found in all ThDP-dependent enzymes. Additionally, the backbone amide NH groups of Ileb472 and Thrb471 form important hydrogen bonding interactions with oxygen atoms of the pyrophosphate group in the holo structure.
Figure 2.

(a) Overall structure of homotetrameric ZmPDC with triazole-ThDP (shown in stick representation) bound. The four protomers are shown in different colors. (b) View of ZmPDC rotated by 90° compared to the view in panel a. (c) Cartoon representation of one protomer of ZmPDC from panel a. The PYR domain (residues 1−188), R domain (residues 189−354), and PP domain (residues 355−566) are colored brown, yellow, and cyan, respectively. The termini of the peptide are labeled with N and C. The color scheme for stick representations in all figures is as follows: red for oxygen, blue for nitrogen, purple for magnesium, and orange for phosphate.
Figure 3.

Active site of the holo structure in stick representation with the triazole derivative of thiamine diphosphate (yellow), a fluoride ion (cyan sphere), and a water molecule (red sphere). Residues from two different protomers of the tetramer are colored wheat and cyan. Black dashed lines represent hydrogen bonds. The electron density is a difference map contoured at 4.5σ using phases that do not include the contribution of the fluoride ion that was subsequently incorporated into the model.
Surprisingly, a strong positive peak in the difference map (4.5σ) was observed in the active site and is most likely due to a fluoride anion derived from the crystallization buffer. The feature is not observed in the apo form, grown in crystallization buffer lacking fluoride, even though the surrounding residues are very similarly positioned. Placing a water molecule instead of the fluoride did not give a good fit to the electron density. The putative fluoride anion is coordinated by the side chains of Aspa27, Hisa114, and Thra72, the backbone NH group of Aspa27, and a water molecule that also contacts the triazole N2 atom and Glub473 (Figure 3). To avoid unfavorable electrostatic interactions with the fluoride anion, it is likely that one or both of the acidic residues, Aspa27 and Glub473, are protonated.
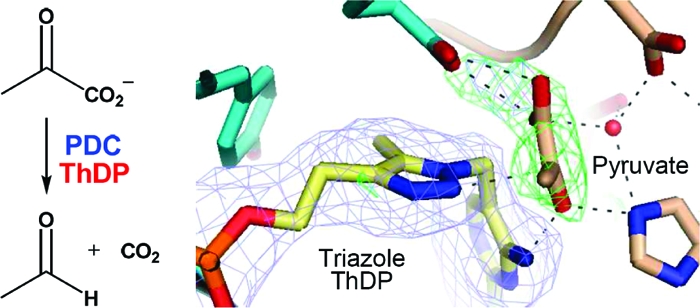
An Image of the Prereaction Complex
In preliminary efforts to obtain cocrystals with pyruvate bound in the active site, crystals of the triazole-ThDP−PDC complex were soaked with pyruvate at 10 mM. However, analysis of the X-ray data from the soaked crystals showed very little electron density anywhere in the structure which could be due to pyruvate. We thought that this might be due to the presence of the fluoride ion in the active site.
Analysis of PDC activity in the presence of potassium fluoride (0.2 M) revealed only 5% of the activity seen in fluoride-free buffer. Replacement of the fluoride with the larger chloride anion in the same assay resulted in almost full activity (92%). The observed inhibition suggests that the fluoride ion might prevent the pyruvate from binding in the active site. Therefore, we cocrystallized PDC with triazole-ThDP and pyruvate in the presence of chloride in place of fluoride.
Two homotetramers occupy the asymmetric unit of the ZmPDC cocrystallized with pyruvate and triazole-ThDP in the fluoride-free crystallization conditions. The overall structure remains similar to the previous native holo structure of ZmPDC (PDB entry 1ZPD), with an rms fit for main chain Cα positions of 0.33 Å. Positive peaks above the 3σ level were observed in the difference map calculated with the initial phases that did not include the cofactor or the pyruvate, showing that the cofactor is present in all eight active sites, but there is only unambiguous electron density for pyruvate in three of the active sites (Figure 4). The electron density for pyruvate was, as expected, roughly disk-shaped, and it was not possible to distinguish the different atoms by electron density alone. However, inspection of the apparent hydrogen bonding clearly reveals the position of the methyl group and of the oxygen atoms. The methyl group points into a pocket lined with hydrophobic residues, particularly the side chain of Ileb472 and the aromatic rings of Tyrb270 and Trpb551, while the oxygen atoms show a large number of potential hydrogen bond interactions. In the final refined structure, the three pyruvate molecules present have very slightly different orientations with respect to the surrounding groups so distances and angles are quoted here as ranges.
Figure 4.

Cartoon and stick representation of one of the active sites of the holo structure in a complex with the substrate pyruvate and the triazole-ThDP cofactor. The two protomers of the enzyme are colored cyan and yellow. The omit map of the triazole-ThDP molecule (blue) was calculated from a difference map at the 3σ level at 2.2 Å resolution. The phases were derived from simulated annealing using a model with triazole-ThDP molecules removed. The omit map of pyruvate (green) was calculated from the difference map at the 3σ level at 2.2 Å resolution. The phases were derived from simulated annealing with pyruvate molecules removed.
Importantly, N2 of the triazole derivative does not have a proton attached which would prevent the close approach of the pyruvate molecule. Instead, it has a lone pair of electrons, just like the ylid it mimics, which could nucleophilically attack C2 of the pyruvate. However, the distances between C2 of pyruvate and N2 of the triazole are 2.9, 3.1, and 3.3 Å in the respective independent active sites of the crystal asymmetric unit, which suggests that the atoms make van der Waals contact but have not progressed very far toward making a covalent bond. As further evidence of this, the angle of approach of the nucleophile (N2−C2=O angle) ranges from 68° to 92°, whereas an angle of 107° (the Bürgi−Dunitz angle) is preferred for nucleophilic attack at a carbonyl (29).
The pyruvate interacts with several residues in the active site through an extensive hydrogen bonding network, involving residues such as Glub473 and Tyrb290 from monomer B and Aspa27, Hisa113, and Hisa114 from monomer A (Figure 4). Hisa114 interacts with both the cofactor (contacting N4′) and the pyruvate (contacting O3). A water molecule, in approximately the same position as the fluoride ion in the holo structure, supports the hydrogen bond network in the substrate binding site through interactions with Aspa27, Thra72, Hisa114, and pyruvate. This water molecule is also present in the active sites that lack pyruvate. The position of pyruvate in the active site probably requires Glub473, Aspa27, or both to be protonated to avoid electrostatic repulsion, as suggested above for the fluoride-bound structure.
Superimposing the holo form and pyruvate-bound structures provides a clue about how fluoride inhibits activity. The water molecule associated with the halide ion occupies the site that would otherwise accommodate the carboxylate group of pyruvate. Thus, binding of pyruvate would be impaired both sterically by the water molecule and electrostatically by mutual repulsion of the negative charges. It is also possible that the water molecule that should be at the site occupied by fluoride plays an important part in the mechanism of the reaction, a point to which we will return in discussions below. As mentioned earlier, chloride is a much less potent inhibitor of PDC activity than fluoride. This is probably due to the different size and/or solvation and binding energies for the two halide ions.
No pyruvate molecules are found anywhere else in the crystal structure other than in the active sites. This is consistent with the fact that, unlike yeast and plant seed PDCs (10,11), the bacterial PDC does not exhibit any known allosteric response to substrate.
Structural Changes Associated with Cofactor Binding
Two homotetramers occupy the asymmetric unit in the crystal of the apo form of PDC, and the electron density map is well-defined for all eight polypeptide chains. No positive peak is observed around the cofactor binding region in the difference map (other than some electron density which has been modeled as water molecules), confirming the absence of the cofactor. The overall structure of the apo form is very similar to the previously reported native holo structure of ZmPDC (PDB entry 1ZPD) (10) except for the position of residues 469−478 of the cofactor-engaging loop (CE loop). The rms fit between apo form and ThDP holo form including these residues is 0.45 Å and excluding them is 0.32 Å.
The loop and adjacent α-helix encompassing residues from Asn467 to Tyr481 make many contacts with the cofactor in the holo form (10). In comparison with the apo structure, the loop undergoes marked conformational changes in the holo structure (Figure 5). In the most obvious change, the side chain of Tyrb470 has flipped to pack onto C5α and the 4-methyl group of the cofactor. This causes the main chain of the loop to move toward the binding site and to fold with a different conformation compared to the apo structure which places the backbone NH groups of Ileb472 and Thrb471 at a good distance and orientation to serve as hydrogen bond donors to oxygen atoms of the pyrophosphate group. Also, the side chain of Glub473 moves ca. 3.8 Å, bringing it into contact with the five-membered ring of the cofactor (which would be a favorable electrostatic interaction for the natural ThDP). This major conformational change is probably the reason why binding of the cofactor is a relatively slow process (30).
Figure 5.

Cartoon and stick representation comparing the cofactor engaging loop (CE loop) conformations in the apo structure (orange), holo structure (cyan and yellow for the two protomers), and pyruvate-bound holo structure (blue). Tyrb290 is not shown for clarity.
Discussion
Conformational Change upon Binding of the Cofactor
The apo structure presented here is one of only a few reported apo forms for ThDP-dependent enzymes. Other examples are transketolase (31), the E1 subunits of pyruvate dehydrogenase (PDB entry 2G67) (32), branched chain 2-oxo acid dehydrogenase (PDB entries 1UM9 and 1V1R) (33,34), and 2-oxoglutarate dehydrogenase (PDB entry 2JGD) (35). In the E1 subunit of PDHc, there is no major change between the apo structure and the holo structure. In all of the other examples, there is a loop adjacent to the cofactor binding site which is disordered in the apo structure and which, in all but the last example, becomes ordered when the cofactor binds. The apo structure reported here is, as far as we know, the first example in which the cofactor-engaging loop is ordered in the apo structure, and a major conformational change occurs when the cofactor binds.
The most significant changes upon the cofactor binding to PDC are in the side chains of Tyrb470, Ileb472, and Glub473. The side chain of Tyrb470 points in quite a different direction in the holo structure compared to the apo structure, a total movement of the OH group of more than 15.5 Å. This brings the aromatic ring to a position where it has a hydrophobic interaction with the 4-methyl group of the cofactor. The movement of the side chain of Glub473 also brings it into contact with the five-membered ring of the cofactor, while Ileb472 moves in to create the hydrophobic pocket for the methyl group of pyruvate. Equally significant are the changes in the backbone conformation, which allow the NH groups of Thrb471 and Ileb472 to form hydrogen bonds with oxygen atoms of the pyrophosphate and the C=O group of Glyb469 to coordinate to the magnesium ion.
Roles of Active Site Residues in the Catalytic Mechanism
The crystal structure of PDC in complex with pyruvate and nonreactive ylid-like analogue, triazole-ThDP 7, provides an approximate image of the prereaction state. We observe that the pyruvate is located in a pocket near the cofactor. The observed binding site for pyruvate is consistent with the finding that fluoride is an inhibitor of PDC, since, in the structure without pyruvate, the halide interacts with a water molecule that occludes the pyruvate-binding site. The pyruvate does not appear to be in the most favorable orientation for bond formation with the ylide to proceed. While the Cα atom of pyruvate is in van der Waals contact (2.9−3.3 Å) with the nitrogen atom of the triazole molecule, corresponding to the C2 atom of ThDP, it will have to move closer (to ∼1.5 Å) as the bond is formed (29). Also, the pyruvate molecule is not in an ideal orientation, because the line from N2 of the cofactor to C2 of the pyruvate is 30.4°, 38.6°, and 30.1° out of the plane of the triazole ring (Figure 4). Thus, a movement of the ThDP and/or the pyruvate molecule would be required before the reaction could take place. We suggest that this movement would cause strain in the cofactor and/or weakening of the hydrogen bonds to the pyruvate molecule. Additionally, the new bond that is formed is likely to be distorted out of the plane of the thiazolium ring, which is seen in a number of crystal structures of other ThDP-dependent enzymes with substrates or substrate analogues bound (36,37). All these factors would tend to raise the energy of the lactyl-ThDP intermediate and thus favor the subsequent decarboxylation step.
The prereaction complex provides some clues about the function of key residues in the catalytic pocket. The proximity of the carboxylate group of the pyruvate and the side chains of Glub473 and Aspa27 is striking and suggests that at least one of these three carboxylates must be protonated in the prereaction state. Of these, only Glub473 is correctly oriented to be a hydrogen bond donor (to the pyruvate carboxyl group) and so is presumably protonated. We believe the likely positions of protons (both in the structure with the triazole ThDP bound and in the ThDP ylid-bound state that it mimics) are as shown in Figure 6A. The carboxylate group of pyruvate is additionally hydrogen bonded to the backbone NH group of Aspa27 and to a water molecule, which bridges between the carboxylate groups of pyruvate and Aspa27. This water molecule is also within hydrogen bonding distance of Hisa114 and Thra72 and appears to have a pivotal role in organizing the enzyme−substrate complex and the hydrogen bond network. The fluoride ion in the holo structure occupies the site of this water molecule, and one reason for the inhibition by fluoride may be the disruption of proton transfer steps that occur via the water molecule. The imidazole group of Hisa114 appears to be protonated because Nδ interacts with a backbone C=O group and Nε interacts with the ketone C=O group of pyruvate (in addition to the water molecule). His113 is also presumably protonated because it bridges Aspa27 and Aspb289.
Figure 6.

Possible mechanism for the enzymic reaction, involving multiple proton transfers between Aspa27/Glub473 and the substrate/cofactor.
The position of the carboxyl group of Glub473 immediately above the five-membered ring of the cofactor suggests that it would prefer to be deprotonated (−CO2−) when the ring is positively charged (as in ThDP) but protonated (−CO2H) when the ring is neutral (as in ylid 3). In accordance with this, it has been suggested on the basis of a molecular dynamics simulation (38) that Glub473 is the likely acceptor of the proton that must be lost from the 4′-NH2 group of ThDP to allow it to deprotonate C2. This would lead to the situation depicted in Figure 6A. The next step, attack of the ylid on the ketone C=O group of the pyruvate, should be accompanied by protonation of the oxygen atom. In the structure reported here, this oxygen is within hydrogen bonding distance of both the 4′-NH2 group of ThDP and Nε of Hisa114. However, the former distance should be shortened and the latter lengthened as the pyruvate molecule moves to within bonding distance of C2 of the cofactor. Therefore, we propose that the initial general acid is the 4′-NH2 group (A → B in Figure 6). This is in accord with the observation that replacement of His114 with Gln decreased the kcat by a factor of only 3 (24), whereas replacement with Ala inactivates the enzyme (39). Clearly, its hydrogen bonding ability is important, but it is not required to be an acid−base catalyst. Studies on yeast PDC have also concluded that the 4′-NH2 group of ThDP is the primary proton donor (40−42).
The orientation of the carboxylate group in the 2-lactyl-ThDP intermediate (Figure 6B), which is dictated by hydrogen bonding to Aspa27 and/or Glub473, allows for maximum resonance stabilization during the decarboxylation step; i.e., it makes the positively charged thiazolium ring optimally oriented to be a sink for the electron pair released since the so-formed p-orbital conjugates with the thiazolium π-system. Indeed, this position of the leaving group, also seen in recent crystal structures of covalently attached reaction intermediates such as xylulose-5P-ThDP in transketolase (36) or 2-lactyl-ThDP in pyruvate oxidase F479W (43), is believed to be a common feature among ThDP-dependent enzymes.
In the lactyl-ThDP intermediate (Figure 6B), the five-membered ring of the cofactor is again positively charged, so Glub473 should now prefer to lose its proton. We propose that a reverse proton transfer now occurs from Glub473 to the 4′-N atom. Several paths for this proton transfer are possible; one is via the carboxylate of pyruvate, the water molecule, and Hisa114, but an alternative pathway is presumably used in the H114Q mutant. The negative charge on Glub473 (Figure 6C) would serve to destabilize the adjacent carboxylate group of pyruvate and facilitate the subsequent decarboxylation step (C → D). This role of Glu473 is consistent with the findings that the E473Q mutant has very low activity (4000-fold lower than that of the wild type) (44) and the E473D mutant (1000-fold less active than the wild type) is mainly deficient in the C−C bond forming step (A → B) and the decarboxylation (C → D) (45).
After the decarboxylation step, the CO2 molecule can be displaced by a water molecule. The stoichiometry of the overall reaction requires a proton, in addition to the pyruvate anion, as a substrate. As Glub473 is now in contact with a neutral thiazole ring of the cofactor, it is likely that this is the residue that accepts the proton. Protonation of the enamine intermediate can then occur from the new water molecule, which in turn receives a proton from Glub473 (E → F). The same protonation of the enamine by the active site Glu residue via an intervening water molecule was proposed for yeast PDC by Lobell and Crout on the basis of their molecular mechanics calculations (42) and by Sergienko and Jordan on the basis of site-directed mutagenesis studies (41). It should be noted that the holo structure presented here has a water molecule at approximately this position in all four active sites. For oxalyl CoA decarboxylase, a structure of the enamine intermediate has been determined, and it has a water molecule in just such a position, poised for protonation of the enamine carbon (46).
For the final step, release of acetaldehyde, deprotonation of the OH group of the hydroxyethyl-ThDP is required. It could be that transfer of a proton from the 4′-NH2 group to Glub473 or Aspa27 now occurs and then the 4′-imino group could be the base. Alternatively, Glub473 or Aspa27 could act directly through the intervening water molecule. This latter possibility is what is shown in Figure 6 (F → G), with Aspa27 acting as the base, because it was found that the D27E mutant was particularly slow in the release of acetaldehyde from the enamine intermediate (45). For yeast PDC also (which has the same active site residues in a very similar arrangement), it has been proposed that the Glu residue participates in enamine protonation while the Asp residue participates in acetaldehyde release (41,42).
We suggest that the mechanism presented in Figure 6 accounts for the effects of all the site-directed mutants studied so far. Mutations of Asp27, Glu473, and His114 have been mentioned already. The one remaining catalytic group that has been mutated, His113 to Gln, Lys, or Arg, which inactivates or severely disables the enzyme (24,39), is explained by the fact that His113 is required to hold the carboxylate group of Asp27 in the correct place and ionization state. Aspa27 is also held in place by hydrogen bonding to Tyrb270, a residue that has not been mutated, to the best of our knowledge. Nevertheless, any assignment of function to the catalytic residues must be treated with caution. For example, benzoylformate decarboxylase catalyzes an reaction equivalent to PDC but lacks Glu and Asp residues equivalent to Asp27 and Glu473(47). Clearly, there are different ways that this reaction can be catalyzed.
Presence of a Water Tunnel between the Cofactors of Two Active Sites
Some form of communication between active sites in ThDP-dependent enzymes has been noted many times, and it has been proposed, at least for the E1 subunit of PDHc, that it is mediated by a water channel that connects the active sites and acts as a proton relay system (48). The structure of ZmPDC also has a water-filled channel between the ThDP pockets, as does yeast PDC. Furthermore, the pattern of the chain of water molecules is almost identical in the bacterial and eukaryotic PDCs (Figure 7). Although conservation of interactions at an interface between subunits is expected in closely related enzymes, the presence of similar water channels in ThDP-dependent enzymes that are not particularly closely related does suggest that the channel and the entrained waters might be functionally important.
Figure 7.

Water tunnel between active sites that is conserved in both Z. mobilis and yeast PDC. PDC from Z. mobilis is colored gray for protomer A and green for protomer B, and waters are represented as red spheres. The hydrogen bonding network of the water tunnel from Z. mobilis is presented as dashed lines. Yeast PDC with pyruvamide (PDB entry 1QPB) is colored cyan and yeast E477Q variant (PDB entry 2VK8) blue. Residues from Z. mobilis involved in the aqueous tunnel are labeled.
We have previously reported the kinetics of binding of triazole-ThDP 7 to apo-ZmPDC (20,21). The data points were fitted poorly with a simple exponential curve but reasonably well using a double-exponential curve, suggesting that there are two distinct reaction rates. This apparent two-stage inhibition might be explained by normal slow-binding inhibition, in which a more rapid initial reversible binding is followed by a slower, essentially irreversible step (Figure 8). The conformational change in the active site of the enzyme could quite possibly account for the second step.
Figure 8.

Possible two-step binding process of the triazole-ThDP analogue (I) to the enzyme (E).
Alternatively, the biphasic inhibition could originate from a form of communication between the subunits in which the presence of the triazole-ThDP in one subunit decreases the rate of binding of the triazole-ThDP in the partner subunit. If this second explanation is correct, the water channel connecting the active sites may well be responsible for the communication between the active sites.
Conclusion
The apo structure presented here shows for the first time the magnitude of the conformational change that can accompany binding of ThDP to an enzyme and provides a good rationale for the relatively slow but tight binding of the cofactor. The holo structure with triazole-ThDP bound that we present is very similar to the previous structure with a degraded form of ThDP bound. This is a good indication that the degraded form of the thiazolium ring in the previous structure was neutral (as in the triazole analogue) rather than positively charged (as in ThDP). The holo structure additionally reveals a binding site for fluoride ion, which we have shown to be an inhibitor of the enzyme in vitro, and suggests that the inhibition arises from the occlusion of the substrate binding site caused by the hydrated halide ion.
Finally, we present a structure of ZmPDC with triazole-ThDP and pyruvate bound. We suggest that this cofactor analogue is the best mimic of the ThDP ylid yet studied, in that it has a lone pair of electrons rather than a hydrogen projecting from the 2-position. This allows a closer approach of the substrate molecule, pyruvate, than has been seen in any previous substrate complex for a ThDP-dependent enzyme.
The structures obtained suggest that a complex hydrogen bonding network is present in the active site and is important in catalysis. This network involves two water molecules, one of which is already present in the pyruvate-bound structure and the second of which replaces the carbon dioxide after decarboxylation. A possible mechanism, which involves these water molecules and is consistent with reported site-directed mutagenesis experiments, is presented in Figure 6.
Acknowledgments
We thank the staff of ESRF and the Diamond Light Source for generous help and use of facilities.
X.y.P. and B.F.L. are supported by the Wellcome Trust, and K.M.E. was funded by the European Commission under a Marie Curie Early Stage Research Training Programme.
The coordinates for the apo structure, the holo structure, and the pyruvate complex have been deposited in the Protein Data Bank as entries 2WVH, 2WVG, and 2WVA, respectively.
Footnotes
Abbreviations: HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; MES, 2-(N-morpholino)ethanesulfonic acid; PDB, Protein Data Bank; PDC, pyruvate decarboxylase; PDHc, pyruvate dehydrogenase multienzyme complex; PEG, polyethylene glycol; rmsd, root-mean-square deviation; ThDP, thiamine diphosphate; ZmPDC, pyruvate decarboxylase from Zymomonas mobilis.
References
- Jordan F. (2003) Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat. Prod. Rep. 20, 184–201. [DOI] [PubMed] [Google Scholar]
- Schellenberger A. (1998) Sixty years of thiamin diphosphate biochemistry. Biochim. Biophys. Acta 1385, 177–186. [DOI] [PubMed] [Google Scholar]
- Kern D.; Kern G.; Neef H.; Tittmann K.; Killenberg-Jabs M.; Wikner C.; Schneider G.; Hübner G. (1997) How Thiamin Diphosphate Is Activated in Enzymes. Science 275, 67–70. [DOI] [PubMed] [Google Scholar]
- Nemeria N.; Korotchkina L.; McLeish M. J.; Kenyon G. L.; Patel M. S.; Jordan F. (2007) Elucidation of the Chemistry of Enzyme-Bound Thiamin Diphosphate Prior to Substrate Binding: Defining Internal Equilibria among Tautomeric and Ionization States. Biochemistry 46, 10739–10744. [DOI] [PubMed] [Google Scholar]
- Nemeria N.; Chakraborty S.; Balakrishnan A.; Jordan F. (2009) Reaction mechanisms of thiamin diphosphate enzymes: Defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J. 276, 2432–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist Y.; Schneider G.; Ermler U.; Sundström M. (1992) Three-dimensional structure of transketolase, a thiamine diphosphate dependent enzyme, at 2.5 Å resolution. EMBO J. 11, 2373–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkola M.; Lindqvist Y.; Schneider G. (1994) Refined Structure of Transketolase from Saccharomyces cerevisiae at 2.0 Å Resolution. J. Mol. Biol. 238, 387–404. [DOI] [PubMed] [Google Scholar]
- Dyda F.; Furey W.; Swaminathan S.; Sax M.; Farrenkopf B.; Jordan F. (1993) Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4 Å resolution. Biochemistry 32, 6165. [DOI] [PubMed] [Google Scholar]
- Arjunan P.; Umland T.; Dyda F.; Swaminathan S.; Furey W.; Sax M.; Farrenkopf B.; Gao Y.; Zhang D.; Jordan F. (1996) Crystal Structure of the Thiamin Diphosphate-dependent Enzyme Pyruvate Decarboxylase from the Yeast Saccharomyces cerevisiae at 2.3 Å Resolution. J. Mol. Biol. 256, 590–600. [DOI] [PubMed] [Google Scholar]
- Dobritzsch D.; König S.; Schneider G.; Lu G. (1998) High Resolution Crystal Structure of Pyruvate Decarboxylase from Zymomonas mobilis: Implications for Substrate Activation in Pyruvate Decarboxylases. J. Biol. Chem. 273, 20196–20204. [DOI] [PubMed] [Google Scholar]
- Lu G.; Dobritzsch D.; Baumann S.; Schneider G.; König S. (2000) The structural basis of substrate activation in yeast pyruvate decarboxylase: A crystallographic and kinetic study. Eur. J. Biochem. 267, 861–868. [DOI] [PubMed] [Google Scholar]
- Kutter S.; Weiss M. S.; Wille G.; Golbik R.; Spinka M.; Konig S. (2009) Covalently Bound Substrate at the Regulatory Site of Yeast Pyruvate Decarboxylases Triggers Allosteric Enzyme Activation. J. Biol. Chem. 284, 12136–12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ævarsson A.; Seger K.; Turley S.; Sokatch J. R.; Hol W. G. J. (1999) Crystal structure of 2-oxoisovalerate and dehydrogenase and the architecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat. Struct. Biol. 6, 785–792. [DOI] [PubMed] [Google Scholar]
- Ciszak E. M.; Korotchkina L. G.; Dominak P. M.; Sidhu S.; Patel M. S. (2003) Structural Basis for Flip-Flop Action of Thiamin Pyrophosphate-dependent Enzymes Revealed by Human Pyruvate Dehydrogenase. J. Biol. Chem. 278, 21240–21246. [DOI] [PubMed] [Google Scholar]
- Muller Y. A.; Schultz G. E. (1993) Structure of the thiamine- and flavin-dependent enzyme pyruvate oxidase. Science 259, 965–967. [DOI] [PubMed] [Google Scholar]
- Muller Y. A.; Schumacher G.; Rudolph R.; Schulz G. E. (1994) The Refined Structures of a Stabilized Mutant and of Wild-type Pyruvate Oxidase from Lactobacillus plantarum. J. Mol. Biol. 237, 315–335. [DOI] [PubMed] [Google Scholar]
- Muller Y. A.; Lindqvist Y.; Furey W.; Schultz G. E.; Jordan F.; Schneider G. (1993) A thiamin diphosphate binding fold revealed by comparison of the crystal structures of transketolase, pyruvate oxidase and pyruvate decarboxylase. Structure 1, 95–103. [DOI] [PubMed] [Google Scholar]
- Versees W.; Spaepen S.; Wood M. D. H.; Leeper F. J.; Vanderleyden J.; Steyaert J. (2007) Molecular Mechanism of Allosteric Substrate Activation in a Thiamine Diphosphate-dependent Decarboxylase. J. Biol. Chem. 282, 35269–35278. [DOI] [PubMed] [Google Scholar]
- Sun S.; Duggleby R. G.; Schowen R. L (1995) Linkage of Catalysis and Regulation in Enzyme Action. Carbon Isotope Effects, Solvent Isotope Effects, and Proton Inventories for the Unregulated Pyruvate Decarboxylase of Zymomonas mobilis. J. Am. Chem. Soc. 117, 7317–7322. [Google Scholar]
- Erixon K. M.; Dabalos C. L.; Leeper F. J. (2007) Inhibition of pyruvate decarboxylase from Z. mobilis by novel analogues of thiamine pyrophosphate: Investigating pyrophosphate mimics. Chem. Commun., 960. [DOI] [PubMed] [Google Scholar]
- Erixon K. M.; Dabalos C. L.; Leeper F. J. (2008) Synthesis and biological evaluation of pyrophosphate mimics of thiamine pyrophosphate based on a triazole scaffold. Org. Biomol. Chem. 6, 3561–3572. [DOI] [PubMed] [Google Scholar]
- Mann S.; Perez Melero C.; Hawksley D.; Leeper F. J. (2004) Inhibition of thiamin diphosphate dependent enzymes by 3-deazathiamin diphosphate. Org. Biomol. Chem 2, 1732–1741. [DOI] [PubMed] [Google Scholar]
- Jordan F.; Li H.; Brown A. (1999) Remarkable Stabilization of Zwitterionic Intermediates May Account for a Billion-fold Rate Acceleration by Thiamin Diphosphate-Dependent Decarboxylases. Biochemistry 38, 6369–6373. [DOI] [PubMed] [Google Scholar]
- Schenk G.; Leeper F. J.; England R.; Nixon P. F.; Duggleby R. G. (1997) The Role of His113 and His114 in Pyruvate Decarboxylase from Zymomonas mobilis. Eur. J. Biochem. 248, 63–71. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Schuettelkopf A. W.; van Aalten D. M. F. (2004) PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Bürgi H. B.; Dunitz J. D.; Lehn J. M.; Wipff G. (1974) Stereochemistry of Reaction Paths at Carbonyl Centres. Tetrahedron 30, 1563–1572. [Google Scholar]
- Diefenbach R. J.; Duggleby R. G. (1991) Pyruvate decarboxylase from Zymomonas mobilis. Structure and re-activation of apoenzyme by the cofactors thiamin diphosphate and magnesium ion. Biochem. J. 276, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström M.; Lindqvist Y.; Schneider G. (1992) Three-dimensional structure of apotransketolase. Flexible loops at the active site enable cofactor binding. FEBS Lett. 313, 229–231. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar K.; Arjunan P.; Sax M.; Nemeria N.; Jordan F.; Furey W. (2006) Active-site changes in the pyruvate dehydrogenase multienzyme complex E1 apoenzyme component from Escherichia coli observed at 2.32 Å resolution. Acta Crystallogr. D62, 1382–1386. [DOI] [PubMed] [Google Scholar]
- Li J.; Wynn R. M.; Machius M.; Chuang J. L.; Karthikeyan S.; Tomchick D. R.; Chuang D. T. (2004) Cross-talk between Thiamin Diphosphate Binding and Phosphorylation Loop Conformation in Human Branched-chain α-Keto Acid Decarboxylase/Dehydrogenase. J. Biol. Chem. 279, 32968–32978. [DOI] [PubMed] [Google Scholar]
- Nakai T.; Nakagawa N.; Maoka N.; Masui R.; Kuramitsu S.; Kamiya N. (2004) Ligand-induced Conformational Changes and a Reaction Intermediate in Branched-chain 2-Oxo Acid Dehydrogenase (E1) from Thermus thermophilus HB8, as Revealed by X-ray Crystallography. J. Mol. Biol. 337, 1011–1033. [DOI] [PubMed] [Google Scholar]
- Frank R. A. W.; Price A. J.; Northrop F. D.; Perham R. N.; Luisi B. F. (2007) Crystal Structure of the E1 Component of the Escherichia coli 2-Oxoglutarate Dehydrogenase Multienzyme Complex. J. Mol. Biol. 368, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztalos P.; Parthier C.; Golbik R.; Kleinschmidt M.; Hübner G.; Weiss M. S.; Friedemann R.; Wille G.; Tittmann K. (2007) Strain and Near Attack Conformers in Enzymic Thiamin Catalysis: X-ray Crystallographic Snapshots of Bacterial Transketolase in Covalent Complex with Donor Ketoses Xylulose 5-phosphate and Fructose 6-phosphate, and in Noncovalent Complex with Acceptor Aldose Ribose 5-phosphate. Biochemistry 46, 12037–12052. [DOI] [PubMed] [Google Scholar]
- Arjunan P.; Sax M.; Brunskill A.; Chandrasekhar K.; Nemeria N.; Zhang S.; Jordan F.; Furey W. (2006) A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 281, 15296–15303. [DOI] [PubMed] [Google Scholar]
- Lie M. A.; Celik L.; Jorgensen K. A.; Schiott B. (2005) Cofactor activation and substrate binding in pyruvate decarboxylase. Insights into the reaction mechanism from molecular dynamics simulations. Biochemistry 44, 14792–14806. [DOI] [PubMed] [Google Scholar]
- Huang C. Y.; Chang A. K.; Nixon P. F.; Duggleby R. G. (2001) Site-directed mutagenesis of the ionizable groups in the active site of Zymomonas mobilis pyruvate decarboxylase: Effect on activity and pH dependence. Eur. J. Biochem. 268, 3558–3565. [DOI] [PubMed] [Google Scholar]
- Liu M.; Sergienko E. A.; Guo F.; Wang J.; Tittmann K.; Hübner G.; Furey W.; Jordan F. (2001) Catalytic Acid-Base Groups in Yeast Pyruvate Decarboxylase. 1. Site-Directed Mutagenesis and Steady-State Kinetic Studies on the Enzyme with the D28A, H114F, H115F, and E477Q Substitutions. Biochemistry 40, 7355–7368. [DOI] [PubMed] [Google Scholar]
- Sergienko E. A.; Jordan F. (2001) Catalytic Acid-Base Groups in Yeast Pyruvate Decarboxylase. 2. Insights into the Specific Roles of D28 and E477 from the Rates and Stereospecificity of Formation of Carboligase Side Products. Biochemistry 40, 7369–7381. [DOI] [PubMed] [Google Scholar]
- Lobell M.; Crout D. H. G. (1996) Pyruvate Decarboxylase: A Molecular Modeling Study of Pyruvate Decarboxylation and Acyloin Formation. J. Am. Chem. Soc. 118, 1867–1873. [Google Scholar]
- Wille G.; Meyer D.; Steinmetz A.; Hinze E.; Golbik R.; Tittmann K. (2006) The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat. Chem. Biol. 2, 324–328. [DOI] [PubMed] [Google Scholar]
- Chang A. K.; Nixon P. F.; Duggleby R. G. (1999) Aspartate-27 and glutamate-473 are involved in catalysis by Zymomonas mobilis pyruvate decarboxylase. Biochem. J. 339, 255–260. [PMC free article] [PubMed] [Google Scholar]
- Tittmann K.; Golbik R.; Uhlemann K.; Khailova L.; Schneider G.; Patel M.; Jordan F.; Chipman D. M.; Duggleby R. G.; Hübner G. (2003) NMR Analysis of Covalent Intermediates in Thiamin Diphosphate Enzymes. Biochemistry 42, 7885–7891. [DOI] [PubMed] [Google Scholar]
- Berthold C. L.; Toyota C. G.; Moussatche P.; Wood M. D.; Leeper F.; Richards N. G. J.; Lindqvist Y. (2007) Crystallographic Snapshots of Oxalyl-CoA Decarboxylase Give Insights into Catalysis by Nonoxidative ThDP-Dependent Decarboxylases. Structure 15, 853–861. [DOI] [PubMed] [Google Scholar]
- Hasson M. S.; Muscate A.; McLeish M. J.; Polovnikova L. S.; Gerlt J. A.; Kenyon G. L.; Petsko G. A.; Ringe D. (1998) The crystal structure of benzoylformate decarboxylase at 1.6 Å resolution: Diversity of catalytic residues in thiamin diphosphate-dependent enzymes. Biochemistry 37, 9918–9930. [DOI] [PubMed] [Google Scholar]
- Frank R. A. W.; Titman C. M.; Pratap J. V.; Luisi B. F.; Perham R. N. (2004) A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 306, 872–876. [DOI] [PubMed] [Google Scholar]