

Abstract
Tumour necrosis factor‐α (TNF‐α) and transforming growth factor‐β1 (TGF‐β1) are peptides with multiple biological activities that influence neoplastic, immunologic and fibroproliferative diseases. There are clear interrelationships and overlap between the actions of TNF‐α and TGF‐β1 in lung fibrosis; therefore, we postulated that TNF‐α may play a significant role in regulating TGF‐β1 expression in lungs. We recently reported that TNF‐α activates the extracellular regulated kinase (ERK)‐specific pathway in fibroblasts resulting in stabilization of TGF‐β1 mRNA and increased expression of TGF‐β1. In the current study, we further investigated the molecular mechanisms involved in TNF‐α regulation of TGF‐β1 expression. Nuclear run‐on assays showed that treatment of Swiss 3T3 fibroblasts with TNF‐α increased transcription of the TGF‐β1 gene in an ERK independent manner. Pre‐treatment with the activator protein‐1 (AP‐1) inhibitor curcumin attenuated TNF‐α induced transcription of the TGF‐β1 gene. TNF‐α induced increased levels of c‐Jun and C‐Fos in the nucleus accompanied by phosphorylation of c‐Jun. In electrophoretic mobility shift assays, AP‐1 binding to an AP‐1 binding site found within the TGF‐β1 promoter was increased in nuclear extracts from Swiss 3T3 fibroblasts treated with TNF‐α. Together, these results suggest that TNF‐α induces expression and DNA binding of AP‐1 resulting in increased transcription of the TGF‐β1 gene. It is essential to know which transcription pathways are activated because of the wide distribution of TNF‐α and TGF‐β1, the general lack of effective treatments for fibroproliferative disease and the possibility that targeting the correct transcription factors could be palliative.
Keywords: transforming growth factor‐β1, tumour necrosis factor‐α, activator‐protein‐1, lung fibrosis
Introduction
Transforming growth factor‐β1 (TGF‐β1) is a multifunctional cytokine involved in a wide variety of biological processes such as embryonic development, neoplastic growth and differentiation, cell proliferation and apoptosis, fibrous tissue deposition and regulation of the immune response [1]. Up‐regulation of TGF‐β1 expression is a consistent feature of most fibrotic diseases. Indeed, patients with pulmonary fibrosis have increased levels of TGF‐β1 in their lungs and the levels correlate with severity of lung disease [2]. In rodent models, expression of active TGF‐β1 in lungs induces a severe fibrotic response, whereas inhibitors of TGF‐β1 expression or signalling afford protection from fibrosis [3]. Consequently, TGF‐β1 has been proposed to be a ‘master switch’ in induction of fibrosis [4]. In the lung, TGF‐β1 can drive epithelial‐to‐mesenchymal transition, fibroblast‐to‐myofibroblast differentiation, and is the most potent inducer of extracellular matrix production characterized to date. Despite intensive investigation into the biological activities of TGF‐β1 in the lung, the mechanisms by which expression of this growth factor are regulated are not yet fully understood.
There is increasing evidence that tumour necrosis factor‐α (TNF‐α) is important in the initiation and perpetuation of the fibrotic process. TNF‐α is a pleiotropic cytokine produced by many cell types in response to infection or injury. In addition to its well‐characterized pro‐inflammatory properties, TNF‐α may modulate cellular differentiation, proliferation and apoptosis. The functions of TNF‐α can be both beneficial and detrimental. TNF‐α expression is increased in the lungs of patients with pulmonary fibrosis, and agents with anti‐TNF‐α properties such as pirfenidone and etanercept have shown promise in treatment of some patients [5]. Furthermore, TNF‐α polymorphisms indicative of high levels of TNF‐α expression correlate with an increased risk of developing interstitial pulmonary fibrosis (IPF) [6, 7]. Consistent with these observations, rodents exposed to fibrogenic agents such as bleomycin, silica or asbestos show increased expression of TNF‐α in their lungs [8, 9]. Infusion of anti‐TNF‐α antibody [10] or soluble TNF receptor [11] reduced bleomycin‐induced lung injury and decreased expression of TGF‐β1 in the treated mice. Likewise, overexpression of TNF‐α in lungs of normal rats induced patchy, interstitial fibrosis and was preceded by a significant increase in TGF‐β1 production [12]. We showed that mice with both the 55 and 75 kD receptors for TNF‐α knocked out (TNFR‐KO) fail to develop significant bleomycin‐induced fibrosis, silicosis or asbestosis [9, 13, 14]. Furthermore, the TNFR‐KO mice had reduced levels of TGF‐β1 in their lungs after exposure to asbestos [13]. We later showed that TGF‐β1 overexpression in TNFR‐KO mice was sufficient to induce pulmonary fibrosis [15].
Since there are clear interrelationships and overlap between the actions of TNF‐α and TGF‐β1 in lung fibrosis, we have postulated that TNF‐α may play a significant role in regulating TGF‐β1 expression in lung. In support of this hypothesis, we showed in vitro that TNF‐α rapidly up‐regulates TGF‐β1 and collagen expression in both primary lung mesenchymal and epithelial cells [16]. In studies designed to investigate a signal transduction pathway by which TNF‐α induces TGF‐β1 expression, we demonstrated that TNF‐α rapidly activates the extracellular regulated kinase (ERK)‐specific mitogen‐activated protein kinase pathway resulting in stabilization of TGF‐β1 mRNA and increased expression of TGF‐β1 protein in primary mouse lung fibroblasts (MLFs) [17]. In the findings presented here, we show that TNF‐α rapidly induces activation of the activator protein‐1 (AP‐1) transcription factor and increases transcription of the TGF‐β1 gene. Together, these results show that TNF‐α up‐regulates TGF‐β1 through a complex mechanism involving both increased transcription and mRNA stability.
Materials and methods
Cell culture
Swiss 3T3 fibroblasts were obtained from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma, St. Louis, MO, USA) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA, USA), L‐glutamine, non‐essential amino acids, penicillin (100 U/ml) and streptomycin (100 μg/ml). Primary cultures of MLFs were established as previously described [16]. In this study, we used MLF cultures at passages 2 and 3. Fresh growth media was added every 3 to 4 days and cells were kept sub‐confluent until used for experiments. Confluent monolayers were rendered quiescent in media containing 1% serum for 48 hrs before use in experiments. All experiments were performed in media containing 1% serum.
Real‐time RT‐PCR
Total RNA was isolated using RNeasy kit (Qiagen, Gaithersburg, MD, USA) and any contaminating DNA removed using TURBO DNA‐free (Ambion, Austin, TX, USA), both according to the manufacturer’s recommendations. Reverse transcription of 250 ng total RNA was performed in a total volume of 20 μl using iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA) according to the manufacturer’s recommendations. One microlitre of cDNA was PCR amplified in 20 μl reactions containing primers at 250 nM in iQ SYBR Green Supermix (Bio‐Rad) as previously described [17]. Primers for TGF‐β1 pre‐mRNA were as follows: 5′‐GCTACCATGCCAACTTCTGTCT‐3′ (sense); and 5′‐CCTACCACCCCAGCCTCTG‐3′ (antisense) resulting in amplification of a 116 bp product. Primers for mature TGF‐β1 mRNA were as follows: 5′‐GGATACCAACTATTGCTTCAGCTCC‐3′ (sense) and 5′‐AGGCTCCAAATATAGGGGCAGGGTC‐3′ (antisense) yielding a 156 bp product. The sense primer was designed to overlap both the fifth and sixth exons to control against amplification of TGF‐β1 pre‐mRNA or DNA. Primers for mouse MMP9 mRNA were: 5′‐ AGACCTGAAAACCTCCAACCTCAC‐3′ (sense) and 5′‐ TGTTATGATGGTCCCACTTGAGGC‐3′ (antisense), resulting in a 262 bp product [18]. Primers for mouse acidic ribosomal phosphoprotein (36B4) mRNA were as follows: 5′‐CGACCTGGAAGTCCAACTAC‐3′ (sense) and 5′‐ATCTGCATCTGCTTG‐3′ (antisense) yielding a 109 bp product [19]. Negative controls, including cDNA reactions without reverse transcriptase or RNA, and PCR mixtures lacking cDNA were included in each PCR. Following amplification, specificity of the reaction was confirmed by melt curve analysis. Relative quantitation was determined using the comparative CT method with data normalized to 36B4 and calibrated to the average CT of untreated controls.
Nuclear run‐on
To determine transcription rate, nuclear run‐on assays were performed. Quiescent Swiss 3T3 cells were treated with TNF‐α (10 ng/ml) or media alone for 2 hrs. Nuclei were then isolated from 1 × 107 cells using Nuclei Isolation Kit (Nuclei PURE Prep, Sigma‐Aldrich, St. Louis, MO, USA). Nuclei were either stored in liquid nitrogen, or used immediately. Traditional nuclear run‐on assays were performed as described [20]. Briefly, 100 μl of nuclei were incubated with 30 μl of 5× reaction buffer (25 mM Tris‐HCl, pH 8, 12.5 mM MgCl2, 750 mM KCl, 1.25 mM each of ATP, GTP and CTP and 100 μCi α‐32P‐UTP) for 30 min. at 30°C, then 15 units of RQ1 DNase (Promega, Madison, WI, USA) were added and incubation continued for another 15 min. Labelled RNA was extracted with Trizol (Invitrogen) and radioactivity was determined by liquid scintillation counting. Each extract was normalized to radioactivity and hybridized to target sequences immobilized on a nylon membrane at 60°C overnight. The membrane was washed extensively to remove excess RNA: 2× saline‐sodium citrate buffer (SSC) (2 × 5 min., room temperature); 2× SSC, 1% SDS (15 min., 65°C); 2× SSC (5 min., room temperature); 2× SSC, 10 μg/ml RNase A (10 min., 37°C); 2× SSC (5 min., room temperature); 0.1× SSC (3 × 5 min., room temperature). After autoradiography, the intensity of hybridized radiolabelled mRNA was determined by Phosphor Imager analysis using ImageGauge software. The alkaline denatured target DNA sequences, murine TGF‐β1[21] and murine 36B4 cDNAs cloned into pGEM7Z were immobilized on nylon membrane by slot blotting followed by UV cross‐linking. To monitor non‐specific hybridization, empty pGEM7Z vector was also blotted on the membrane.
In some experiments, PCR‐based nuclear run‐on assays were performed as previously described [22, 23]. Briefly, fresh or thawed nuclei (1 × 107) were split into two aliquots. The first aliquot was incubated with 2× reaction buffer (20% (v/v) glycerol, 100 mM KCl, 10 mM MgCl2, 4.5 mM dithiothreitol (DTT), 1.2 mM adenosine 5′‐triphosphate (ATP), 0.6 mM each of cytosine 5′‐triphosphate (CTP), guanidine 5′‐triphosphate (GTP) and uridine 5′‐triphosphate (UTP), 0.5 mM spermidine, 0.15 mM spermine and 80 U/ml RNase inhibitor) for 15 min. at 22°C. The second aliquot was treated the same as the first, except that no ribonucleotides (rNTPs) were added. Nuclei were then subjected to RQ1 DNase I (20 units, Promega) digestion in the presence of 1 mM CaCl2 for 15 min. at 37°C. Nuclear proteins were digested by adding 10× digestion buffer (100 mM Tris, pH 8.0, 10 mM ethylenediaminetetraacetic acid, 5% SDS and 100 μg/ml proteinase K) and incubation for 30 min. at 37°C. Nuclear RNA was then purified using RNeasy kit (Qiagen). Isolated RNA was treated with TURBO DNA‐free (Ambion) to ensure no contaminating DNA. Real‐time RT‐PCR was carried out using primers that amplify TGF‐β1 pre‐mRNA as described above. To quantitate the nascent TGF‐β1 pre‐mRNA transcripts, values obtained from nuclear run on reactions with no rNTPs added were subtracted from reactions with rNTPs added.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed with the Gel Shift Assay System and AP‐1 consensus oligonucleotides (5′‐CGCTTGATGAGTCAGCCGGAA‐3′ (Promega). The double‐stranded oligonucleotides were labelled with [γ‐32P]ATP (Perkin Elmer, Waltham, MA, USA). Binding reactions were carried out with nuclear extracts prepared from TNF‐α treated or untreated Swiss 3T3 cells at the indicated times using the Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s instructions. One microlitre of 32P‐labelled probe was incubated with 5 μg of Swiss 3T3 nuclear extract for 10 min. at 37°C. DNA‐protein binding specificity was tested by competition assays in which the binding reactions were pre‐incubated with 100‐fold excess of unlabelled specific or non‐specific competitor oligonucleotides prior to the addition of the labelled probe. Positive controls consisted of HeLa extracts provided with the Gel Shift Assay System (Promega). For the supershift experiment, 2 μg of each antibody were added to the reaction mixtures that already contained the probe and 10 μg nuclear lysates. The entire mixture was incubated for an additional 30 min. at room temperature. The antibodies used were as follows: anti‐c‐Jun (sc‐44X, Santa Cruz, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐phospho‐c‐Jun (sc‐822X, Santa Cruz), anti‐c‐Fos (sc‐253X, Santa Cruz) and normal mouse IgG (PP100, Chemicon, Temecula, CA, USA). After the binding reaction was complete, the DNA‐protein complexes were resolved by electrophoresis in a 6% non‐denaturing acrylamide gel (Invitrogen). After electrophoresis, the gels were transferred onto 3M Whatman paper, dried and subjected to autoradiography.
Western immunoblotting
Nuclear extracts prepared as described above were separated by SDS‐PAGE (12% pre‐cast gels from NuPAGE, Invitrogen) followed by semi‐wet electrophoretic transfer to 0.45 μm polyvinyl difluoride (PVDF) membrane as previously described [17]. The membranes were blocked for 1 hr in tris‐buffered saline with tween 20 (TBST) (20 mM Tris pH 7.6, 137 mM NaCl, 0.1% Tween 20) containing 5% bovine serum albumin and then hybridized overnight with one of the following primary antibodies: rabbit anti‐c‐Fos, rabbit anti‐c‐Jun, rabbit anti‐phospho‐c‐Jun (Ser63) (all from Cell Signaling, Danvers, MA, USA) or mouse anti‐β‐actin (Abcam, Cambridge, MA, USA). After three washes in TBST, membranes were probed with the appropriate secondary antibody: sheep antimouse (Amersham, Piscataway, NJ, USA) or donkey anti‐rabbit IgG (Amersham), each conjugated to horseradish peroxidase (HRP). The HRP‐catalysed chemiluminescence reaction was developed with Enhanced Chemiluminescense System (ECL, Amersham). The ECL‐treated membranes were exposed to X‐ray film to visualize the immunoreactive protein bands. The membranes were stripped and reprobed with β‐actin to correct for differences in protein loading.
Statistics
All values are expressed as means ± S.E.M., where n= 3 or 4. An unpaired Student’s t‐test was used to assess the difference between two groups. anova was performed when more than two groups were compared to a single control and Tukey post‐tests were used to assess differences between individual groups within the set. Differences were considered statistically significant when P < 0.05.
Results
De novo transcription is required for TNF‐α induction of TGF‐β1
In a recent study [17] we showed that treatment of fibroblasts isolated from the lungs of adult C57 mice, or Swiss 3T3 mouse embryo fibroblasts, with recombinant, murine TNF‐α resulted in a dose‐ and time‐dependent increase in levels of TGF‐β1 mRNA through a mechanism involving mRNA stabilization. The increase in TGF‐β1 message could be detected as early as 2 hrs after addition of TNF‐α suggesting that transcription of the TGF‐β1 gene may also be up‐regulated. To determine if TNF‐α‐induction of TGF‐β1 mRNA expression requires de novo transcription, Swiss 3T3 fibroblasts were treated with 50 μM 5,6‐dichloro‐1‐β‐D‐ribofuranosylbenzimidazole (DRB), an inhibitor of RNA polymerase II transcription, for 1 hr prior to addition of TNF‐α (10 ng/ml). In our previous study [17], treatment of cells with 10 ng/ml of TNF‐α produced the strongest induction of TGF‐β1 expression. After 12 hrs, TGF‐β1 mRNA levels were measured by real‐time RT‐PCR. As a control, we also measured the effect of DRB on TNF‐α induction of MMP‐9 mRNA, which is known to be transcriptionally up‐regulated by TNF‐α in fibroblasts [24]. Figure 1 shows that DRB reduced the TNF‐α induction of TGF‐β1 by 37.5% whereas TNF‐α induction of MMP9 mRNA was completely blocked. These results suggest that TNF‐α induces TGF‐β1 mRNA expression at least partially via a transcriptional mechanism.
Figure 1.
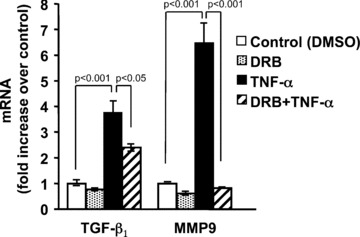
Inhibition of transcription partially blocks TNF‐α induction of TGF‐β1 mRNA. Quiescent Swiss 3T3 fibroblasts were pre‐treated with 50 μM 5,6‐dichloro‐1‐β‐D‐ ribofuranosylbenzimidazole (DRB), or vehicle (DMSO) as control, for 1 hr followed by addition of 10 ng/ml TNF‐α for another 12 hrs. At the indicated times, total RNA was isolated and TGF‐β1 mRNA was measured by quantitative real‐time RT‐PCR. As a control, MMP9 (a gene known to be transcriptionally up‐regulated by TNF‐α) mRNA was also measured. Relative quantitation was determined using the comparative CT method with data normalized to 36B4 riboprotein mRNA and calibrated to the average ΔCT of untreated controls (fold induction = 2−ΔΔCT). Data are represented as the mean ± S.E.M., of three independent experiments.
TNF‐α increases transcription of the TGF‐β1 gene
To determine if TNF‐α increased transcription of TGF‐β1, we first measured the levels of pre‐mRNA since these newly synthesized and unspliced RNAs have been shown to reflect transcriptional activity [25]. To measure TGF‐β1 pre‐mRNA, we designed PCR primers such that the sense primer is located in exon 6 and the antisense primer is located within intron 6. Processed or mature mRNA levels were measured by using a sense primer that spanned the junction of exons 5 and 6 and an antisense primer in exon 6. As shown in Fig. 2A, TGF‐β1 pre‐mRNA increased dramatically between 1 and 2 hrs after treatment with TNF‐α and preceded the increase of TGF‐β1 mature mRNA. Pre‐treatment of the cells with DRB to inhibit transcription completely blocked the increase in TGF‐β1 pre‐mRNA (Fig. 2B) suggesting that TNF‐α treatment increased transcriptional activity of the TGF‐β1 promoter. Experiments using actinomycin D yielded similar results at the 1 and 2 hrs time‐points; however, pronounced toxicity was observed in cultures receiving TNF‐α and actinomycin D at the 4‐hr time‐point.
Figure 2.
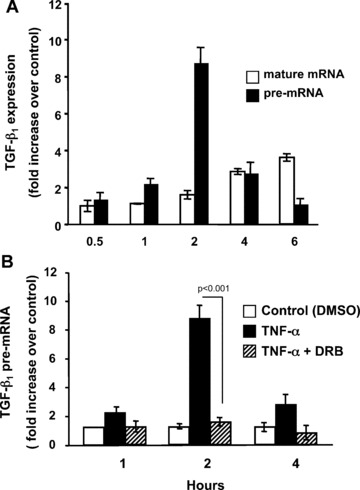
TNF‐α induction of TGF‐β1 mRNA is preceded by an increase in TGF‐β1 pre‐mRNA and requires de novo transcription. (A) Quiescent Swiss 3T3 fibroblasts were treated with 10 ng/ml TNF‐α for the indicated times. Expression of TGF‐β1 pre‐mRNA and mature mRNA were determined by quantitative real‐time RT‐PCR using primers designed to amplify unprocessed mRNA (pre‐mRNA) or spliced mRNA (mature mRNA) after extensive DNase treatment. (B) Quiescent Swiss 3T3 fibroblasts were pre‐treated with 50 μM DRB or vehicle (DMSO) as control for 1 hr followed by addition of 10 ng/ml TNF‐α. At the indicated times TGF‐β1 pre‐mRNA was measured as described above. (Mean ± S.E.M., relative units, n= 3.)
We previously reported that TNF‐α failed to activate a TGF‐β1 promoter‐luciferase construct (phTG −1362/+819) transfected into Swiss 3T3 cells [17]. We considered that the promoter–reporter assay may not accurately reflect regulation of the endogenous TGF‐β1 gene where chromatin effects may represent another level of control. Therefore we performed nuclear run‐on assays to determine if TNF‐α increases the rate of TGF‐β1 transcription. Addition of TNF‐α resulted in a greater than twofold increase in the rate of TGF‐β1 transcription when compared to addition of media alone (Fig. 3). Together, these results strongly suggest that TNF‐α induces transcription of the TGF‐β1 gene.
Figure 3.
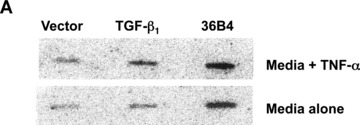
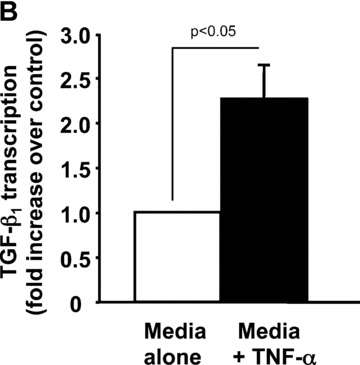
TNF‐α stimulates transcription of the TGF‐β1 gene in fibroblasts. Quiescent Swiss 3T3 cells were treated with TNF‐α (10 ng/ml) or media alone for 2 hrs. Nuclei were then isolated and nuclear run‐on transcription reactions performed in the presence of α‐[32P] labelled UTP. Radiolabelled RNA was hybridized to plasmid DNAs carrying cDNAs for murine TGF‐β1, murine 36B4, or vector alone under stringent conditions. (A) Autoradiogram. (B) Densitometric measurements of TGF‐β1 transcription rate were performed with ImageGauge software and are expressed relative to 36B4 transcription rate after subtracting background hybridization to the vector control. In some experiments, PCR‐based nuclear run‐on reactions were performed. Data are representative of four experiments.
ERK signalling is not required for TNF‐α induced transcription of the TGF‐β1 gene
We recently showed that TNF‐α rapidly activates the ERK signalling pathway resulting in stabilization of TGF‐β1 mRNA [17]. Here, we asked if TNF‐α activation of ERK signalling also leads to increased transcription of the TGF‐β1 gene. Quiescent Swiss 3T3 cells were pre‐treated with U0126, a highly selective inhibitor of ERK activation, followed by addition of TNF‐α for 2 hrs. Nuclear run‐on assays were then performed as described above. As seen in Fig. 4, inhibition of ERK activation had no effect on TNF‐α induced transcription of the TGF‐β1 gene. In similar experiments, TNF‐α induced increases in TGF‐β1 pre‐mRNA levels were also unaffected by pre‐treatment with U0126 (data not shown). These results suggest that ERK signalling is not required for TNF‐α induction of TGF‐β1 transcription.
Figure 4.
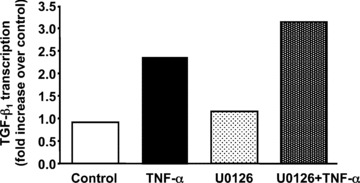
Inhibition of the MEK/ERK signalling pathway does not block TNF‐α stimulated transcription of the TGF‐β1 gene. Quiescent Swiss 3T3 cells were treated with 10 μM U0126 or DMSO as vehicle control for 30 min. before stimulation with TNF‐α (10 ng/ml) for an additional 2 hrs. Nuclei were then isolated and PCR‐based nuclear run‐on assays were performed. This experiment was repeated with similar results.
AP‐1 activity is required for TNF‐α induction of TGF‐β1
Transcription of the human TGF‐β1 gene has been shown to be regulated primarily by AP‐1 [26, 27, 28] and selective promoter factor (Sp1) [29]. To determine if either of these transcription factors is involved in TNF‐α induced TGF‐β1 transcription, we pre‐treated Swiss 3T3 cells with curcumin, an inhibitor of AP‐1 or mithramycin, an inhibitor of Sp1. Curcumin, completely blocked TNF‐α induction of TGF‐β1 pre‐mRNA (Fig. 5A) and reduced mature mRNAs by 60% (Fig. 5B). In contrast, mithramycin had no effect on TGF‐β1 pre‐mRNA (Fig. 5A) or mature mRNA (Fig. 5B) levels. Curcumin has been reported to block NF‐κB activation [30], therefore we also pre‐treated cells with SC‐514 (Calbiochem, San Diego, CA, USA), a highly selective inhibitor of IKK‐2 that has been shown to specifically block NF‐κB‐dependent gene expression. As shown in Fig. 6, SC‐514 failed to block TNF‐α induction by TGF‐β1, even when added at 100 μM. These results were confirmed by transfection of Swiss 3T3 cells with a cDNA encoding a dominant negative IκB (IκBΔN), a cellular inhibitor of NF‐κB activation, prior to treatment with TNF‐α (data not shown). Together, these results implicate AP‐1 in TNF‐α up‐regulation of TGF‐β1 transcription.
Figure 5.
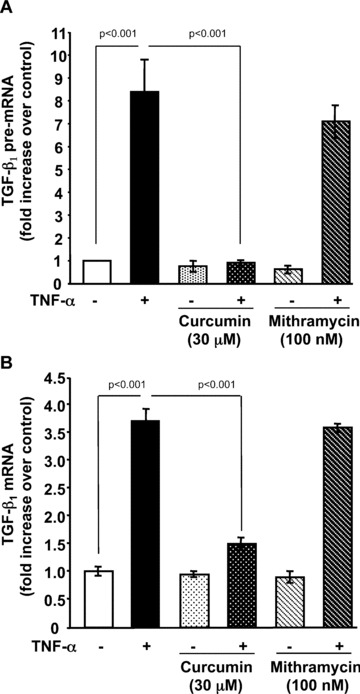
Curcumin, an AP‐1 inhibitor, abrogates TNF‐α induction of TGF‐β1 pre‐mRNA and mature mRNA. Quiescent Swiss 3T3 fibroblasts were treated with TNF‐α (10 ng/ml) or media alone in the presence (+) or absence (–) of curcumin (30 μM) or mithramycin (100 nM). (A) After 2 hrs, TGF‐β1 pre‐mRNA was measured by quantitative real‐time RT‐PCR as described above. (B) After 6 hrs, mature TGF‐β1 mRNA was measured by quantitative real‐time RT‐PCR as described above. (Mean ± S.E.M., relative units, n= 4.)
Figure 6.
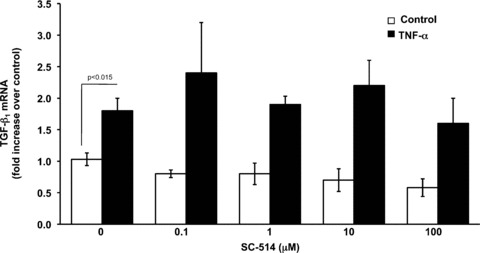
NF‐κB activation is not required for TNF‐α induction of TGF‐β1. Quiescent Swiss 3T3 fibroblasts were pre‐treated with increasing doses (0.1–100 μM) of the IKK‐2 inhibitor SC‐514 or vehicle (DMSO) for 1 hr and then treated with TNF‐α (10 ng/ml) or media alone. After 6 hrs, mature TGF‐β1 mRNA was measured by quantitative real‐time RT‐PCR as described above. (Mean ± S.E.M., relative units, n= 3.)
TNF‐α activates AP‐1 binding to DNA
To determine whether TNF‐α activated AP‐1 in our cells, we performed EMSAs. The mouse TGF‐β1 promoter contains a canonical AP‐1 binding site (TGAGTCA) at nucleotide 445–451 (M57902). Therefore, nuclear protein extracts prepared from Swiss 3T3 cells treated with TNF‐α, or media alone as control, were incubated with radiolabelled double‐stranded oligonucleotides representing the consensus binding site for AP‐1 (TGAGTCA) as a probe. As shown in Fig. 7A and B, TNF‐α increased DNA binding of AP‐1 in a time‐dependent manner reaching approximately twofold over control after 2 hrs. To determine if the observed binding was specific to the AP‐1 site, we used unlabelled probe and a non‐specific oligonucleotide as competitors. The addition of 100‐fold molar excess of unlabelled probe completely blocked binding to the labelled probe whereas addition of an equal concentration of a non‐specific oligonucleotide only slightly reduced binding. To confirm that AP‐1 was a component of the complexes observed, we performed supershift assays using antibodies to c‐Jun and c‐Fos, components of the AP‐1 transcription factor. As seen in Fig. 7C, addition of a monoclonal antibody that specifically recognizes phosphorylated c‐Jun elicited a strong supershift in the nuclear extract from Swiss 3T3 fibroblasts 2 hrs after treatment with TNF‐α. Polyclonal antibodies to total c‐Jun and c‐Fos reduced DNA binding and caused a supershift of less intensity. This is consistent with some antibody binding to the DNA binding regions of c‐Jun and c‐Fos. These data show that TNF‐α induces specific binding of AP‐1 to DNA.
Figure 7.
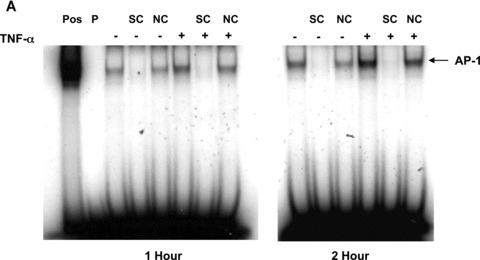
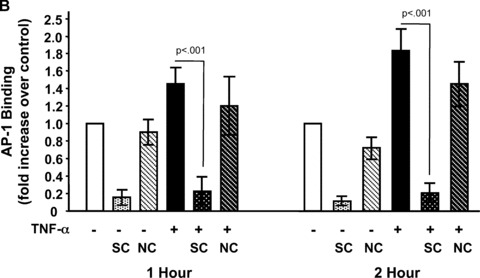

TNF‐α increases binding of AP‐1 to DNA. (A) Quiescent Swiss 3T3 fibroblasts were treated with TNF‐α (10 ng/ml) or media alone. At the indicated times, nuclear extracts were prepared and EMSA was performed with [32P]‐labelled oligonucleotides representing the AP‐1 consensus sequence as a probe (P). HeLa extract was used as a positive control (Pos). The specificity of binding was confirmed using 100‐fold molar excess of unlabelled probe as a specific competitor (SC) or 100‐fold molar excess of an unrelated oligonucleotide as non‐specific competitor (NC). (B) AP‐1 binding was quantitated after densitometry using ImageGauge software. The results represent the average of 3 independent experiments. (mean ± S.E.M., relative units, n= 3). (C) Antibodies against phosphorylated c‐Jun, c‐Jun and c‐Fos were added to binding reactions containing nuclear extracts collected 2 hrs after TNF‐α treatment and EMSA performed as described above. The arrows indicate super shifted bands.
To determine if TNF‐α increases the levels of c‐Jun and c‐Fos in the nucleus, we performed Western blot analyses of nuclear extracts from Swiss 3T3 cells treated with TNF‐α or media alone. Incubation of quiescent Swiss 3T3 fibroblasts with TNF‐α resulted in a rapid increase in nuclear c‐Jun levels that was further increased at 2 hrs (Fig. 8). Moreover, TNF‐α induced rapid phosphorylation of c‐Jun within 1 hr of treatment (Fig. 8). Similarly, the nuclear levels of c‐Fos were significantly increased in TNF‐α treated cells after 1 hr and returned to near control levels by 2 hrs (Fig. 8). Together these results clearly demonstrate that TNF‐α increases nuclear levels of c‐Jun and c‐Fos and that these transcription factors specifically bind to an AP‐1 site found within the TGF‐β1 promoter.
Figure 8.
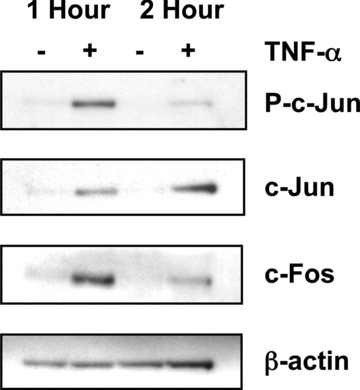
TNF‐α increases nuclear levels of c‐Jun and c‐Fos. Quiescent Swiss 3T3 fibroblasts were treated with TNF‐α (10 ng/ml) or media alone for the indicated times. Nuclear extracts were prepared and analysed by Western blot for c‐Jun phosphorylated at Ser63 (P‐c‐Jun). The blot was stripped and reprobed for total c‐Jun and then c‐Fos. Human β‐actin was used as an internal loading control after stripping the blot a third time. The results presented are representative of three independent experiments.
TNF‐α up‐regulates transcription of TGF‐β1 mRNA in an AP‐1 dependent manner in primary mouse lung fibroblasts
We previously demonstrated that the Swiss 3T3 fibroblast cell line is very similar to primary MLFs in their response to TNF‐α[17]. To confirm that TNF‐α induces transcription of TGF‐β1 in an AP‐1 dependent manner in MLFs, we next asked whether curcumin could block TNF‐α induced expression of TGF‐β1 pre‐mRNA in primary lung fibroblasts isolated from adult C57Bl6 mice. As shown in Fig. 9, TNF‐α induced a rapid increase in TGF‐β1 pre‐mRNA that was completely blocked by the AP‐1 inhibitor, curcumin. These results confirm the biological relevance of our findings in primary lung cells.
Figure 9.
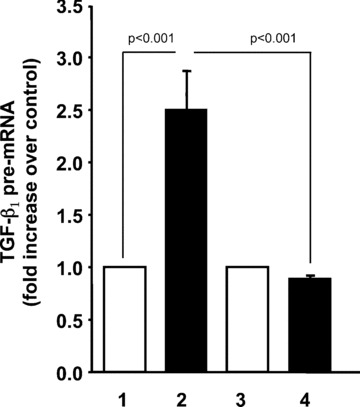
TNF‐α induces transcription of TGF‐β1 in primary mouse lung fibroblasts by an AP‐1 dependent mechanism. Fibroblasts isolated from the lungs of adult C57 mice were rendered quiescent and treated with 10 ng/ml TNF‐α (lanes 2 and 4) or media alone (lanes 1 and 3) for 2 hrs. Cells in lanes 3 and 4 were incubated with 30 μM curcumin for 1 hr prior to treatment. TGF‐β1 pre‐mRNA was measured by quantitative real‐time RT‐PCR as described above. (mean ± S.E.M., relative units, n= 4).
Discussion
In the present study, we show that TNF‐α doubles the rate of transcription of the TGF‐β1 gene in an AP‐1 dependent manner. We previously demonstrated that TNF‐α also stabilizes TGF‐β1 mRNA via an ERK dependent pathway. Together these results demonstrate that the mechanisms by which TNF‐α up‐regulate TGF‐β1 are complex; they involve a combination of transcriptional and post‐transcriptional events that lead to the observed three‐ to fourfold increase in steady‐state TGF‐β1 mRNA 6–12 hrs after exposure to TNF‐α. It is essential to have this information because TNF‐α and TGF‐β1 are potent peptide growth factors that have been implicated in playing important roles in the development of a variety of diseases including neoplastic and fibrogenic processes. Moreover, there is increasing evidence that TNF‐α influences TGF‐β1 expression. TNF‐α has been shown to induce the expression of TGF‐β1 in many cell types including microglial cells [31], mature adipocytes [32], human proximal tubular cells [33] and rat pulmonary artery endothelial cells [34]in vitro. Adenovirus‐mediated expression of TNF‐α in lungs of normal rats was shown to induce TGF‐β1 production and interstitial fibrogenesis [12], suggesting a functional importance of TNF‐α‐induced TGF‐β1 expression in vivo. However, there is clear evidence that TNF‐α has antifibrotic properties as well. In addition to its well known pro‐inflammatory effects, TNF‐α also inhibits synthesis of structural components such as type I collagen and induces production of stromal collagenases [35]. Moreover, overexpression of TNF‐α diminished pulmonary fibrosis induced by bleomycin and TGF‐β1 in vivo[36]. A possible explanation for the contradictory effects of TNF‐α was recently provided by Oikonomou et al.[37]. They showed that in bleomycin‐induced fibrosis, expression of the membrane‐bound form of TNF‐α is sufficient to elicit an inflammatory response, but does not lead to fibrosis, whereas expression of the soluble form of TNF‐α is crucial for lymphocyte recruitment and a prerequisite for TGF‐β1 expression and the development of fibrosis [37]. We propose that soluble TNF‐α directly up‐regulates expression of TGF‐β1 in the lung thereby initiating or perpetuating a cytokine cascade that leads to fibrosis. The present study provides new information that clarifies the intracellular signalling mechanisms involved in TNF‐α up‐regulation of TGF‐β1. This pathway is not unique to mouse fibroblasts as we have shown that fibroblasts and alveolar epithelial cells from the lungs of both mice and human beings exhibit up‐regulated TGF‐β1 expression in response to TNF‐α[16, 38].
Interstitial pulmonary fibrosis afflicts millions of individuals worldwide, yet there are no effective treatments and few therapeutic approaches (5). Our findings are consistent with the thinking that blunting TNF‐α expression or signalling pharmacologically may be useful in preventing or treating pulmonary fibrosis. As mentioned above, anti‐TNF‐α treatment with agents such as pirfenidone and etanercept have shown promise in treatment of some patients [5]. Interestingly, the AP‐1 inhibitor, curcumin has been shown to inhibit the development of pulmonary fibrosis by either bleomycin [39] or amiodarone [40]. Further studies aimed at understanding the mechanisms involved in TNF‐α regulation of TGF‐β1 may lead to new strategies for the treatment of IPF.
Numerous studies have shown that TGF‐β1 expression may be controlled at the levels of transcription, mRNA stability, mRNA translation or secretion of preformed protein [33, 41, 42, 43]. Our results showed that TNF‐α induced a rapid increase in TGF‐β1 mRNA levels detected as early as 2 hrs after treatment (Fig. 2A). Although our previous study showed that TNF‐α increased the half‐life of TGF‐β1 mRNA to more than 24 hrs, the rapid increase in TNF‐α mRNA levels suggested that TNF‐α may also increase transcription of the TGF‐β1 gene. In this study, we show that de novo transcription is required for maximal TNF‐α induction of TGF‐β1. In an effort to separate the effects of TNF‐α on TGF‐β1 mRNA stability and transcriptional activation, we measured levels of newly transcribed unprocessed pre‐mRNA, also known as heterogeneous nuclear RNA (hnRNA), by real‐time RT‐PCR [25] and found that TNF‐α induced a significant increase in TGF‐β1 pre‐mRNA that preceded the increase in mature mRNA (Fig. 2A). Because changes in the amount of unprocessed pre‐mRNA may reflect not only altered transcription but also altered processing, we confirmed these results using traditional nuclear run‐on analyses (Fig. 3). Together these results show that TNF‐α treatment does indeed increase the rate of transcription of the TGF‐β1 gene as well as stabilize the mRNA.
That TNF‐α regulation of TGF‐β1 involves both transcriptional and post‐transcriptional mechanisms is not surprising since several factors, including TGF‐β2[44] and nerve growth factor [45], regulate TGF‐β1 in a similar manner. It is becoming apparent that carefully orchestrated transcriptional and post‐transcriptional processes allow precise and rapid changes in the production of TGF‐β1. The relative contribution of gene transcription and mRNA turnover to TGF‐β1 expression are likely to differ according to the stimulus and the cell type involved. In our studies, TNF‐α induced TGF‐β1 expression in lung fibroblasts was inhibited ∼37% when RNA polymerase II transcription was inhibited (Fig. 4), and more than 60% when ERK signalling was inhibited [17]. Thus, in lung fibroblasts, post‐ transcriptional events initiated by TNF‐α appear to be a major contributor for the increased expression of TGF‐β1; however, increased transcription contributes to the over‐all increase in TGF‐β1.
The mouse and human TGF‐β1 promoters are well conserved in sequence and general structure. Both the human [46] and mouse [47] TGF‐β1 genes have two promoter regions, one located 5′ to the most upstream transcriptional start site, and another located between the two major transcriptional initiation sites. Both human and mouse TGF‐β1 promoter regions contain consensus sequences for AP‐1, NF‐κB and Sp1. Curcumin, an AP‐1 inhibitor blocked TNF‐α induced TGF‐β1 transcription (Fig. 5A) resulting in a 60% reduction in the steady state levels of TGF‐β1 mRNA, whereas inhibitors of Sp1 (Fig. 5B) or NF‐κB (Fig. 6) had no effect. These results strongly suggest that AP‐1 mediates TNF‐α induced TGF‐β1 transcription. Consistent with these results, TNF‐α induced AP‐1 binding (Fig. 7A) that was preceded by phosphorylation of c‐Jun within the transactivation domain (Ser63) (Fig. 8). Activation of c‐Jun is known to initiate dimerization of c‐Jun with other AP‐1 binding partners, such as c‐Fos [48], which was also up‐regulated by TNF‐α in our experiments. Supershift assays confirmed that TNF‐α induced binding of c‐Jun and c‐Fos to a sequence found in the TGF‐β1 promoter (Fig. 6C). Additional studies using chromatin immunoprecipitation will be required to demonstrate increased binding of AP‐1 to the endogenous TGF‐β1 promoter in response to TNF‐α and whether other transcription factors are involved.
Our previous results show that TNF‐α activation of ERK signalling leads to stabilization of the TGF‐β1 mRNA in lung fibroblasts [17]. ERK has been shown to phosphorylate residues of the c‐Jun transactivation domain, thereby facilitating AP‐1 transcription factor dimerization, DNA binding, and transcriptional regulation [49]. Therefore, we postulated that TNF‐α activation of the ERK pathway may mediate both AP‐1 dependent induction of TGF‐β1 transcription and mRNA stabilization. However, inhibition of ERK signalling had no effect on TNF‐α induction of TGF‐β1 transcription (Fig. 4), suggesting that independent signalling pathways are likely involved in up‐regulation of TGF‐β1 expression via these two mechanisms. Although our results do not rule out the possibility that TNF‐α activation of ERK signalling and AP‐1 induces transcription of an intermediate that in turn up‐regulates TGF‐β1 expression. Additional studies will be required to answer these fundamental questions.
Acknowledgements
This work was supported by National Institutes of Health Grants ES06766 (A.R.B.) and HL60532 (A.R.B.) and the Louisiana Cancer Research Consortium grant (D.E.S.). We are grateful to Dr. Gilbert Morris for helpful discussions and advice.
References
- 1. Dennler S, Goumans MJ, ten Dijke P. Transforming growth factor beta signal transduction. J Leukoc Biol . 2002; 71: 731–40. [PubMed] [Google Scholar]
- 2. Bartram U, Speer CP. The role of transforming growth factor β in lung development and disease. Chest . 2004; 125: 754–65. [DOI] [PubMed] [Google Scholar]
- 3. Zhao J, Shi W, Wang YL, et al . Smad3 deficiency attenuates bleomycin‐induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol . 2002; 282: L585–93. [DOI] [PubMed] [Google Scholar]
- 4. Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol . 2001; 99: 308–19. [DOI] [PubMed] [Google Scholar]
- 5. Selman M, Thannickal VJ, Pardo A, et al . Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs . 2004; 64: 405–30. [DOI] [PubMed] [Google Scholar]
- 6. Riha RL, Yang IA, Rabnott GC, et al . Cytokine gene polymorphisms in idiopathic pulmonary fibrosis. Intern Med J . 2004; 34: 126–29. [DOI] [PubMed] [Google Scholar]
- 7. Whyte M, Hubbard R, Meliconi R, et al . Increased risk of fibrosing alveolitis associated with interleukin‐1 receptor antagonist and tumor necrosis factor‐alpha gene polymorphisms. Am J Respir Crit Care Med . 2000; 162: 755–58. [DOI] [PubMed] [Google Scholar]
- 8. Brass D, Hoyle G, Poovey H, et al . Reduced TNF‐α and TGF‐β1 expression in the lungs of inbred mice that fail to develop fibroproliferative lesions consequent to asbestos exposure. Am J Pathol . 1999; 154: 853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ortiz LA, Lasky J, Hamilton RF Jr, et al . Expression of TNF and the necessity of TNF receptors in bleomycin‐induced lung injury in mice. Exp Lung Res . 1998; 24: 721–43. [DOI] [PubMed] [Google Scholar]
- 10. Phan SH, Kunkel SL. Lung cytokine production in bleomycin‐induced pulmonary fibrosis. Exp Lung Res . 1992; 18: 29–43. [DOI] [PubMed] [Google Scholar]
- 11. Piguet PF, Vesin C. Treatment by human recombinant soluble TNF receptor of pulmonary fibrosis induced by bleomycin or silica in mice. Eur Respir J . 1994; 7: 515–18. [DOI] [PubMed] [Google Scholar]
- 12. Sime PJ, Marr RA, Gauldie D, et al . Transfer of tumor necrosis factor‐α to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor‐β1 and myofibroblasts. Am J Pathol . 1998; 153: 825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu J‐Y, Brass DM, Hoyle GW, et al . TNF‐α receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol . 1998; 153: 1839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ortiz LA, Lasky J, Lungarella G, et al . Upregulation of the p75 but not the p55 TNF‐α receptor mRNA after silica and bleomycin exposure and protection from lung injury in double receptor knockout mice. Am J Respir Cell Mol Biol . 1999; 20: 825–33. [DOI] [PubMed] [Google Scholar]
- 15. Liu JY, Sime PJ, Wu T, et al . Transforming growth factor‐beta(1) overexpression in tumor necrosis factor‐alpha receptor knockout mice induces fibroproliferative lung disease. Am J Respir Cell Mol Biol . 2001; 25: 3–7. [DOI] [PubMed] [Google Scholar]
- 16. Warshamana GS, Corti M, Brody AR. TNF‐alpha, PDGF, and TGF‐beta(1) expression by primary mouse bronchiolar‐alveolar epithelial and mesenchymal cells: TNF‐alpha induces TGF‐beta(1). Exp Mol Pathol . 2001; 71: 13–33. [DOI] [PubMed] [Google Scholar]
- 17. Sullivan DE, Ferris M, Pociask D, et al . Tumor necrosis factor‐alpha induces transforming growth factor‐beta1 expression in lung fibroblasts through the extracellular signal‐regulated kinase pathway. Am J Respir Cell Mol Biol . 2005; 32: 342–9. [DOI] [PubMed] [Google Scholar]
- 18. Lambert V, Munaut C, Jost M, et al . Matrix metalloproteinase‐9 contributes to choroidal neovascularization. Am J Pathol . 2002; 161: 1247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simpson DA, Feeney S, Boyle C, et al . Retinal VEGF mRNA measured by SYBR green I fluorescence: a versatile approach to quantitative PCR. Mol Vis . 2000; 6: 178–83. [PubMed] [Google Scholar]
- 20. Schubeler D, Bode J. A sensitive transcription assay based on a simplified nuclear runon protocol. http://jbode.homepage.t‐online.de/t01176.htm. [last accessed 23 January 2009]
- 21. Pelton RW, Johnson MD, Perkett EA, et al . Expression of transforming growth factor‐beta 1, ‐beta 2, and ‐beta 3 mRNA and protein in the murine lung. Am J Respir Cell Mol Biol . 1991; 5: 522–30. [DOI] [PubMed] [Google Scholar]
- 22. Hildebrandt AL, Neufer PD. Exercise attenuates the fasting‐induced transcriptional activation of metabolic genes in skeletal muscle. Am J Physiol Endocrinol Metab . 2000; 278: E1078–86. [DOI] [PubMed] [Google Scholar]
- 23. Rolfe FG, Sewell WA. Analysis of human interleukin‐5 gene transcription by a novel nuclear run on method based on the polymerase chain reaction. J Immunol Methods . 1997; 202: 143–51. [DOI] [PubMed] [Google Scholar]
- 24. Hozumi A, Nishimura Y, Nishiuma T, et al . Induction of MMP‐9 in normal human bronchial epithelial cells by TNF‐alpha via NF‐kappa B‐mediated pathway. Am J Physiol Lung Cell Mol Physiol . 2001; 281: L1444–52. [DOI] [PubMed] [Google Scholar]
- 25. Elferink CJ, Reiners JJ Jr. Quantitative RT‐PCR on CYP1A1 heterogeneous nuclear RNA: a surrogate for the in vitro transcription run‐on assay. Biotechniques . 1996; 20: 470–7. [DOI] [PubMed] [Google Scholar]
- 26. Kim SJ, Angel P, Lafyatis R, et al . Autoinduction of transforming growth factor beta 1 is mediated by the AP‐1 complex. Mol Cell Biol . 1990; 10: 1492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee KY, Ito K, Hayashi R, et al . NF‐kappaB and activator protein 1 response elements and the role of histone modifications in IL‐1beta‐induced TGF‐beta1 gene transcription. J Immunol . 2006; 176: 603–15. [DOI] [PubMed] [Google Scholar]
- 28. Weigert C, Sauer U, Brodbeck K, et al . AP‐1 proteins mediate hyperglycemia‐induced activation of the human TGF‐beta1 promoter in mesangial cells. J Am Soc Nephrol . 2000; 11: 2007–16. [DOI] [PubMed] [Google Scholar]
- 29. Wolf G, Hannken T, Schroeder R, et al . Antioxidant treatment induces transcription and expression of transforming growth factor beta in cultured renal proximal tubular cells. FEBS Lett . 2001; 488: 154–9. [DOI] [PubMed] [Google Scholar]
- 30. Singh S, Aggarwal BB. Activation of transcription factor NF‐kappa B is suppressed by curcumin (diferuloylmethane). J Biol Chem . 1995; 270: 24995–5000. [DOI] [PubMed] [Google Scholar]
- 31. Chao CC, Hu S, Sheng WS, et al . Tumor necrosis factor‐α mediates the release of bioactive transforming growth factor‐β in murine microglial cell cultures. Clin Immunol Immunopathol . 1995; 77: 358–65. [DOI] [PubMed] [Google Scholar]
- 32. Samad F, Uysal KT, Wiesbrock SM, et al . Tumor necrosis factor alpha is a key component in the obesity‐linked elevation of plasminogen activator inhibitor 1. Proc Natl Acad Sci USA . 1999; 96: 6902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Phillips AO, Topley N, Steadman R, et al . Induction of TGF‐β1 synthesis in D‐glucose primed human proximal tubular cells: differential stimulation by the macrophage derived pro‐inflammatory cytokines IL‐1β and TNF‐α. Kidney Int . 1996; 50: 1546–54. [DOI] [PubMed] [Google Scholar]
- 34. Phan SH, Gharaee‐Kermani M, McGarry B, et al . Regulation of rat pulmonary artery endothelial cell transforming growth factor‐β production by IL‐1β and tumor necrosis factor‐α. J Immunol . 1992; 149: 103–6. [PubMed] [Google Scholar]
- 35. Verrecchia F, Mauviel A. TGF‐beta and TNF‐alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal . 2004; 16: 873–80. [DOI] [PubMed] [Google Scholar]
- 36. Fujita M, Shannon JM, Morikawa O, et al . Overexpression of tumor necrosis factor‐alpha diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor‐beta. Am J Respir Cell Mol Biol . 2003; 29: 669–76. [DOI] [PubMed] [Google Scholar]
- 37. Oikonomou N, Harokopos V, Zalevsky J, et al . Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS ONE . 2006; 1: e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sullivan DE, Pociask D, Ferris MB, et al . The latent form of TGF‐beta1 is induced by TNF‐alpha through an ERK specific pathway and is activated by asbestos‐derived reactive oxygen species in vitro and in vivo . J Immunotox . 2008; 5: 145–9. [DOI] [PubMed] [Google Scholar]
- 39. Punithavathi D, Venkatesan N, Babu M. Curcumin inhibition of bleomycin‐induced pulmonary fibrosis in rats. Br J Pharmacol . 2000; 131: 169–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Punithavathi D, Venkatesan N, Babu M. Protective effects of curcumin against amiodarone‐induced pulmonary fibrosis in rats. Br J Pharmacol . 2003; 139: 1342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Phillips AO, Steadman R, Topley N, et al . Elevated D‐glucose concentrations modulate TGF‐β1 synthesis by human cultured renal proximal tubular cells: the permissive role of platelet‐derived growth factor. Am J Pathol . 1995; 147: 362–74. [PMC free article] [PubMed] [Google Scholar]
- 42. Phillips AO, Topley N, Morrisey K, et al . Basic fibroblast growth factor stimulates the release of pre‐formed TGF‐β1 from human proximal tubular cells in the absence of de‐novo gene transcription of mRNA translation. Lab Invest . 1997; 76: 591–600. [PubMed] [Google Scholar]
- 43. Morrisey K, Evans RA, Wakefield L, et al . Translational regulation of renal proximal tubular epithelial cell transforming growth factor‐β1 generation by insulin. Am J Pathol . 2001; 159: 1905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bascom CC, Wolfshohl JR, Coffey RJ Jr, et al . Complex regulation of transforming growth factor beta 1, beta 2, and beta 3 mRNA expression in mouse fibroblasts and keratinocytes by transforming growth factors beta 1 and beta 2. Mol Cell Biol. 1989; 9: 5508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cosgaya JM, Aranda A. Nerve growth factor regulates transforming growth factor‐beta 1 gene expression by both transcriptional and posttranscriptional mechanisms in PC12 cells. J Neurochem . 1995; 65: 2484–90. [DOI] [PubMed] [Google Scholar]
- 46. Kim SJ, Glick A, Sporn MB, et al . Characterization of the promoter region of the human transforming growth factor‐beta 1 gene. J Biol Chem . 1989; 264: 402–8. [PubMed] [Google Scholar]
- 47. Geiser AG, Kim SJ, Roberts AB, et al . Characterization of the mouse transforming growth factor‐beta 1 promoter and activation by the Ha‐ras oncogene. Mol Cell Biol . 1991; 11: 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Halazonetis TD, Georgopoulos K, Greenberg ME, et al . c‐Jun dimerizes with itself and with c‐Fos, forming complexes of different DNA binding affinities. Cell . 1988; 55: 917–24. [DOI] [PubMed] [Google Scholar]
- 49. Dhandapani KM, Khan MM, Wade FM, et al . Induction of transforming growth factor‐beta1 by basic fibroblast growth factor in rat C6 glioma cells and astrocytes is mediated by MEK/ERK signaling and AP‐1 activation. J Neurosci Res . 2007; 85: 1033–45. [DOI] [PubMed] [Google Scholar]