

Abstract
Summary
The serine/threonine kinase, B-RAF, is frequently mutated in melanoma and is required for cell proliferation. Proteasomal turnover of cyclins and cyclin-dependent kinase inhibitors via E3 ubiquitin ligases regulates cell cycle progression. We previously showed that B-RAF regulates Cks1, a co-factor for the F-box protein Skp2. Recently, a second F-box protein cofactor was identified, αB-crystallin, that binds Fbx4 and promotes cyclin D1 degradation. Here, we demonstrate that αB-crystallin is down-regulated in mutant B-RAF melanoma cells compared to melanocytes in a B-RAF and MEK-dependent manner. In a subset of lines, MEK inhibition was sufficient to up-regulate αB-crystallin protein levels; whereas in other lines combined MEK and proteasome inhibition was required. αB-crystallin knockdown partially stabilized cyclin D1 in melanocytes. Expression of αB-crystallin in mutant B-RAF melanoma cells did not promote cyclin D1 turnover under normal conditions, but did enhance turnover following etoposide-induced DNA damage. Together, these data show that αB-crystallin is highly expressed in melanocytes contributing, in part, to cyclin D1 turnover. Furthermore, αB-crystallin is down-regulated in a B-RAF-dependent manner in melanoma cells and its re-expression regulates cyclin D1 turnover after DNA damage.
Significance
αB-crystallin has been implicated in cellular functions as a heat shock protein and, more recently, as a cofactor for an E3 ligase ubiquitin ligase complex that degrades the cell cycle protein, cyclin D1. In this study we identify αB-crystallin as a target of aberrant B-RAF-MEK signaling that is hyper-activated in the majority of melanomas through mutation of B-RAF. Furthermore, we provide evidence for a functional role of αB-crystallin in contributing to the turnover of cyclin D1 in melanocytes and in melanoma cells following DNA damage inducing signals. These findings further our understanding of the regulation of cyclin D1 in melanocytic cells.
Keywords: αB-crystallin, B-RAF, cyclin D1, Fbx4, melanoma
Introduction
Ubiquitin-mediated proteolysis of cell cycle proteins is critical for tight control of normal cell proliferation and is often deregulated in human cancers (Cardozo and Pagano, 2004). The rate limiting step in ubiquitin-mediated proteolysis is substrate recognition by E3 ubiquitin ligases. Two major types of E3 ligase exist: HECT-type and RING-finger E3 ligases. RING-finger like proteins can be either monomeric or members of multi-subunit E3 ligase complexes. One major class of multi-subunit E3 ubiquitin ligases is Skp1/Cullin/F-box protein (SCF) complexes. In these complexes, the Cullin protein serves as a scaffold for the E2 ubiquitin-conjugating enzyme and Skp1/F-box proteins, whereas the F-box protein determines the substrate specificity. SCF complexes target many cell cycle regulators and their deregulation contributes to neoplastic transformation.
The most well studied SCF complex contains the F-box protein Skp2 and is regulated by a cofactor, Cks1 (Spruck et al., 2001). Cks1 is critical for the interaction between Skp2 and one of its degradation targets, phosphorylated p27Kip1. Phosphorylated threonine-187 of p27Kip1 is recognized by Cks1 and the side chain of glutamic acid 185 in p27Kip1 inserts into the interface between Skp2 and Cks1 (Hao et al., 2005). We have shown that mutant B-RAF-MEK signaling controls the expression of Cks1 in melanoma cells (Bhatt et al., 2007). Recently, a second SCF co-factor was identified. αB-crystallin, previously shown to be a small heat shock protein, functions as a cofactor for the SCFFbx4 complex (Lin et al., 2006). Expression of αB-crystallin is required for the rapid ubiquitylation and degradation of threonine 286 phosphorylated cyclin D1 and depletion of either Fbx4 or αB-crystallin results in the increased stability and accumulation of cyclin D1 (Lin et al., 2006). αB-crystallin can be phosphorylated at three serine sites: 19, 45 and 59. Serine 19 and serine 45 phosphorylation promotes the interaction between αB-crystallin and Fbx4 (den Engelsman et al., 2003).
Cyclin D1 is an important G1/S cell cycle regulator that is often up-regulated in many cancers (Alao, 2007). Cyclin D1 expression is induced during G1 phase for cells to initiate DNA synthesis. Subsequently, cyclin D1 must be suppressed since sustained high levels of cyclin D1 during S phase inhibit DNA synthesis (Pagano et al., 1994, Guo et al., 2005). Cyclin D1 undergoes proteasomal degradation in a phosphothreonine 286-dependent manner (Diehl et al., 1997); however, the regulatory machinery remains unclear. Multiple kinases (GSK3β, ERK2 and IκBα) and E3 ubiquitin ligases (SCFFbx4-αB-crystallin, SCFFbxw8, SCFβ-TrCP and SCFFbxo31) have been implicated in targeting cyclin D1 for phosphorylation and degradation under various cellular conditions (Lin et al., 2006, Okabe et al., 2006, Wei et al., 2008, Santra et al., 2009).
We and others have shown that cyclin D1 is highly expressed in melanoma cells and is required for G1/S progression (Sauter et al., 2002, Bhatt et al., 2005). Since αB-crystallin is a co-factor for the SCFFbx4 complex that regulates cyclin D1 stability, we analyzed the expression and regulation of αB-crystallin, and its role in cyclin D1 turnover in human melanocytes and melanoma cells. We show that αB-crystallin is highly expressed in melanocytes and is down-regulated in melanoma cells. The repression of αB-crystallin at the mRNA level is B-RAF dependent in mutant B-RAF melanoma and thyroid cancer lines. In a subset of mutant B-RAF lines, proteasomal degradation also contributed to the down-regulation of αB-crystallin. Finally, we demonstrate that αB-crystallin expression contributes, in part, to basal cyclin D1 turnover in melanocytes, and cyclin D1 turnover in melanoma cells after DNA damage.
Results
αB-crystallin is down-regulated in melanoma cells
αB-crystallin has recently been shown to act as a co-factor for Fbx4 in the proteasomal turnover of cyclin D1 (Lin et al., 2006). We hypothesized that the expression of αB-crystallin would inversely correlate with cyclin D1 expression in tumor cells. To test this, we analyzed αB-crystallin levels in human melanocytes and a panel of human melanoma cell lines that display characteristics of different stages of melanoma progression (Satyamoorthy et al., 1997). By Western blot analysis, αB-crystallin was readily detected in lysates from human melanocytes that express low levels of cyclin D1 (Fig. 1A). In contrast, αB-crystallin was barely detectable in the radial growth phase cell lines, WM35 and Sbcl2; the vertical growth phase cell lines, WM278, WM793; and metastatic SK-MEL-5 and SK-MEL-28 cells. In two lines, WM115 and 1205-Lu, αB-crystallin was detectable but at low levels. All melanoma cell lines used harbor mutant B-RAF with the exception of Sbcl2 that are wild type for B-RAF but contain an N-RAS mutation. Importantly, high αB-crystallin expression was detected in multiple independent melanocyte cultures from distinct donors, ruling out donor-to-donor differences (Fig. 1B). All of the melanoma cell lines analyzed showed cyclin D1 expression markedly above that in melanocytes, consistent with our previous observations (Bhatt et al., 2005). These data show that αB-crystallin is highly expressed in normal melanocytes, is down-regulated in melanoma cell lines, and shows an inverse correlation with cyclin D1 expression.
Figure 1.
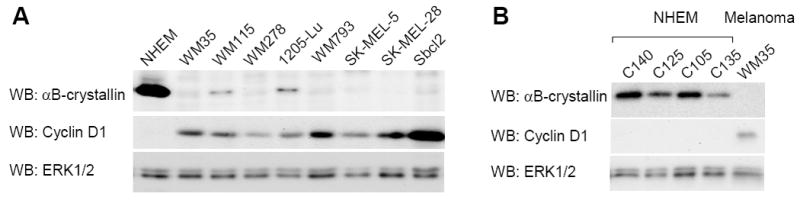
αB-crystallin expression is down-regulated in melanoma cells. (A) Serum-starved cultures of primary human melanocytes (NHEM) and eight human melanoma cell lines (WM35, WM115, WM278, 1205-Lu, WM793, SK-MEL-5, SK-MEL-28 and Sbcl2) were analyzed by Western blotting for αB-crystallin, cyclin D1 and ERK1/2 (loading control). All melanoma cell lines harbor mutant B-RAF with the exception of Sbcl2 that are wild-type for B-RAF but carry an N-RAS mutation. (B) As for A, except that four different batches of NHEM were analyzed.
Up-regulation of αB-crystallin following B-RAF-MEK inhibition in a subset of melanoma cell lines
B-RAF is frequently mutated in melanoma and in the majority of cell lines analyzed above (Davies et al., 2002); therefore, we tested whether αB-crystallin expression was regulated by B-RAF and its downstream target, MEK. Treatment with the MEK inhibitor, U0126, potently inhibited ERK1/2 phosphorylation in all cell lines tested (Fig. 2A). In WM115, 1205-Lu and WM983A cells, αB-crystallin protein expression was increased following U0126 treatment (Fig. 2A). However, no increase in αB-crystallin protein level was detected in U0126-treated WM793, WM35, Sbcl2, WM278 and A375 cells (Fig. 2A). B-RAF is also frequently mutated in thyroid carcinomas (Kimura et al., 2003, Mineva et al., 2005), which also show low levels of αB-crystallin. In the thyroid carcinoma cell lines NPA and ARO, both of which harbor the B-RAFV600E mutation, αB-crystallin protein level was increased following U0126 treatment (Fig. 2B). In responsive WM115 cells, up-regulation of αB-crystallin was not detected within 9 hours of MEK inhibition suggesting that is not an early response (Supplemental Fig. 1A). Furthermore, real-time quantitative RT-PCR (qRT-PCR) experiments showed that αB-crystallin up-regulation occurred at the mRNA level in WM115 cells (Fig. 2C). As a parallel approach, we inhibited MEK-ERK1/2 signaling through the knockdown of B-RAF with siRNA. Up-regulation of αB-crystallin was observed in B-RAF knockdown WM115 cells, but not WM793 cells (Fig. 2D). Knockdown of B-RAF with three independent siRNA sequences led to similar effects (Fig. 2E), arguing against off-target effects of siRNAs. We have previously utilized a tamoxifen-inducible B-RAF-estrogen receptor fusion (delta B-RAF-ER*) adenoviral system to efficiently activate B-RAF signaling in primary human melanocytes (Bhatt et al., 2005). Activation of B-RAF-ER and downstream ERK1/2 signaling was not sufficient to down-regulate αB-crystallin in melanocytes (Supplemental Fig. 1B). These data show that inhibition of B-RAF-MEK signaling leads to up-regulation of αB-crystallin protein levels in a subset of melanoma cells and B-RAFV600E containing thyroid cancer cells.
Figure 2.
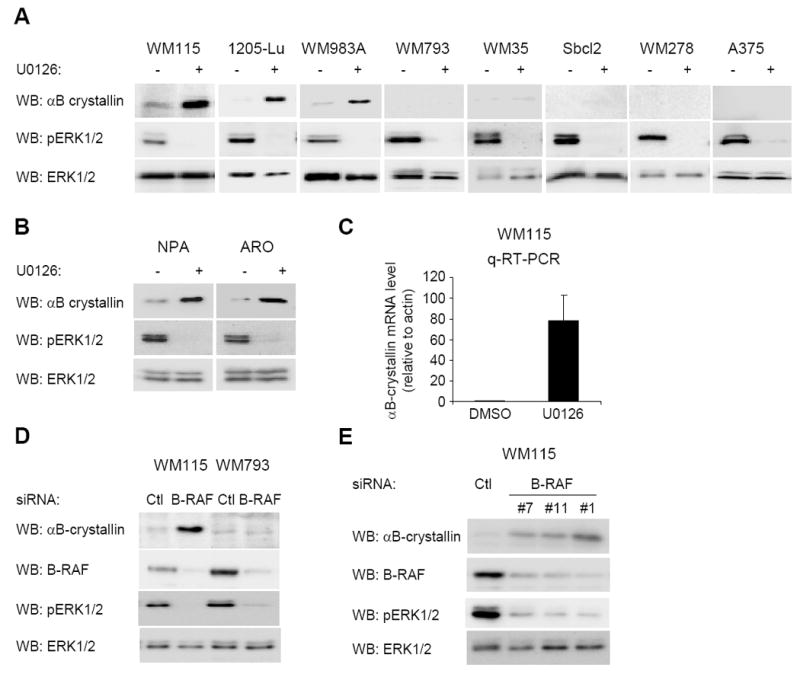
Down-regulation of αB-crystallin is B-RAF-MEK-dependent in melanoma cell lines. (A) Human melanoma cells (WM115, 1205-Lu, WM983A, WM793, WM35, Sbcl2, WM278 and A375) were treated with 10 μM U0126 for 16 hours. Cell lysates were analyzed by Western blotting for αB-crystallin, phosphorylated ERK (pERK1/2) and ERK1/2 (loading control). (B) Thyroid cancer cells (NPA and ARO) were analyzed as in A. (C) qRT-PCR analysis of αB-crystallin mRNA levels in U0126-treated (16 hours) or control (DMSO) treated WM115 cells. The graph represents the mean and s.d. for the percentage change in αB-crystallin mRNA level relative to actin from three independent experiments. (D) WM115 and WM793 cells were transfected with control and B-RAF#1 siRNA. Seventy-two hours post-transfection, cells were lysed and αB-crystallin, B-RAF, pERK1/2 and ERK1/2 levels were determined by Western blot analysis. (E) As for D, except with B-RAF siRNA duplexes #7, #11 and #1 were utilized in WM115 cells.
Expression of αB-crystallin is regulated at both transcriptional and degradation level
Despite our data showing that αB-crystallin protein levels are up-regulated following inhibition of B-RAF-MEK signaling in a subset of melanoma cell lines, qRT-PCR results revealed that αB-crystallin mRNA was up-regulated in the presence of the MEK inhibitor, U0126, in cell lines that showed little/no up-regulated αB-crystallin protein in these conditions (Fig. 3A, 3B and Supplemental Fig. 1C). Indeed, all melanoma cell lines tested showed greater than a 2-fold increase of αB-crystallin mRNA following MEK inhibition with the exception of WM35 cells. These results raised the possibility that in melanoma cells, αB-crystallin might be rapidly turned over by degradation. To test this notion, we treated melanoma A375, SK-MEL-24 and SK-MEL-5 cells with U0126 in combination with the proteasomal inhibitor, MG132. Treatment with MG132 alone did not induce αB-crystallin protein accumulation in A375, SK-MEL-24 and SK-MEL-5 cells (Fig. 3B). However, the combination of the two drugs led to detectable levels of αB-crystallin in all three cell lines. In WM115 and WM983A cells, in which αB-crystallin was detected following U0126 treatment, additional treatment with MG132 for 6 hours did not further enhance αB-crystallin levels (data not shown). Together, these results indicate that multiple mechanisms contribute to regulation of αB-crystallin. The mRNA levels of αB-crystallin are repressed by B-RAF-MEK signaling and αB-crystallin is additionally regulated by proteasomal turnover in a subset of cell lines.
Figure 3.
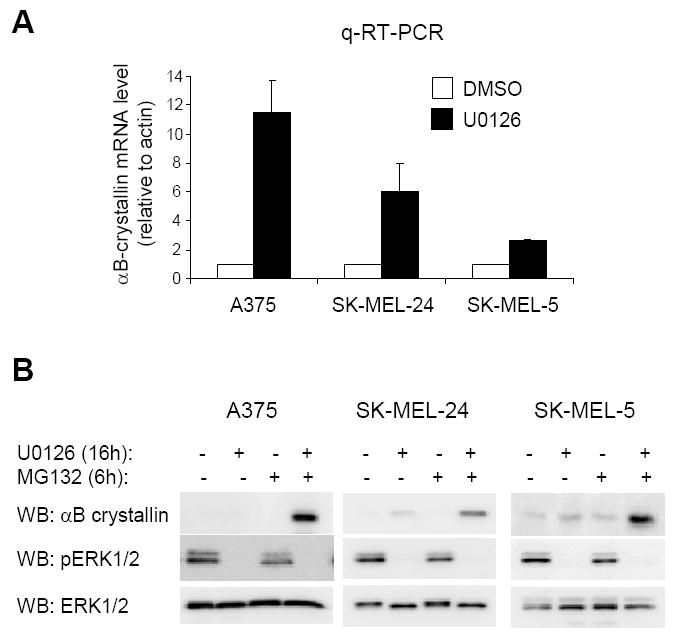
αB-crystallin expression is regulated by both B-RAF signaling and proteasomal degradation. (A) qRT-PCR analysis of αB-crystallin mRNA in U0126 (16 hours) or control (DMSO) treated A375, SK-MEL-24, SK-MEL-5 cells. The graph represents the mean and s.d. for the percentage change in αB-crystallin mRNA level relative to actin from three independent experiments. (B) A375, SK-MEL-24, and SK-MEL-5 cells were treated with 10 μM U0126 for 16 hours followed by 10 μM MG132 treatment for 6 hours. Cell lystates were analyzed by Western blotting for αB-crystallin, pERK1/2 and ERK1/2 (loading control).
Cyclin D1 is regulated by the proteasome in melanoma cells
Since αB-crystallin is implicated in cyclin D1 turnover, we next analyzed whether cyclin D1 levels were regulated by the proteasome in human melanoma cells and melanocytes. Treatment of WM115 and WM793 cells with the proteasomal inhibitor, MG132, led to up-regulation of cyclin D1 (Fig. 4A). Levels of threonine-286 phosphorylated cyclin D1 increased accordingly in MG132-treated cells, indicating that the cyclin D1 phosphorylation pathway is active in melanoma cells. Similarly, cyclin D1 levels were increased in normal human melanocytes (Fig. 4A). Cyclin D1 mRNA abundance was unaltered following the same duration of MG132 (data not shown) and ectopic CMV-driven cyclin D1 was similarly up-regulated following short-term treatment (6 hours) with MG132 (Fig. 4B). ERK1/2-mediated phosphorylation of threonine 286 has been reported (Okabe et al., 2006); however, in MG132-treated melanoma cells, inhibition of MEK did not dramatically inhibit cyclin D1 phosphorylation at threonine 286 (Fig. 4C). These data show that cyclin D1 is proteasomally regulated in human melanoma cells and melanocytes.
Figure 4.
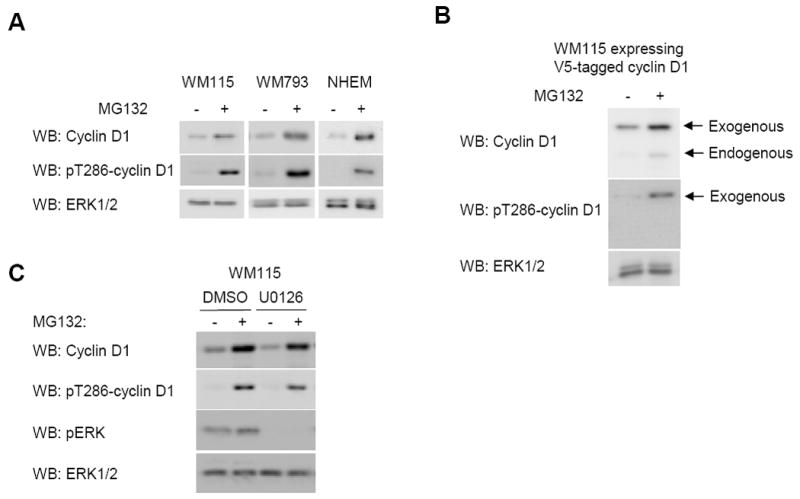
Cyclin D1 is regulated by the proteasome in melanoma cells but threonine 286 phosphorylation is MEK-independent. (A) WM115, WM793, and melanocytes (NHEM) were treated with MG132 for 24 hours. Cell lysates were analyzed by Western blotting for cyclin D1, threonine-286 phosphorylated cyclin D1 (pT286-cyclin D1) and ERK1/2 (loading control). (B) WM115 cells expressing V5-tagged cyclin D1 were treated with MG132 for 24 hours. Cell lysates were analyzed by Western blotting for cyclin D1, pT286-cyclin D1 and ERK1/2. Exogenous and endogenous cyclin D1 are indicated. (C) WM115 cells were treated with MG132 for 6 hours after which U0126 (10 μM) was added for a further 1 hour. Cell lysates were analyzed by Western blotting for pT286-cyclin D1, cyclin D1, pERK1/2 and ERK1/2.
αB-crystallin regulates cyclin D1 turnover in melanocytes
To determine whether αB-crystallin regulated cyclin D1 turnover in human melanocytes, we knocked down αB-crystallin with siRNA and measured cyclin D1 levels in cells treated with cycloheximide to inhibit new protein synthesis. Depletion of αB-crystallin decreased cyclin D1 degradation rates in melanocytes at early time points followed by a return to control levels at later time points (Supplemental Fig. 2). Therefore, we focused our examination on cyclin D1 degradation at earlier time points. Knockdown of αB-crystallin decreased the rate of cyclin D1 degradation in melanocytes during the initial 25 minutes of turnover but did not completely block cyclin D1 turnover (Fig. 5A & 5B). Notably, the t1/2 of cyclin D1 increased from 48 mins to 2 hrs 7 mins in αB-crystallin-depleted melanocytes. These data indicate αB-crystallin expression in melanocytes contributes, in part, to cyclin D1 turnover.
Figure 5.
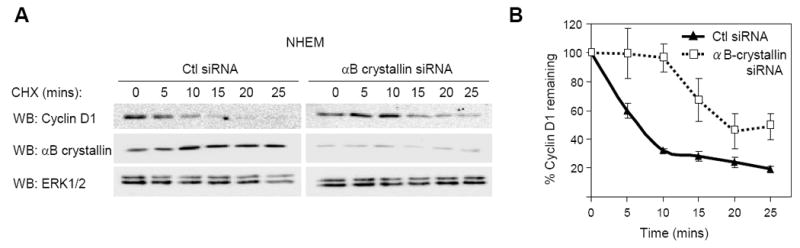
αB-crystallin regulates cyclin D1 turnover in melanocytes. (A) NHEMs were transfected with control or αB-crystallin siRNA. Seventy-two hours post-transfection, serum starved cells were treated with medium containing fresh growth factors to stimulate proliferation for 6 hours following which medium was replaced with serum free medium containing cycloheximide (10 μg/ml) for the indicated times. Cell lysates were analyzed by Western blotting for cyclin D1, αB-crystallin and ERK1/2. (B) The graph represents the mean and s.d. for the percentage change in cyclin D1 protein level relative to ERK1/2 over time from five independent experiments.
αB-crystallin does not regulate basal cyclin D1 turnover in melanoma cells
We also tested whether levels of αB-crystallin altered cyclin D1 turnover in human melanoma cells. We generated doxycycline inducible αB-crystallin expressing melanoma cells and treated these cells with cycloheximide. Induction of wild-type αB-crystallin in WM115 cells did not alter the turnover rate of cyclin D1 over a 6 hour time period (Fig. 6A & 6B). The t1/2 of cyclin D1 was 1 hr 27 min and 1 hr 26 min with or without induction, respectively. Additionally, induction of αB-crystallin did not alter the melanoma cell proliferation (data not shown). Next, we utilized αB-crystallin containing S19D/S45D mutations, a phosphorylation-mimic mutant form of αB-crystallin that has a higher binding affinity for Fbx4 (den Engelsman et al., 2003). Induction of S19D/S45D αB-crystallin also did not alter cyclin D1 turnover rate in WM115 cells (Fig. 6C & 6D). The t1/2 of cyclin D1 was 1 hr 30 min and 1 hr 24 min with or without induction, respectively. To ensure that these results were not limited to WM115 cells, we performed similar experiments in WM793 cells; however, expression of wild-type and S19D/S45D αB-crystallin in WM793 cells did not promote cyclin D1 turnover (Supplemental Fig. 3). Since cyclin D1 turnover depends on the stage of the cell cycle, we synchronized cells by nocodozole treatment and mitotic shake-off and analyzed cyclin D1 turnover at 18 hours after release from nocodozole when cyclin D1 levels were elevated. In synchronized WM793 cells, cyclin D1 turnover rate was not altered by expression of αB-crystallin (Supplemental Fig. 4). These data show that expression of αB-crystallin in melanoma cells is not sufficient to promote cyclin D1 degradation under basal culture conditions.
Figure 6.
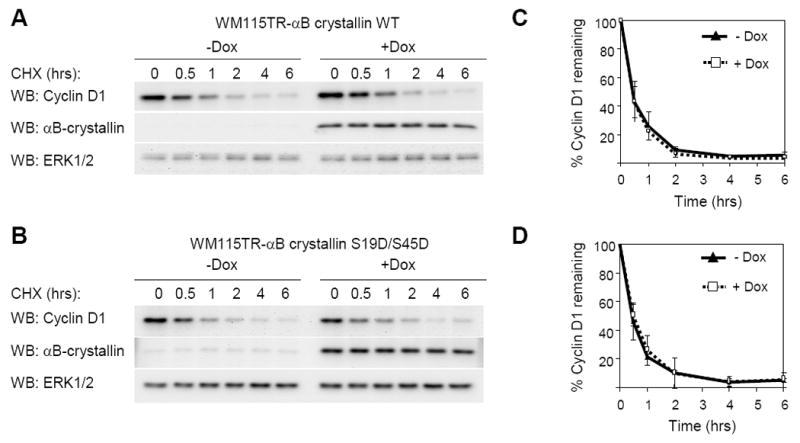
αB-crystallin expression is not sufficient to regulate cyclin D1 turnover in melanoma cells. (A) WM115TR wild type αB-crystallin cells were treated with or without 0.1 μg/ml doxycycline for 72 hours. Cycloheximide (10 μg/ml) was added to cells for indicated times and cell lysates were analyzed by Western blotting for cyclin D1, αB-crystallin and ERK1/2. (B) The same as A, except that WM115TR S19D/S45D αB-crystallin cells were utilized. Graphs represent the mean and s.d. for the percentage change in cyclin D1 protein level relative to ERK1/2 from three independent experiments in (C) wild-type αB-crystallin expressing WM115 cells, and (D) S19D/S45D αB-crystallin expressing WM115 cells.
It has been shown that αB-crystallin functions as a co-factor with Fbx4 in the turnover of cyclin D1 (Lin et al., 2006, den Engelsman et al., 2003) and mutations in FBX4 have been reported in esophageal carcinomas (Barbash et al., 2008). Thus, a possible explanation for the lack of αB-crystallin effect on basal cyclin D1 turnover was that FBX4 was mutated in melanoma cells. We sequenced exon 1 that harbors the majority of reported mutations but FBX4 was wild-type at the reported exon 1 mutation sites in WM115 and WM793 (Supplemental Fig. 5). Additional sequencing did not identify any mutations in the remaining FBX4 exons in WM115.
αB-crystallin regulates cyclin D1 turnover in melanoma cells in the presence of DNA damaging drug
Both cyclin D1 and F-box proteins are known to be regulated under various stress conditions including DNA damage (Alao, 2007, Santra et al., 2009). To test whether αB-crystallin regulates cyclin D1 turnover in the presence of DNA damage in melanoma cells, we treated αB-crystallin over-expressing WM115 cells with etoposide before the cycloheximide treatment and cyclin D1 analysis. Etoposide is a DNA topoisomerase II inhibitor that leads to accumulation of DNA strand breaks in cells (Baldwin and Osheroff, 2005). Following overnight treatment, the turnover of cyclin D1 was slightly slower than in non-etoposide treated cells. Over-expression of wild-type αB-crystallin modestly increased cyclin D1 turnover rates in etoposide-treated WM115 cells (Fig. 7A & 7B). The t1/2 of cyclin D1 decreased from 2 hrs 12 min to 1 hr 48 min after induction of wild-type αB-crystallin. However, S19D/S45D αB-crystallin over-expression more efficiently accelerated cyclin D1 turnover in etoposide-treated cells (Fig. 7C & 7D). The t1/2 of cyclin D1 decreased from 1 hr 53 min to 1 hr 14 min after induction of S19/S45D αB-crystallin. These data show that expression of αB-crystallin in melanoma cells promotes cyclin D1 degradation in the presence of DNA damaging reagents.
Figure 7.
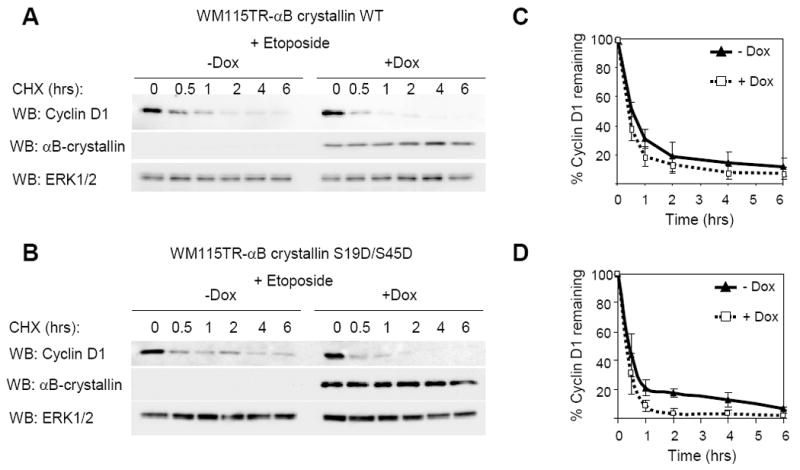
αB-crystallin expression regulates cyclin D1 turnover in melanoma cells in the presence of DNA damaging drug etoposide. (A) WM115TR wild type αB-crystallin cells were treated with or without 0.1 μg/ml doxycycline for 56 hours, followed by treatment of 25 μg/ml etoposide for 16 hours. Next, cycloheximide (10 μg/ml) was added to cultures and cells lysed after the indicated times for indicated times. Cell lysates were analyzed by Western blotting for cyclin D1, αB-crystallin and ERK1/2. (B) The same as A, except that WM115TR S19D/S45D αB-crystallin cells were utilized. Graphs represent the mean and s.d. for the percentage change in cyclin D1 protein level relative to ERK1/2 from three independent experiments in (C) wild-type αB-crystallin expressing WM115 cells, and (D) S19D/S45D αB-crystallin expressing WM115 cells.
Discussion
Expression of αB-crystallin has been described as being up-regulated in some human cancers but down-regulated in others (Chelouche-Lev et al., 2004, Moyano et al., 2006, Lin et al., 2006, Mineva et al., 2005). Its expression in melanoma remains unknown. In this study, we demonstrate that αB-crystallin is highly expressed in primary melanocytes, whereas its expression level is down-regulated in human melanoma cell lines. B-RAF-MEK signaling, which is elevated in the majority of melanomas, down-regulates αB-crystallin in mutant B-RAF-expressing melanomas at the mRNA level. Our findings are not limited to melanoma cells since αB-crystallin expression can be regulated by B-RAF-MEK signaling in papillary and anaplastic thyroid carcinoma cells, which also frequently contain B-RAFV600E mutations (Kimura et al., 2003, Mineva et al., 2005). We also find that B-RAF-MEK signaling is not the only mechanism leading to αB-crystallin down-regulation. αB-crystallin is also controlled by the proteasomal degradation pathway in a manner that appears independent of ERK1/2 signaling. Proteasomal regulation of αB-crystallin was not detected in all cell lines. Thus, expression of αB-crystallin requires inhibition of the B-RAF-MEK pathway and, in a subset of cell lines, proteasomal degradation additionally needs to be repressed. B-RAF activation in melanocytes was not sufficient to down-regulate αB-crystallin; thus, alterations in additional pathways, possibly involved in proteasomal regulation, are likely necessary for αB-crystallin down-regulation in melanocytes.
Down-regulation of αB-crystallin in melanoma cells through multiple mechanisms suggests an important cellular function. We explored the possibility that αB-crystallin down-regulation contributed to regulation of cyclin D1 levels. αB-crystallin contributes to cyclin D1 turnover in primary human melanocytes; however, since melanocytes depleted of αB-crystallin are still able to degrade cyclin D1, additional mechanisms clearly exist. Enhancing the expression of αB-crystallin in melanoma cells under normal cell culture conditions did not increase the cyclin D1 turnover suggesting that down-regulation of αB-crystallin is not the limiting component of cyclin D1 degradation. Besides αB-crystallin expression level, other components of the cyclin D1 degradation pathway might be altered in melanoma cells. The finding that MG132 treatment induces accumulation of threonine 286 phosphorylated cyclin D1 suggests that the targeting of cyclin D1 for recognition by SCF complexes remains active in these cells. However, we cannot rule out that cyclin D1 may be phosphorylated at sub-optimal levels to observe effects on its turnover following αB-crystallin expression. Given the recent identification of Fbx4 mutations that inhibit SCF complex dimerization in esophageal tumors, an alternative possibility was that Fbx4 is mutated and non-functional in cutaneous melanomas. Arguing against this possibility, our initial studies showing that Fbx4 is wild-type in all exons in WM115 cells and does not harbor exon 1 mutations in WM793 cells. A further possible mechanism is that the overexpressed αB-crystallin and Fbx4 lack the signal to be activated and recognize cyclin D1 for degradation in basal conditions, but exposure to DNA damaging reagents initiates signaling that is required for Fbx4-αB-crystallin activation. Our results showing overexpressed αB-crystallin promoted cyclin D1 turnover following DNA damage supports this hypothesis. However, the underlying mechanism of how DNA damage regulates this process needs further investigation. Additionally, given its role as a heat shock protein in other cell systems, it is likely that αB-crystallin may perform cyclin D1 independent functions in melanocytes and melanomas.
While Fbx4 has been shown to act in concert with αB-crystallin, it is possible that a distinct F-box protein recognizes phosphorylated cyclin D1 in melanoma cells. Fbxw8, β-TrCP and recently Fbxo31 have also been reported as E3 ubiquitin ligases for cyclin D1 (Okabe et al., 2006, Wei et al., 2008, Santra et al., 2009). The expression level of Fbxw8 in melanoma is unknown and whether it controls cyclin D1 turnover in melanoma cells warrants further investigation. β-TrCP expression is enhanced in melanoma cells (Liu et al., 2007), making it less likely to be the main E3 ubiquitin ligase for cyclin D1. Fbxo31 has been shown to be expressed at very low levels in the melanoma cell line SK-MEL-28, but upon γ-irradiation, accumulates rapidly and controls cyclin D1 turnover (Santra et al., 2009). However, similar to αB-crystallin in our studies herein, Fbxo31 seems play minimal role in regulating cyclin D1 turnover under normal conditions.
In summary, our results show that αB-crystallin is highly expressed in melanocytes, and is down-regulated in melanoma cells. Multiple mechanisms are involved in down-regulating αB-crystallin in melanoma cells. The mRNA level of αB-crystallin is dependent on B-RAF signaling in melanoma cells, and αB-crystallin protein is rapidly turnover by proteasomal degradation. αB-crystallin plays a role in regulating cyclin D1 turnover in melanocytes. αB-crystallin is not the limiting factor for cyclin D1 turnover in melanoma cells in normal conditions; however, its expression of αB-crystallin promotes cyclin D1 degradation after DNA damage in melanoma cells.
Materials and Methods
Cell culture
Normal human epidermal melanocytes were isolated from neonatal foreskins, as previously described (Conner et al., 2003). Human melanoma SK-MEL cell lines and A375 cells were purchased from ATCC (Manassas, VA) and other melanoma cell lines were kindly donated by Dr. Meenhard Herlyn (Wistar Institute, Philadelphia). Papillary (NPA) and anaplastic thyroid (ARO) carcinoma cell lines were kindly supplied by Dr. Jeffrey Knauf (Memorial Sloan Kettering Cancer Center, NY). Human melanoma cell lines, Sbcl2, WM35, WM115, 1205-Lu, WM278, WM793, WM983A and SK-MEL-28, were cultured in modified chemically defined medium (MCDB) 153 medium containing 20% Leibovitz L-15 medium, 2% (v/v) FBS, 0.2% (w/v) sodium bicarbonate, and 5 μg/ml insulin. Melanoma cell lines, SK-MEL-5 and SK-MEL-24 were maintained in MEM containing Earle’s salts, 2 mM glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5% (w/v) sodium bicarbonate, and 10% FBS. A375 cells were maintained in DMEM containing 10% FBS. NPA and ARO were cultured in RPMI 1640 supplemented with 10% FBS (Namba et al., 2003). All melanoma and thyroid cancer cell lines harbor mutant B-RAF with the exception of Sbcl2 that are wild-type for B-RAF but carry an N-RAS mutation (Davies et al., 2002).
Inhibitors
The MEK inhibitor, U0126 was purchased from Cell Signaling Technology (Danvers, MA). MG132 was purchased from Calbiochem (La Jolla, CA). Cycloheximide and etoposide were obtained from Sigma (St. Louis, MO). Tetracycline, blasticidin and zeocin were purchased from Invitrogen (Carlsbad, MA).
siRNA transfection
Melanoma cells (2 ×105) were transfected with siRNA (Dharmacon, Lafayette, CO, USA) at a concentration of 25 nM using Oligofectamine (Invitrogen). Melanocytes (2 ×105) were transfected using Lipofectamine 2000 (Invitrogen). The sequences of the individual siRNAs used were αB-crystallin: GAAGAGAAGCCUGCUGUCAUU; B-RAF#1: ACAGAGACCUCAAGAGUAAUU; B-RAF#7: GAGAUGAUCAAACUUAUAGUU; B-RAF#11: AAGUGGCAUGGUGAUGUGGCA. A non-targeting siRNA from Dharmacon was used as a control. Cells were transfected for 4 hours in serum free medium following which 1.5 ml of cell culture medium was added. Cells were harvested after 72 hours, unless otherwise indicated.
Western blotting
Melanoma and melanocyte cell lysates were analyzed for protein expression, as previously described (Bhatt et al., 2007). The following primary antibodies were utilized: αB-crystallin (SPA-222; Stressgen), cyclin D1 (DCS-6; BD-Pharmingen), phospho-cyclin D1 Thr286 (2921; Cell Signaling), B-RAF (F-7; Santa Cruz), ERK1/2 (K-23; Santa Cruz), and pERK1/2 (197G2; Cell Signaling). Western blots were developed using SuperSignal chemiluminescent substrate (Pierce, Rockford, IL, USA) and quantitated with a Versadoc Imager and Quantity-One software (BioRad, Hercules, CA).
qRT-PCR
Seventy-two hours post-knockdown, total RNA was extracted from melanoma cells using the Versagene RNA isolation kit (Gentra System, Minneapolis, MN, USA). RNA (1 μg) was reverse transcribed and 1/20th of the resulting complementary DNA (cDNA) was utilized to detect mRNA abundance with primers for actin (forward: TACCTCATGAAGATCCTCACC, reverse: TTTCGTGGATGCCACAGGAC), αB-crystallin (forward: TGGTTTGACACTGGACTCTC, reverse: ATGTTCATCCTGGCGCT). Product sizes were 268 bp and 156 bp for actin and αB-crystallin, respectively. Specificity was confirmed by melt curve analysis. Reactions were performed using SYBR Green mix and MyiQ real-time PCR detection system (BioRad). Relative mRNA levels were calculated using the Comparative Ct method (ΔCt) (Pfaffl, 2001).
Inducible cell lines
Wild type and S19/45D-αB-crystallin cDNAs were kind gifts from Dr. Wilbert Boelens (Radboud University, Netherlands) (den Engelsman et al., 2003). cDNAs were cloned into pENTR/3C vector and, after verification by DNA sequencing, were recombined with pLenti4/TO/DEST using the LR Clonase II kit (Invitrogen). The generated pLenti4/TO/WT-αB-crystallin and pLenti4/TO/S19/45D-αB-crystallin were transfected along with the packaging plasmids pLP1, pLP2, and pLP/VSVG into 293FT cells using Fugene HD (Roche Diagnostic Co., Indianapolis, IN). Harvested lentiviruses were used to infect cell lines expressing the tetracycline repressor (WM115TR and WM793TR) (Hu and Aplin, 2008). Infected cells were selected with 50 μg/ml zeocin for 2 weeks. Doxycyclin was used to induce gene expression at a final concentration of 0.1 μg/ml.
Calculation of t1/2
The graph of cyclin D1 degradation was plotted on exponential scale. The exponential curve and formula was obtained with Microsoft Excel software. The t1/2 was calculated according to
Supplementary Material
Acknowledgments
Grant support: National Institutes of Health grants, GM067893 and CA125103 to A.E.A, and a National Cancer Center pre-doctoral fellowship to R.H. We thank Dr. Wilbert Boelens (Radboud University, Netherlands) for αB-crystallin cDNAs, Dr. Meenhard Herlyn (Wister Institute, Philadelphia) for the melanoma cell lines and Dr. Jeffrey Knauf (Memorial Sloan Kettering Cancer Center, New York) for the thyroid cancer lines. We thank the Cancer Genomic facility in the Kimmel Cancer Center for DNA sequencing.
Abbreviations
- qRT-PCR
real-time quantitative RT-PCR
- SCF
Skp1/Cullin/F-box protein
- siRNA
short-interfering RNA
References
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:1–16. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, Lu F, Rustgi AK, Diehl JA. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008;14:68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt KV, Hu R, Spofford LS, Aplin AE. Mutant B-RAF signaling and cyclin D1 regulate Cks1/S-phase kinase-associated protein 2-mediated degradation of p27(Kip1) in human melanoma cells. Oncogene. 2007;26:1056–1066. doi: 10.1038/sj.onc.1209861. [DOI] [PubMed] [Google Scholar]
- Bhatt KV, Spofford LS, Aram G, Mcmullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;12:3459–3471. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Chelouche-Lev D, Kluger HM, Berger AJ, Rimm DL, Price JE. alphaB-crystallin as a marker of lymph node involvement in breast carcinoma. Cancer. 2004;100:2543–2548. doi: 10.1002/cncr.20304. [DOI] [PubMed] [Google Scholar]
- Conner SR, Scott G, Aplin AE. Adhesion-dependent activation of the ERK1/2 cascade is by-passed in melanoma cells. J Biol Chem. 2003;278:34548–34554. doi: 10.1074/jbc.M305797200. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Den Engelsman J, Keijsers V, De Jong WW, Boelens WC. The small heat-shock protein alphaB-crystallin promotes FBX4-dependent ubiquitination. J Biol Chem. 2003;278:4699–4704. doi: 10.1074/jbc.M211403200. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Guo Y, Yang K, Harwalkar J, Nye JM, Mason DR, Garrett MD, Hitomi M, Stacey DW. Phosphorylation of cyclin D1 at Thr 286 during S phase leads to its proteasomal degradation and allows efficient DNA synthesis. Oncogene. 2005;24:2599–2612. doi: 10.1038/sj.onc.1208326. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Hu R, Aplin A. Skp2 regulates G2/M progression in a p53-dependent manner. Mol Biol Cell. 2008;19:4602–4610. doi: 10.1091/mbc.E07-11-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Suresh Kumar KG, Yu D, Molton SA, Mcmahon M, Herlyn M, Thomas-Tikhonenko A, Fuchs SY. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007;26:1954–1958. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineva I, Gartner W, Hauser P, Kainz A, Loffler M, Wolf G, Oberbauer R, Weissel M, Wagner L. Differential expression of alphaB-crystallin and Hsp27-1 in anaplastic thyroid carcinomas because of tumor-specific alphaB-crystallin gene (CRYAB) silencing. Cell Stress Chaperones. 2005;10:171–184. doi: 10.1379/CSC-107R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyano JV, Evans JR, Chen F, Lu M, Werner ME, Yehiely F, Diaz LK, Turbin D, Karaca G, Wiley E, et al. Alpha B-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest. 2006;116:261–270. doi: 10.1172/JCI25888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T, Yamashita S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab. 2003;88:4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- Okabe H, Lee SH, Phuchareon J, Albertson DG, Mccormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Theodoras AM, Tam SW, Draetta GF. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994;8:1627–1639. doi: 10.1101/gad.8.14.1627. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy K, Dejesus E, Linnenbach AJ, Kraj B, Kornreich DL, Rendle S, Elder DE, Herlyn M. Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res. 1997;7:S35–42. [PubMed] [Google Scholar]
- Sauter ER, Yeo U-C, Von Stemm A, Zhu W, Litwin S, Tichansky DS, Pistritto G, Nesbit M, Pinkel D, Herlyn M, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62:3200–3206. [PubMed] [Google Scholar]
- Spruck C, Strohmaier H, Watson M, Smith APL, Ryan A, Krek W, Reed SI. A CDK-independent function of mammalian Cks1: targeting of SCFSkp2 to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- Wei S, Yang H-C, Chuang H-C, Yang J, Kulp SK, Lu P-J, Lai M-D, Chen C-S. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008;283:26759–26770. doi: 10.1074/jbc.M802160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.