

Abstract
Neuroblastoma is the most common extracranial solid tumor of childhood. Focal adhesion kinase (FAK) is an intracellular kinase that regulates both cellular adhesion and apoptosis. FAK is overexpressed in a number of human tumors including neuroblastoma. Previously, we have shown that the MYCN oncogene, the primary adverse prognostic indicator in neuroblastoma, regulates the expression of FAK in neuroblastoma. In this study, we have examined the effects of FAK inhibition upon neuroblastoma using a small molecule [1,2,4,5-benzenetetraamine tetrahydrochloride (Y15)] to inhibit FAK expression and the phosphorylation of FAK at the Y397 site. Utilizing both non-isogenic and isogenic MYCN+/MYCN− neuroblastoma cell lines, we found that Y15 effectively diminished phosphorylation of the Y397 site of FAK. Treatment with Y15 resulted in increased detachment, decreased cell viability and increased apoptosis in the neuroblastoma cell lines. We also found that the cell lines with higher MYCN are more sensitive to Y15 treatment than their MYCN negative counterparts. In addition, we have shown that treatment with Y15 in vivo leads to less tumor growth in nude mouse xenograft models, again with the greatest effects seen in MYCN+ tumor xenografts. The results of the current study suggest that FAK and phosphorylation at the Y397 site plays a role in neuroblastoma cell survival, and that the FAK Y397 phosphorylation site is a potential therapeutic target for this childhood tumor.
Keywords: neuroblastoma, focal adhesion kinase, FAK, MYCN, pediatric, Y397, apoptosis
Introduction
Neuroblastoma is the most common extracranial solid tumor of childhood. This tumor of neural crest origin is responsible for over 15% of pediatric cancer deaths.1 Despite recent advances in chemotherapeutic and surgical care, this tumor continues to carry a dismal prognosis for children presenting with advanced or metastatic disease, with a survival of only 18–30%.2 The primary adverse prognostic factor for neuroblastoma is amplification of the MYCN oncogene.3, 4 Amplification of MYCN has been shown to be associated with increased proliferation and cell survival in neuroblastoma, and knockdown of MYCN with siRNA results in cell death and apoptosis in neuroblastoma cell lines. 5–7
Focal adhesion kinase (FAK) is a nonreceptor protein tyrosine kinase that localizes to focal adhesions, and controls a number of cell signaling pathways including proliferation, viability and survival. 8–11 Tyrosine 397 is an autophosphorylation site of FAK and is important in these downstream signaling functions. Phosphorylation of FAK at the tyrosine 397 (Y397) site results in a high affinity binding site for the SH2 domain of the Src family kinases that results in the activation of pathways leading to cellular proliferation and survival. 12, 13 In addition, Y397 is also a binding site for PI3 kinase, resulting in activation of a number of inhibitor-of-apoptosis proteins.14 The inhibition of FAK activation has been found to affect a number of cellular pathways. FAK antisense oligonucleotides or a dominant-negative FAK protein (FAK-CD) has been shown to cause decreased growth in human breast cancer cells and melanoma cells.15–18 Silencing FAK expression with small interfering RNAs resulted in decreased migration of lung cancer cells and glioblastoma cells. 19, 20 Finally, small molecule inhibitors of FAK kinase have been reported in the literature. These inhibitors were able to increase apoptosis in breast cancer cells in vitro and to decrease the in vivo growth of gliomas and ovarian tumors.21–23 Recently, a small molecule FAK inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15), has been reported to inhibit the growth of breast and pancreatic cancers. 24, 25
Initial studies from our laboratory have revealed that both the abundance of FAK mRNA and the expression of FAK protein are significantly increased in aggressive human neuroblastoma tumors. 26 In addition, we demonstrated that MYCN regulates the expression of FAK through its promoter in human neuroblastoma cell lines and that MYCN+ cell lines have increased FAK expression. 27 Since FAK is overexpressed in MYCN+ neuroblastoma cell lines, we hypothesized that inhibition of FAK may result in decreased cell viability and apoptosis in these cells. In the current study, we show that Y15 treatment leads to decreased cellular viability, increased cellular detachment and increased apoptosis that is more marked in the neuroblastoma cell lines with greater MYCN. In addition, we show that Y15 inhibits growth of MYCN+ neuroblastoma tumors in vivo. We believe that targeting FAK and the phosphorylation of the Y397 site of FAK may be an effective strategy when designing therapeutic interventions for aggressive neuroblastomas.
Results
1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) causes dephosphorylation of FAK in MYCN+ (Tet−) / MYCN− (Tet+) isogenic neuroblastoma cells
Previous experiments from our laboratory have shown that FAK is associated with MYCN expression in human neuroblastomas.26, 27 In addition, previous data showed that MYCN expressing neuroblastoma cell lines may be more dependent upon FAK for survival than non-MYCN expressing neuroblastoma cell lines.27 Therefore, we wished to define the biologic significance of interruption of FAK function (phosphorylation) in human neuroblastoma cell lines with varying status of MYCN. To perform these studies, we utilized an isogenic neuroblastoma cell line that has a tetracycline repressible MYCN expression vector; that is, when tetracycline is present, MYCN is silenced (MYCN−, Tet+), and MYCN is expressed when tetracycline is removed from the media (MYCN+, Tet−).
As demonstrated in Fig. 1A, the MYCN+ (Tet−) cells have increased MYCN, total FAK and increased phosphorylation of FAK Y397 when compared to the isogenic MYCN− (Tet+) cell line. To inhibit phosphorylation of Y397 FAK, we utilized a small molecule, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15), which has been shown to inhibit the level of phosphorylation of the tyrosine 397 site of FAK. We treated the MYCN+ (Tet−) / MYCN− (Tet+) cells with Y15 at various concentrations and performed Western blotting with Y397 FAK specific antibody (Fig. 1B). We found that the Y15 compound decreased the phosphorylation of Y397 FAK. At 1 μM concentration, there was a greater decrease in the phosphorylation of Y397 FAK in the MYCN+ cells than in the MYCN− cells (Fig. 1B). At Y15 10 μM concentration, there is almost a complete loss of phosphorylation of Y397 FAK in both the MYCN+ and MYCN− cells at (Fig. 1B).
Fig. 1.
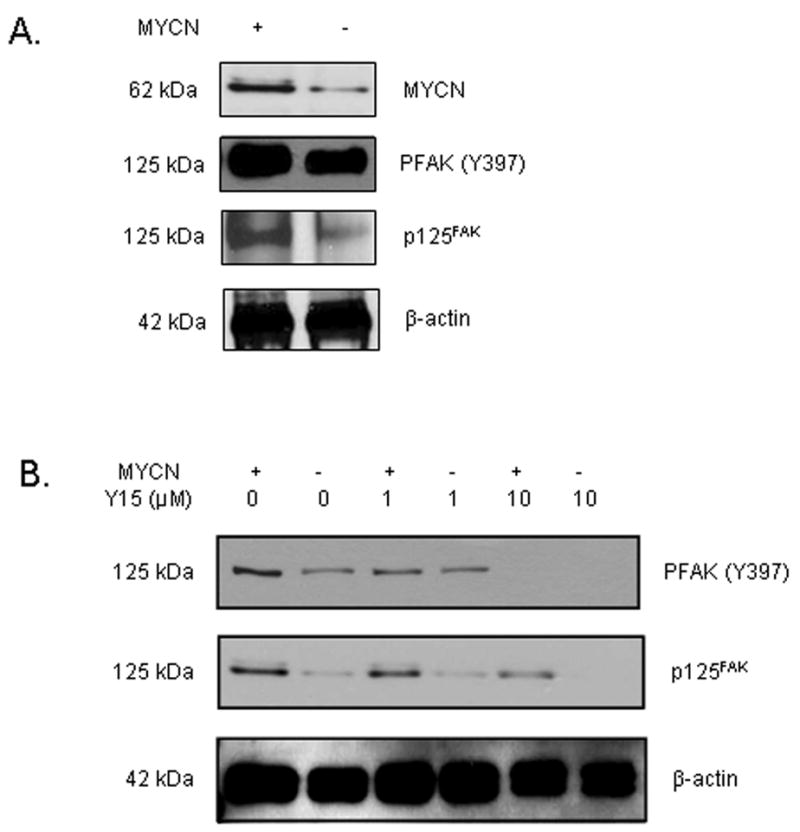
The phosphorylation of Y397 FAK is decreased after treatment of MYCN+ (Tet−)/MYCN− (Tet+) isogenic neuroblastoma cell lines with 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15). A, The isogenic MYCN+ (Tet−) and MYCN− (Tet+) neuroblastoma cell lines were examined with immunoblotting. There is increased MYCN, FAK and phosphorylation of Y397 FAK in the MYCN+ (Tet−) compared to the isogenic MYCN− (TET+) cells. B, The isogenic MYCN+ (Tet−) and MYCN− (Tet+) cells were treated with various concentrations of Y15 and cell lysates were examined with immunoblotting. There was a decrease in phosphorylation of Y397 FAK with Y15 treatment. At 1 μM concentration of Y15, there was a greater decrease in phosphorylation of Y397 FAK in the MYCN+ cells. At concentrations of 10 μM, Y15 treatment results in almost complete loss of Y397 phosphorylation in both cell lines.
1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment causes cellular detachment in MYCN+ (Tet−) / MYCN− (Tet+) isogenic neuroblastoma cell lines
Next, we wished to determine the biologic effects of 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) on human neuroblastoma cells with varying FAK expression. Since FAK and the phosphorylation of Y397 FAK are known to play an important role in cellular attachment, we treated the isogenic MYCN+ (Tet−) and MYCN− (Tet+) neuroblastoma cell lines with Y15 and measured the percentage of cells that were detached after treatment. As early as 24 hours after treatment with Y15, there was significantly more detachment in the MYCN+ (Tet−) cells than in the MYCN− (Tet+) cells (23 ± 2% vs. 5.6 ± 2.6%, mean ± SEM, p=0.03, Fig. 2A). These data correspond to the decreased phosphorylation of Y397 FAK noted in the MYCN+ (Tet−) cells after Y15 treatment compared to the MYCN− (Tet+) cells (Fig. 1B), implying that MYCN+ cells are more dependent upon FAK as a survival mechanism that their isogenic MYCN− counterparts.
Fig. 2.
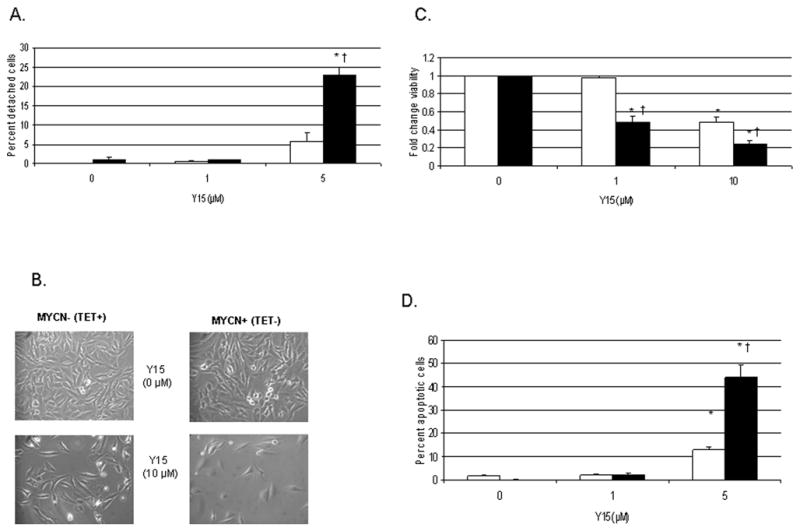
1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment causes cellular detachment, decreased viability and increased apoptosis in human neuroblastoma cell lines. We treated the MYCN+ (Tet−)/MYCN− (Tet+) cells with 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) to test its effects upon human neuroblastoma cells relative to MYCN status. A, Cellular detachment is measured by counting the number of detached cells and expressing it as a ratio to the total number of cells present, that is detached plus attached cells (mean ± SEM). MYCN− (Tet+) cells are represented by the open bars (□) and MYCN+ (Tet−) cells are represented by the black bars (■). After only 24 hours of treatment with Y15, there is a significant increase in the number of MYCN+ (■) neuroblastoma cells that are detached compared to the MYCN− (□) cells [23 ± 2% vs. 5.6 ± 2.6%; *p≤0.01 control vs. Y15; †p≤0.05 MYCN+ (■) vs. MYCN− (□)]. B, MYCN+ (Tet−) and MYCN− (Tet+) cells are treated with Y15 for 48 hours and cells are examined by phase contrast microscopy. There are significantly fewer cells present after Y15 treatment, with the decrease in cell number being more marked in the MYCN+ (Tet−) cell line (bottom right panel). C, MYCN+ (Tet−) (■) and MYCN− (Tet+) (□) cells are treated with Y15 for 48 hours and viability is measured with MTT assay, expressed as fold change (mean ± SEM). There is a significant decrease in viability in both cell lines compared to non-treated controls, but the decrease is more pronounced in the MYCN+ (■) cell line, which reaches 50% decrease in viability at 1 μM treatment [*p≤0.01 control vs. Y15; †p≤0.05 MYCN+ (■)vs. MYCN− (□)]. D, MYCN+ (Tet−) (■) and MYCN− (Tet+) (□) cells are treated with Y15 for 24 hours and Hoechst staining is used to detect apoptosis, expressed as a percentage of apoptotic cells to non-apoptotic cells (mean ± SEM). Y15 resulted in a significant increase in apoptosis in both cell lines after only 24 hours of treatment, and similar to the results with viability, Y15 had a greater effect upon apoptosis in the MYCN+ (■) cell line compared to the MYCN− (□) cells [44.3 ± 4.8% vs. 13 ± 1.2%, MYCN+ (■) vs. MYCN− (□) at Y15 5 μM; *p≤0.01 control vs. Y15; †p≤0.01 MYCN+ (■) vs. MYCN− (□)].
Inhibition of Y397 FAK with 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) leads to decreased cellular viability and increased cellular apoptosis in MYCN+ (Tet−) / MYCN− (Tet+) isogenic neuroblastoma cell lines
FAK and phosphorylation at the Y397 site of FAK are also important for cellular survival. Therefore, we wished to determine the effects Y15 upon viability and apoptosis in neuroblastoma cell lines. Using phase contrast microscopy to evaluate the cells, we found that there was a decrease in neuroblastoma cell number following Y15 treatment, and the amount of decrease was related to the MYCN status of the cells (Fig. 2B). After 48 hours of Y15 treatment, there was an obvious change in cell numbers, most marked in the MYCN+ (Tet−) cells (Fig. 2B, bottom right panel).
Cell viability was measured with MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assays. We found that after 48 hours of 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment, there is a significant decrease in viability in both the MYCN+ (Tet−) and MYCN− (Tet+) cell lines compared to non-treated controls (Fig. 2C), but the decrease is more pronounced in the MYCN+ (Tet−) cell line. The MYCN+ (Tet−) cells reached 50% of their viability at 1 μM Y15, versus the 10 μM Y15 required for the MYCN− (Tet+) cell line to reach 50% (0.49 ± .06 vs. 0.98 ± .02, MYCN+ (Tet−) vs. MYCN− (Tet+) at Y15 1 μM, p=0.01) (Fig. 2C).
We next examined the effects of Y15 upon neuroblastoma apoptosis in these isogenic cell lines. Using Hoechst staining, we found that treatment with Y15 resulted in a significant increase in apoptosis in both the MYCN+ (Tet−) and MYCN− (Tet+) cell lines after only 24 hours of treatment (Fig. 2D). In addition, similar to the results with viability, Y15 had a greater effect upon apoptosis in the MYCN+ (Tet−) cell line compared to the MYCN− (Tet+) cells (44.3 ± 4.8% vs. 13 ± 1.2%, MYCN+ (Tet−) vs. MYCN− (Tet+) at Y15 5 μM, p=0.003). These data demonstrate the effects of Y15 upon the viability and survival of neuroblastoma cells, and show that the decrease in viability and increase in apoptosis are more marked in the MYCN+ (Tet−) cells with more FAK and phosphorylation of Y397 FAK.
1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment leads to decreased phosphorylation of Y397 FAK, cellular detachment and viability, and increased apoptosis in other neuroblastoma cell lines
Since the MYCN+ (Tet−) / MYCN− (Tet+) isogenic cell lines are not tumorigenic in a nude mouse xenograft model, we began investigations with other neuroblastoma cell lines with differing MYCN status for our future plans for in vivo experiments. We treated SK-N-AS (non-amplified MYCN) and SK-N-BE(2) (amplified MYCN) human neuroblastoma cell lines (non-isogenic) with 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) to determine what biologic effects that it may have upon these cell lines. Western blotting confirmed that these two neuroblastoma cell lines differ in MYCN, total FAK, and phosphorylation of Y397 FAK, with the SK-N-BE(2) (MYCN+) cell line having more total FAK and increased phosphorylated Y397 FAK (Fig. 3A). Treatment with Y15 leads to cellular detachment in both cell lines, but the SK-N-BE(2) (MYCN+) cells are more sensitive to Y15 treatment than the SK-N-AS (MYCN−) cells (Fig. 3B). Y15 treatment also leads to decreased cell viability (Fig. 3C) which are most marked in the MYCN+ cell line. Western blotting was used to detect FAK and the phosphorylation of Y397 FAK. In the SK-N-AS (MYCN−) cell line, Y15 treatment does result in a loss of Y397 FAK phosphorylation at 25 μM (Fig. 3D, second panel), and at this concentration, there is biochemical evidence of apoptosis with a decrease in total PARP and an increase in cleaved PARP (Fig. 3D, top panel). For the SK-N-BE(2) (MYCN+ ) cell line, Y15 treatment also leads to a loss of Y397 FAK phosphorylation (Fig. 3E), but these changes are seen at a lower concentration (10 μM) than the SK-N-AS (MYCN−) cell line (Fig. 3D). In addition, at only 10 μM Y15 treatment, there is evidence that apoptosis is occurring with a loss of total PARP and an increase in cleaved PARP in the SK-N-BE(2) cell line (Fig. 3E). Therefore, Y15 results in a decrease in FAK and Y397 FAK phosphorylation, an increase in cellular detachment, a decrease in cellular viability and an increase in apoptosis in both SK-N-AS and SK-N-BE(2) human neuroblastoma cell lines, but these effects are most marked in the SK-N-BE(2) (MYCN+) cells. These findings suggest that the SK-N-BE(2) cell line relies more upon FAK and the phosphorylation of Y397 FAK as a survival signal than the non-isogenic, SK-N-AS (MYCN−) cells.
Fig. 3.

1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment leads to decreased phosphorylation of Y397 FAK, cellular detachment and viability, and increased apoptosis in SK-N-AS and SK-N-BE(2) neuroblastoma cell lines. A, The SK-N-BE(2) (MYCN+) neuroblastoma cells have more total FAK expression and more phosphorylation of Y397 FAK that the SK-N-AS (MYCN−) cell line as shown by immunoblotting. B, Detachment is measured by counting the detached cells and expressing as a ratio of total cells present, that is, detached plus attached cells (mean ± SEM). SK-N-AS (MYCN−) cells are represented by the open bars (□) and SK-N-BE(2) (MYCN+) cells are represented by the black bars (■). Treatment with Y15 leads to cellular detachment in both cell lines, but the SK-N-BE(2) (MYCN+) (■) cells are more sensitive to treatment and have more detachment than the SK-N-AS (MYCN−) (□) cells [*p≤0.01 control vs. Y15; †p≤0.05 SK-N-AS (□) vs. SK-N-BE(2) (■)]. C, MTT assay for cell viability shows that Y15 treatment leads to decreased cell viability, and again, the SK-N-BE(2) (■) cell line is more sensitive to Y15 treatment, reaching a 50% decrease in viability at only 10 μM Y15 compared to the 25 μM required for the SK-N-AS (□) cells to reach 50% viability (*p≤0.01 control vs. Y15). D, Western blotting for evaluation of FAK and apoptosis in the SK-N-AS (MYCN−) cell line reveals some loss of Y397 FAK phosphorylation with Y15 treatment at 25 μM (second panel). In addition, at this highest concentration, there is biochemical evidence of apoptosis with a decrease in total PARP and an increase in cleaved PARP (top panel). E, Western blotting for evaluation of FAK and apoptosis in the SK-N-BE(2) (MYCN+ ) cell line reveals a marked loss of Y397 FAK phosphorylation with Y15 treatment at 10 μM with significant loss of total PARP and increase in cleaved PARP (top panel) at 10 μM as biochemical evidence of apoptosis. These data correspond to the detachment and viability data showing that the SK-N-BE(2) (MYCN+) cell line is more sensitive to inhibition of FAK phosphorylation than the SK-N-AS (MYCN−) cell line.
Next, we chose a second isogenic MYCN overexpressing neuroblastoma cell line. The parent cell line, SH-EP, is a MYCN non-amplified cell line. This cell line has been stably transfected with a MYCN overexpressing vector and has been labeled as WAC2 cell line. These cell lines have been extensively described previously (29). Western blotting confirmed that these two neuroblastoma cell lines differ in MYCN, total FAK, and Y397 FAK, with the WAC2 (MYCN+) cell line having more MYCN, total FAK and more phosphorylated Y397 FAK than the SH-EP (MYCN−) cell line (Fig. 4A). Treatment with Y15 leads to decreased cell viability as measured by trypan blue exclusion, and these effects are noted at a much lower concentration in the WAC2 (MYCN+) cell line. The WAC2 cells reach 50% viability at only 2 μM concentration of Y15. The SH-EP cell line required over twice the concentration of Y15 to reach a 50% decrease in viability (Fig. 4B). Treatment with Y15 leads to loss of Y397 FAK phosphorylation in the WAC2 cell line at 2.5 μM and in the SH-EP cell line at 5 μM (Fig. 4C). Fig. 4D demonstrates apoptotic cells as noted on Hoechst stain, with condensation of nuclei (white arrows). These data are presented in quantitative form in Fig. 4E. Y15 treatment of SH-EP (MYCN−) cell lines does result in apoptosis, but requires twice the concentration of Y15 than do the isogenic WAC2 (MYCN+) cells to achieve the same effect (Fig. 4E). Biochemical confirmation of apoptosis in the SH-EP and WAC2 cell lines is noted in Fig. 4F, with an increase in cleaved PARP accompanied by a decrease in total PARP. Note again that the WAC2 (MYCN+) cells show evidence of apoptosis at concentration of Y15 (2.5 μM) that is almost half of the amount required to cause apoptosis (5 μM) in the isogenic, SH-EP (MYCN−) cells (Fig. 4F). This increase in apoptosis in the WAC2 cell line corresponds to the loss of Y397 FAK phosphorylation seen in these cells after treatment with Y15 at 2.5 μM. These data using the isogenic WAC2 and SH-EP cell lines show that the WAC2 (MYCN+) cell line is more reliant upon FAK and phosphorylation of Y397 FAK as a survival signal than their SH-EP (MYCN−), counterparts.
Fig. 4.

1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) treatment leads to decreased phosphorylation of Y397 FAK, cellular detachment and viability, and increased apoptosis in the isogenic WAC2 (MYCN+) and SH-EP (MYCN−) neuroblastoma cell lines. A, Western blotting confirms that these isogenic neuroblastoma cell lines differ in MYCN, total FAK, and Y397 FAK, with the WAC2 cell line having more MYCN, total FAK and more phosphorylation at the Y397 site of FAK than the SH-EP cell line. B, MTT assay was used to measure cell viability after Y15 treatment. Viability is expressed as mean fold change ± SEM. SH-EP (MYCN−) cells are represented by the open bars (□) and WAC2 (MYCN+) cells are represented by the black bars (■). Treatment of the WAC2 (MYCN+) (■) cell line with Y15 leads to 50% decrease in cell viability at 2 μM concentration. The SH-EP (□) cell line required over twice that concentration of Y15 to reach a 50% decrease in viability [*p≤0.01 control vs. Y15; †p≤0.01 SH-EP (□) vs. WAC2 (■)]. C, Western blotting is utilized to detect FAK and Y397 FAK phosphorylation. Treatment with Y15 leads to loss of Y397 FAK phosphorylation in the WAC2 cell line at 2.5 μM (lane 7) and in the SH-EP cell line at 5 μM (lane 4). D, Hoechst staining is utilized to detect apoptosis. The condensed and fragmented nuclei (white arrows) are easily detected and counted. The loss of Y397 FAK phosphorylation corresponds to increased apoptosis seen in the WAC2 cell line after treatment with Y15 at 2.5 μM (white arrows, middle right panel) and in the SH-EP cells after 5 μM (white arrows, lower left panel). E, Graphical representation of Hoechst staining data. There is a significant increase in the percentage of apoptotic cells in the WAC2 cell line (■) after Y15 treatment at 2.5 μM, and in the SH-EP cells (□) at 5 μM [*p≤0.01 control vs. Y15; †p≤0.01 SH-EP (□) vs. WAC2 (■)]. F, Western blotting to detect total and cleaved PARP shows biochemical corroboration of apoptosis in the SH-EP and WAC2 cell lines consistent with the Hoechst staining data. Again, the MYCN+ WAC2 cells are more sensitive to Y15 treatment than their isogenic MYCN− SH-EP counterparts and show significant changes in cleaved PARP at 2.5 μM Y15 treatment.
1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) inhibits human neuroblastoma tumor growth in vivo
To determine the in vivo effect of 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15), we utilized the human neuroblastoma cell lines SK-N-AS and SK-N-BE(2). The reason for choosing these cell lines was to determine if MYCN status (and FAK expression) affects the response of neuroblastoma to Y15 treatment in vivo. We injected SK-N-AS (non-amplified MYCN) or SK-N-BE(2) (amplified MYCN) cells subcutaneously into the flank of 6 week old female nude mice. Once the tumors were palpable, we began treatment with Y15 or control vehicle. Animals were treated daily for 23 days with 30 mg/kg/day IP injections. Previous reports determined this amount of Y15 to be the optimal treatment.24 Treatment was discontinued at 23 days since this is the time point that the control tumors reached maximal size allowed by protocol. Treatment with Y15 had no significant effect upon tumor volume or weight in the SK-N-AS (MYCN−) tumors (Fig. 5A, C). Conversely, treatment of the SK-N-BE(2) (MYCN+) neuroblastoma xenografts with Y15 resulted in a significant decrease in tumor volume and weight (Fig. 5B, C).
Fig. 5.

1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) inhibits human neuroblastoma SK-N-BE(2) neuroblastoma xenografts. A, SK-N-AS tumor cells were injected into the flank of female nude mice, mice were treated daily with control vehicle (normal saline, N=8) or Y15 (30 mg/kg/day, N=12), and tumor volumes were measured. In the Figure, vehicle treated mice are represented by the white squares (□), and Y15 treated mice are represented by the black squares (■). Treatment with Y15 had no significant effect upon SK-N-AS tumor volume. Black bars in the photographs represent 1 cm. B, SK-N-BE(2) tumor cells were injected into the flank of female nude mice, mice were treated daily with control vehicle (normal saline, N=7) or Y15 (30 mg/kg/day, N=13) and tumor volumes were measured. In the Figure, vehicle treated mice are represented by the white squares (□), and Y15 treated mice are represented by the black squares (■). Treatment with Y15 resulted in a significant decrease in tumor volumes in these animals [*p≤0.05 control (□) vs. Y15 (■)]. Black bars in the photograph represent 1cm. C, In the Figure, vehicle treated mice are represented by the white bars (□) and Y15 treated animals by the black bars (■). Tumors were weighed, and there was a significant decrease in tumor weight in the Y15 treated SK-N-BE(2) (MYCN+) (■) neuroblastoma xenografts [*p≤0.01 control (□) vs. Y15 (■)]. Treatment with Y15 did not affect the weights of the SK-N-AS xenografts. D, Tumor xenografts from the SK-N-AS tumors were homogenized and protein was harvested. Western blotting was performed to detect FAK and phosphorylation of Y397 FAK. Y15 treatment results in little change in phosphorylation of Y397 or FAK in the SK-N-AS (MYCN−) tumor xenografts. E, Tumor xenografts from the SK-N-BE(2) tumors were homogenized and protein was harvested. Western blotting was performed to detect FAK and phosphorylation of Y397 FAK. In contrast to the SK-N-AS (MYCN−) tumor xenografts, in the SK-N-BE(2) (MYCN+) tumor xenografts, Y15 treatment results in a decrease in FAK and in Y397 FAK phosphorylation.
To evaluate whether Y15 treatment resulted in decreased expression of FAK and decreased phosphorylation of Y397 FAK in vivo, tumor specimens were homogenized and protein extracted. Immmunoblotting of the proteins was performed for FAK and Y397 FAK. In the SK-N-AS tumors, Y15 treatment results in little change in FAK expression or Y397 FAK phosphorylation (Fig. 5D). However, in the SK-N-BE(2) tumors, immunoblotting reveals a decrease in Y397 FAK phosphorylation and a decrease in FAK in those tumors treated with Y15 (Fig. 5E).
In addition, we performed in vivo experiments with the isogenic cell lines SH-EP and WAC2 to corroborate our in vivo findings with the non-isogenic SK-N-AS and SK-N-BE(2) tumors. SH-EP (MYCN−) or WAC2 (MYCN+) cells were injected subcutaneously into the flank of 6 week old female nude mice. Once the tumors were palpable, we began treatment with Y15 or control vehicle. Animals were treated daily with IP injection of 30 mg/kg/day for 20 days. Treatment was discontinued at 20 days since that is the time point that the control tumors reached maximal size allowed by protocol. Treatment with Y15 had no significant effect upon tumor volume in the SH-EP (MYCN−) tumors (Fig. 6A), but there is a significant reduction in tumor volume in the animals with WAC2 xenograft tumors (Fig. 6B). Therefore, Y15 significantly decreased tumorigenesis of the MYCN+ neuroblastoma xenografts in vivo, both the non-isogenic SK-N-BE(2) (MYCN+) and the isogenic WAC2 (MYCN+) xenografts, compared to their MYCN− counterparts (SK-N-AS and SH-EP, respectively). These in vivo findings correspond to those of our in vitro studies.
Fig. 6.

1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) inhibits human neuroblastoma WAC2 (MYCN+) neuroblastoma xenografts, but has no effect upon SH-EP (MYCN−) xenografts. A, SH-EP (MYCN−) human neuroblastoma tumor cells were injected into the flank of female nude mice, mice were treated daily with control vehicle (normal saline, N=11) or Y15 (30 mg/kg/day, N=15), and tumor volumes were measured. Vehicle treated mice are represented by the white squares (□), and Y15 treated mice are represented by the black squares (■). Treatment with Y15 had no significant effect upon SH-EP tumor volume. Black bars in the Figure represent 100 mm. B, WAC2 (MYCN+) human neuroblastoma tumor cells were injected into the flank of female nude mice, the mice were treated with control vehicle (normal saline, N=10) or Y15 (30 mg/kg/day, N=15) daily, and tumor volumes were measured. In the Figure, vehicle treated mice are represented by the white squares (□), and Y15 treated mice are represented by the black squares (■). Treatment of the WAC2 tumor xenografts with Y15 resulted in a significant decrease in tumor volume [*p≤0.01 control (□) vs. Y15 (■)]. Black bars in the Figure represent 1cm.
Discussion
Since neuroblastoma carries such a dismal prognosis and so little progress has been made in the treatment of this tumor, and it is apparent that novel therapeutic strategies will be required. Focal adhesion kinase (FAK) has been shown to be overexpressed in a number of human tumors including colon, breast, ovarian, and thyroid cancers, and is thought to be a key factor in tumorigenesis.28–35 We have shown that FAK is also overexpressed in neuroblastoma tumors and cell lines, and that this overexpression is associated with MYCN.26, 27 This finding led us to hypothesize that FAK and the phosphorylation of Y397 FAK is important in cell survival in aggressive, MYCN amplified tumors. To evaluate this hypothesis, we undertook the current study using neuroblastoma cell lines with documented differences in MYCN status. We utilized a well-characterized isogenic neuroblastoma cell model to minimize cell type-specific differences. The first cell lines, the MYCN+/− (Tet−/+), are SH-EP non-amplified MYCN neuroblastoma cells that have been stably transfected with an N-myc tetracycline-repressible expression vector.36–38 The MYCN+/− (Tet−/+) isogenic cell lines express minimal amounts of MYCN when cultured in the presence of tetracycline, however, in the absence of tetracycline, the MCYN expression vector is activated, significantly increasing the expression of MYCN. Since these cells are not tumorigenic in mice, we included another pair of neuroblastoma cell lines that are not isogenic, but have differing expression of MYCN and are tumorigenic in xenograft models, SK-N-AS (non-amplified MYCN) and SK-N-BE(2) (amplified MYCN) neuroblastoma cell lines.38–40 Finally, a second isogenic cell line became available that has been reported to be tumorigenic in mice and, therefore, was also studied. The SH-EP and WAC2 cell lines have been previously characterized in the literature.41 In these cells, the parental SH-EP neuroblastoma cells (MYCN non-amplified) have been stably transfected with a MYCN expression vector to create neuroblastoma cells that express MYCN, named WAC2 cells.
In this study, we demonstrated that 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) decreases FAK Y397 phosphorylation in a number of human neuroblastoma cell lines. Treatment with Y15 resulted in cellular rounding and increased cellular detachment, most notably in the neuroblastoma cell lines with increased MYCN and FAK. Phosphorylation of Y397 FAK at focal adhesion sites enhances cellular adhesion and survival.42, 43 Owen et al reported that phosphorylation of FAK at the Y397 site is critical for cell spreading. Utilizing phosphorylation site mutants in FAK−/− mouse embryo fibroblasts, they demonstrated that FAK Y397 mutants showed more cell rounding and less spreading than their normal Y397 counterparts.44 Inhibition of FAK Y397 phosphorylation through a number of different mechanisms has been shown to result in cellular detachment in other tumor cell lines. Xu et al reported that a dominant-negative to the carboxy-terminal FAK, FAK-CD, resulted in a loss of Y397 phosphorylation in breast cancer cells, leading to cellular rounding and increased cellular detachment.15, 16 In addition, FAK Y397 attenuation with antisense oligonucleotides has also been shown to result in cellular detachment. Treatment with FAK antisense oligonucleotides led to marked cellular detachment in human melanoma, sarcoma, and breast cancer cell lines, but had no effect upon normal human fibroblasts.17 Finally, and even more specifically, inhibition of FAK Y397 phosphorylation levels through siRNA technology has been reported to lead to decreased cellular migration in H1299 lung cancer cells.19
In the current study we also show that 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) leads to apoptosis in human neuroblastoma cell lines, which may be attributed to the decrease in phosphorylation of Y397 FAK. Tyr-397 is the major autophosphorylation site of FAK, and activation at this site results in the binding and activation of other signaling molecules that ultimately results in decreased apoptosis.12, 45,46 There are a number of reports in the literature discussing the effects of dephosphorylation of FAK Tyr-397 upon apoptosis.42, 47 Kasahara and others showed that human leukemia HL-60 cells transfected with FAK (HL-60/FAK) are resistant to ionizing radiation-induced apoptosis, and that Y397 was critical to the anti-apoptotic properties seen in these HL-60/FAK cells.48 Inhibition of Y397 phosphorylation with FAK dominant-negative protein (FAK-CD) led to increased apoptosis in BT474 breast cancer cells.49 Beviglia and others also showed that the inhibition of Y397 phosphorylation with overexpression of the amino-terminus of FAK protein (FAK-NT) resulted in apoptosis in MCF-7 breast cancer cells, but MCF-10A, non-malignant breast epithelial cells, were not affected, suggesting that phosphorylation at this site is important in breast tumorigenesis.50
Other authors have utilized small molecules to show that dephosphorylation of FAK at Tyr-397 results in cellular apoptosis and decreased tumor survival.51, 52 For example, Halder et al found that TAE226, a FAK kinase inhibitor, resulted in FAK Y397 dephosphorylation and a significant decrease in ovarian tumor cell growth in vitro and decreased tumor burden in vivo.23 Using the same small molecule, Shi and others demonstrated a significant decrease in Y397 phosphorylation accompanied by decreased cell proliferation and increased cellular apoptosis in human glioma cell lines.53 Golubovskaya and others showed that TAE226 resulted in FAK (Y397) dephosphorylation and apoptosis in breast cancer cells.21 This small molecule has been shown to cause dephosphorylation of Y397 FAK and apoptosis in a number of human tumor cell lines including esophageal, pancreatic and gastrointestinal stromal tumor cells.54–56 One confounding variable associated with TAE226 is that it may not be entirely FAK specific. TAE226 has been shown in a number of studies to also inhibit IGF-IR in some cell lines at certain concentrations.22, 54, 55 These limitations do not, however, negate their usefulness as tools to provide preliminary studies of the effects of small molecule-dephosphorylation of Y397 FAK upon tumor cell survival.
In this study we have shown that decreased phosphorylation of Y397 FAK results in neuroblastoma cell detachment and apoptosis. This study and previous work leads us to believe that decreasing phosphorylation of FAK Y397 is a valid target in neuroblastoma.57 Other authors have demonstrated the potential for inhibition of FAK Tyr-397 phosphorylation to decrease neuroblastoma tumorigenesis. Kim et al showed that treatment of neuroblastoma cells with okadaic acid, a serine phosphatase inhibitor, resulted in decreased phosphorylation of FAK Tyr-397 and increased cellular detachment and apoptosis, leading those authors to suggest that agents causing FAK dephosphorylation may have utility in the treatment of neuroblastoma.58
Finally, in the current study, we have compared the differences in response to decreased phosphorylation of Y397 FAK in neuroblastoma cell lines with differing MYCN expression. Amplification of the MYCN oncogene is the primary adverse prognostic factor for neuroblastoma survival. Previous studies from our laboratory have shown that FAK expression is increased in MYCN amplified, higher grade neuroblastoma tumors, and that MYCN regulates FAK expression at the promoter level.26, 27 We have also noted that in an isogenic MYCN neuroblastoma cell line, inhibition of FAK with siRNA or AdFAK-CD results in greater decrease in survival in the MYCN+ cells (high FAK) compared to their isogenic MYCN− (low FAK) counterparts.27 The data from the current study confirm that total FAK and phosphorylation at the Y397 site are increased in the MYCN+ neuroblastoma cell lines compared to those that are MYCN−, and that inhibition of Tyr-397 phosphorylation results in more apoptosis in the MYCN+ cell lines. These findings suggest that the MYCN+ cell lines have a greater dependence upon phosphorylated Y397 for survival than their MYCN− counterparts.
Neuroblastoma is an aggressive childhood cancer that is often resistant to the most aggressive chemotherapeutic and surgical interventions. An important finding in this study is that inhibition of Y397 FAK phosphorylation resulted in decreased neuroblastoma cell adhesion and survival in vitro and inhibited neuroblastoma tumorigenesis in vivo. The ability to directly target the FAK autophosphorylation site at Y397 may prove to be an important tool for therapy for neuroblastoma in the future.
Experimental Procedures
Cells and Cell Culture
The Tet-off MYCN +/− cell line, (Tet-21/N or SHEP-21/N), was generously provided by Dr. S. L. Cohn (Northwestern University’s Feinberg School of Medicine, Chicago, IL) with permission from Dr. M. Schwab (Deutsches Krebsforschungszentrum, Heidelberg, Germany).36 These cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 μg/mL penicillin and 1 μg/mL streptomycin, and grown in the presence or absence of tetracycline (1 μg/mL) for 48–72 hours for MYCN− (Tet+) and MYCN+ (Tet−) cells, respectively.
SK-N-AS, MYCN non-amplified, human neuroblastoma cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 1 μg/mL penicillin and 1 μg/mL streptomycin. SK-N-BE(2), MYCN amplified, human neuroblastoma cells were maintained in a 1:1 mixture of Eagle’s Minimum Essential Medium and F12 with 10% fetal bovine serum, 1 μg/mL penicillin and 1 μg/mL streptomycin.
The SH-EP (MYCN−) and the isogenic WAC2 (MYCN+) cell lines were generously provided by Dr. M. Schwab (Deutsches Krebsforschungszentrum, Heidelberg, Germany). These cells have been described in detail previously.41 Briefly, the parent cell line, SH-EP, is a MYCN non-amplified cell line. This cell line was stably transfected with a vector containing MYCN to create the WAC2 MYCN overexpressing neuroblastoma cell line. These two cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 μg/mL penicillin and 1 μg/mL streptomycin.
Antibodies and Reagents
Monoclonal anti-FAK (4.47) and polyclonal anti-phospho-FAK (Y397) antibodies were obtained from Upstate Biotechnology, Inc. (Upstate, NY), and Invitrogen (Carlsbad, CA), respectively. Monoclonal antibody for cleaved PARP (9532) and polyclonal antibody for MYCN (9405) was obtained from Cell Signaling Technology, Inc. (Danvers, MA). Monoclonal antibodies against GAPDH (6C5) and β-actin were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The small molecule Y15 (C6H10N4·4ClH, 1,2,4,5-benzenetetraamine tetrahydrochloride) was obtained from Sigma-Aldrich Corp. (St. Louis, MO).
Western Blotting
Cells or homogenized tumor samples were washed twice with cold 1× PBS and lysed on ice for 30 min in a buffer containing 50 mM Tris-HCL, (pH 7.5), 150 mM NaCl, 1% Triton-X, 0.5% NaDOC, 0.1% SDS, 5 m MEDTA, 50 mM NaF, 1 mM NaVO3, 10% glycerol, and protease inhibitors: 10 μg/mL leupeptin, 10 μg/mL PMSFm and 1 μg/mL aprotinin. The lysates were cleared by centrifugation at 10 000 rpm for 30 min at 4 °C. Protein concentrations were determined using a Bio-Rad kit. The boiled samples were loaded on Ready SDS-10% PAGE gels (BioRad, Inc., Hercules, CA). Western blots were performed as previously described.27, 50, 59 Briefly, antibodies were used according to manufacturer’s recommended conditions. Molecular weight markers were used to confirm the expected size of the target proteins. Immunoblots were developed with chemiluminescence Renaissance Reagent (PerkinElmer Life Sciences, Waltham, MA). Blots were stripped with stripping solution (Bio-Rad, Inc.) at 37 °C for 15 minutes and then reprobed with selected antibodies. Immunoblotting with antibody to β-actin or GAPDH provided an internal control for equal protein loading.
Detachment Assay
Cells were treated with and without 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) inhibitor for 24 or 48 hours. Detached and attached cells were collected and counted with a hemacytometer. We calculated the percent detached cells by dividing the number of detached cells by the total number of cells (detached plus attached).
Cell Viability Assays
Cells were treated with and without 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) for 48 hours and cellular viability was measured using trypan blue exclusion and cell counting with a hemacytometer. Viability was further measured with Cell Titre 96 AQeous One solution assay kit (Promega, Madison, WI). In brief, cells were plated 5 × 103 cells per well on 96-well culture plates and allowed to attach. Following 48 hours of Y15 treatment, 20 μL of Cell Titer 96 AQueous One solution reagent was added to 100 μL of cell medium. After 1–4 h, the absorbance at 490 nm was measured using a kinetic microplate reader (Vmax, Molecular Devices). Data are expressed as fold change in viability compared to untreated control cells.
Apoptosis Assays
Apoptosis was determined by two methods. Following treatment with and without 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15) for 48 hours, cells were stained with Hoechst 33258 as previously described.60, 61 Cells undergoing apoptosis have condensation and fragmentation of nuclei. Hoechst stain binds to DNA and demonstrates condensed chromatin or micronuclei in cells that are undergoing apoptosis. The cells are harvested, fixed to a glass slide, stained with Hoechst 33258, positive cells counted with fluorescence microscopy, and a percentage of apoptotic cells calculated in three independent fields with 100 nuclei per field.
Apoptosis was also detected by immunoblotting for PARP expression. During apoptosis, poly (ADP-ribose) polymerase (PARP) is cleaved. The disappearance of the total protein or the accumulation of cleaved protein as detected by immunoblotting is a method utilized to detect apoptosis. Cells are treated as described, lysates are collected, and immunoblotting is performed. Bands are detected by chemiluminescence with β-actin or GAPDH serving as internal controls.
Tumor Growth In Vivo
Female nude mice, 6 weeks old, were purchased from Harlan Laboratories, Inc. (Indianapolis, IN). The mice were maintained in the SPF animal facility with standard 12 hour light / dark cycles and allowed chow and water ad libitum. All experiments were performed after obtaining protocol approval by the Institutional Animal Care and Use Committee (200801260), and in compliance with the NIH animal use guidelines. Human neuroblastoma cells [SK-N-AS, SK-N-BE(2), SH-EP, WAC2], 2 ×106, in Matrigel (BD Biosciences, San Jose, CA) were injected subcutaneously into the right flank. When the tumors reached palpable size (100 mm3), animals were treated daily with intraperitoneal injections of control vehicle (normal saline) or 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15, 30 mg/kg/day). Previous experiments with various doses and dosing schedules of the compound proved this to be the optimal, nontoxic dose.24 Tumors were measured twice weekly with a caliper and tumor volume in mm3 was calculated using the standard formula [(width) 2 × length]/2, where width is the smaller diameter. Tumor weight was determined at the completion of the experiment.
Data Analysis
Experiments were repeated at least in triplicate, and data are reported as mean ± standard error of the mean. An ANOVA or student’s t-test was used as appropriate to compare data between groups. Statistical significance was determined at the P<0.05 level.
Acknowledgments
We thank Dr. S. L. Cohn and Dr. M. Schwab for their kind gift of the N-myc+/− cells (Tet-21/N or SH-EP-21/N). We also thank Dr. M. Schwab for his kind gift of the SH-EP and WAC2 tumor cells. This work was sponsored in part by a grant from St. Baldrick’s Foundation (EAB) and by a grant from the National Cancer Institute, Grant Number K08CA118178 (EAB).
Abbreviations
- FAK
focal adhesion kinase
- Y15
1,2,4,5-benzenetetraamine tetrahydrochloride
- Tet
tetracycline
Contributor Information
Elizabeth A. Beierle, Email: elizabeth.beierle@ccc.uab.edu.
Xiaojie Ma, Email: xiaojie.ma@surgery.ufl.edu.
Jerry Stewart, Email: jessy@uab.edu.
Carl Nyberg, Email: cdnyberg@ufl.edu.
Angelica Trujillo, Email: pangietron@hotmail.com.
William G. Cance, Email: William.Cance@roswellpark.org.
Vita M. Golubovskaya, Email: Vita.Golubovskaya@roswellpark.org.
References
- 1.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17(7):2263–79. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 2.Cotterill SJ, Parker L, More L, Craft AW. Neuroblastoma: changing incidence and survival in young people aged 0–24 years. A report from the North of England Young Persons’ Malignant Disease Registry. Med Pediatr Oncol. 2001;36(1):231–4. doi: 10.1002/1096-911X(20010101)36:1<231::AID-MPO1056>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced stage disease. Science. 1984;224(4653):1121–4. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 4.Seeger RC, Brodeur GM, Sather H, Dalton A, Siegle SE, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313(18):1111–6. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 5.Zaizen Y, Taniguchi S, Noguchi S, Suita S. The effect of N-myc amplification and expression on invasiveness of neuroblastoma cells. J Pediatr Surg. 1993;28(6):766–9. doi: 10.1016/0022-3468(93)90321-b. [DOI] [PubMed] [Google Scholar]
- 6.Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM, Chung DH. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem Biophys Res Commun. 2006;351(1):192–7. doi: 10.1016/j.bbrc.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nara K, Kusafuka T, Yoneda A, Oue T, Sangkhathat S, Fukuzawa M. Silencing of MYCN by RNA interference induces growth inhibition, apoptotic activity and cell differentiation in a neuroblastoma cell line with MYCN amplification. Int J Oncol. 2007;30(5):1189–96. [PubMed] [Google Scholar]
- 8.Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89(11):5192–6. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanks SK, Polte TR. Signaling through focal adhesion kinase. BioEssays. 1997;19(2):137–45. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 10.Zachary I. Focal adhesion kinase. Int J Biochem Cell Biol. 1997;29(7):929–34. doi: 10.1016/s1357-2725(97)00008-3. [DOI] [PubMed] [Google Scholar]
- 11.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22(4):359–74. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 12.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein tyrosine kinase, pp125FAK. Mol Cell Biol. 1994;14(1):147–55. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor of apoptosis proteins and apoptosis suppression by the overexpression of Fak in a human leukemic cell line, HL60. J Biol Chem. 2000;275(21):16309–15. doi: 10.1074/jbc.275.21.16309. [DOI] [PubMed] [Google Scholar]
- 15.Xu LH, Yang XH, Bradham CA, Brenner DA, Baldwin AS, Craven RJ, et al. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275(39):30597–604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 16.Xu LH, Yang XH, Craven RJ, Cance WG. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998;9(12):999–1005. [PubMed] [Google Scholar]
- 17.Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ, et al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Diff. 1996;7(4):413–8. [PubMed] [Google Scholar]
- 18.Golubovskaya VM, Beviglia L, Xu LH, Earp HS, Craven RJ, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;277(41):38978–87. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- 19.Han EK, Mcgonigal T, Wang J, Giranda VL, Luo Y. Functional analysis of focal adhesion kinase (FAK) reduction by small inhibitory RNAs. Anticancer Res. 2004;24(6):3899–905. [PubMed] [Google Scholar]
- 20.Lipinski CA, Tran NL, Menashi E, Rohl C, Kloss J, Bay RC, et al. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia. 2005;7(5):435–5. doi: 10.1593/neo.04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golubovskaya VM, Virnig C, Cance WG. TAE226-induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol Carcinog. 2008;47(3):222–34. doi: 10.1002/mc.20380. [DOI] [PubMed] [Google Scholar]
- 22.Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, et al. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6(4):1357–67. doi: 10.1158/1535-7163.MCT-06-0476. [DOI] [PubMed] [Google Scholar]
- 23.Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67(22):10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 24.Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the Y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51(23):7405–16. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–43. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beierle EA, Massoll NA, Hartwich J, Kurenova EV, Golubovskaya VM, Cance WG, et al. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res. 2008;14(11):3299–305. doi: 10.1158/1078-0432.CCR-07-1511. [DOI] [PubMed] [Google Scholar]
- 27.Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, et al. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282(17):12503–16. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- 28.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of focal adhesion kinase gene and invasive cancer. Lancet. 1993;342(8878):1024–25. doi: 10.1016/0140-6736(93)92881-s. [DOI] [PubMed] [Google Scholar]
- 29.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of growth factor receptors, the focal adhesion kinase, and other tyrosine kinases in human soft tissue tumors. Ann Surg Oncol. 1994;1(1):18–27. doi: 10.1007/BF02303537. [DOI] [PubMed] [Google Scholar]
- 30.Agochiya M, Brunton VG, Owens DW, Parkinson EK, Paraskeva C, Keith WN, et al. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene. 1999;18(41):5646–53. doi: 10.1038/sj.onc.1202957. [DOI] [PubMed] [Google Scholar]
- 31.Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55(13):2752–5. [PubMed] [Google Scholar]
- 32.Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6(6):2417–23. [PubMed] [Google Scholar]
- 33.Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X, et al. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003;9(1):215–22. [PubMed] [Google Scholar]
- 34.Judson PL, He X, Cance WG, Van Le L. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 1999;86(8):1551–6. doi: 10.1002/(sici)1097-0142(19991015)86:6<1551::aid-cncr23>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 35.Owens LV, Xu L, Dent GA, Yang X, Sturge GC, Craven RJ, et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann Surg Oncol. 1996;3(1):100–5. doi: 10.1007/BF02409059. [DOI] [PubMed] [Google Scholar]
- 36.Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A, Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996;13(4):803–12. [PubMed] [Google Scholar]
- 37.Zhu X, Wimmer K, Kuick R, Lamb BJ, Motyka S, Jasty R, et al. N-myc modulates expression of p73 in neuroblastoma. Neoplasia. 2002;4(5):432–9. doi: 10.1038/sj.neo.7900255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen T, Hocker JE, Thomas W, Smith SA, Norris MD, Haber M, et al. Combined RARα- and RXR-specific ligands overcome N-myc-associated retinoid resistance in neuroblastoma cells. Biochem Biophys Res Comm. 2003;302(3):462–8. doi: 10.1016/s0006-291x(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 39.Ucar K, Seeger RC, Challita PM, Watanabe CT, Yen TL, Morgan JP, et al. Sustained cytokine production and immunophenotypic changes in human neuroblastoma cell lines transduced with a human gamma interferon vector. Cancer Gene Therapy. 1995;2(3):171–81. [PubMed] [Google Scholar]
- 40.Livingstone A, Mairs RJ, Russell J, O’Donoghue J, Gaze MN, Wheldon TE. N-myc gene copy number in neuroblastoma cell lines and resistance to experimental treatment. Eur J Cancer. 1994;30A(3):382–9. doi: 10.1016/0959-8049(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 41.Schweigerer L, Breit S, Wenzel A, Tsunamoto K, Ludwig R, Schwab M. Augmented MYCN expression enhances the malignant phenotype of human neuroblastoma cells: evidence for induction of autocrine growth factor activity. Cancer Res. 1990;50(14):411–16. [PubMed] [Google Scholar]
- 42.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134(3):793–9. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 44.Owen JS, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto-and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19(7):4806–18. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954–63. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91(21):10148–52. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hungerford J, Compton J, Matter J, Hoffstrom B, Otey C. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135(5):1383–90. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasahara T, Koguchi E, Funakoshi M, Aizu-Yokota E, Sonoda Y. Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid Redox Signal. 2002;4(3):491–9. doi: 10.1089/15230860260196290. [DOI] [PubMed] [Google Scholar]
- 49.Park HB, Golubovskaya VM, Xu L, Yang X, Lee JW, Scully S, et al. Activated Src increases adhesion, survival and alpha2-integrin expression in human breast cancer cells. Biochem J. 2004;378(Pt 2):559–67. doi: 10.1042/BJ20031392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beviglia L, Golubovskaya V, Xu LH, Yang X, Craven RJ, Cance WG. Focal adhesion kinase N-terminus in breast carcinoma cells induces rounding, detachment and apoptosis. Biochem J. 2003;373(Pt 1):201–10. doi: 10.1042/BJ20021846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282(20):14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 52.Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68(6):1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 53.Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Molec Carcinog. 2007;46(6):488–96. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 54.Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol Reports. 2009;20(6):1473–7. [PubMed] [Google Scholar]
- 55.Lui W, Bloom DA, Cance WG, Kurenova EV, Golubovskaya VM, Hochwald SN. FAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cells. Carcinogenesis. 2008;29(6):1096–107. doi: 10.1093/carcin/bgn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sakurama K, Noma K, Takaoka M, Tomono Y, Watanabe N, Hatakeyama S, et al. Inhibition of focal adhesion kinase as a potential therapeutic strategy for imatinib-resistant gastrointestinal stromal tumor. Mol Cancer Ther. 2009;8(1):127–34. doi: 10.1158/1535-7163.MCT-08-0884. [DOI] [PubMed] [Google Scholar]
- 57.Beierle EA, Trujillo A, Nagaram A, Golubovskaya VM, Cance WG, Kurenova EV. TAE226 inhibits neuroblastoma cell survival. Cancer Invest. 2008;26(2):145–51. doi: 10.1080/07357900701577475. [DOI] [PubMed] [Google Scholar]
- 58.Kim B, van Golen CM, Feldman EL. Degradation and dephosphorylation of focal adhesion kinase during okadaic acid-induced apoptosis in human neuroblastoma cells. Neoplasia. 2003;5(5):405–16. doi: 10.1016/s1476-5586(03)80043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurenova E, Xu LH, Yang X, Baldwin AS, Craven RJ, Hanks SK, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24(10):4361–71. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang XH, et al. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol Cancer Res. 2003;1(10):755–64. [PubMed] [Google Scholar]
- 61.Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66(3):1446–54. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]