

Abstract
Rationale
NADPH oxidases are a major source of superoxide (O2−) in the cardiovascular system. The function of Nox4, a member of the Nox family of NADPH oxidases, in the heart is poorly understood.
Objective
The goal of this study was to elucidate the role of Nox4 in mediating oxidative stress and growth/death in the heart.
Methods and Results
Expression of Nox4 in the heart was increased in response to hypertrophic stimuli and aging. Neither transgenic mice with cardiac specific overexpression of Nox4 (Tg-Nox4) nor those with catalytically inactive Nox4 (Tg-Nox4-P437H) showed an obvious baseline cardiac phenotype at young ages. Tg-Nox4 gradually displayed decreased left ventricular (LV) function with enhanced O2− production in the heart, which was accompanied by increased apoptosis and fibrosis at 13–14 months of age. On the other hand, the level of oxidative stress was attenuated in Tg-Nox4-P437H. Although the size of cardiac myocytes was significantly greater in Tg-Nox4 than in NTg, the LV weight/tibial length was not significantly altered in Tg-Nox4 mice. Overexpression of Nox4 in cultured cardiac myocytes induced apoptotic cell death but not hypertrophy. Nox4 is primarily localized in mitochondria and upregulation of Nox4 enhanced both rotenone- and diphenyleneiodonium-sensitive O2− production in mitochondria. Cysteine residues in mitochondrial proteins, including aconitase and NADH dehydrogenases, were oxidized and their activities decreased in Tg-Nox4.
Conclusions
Upregulation of Nox4 by hypertrophic stimuli and aging induces oxidative stress, apoptosis and LV dysfunction, in part due to mitochondrial insufficiency caused by increased O2− production and consequent cysteine oxidation in mitochondrial proteins.
Keywords: Reactive oxygen species, oxidative stress, superoxide, hypertrophy, apoptosis, aging
Introduction
Reactive oxygen species (ROS), such as O2− and H2O2, play an important role in regulating cell growth and death of cardiac myocytes 1–3. In the heart under pathological conditions, mitochondria are the major source of ROS, which are generated primarily through electron leakage from the electron transport chain 4. The leakage of electrons is a passive process due to damage and/or downregulation of mitochondrial proteins, and does not appear to be tightly regulated 5. ROS are also produced through O2−-producing enzymes, such as NADPH oxidases and xanthine oxidase. Although NADPH oxidases are the major source of O2− production, their contribution to overall increases in ROS and myocardial responses under stress is not fully understood.
Thus far, seven members of the NADPH oxidase (Nox) family of proteins (Nox1 to Nox5 and Duox1 and 2) have been identified 6–8. All Nox proteins possess 6 membrane-spanning domains and a cytoplasmic region containing NAD(P)H- and FAD-binding domains in their C-terminal regions. Nox1, 2, 3 and 4 form a heterodimer with p22phox, another catalytic core component of NADPH oxidases which stabilizes Nox proteins. Nox proteins accept electrons from either NADPH or NADH 8, 9, and transfer them to molecular oxygen to generate O2−.
Nox4 is ubiquitously expressed in various cell types and tissues, including kidneys, the heart, and blood vessels 10, 11. Distinct from other members of the Nox family, Nox4 is believed to be constitutively active and does not require cytosolic factors, such as p47phox, p67phox and the small GTPase Rac, for its activation. Therefore, its expression level essentially determines the amount of O2− production in cells. Importantly, it is currently unclear to what extent Nox4 plays an important role in mediating the production of ROS in the heart.
Accumulating lines of evidence suggest that NADPH oxidases play an important role in mediating the development of cardiac hypertrophy and the progression of heart failure 11. For example, Nox2 mediates angiotensin II-induced cardiac hypertrophy 12. However, neither oxidative stress nor cardiac hypertrophy is suppressed in Nox2 knockout mice under pressure overload 13, 14. On the other hand, expression of Nox4 is upregulated during cardiac hypertrophy induced by pressure overload 13. Thus, Nox4 may play an important role in mediating ROS generation and the development of cardiac hypertrophy and heart failure. However, the role of Nox4 in mediating cardiac hypertrophy and LV dysfunction has not been clearly demonstrated due to a lack of an animal model in which the function of Nox4 in vivo can be elucidated in an isoform specific manner.
Thus, the major goal in this investigation was to elucidate the function of Nox4 in the heart and in the cardiac myocytes therein. To this end, we have generated a specific anti-Nox4 antibody and transgenic mouse models in which Nox4 in the heart is either stimulated or inhibited in an isoform specific manner. In particular, we evaluated 1) how expression of Nox4 is regulated in response to hypertrophic stimuli and aging, 2) whether Nox4 affects growth and death of cardiac myocytes in the heart, and 3) subcellular localization of Nox4 and O2− generation in cardiac myocytes.
Methods
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Monoclonal antibodies against Nox4
We made mouse monoclonal antibodies against Nox4 using a recombinant protein encoding the C-terminal cytoplasmic region of Nox4 (residues 307–578) tagged with His6. We solubilized the recombinant Nox4 with 6M urea and injected it into mice after diluting it in PBS containing 0.6M urea. After screening more than 1,000 clones by ELISA, we chose clones that detect the recombinant Nox4 protein used as an antigen, endogenous Nox4 in mouse tissues, including the heart and kidney (positive control), and endogenous Nox4 in neonatal rat cardiac myocytes by immunoblot. We found that some antibodies cross-react with Nox2. We selected antibodies which specifically react with Nox4, but not with Nox2.
Transgenic mice
All transgenic mice used in this study were generated on an FVB background with the α-myosin heavy chain promoter (courtesy of Dr. J Robbins). All protocols concerning the use of animals were approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey.
Isotope-coded affinity tag (ICAT) labeling and multidimensional chromatography
The ICAT analysis was conducted as described previously 15.
Statistical analysis
All values are expressed as mean ± SEM. Statistical analyses between groups were done by unpaired Student's t test or one-way ANOVA followed by a post hoc Fisher's comparison test. A value of p < 0.05 was accepted as significant.
Results
Nox4 is expressed in cardiac myocytes and is upregulated by hypertrophic stimuli
In order to characterize protein expression of Nox4 in the heart, we made Nox4 monoclonal antibodies, using the C-terminal cytoplasmic region of Nox4 as an antigen. Clone 3D2 was identified through ELISA and immunoblot analyses as a specific Nox4 antibody, which reacts with Nox4 but not with Nox2 (Fig. 1A). Immunoblot analyses using the Nox4 antibody (3D2) confirmed that Nox4 is expressed in the mouse heart and in cultured neonatal rat cardiac myocytes. Expression of Nox4 in the mouse kidney, where Nox4 was originally identified 16, 17 , is shown as a positive control (Fig. 1B).
Figure 1.
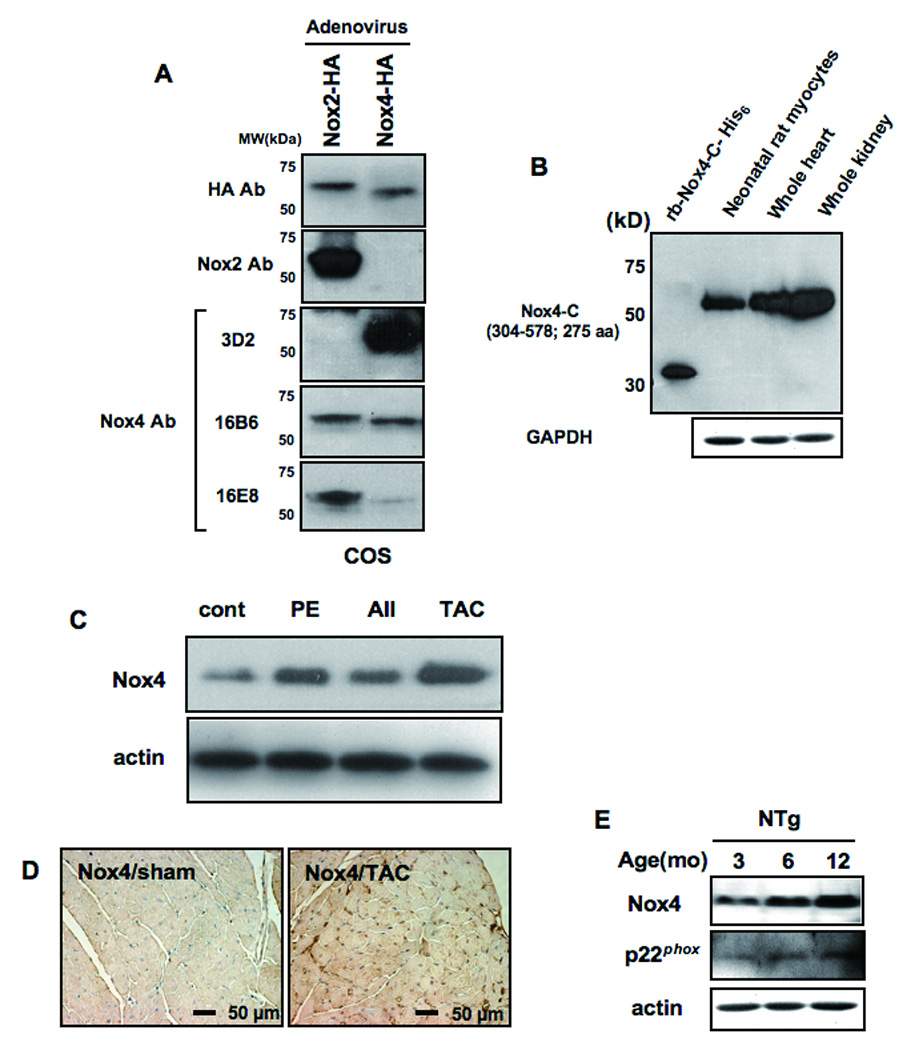
A) COS7 cells were transduced with adenovirus harboring either Nox2-HA or Nox4-HA. Immunoblot analyses were conducted with the indicated antibodies. Although Nox4 has a greater molecular weight than Nox2, it migrated faster on SDS-PAGE gel, possibly due to cleavage at the N-terminal mitochondrial localization signal (MLS). B) An immunoblot showing expression of Nox4 in the heart and cardiac myocytes. Recombinant protein used as an antigen for 3D2 and mouse kidney homogenate were used as positive controls. C) The effect of hypertrophic stimuli on expression of Nox4 in the mouse heart. Mice were subjected to continuous infusion with phenylephrine (PE) or angiotensin II (AII) or to transverse aortic constriction (TAC) for 2 weeks. N=4–5. D) Nox4 expression in the mouse heart subjected to sham or TAC. Immunostaining was conducted with anti-Nox4 antibody (3D2). E) The effect of aging upon Nox4, p22phox, and actin expression in the control mouse heart was evaluated by immunoblot analyses.
We next examined the effects of hypertrophic stimuli on expression of Nox4 in the mouse heart. We treated mice with either angiotensin II (AII) (200 ng/kg/min) or phenylephrine (75mg/kg/day), using osmotic pumps, or with transverse aortic constriction (TAC) for 14 days. Immunoblot analyses (Fig. 1C) showed that Nox4 is upregulated 1.5-, 1.9- and 3.4-fold by AII, phenylephrine and TAC, respectively (all p<0.05 vs control). Immunostaining of mouse hearts with the anti-Nox4 antibody confirmed that myocardial expression of Nox4 is upregulated in hearts subjected to TAC (Fig. 1D). PE-induced upregulation of Nox4 was observed in myocyte-rich cultures but not in non-myocyte-rich ones, suggesting that Nox4 upregulation occurs in cardiac myocytes (Online Fig. IA).
We also examined the effect of aging on Nox4 expression. Nox4 expression increased gradually between 3 and 12 months. Expression of p22phox, a component of the NADPH oxidase complex known to associate with Nox4, was also increased with aging (Fig. 1E). Staining with the anti-Nox4 antibody showed that upregulation of Nox4 is diffusely observed in the myocardium, including in cardiac myocytes, in response to aging (Online Fig. IB).
Establishment of transgenic mice with cardiac specific overexpression of Nox4
In order to elucidate the functional consequence of Nox4 upregulation in the heart, we generated mice with cardiac-specific overexpression of wild-type Nox4 (Tg-Nox4) or a Nox4 mutant, in which proline-437 in the NADPH binding domain is substituted with a histidine residue (Tg-Nox4-P437H), using the α-myosin heavy chain promoter. Since a proline to histidine substitution in the NADPH binding domain in other Nox family proteins has been shown to abolish O2− production 18, we expect that the Nox4-P437H mutant is catalytically inactive. Furthermore, Nox4-P437H could function as a dominant negative against endogenous Nox4 by competing for interaction with p22phox.
We established transgenic lines from 10 founders in Tg-Nox4 and 3 founders in Tg-Nox4-P437H. All of them were fertile, and expression levels of the transgenes were up to 4-fold higher than endogenous Nox4 in non-transgenic (NTg) mice (Fig. 2A and data not shown). Transgenic expression of Nox4 or Nox4-P437H did not affect the level of Nox2 (Online Fig. IIA). Although expression of antioxidant genes, such as MnSOD and thioredoxin1, was not affected, catalase was modestly upregulated (1.7 fold) in Tg-Nox4 at 3–4 months of age (Online Fig. IID). None of the Tg-Nox4 or Tg-Nox4-P437H mice showed cardiac hypertrophy or abnormal LV function at baseline at 2–3 months of age (Online Fig. IIBC), although Tg-Nox4 mice had a tendency to show a slightly smaller left ventricular weight/body weight (LVW/BW) (Online Fig. IB). Among the transgenic mouse lines generated, we chose founders 3 (1.3-fold overexpression) and 13 (2.1-fold) in Tg-Nox4 and founders 11 (2.0-fold) and 14 (3.5-fold) in Tg-Nox4-P437H for further experiments.
Figure 2.
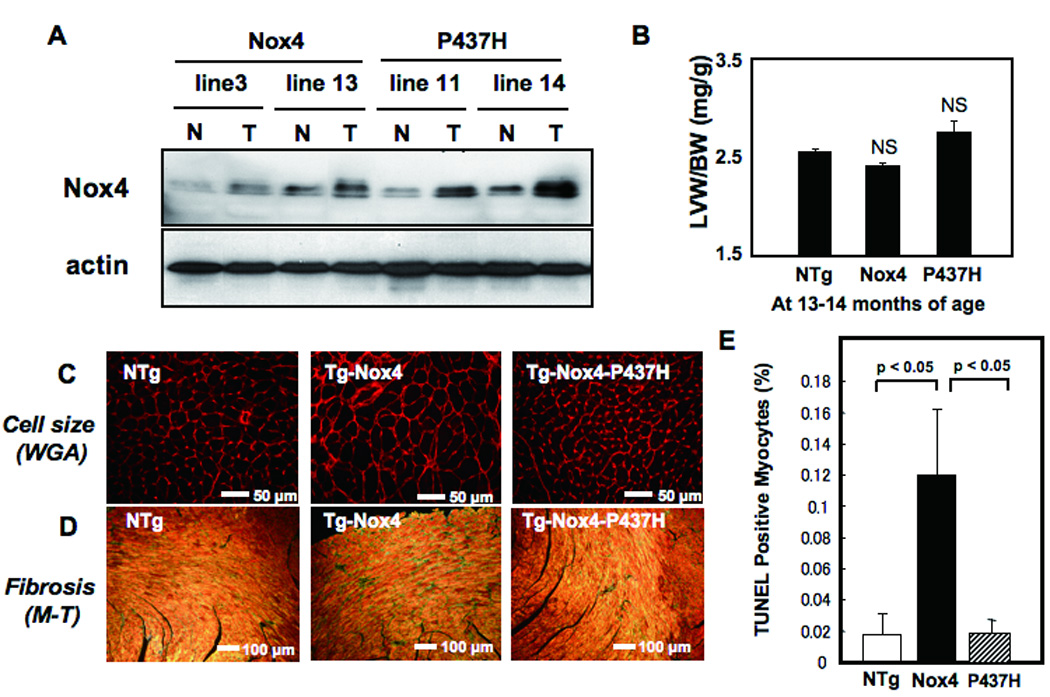
A) Expression of Nox4 in Tg-Nox4 and Tg-Nox4-P437H. N, non-transgenic; T, transgenic. B) Left ventricular weight/body weight (LVW/BW) at 13–14 months of age. NS, not significant vs NTg. N=8–9. (C–E) Age-dependent changes in histology in Tg-Nox4, Tg-Nox4-P437H and NTg. C) LV myocyte cross sectional area as evaluated by WGA staining, D) LV fibrosis as determined by Masson Trichrome staining, and E) apoptosis as determined by TUNEL staining.
Aged Tg-Nox4 mice display decreased cardiac function without cardiac hypertrophy
Although the cardiac phenotypes of young Tg-Nox4 and Tg-Nox4-P437H were not significantly different from NTg, Tg-Nox4 mice at the age of 13–14 months showed modestly but significantly increased LV end diastolic dimension and reduced LV ejection fraction (LVEF), an index of LV systolic function, compared to NTg and Tg-Nox4-P437H, as determined by echocardiographic measurement (Online Table I). Although LVW/BW at 13–14 months old in Tg-Nox4 was not significantly different from that in NTg or Tg-Nox4-P437H (Fig. 2B), histological analyses showed that the LV cardiac myocyte cross-sectional area was significantly greater (Fig. 2C and Online Fig. IIIA) in Tg-Nox4 than in NTg or Tg-Nox4-P437H mice. Cardiac fibrosis (Fig. 2D and Online Fig. IIB) and the number of TUNEL positive cells (Fig. 2E and Online Fig. IIIC) were greater in Tg-Nox4 than in NTg and Tg-Nox4-P437H mice. The level of oxidative stress, including oxidative DNA damage and O2−, as evaluated by immunostaining of 8-OHdG and DHE, respectively, was greater in Tg-Nox4 than in NTg and Tg-Nox4-P437H mice (Fig. 3). Quantitative analysis showed that the intensity of DHE staining was significantly greater in Tg-Nox4 and significantly lower in Tg-Nox4-P437H, than in NTg (Fig. 3B). These results suggest that increased expression of Nox4 enhances cardiac dysfunction and pathological changes in an age-dependent manner.
Figure 3.
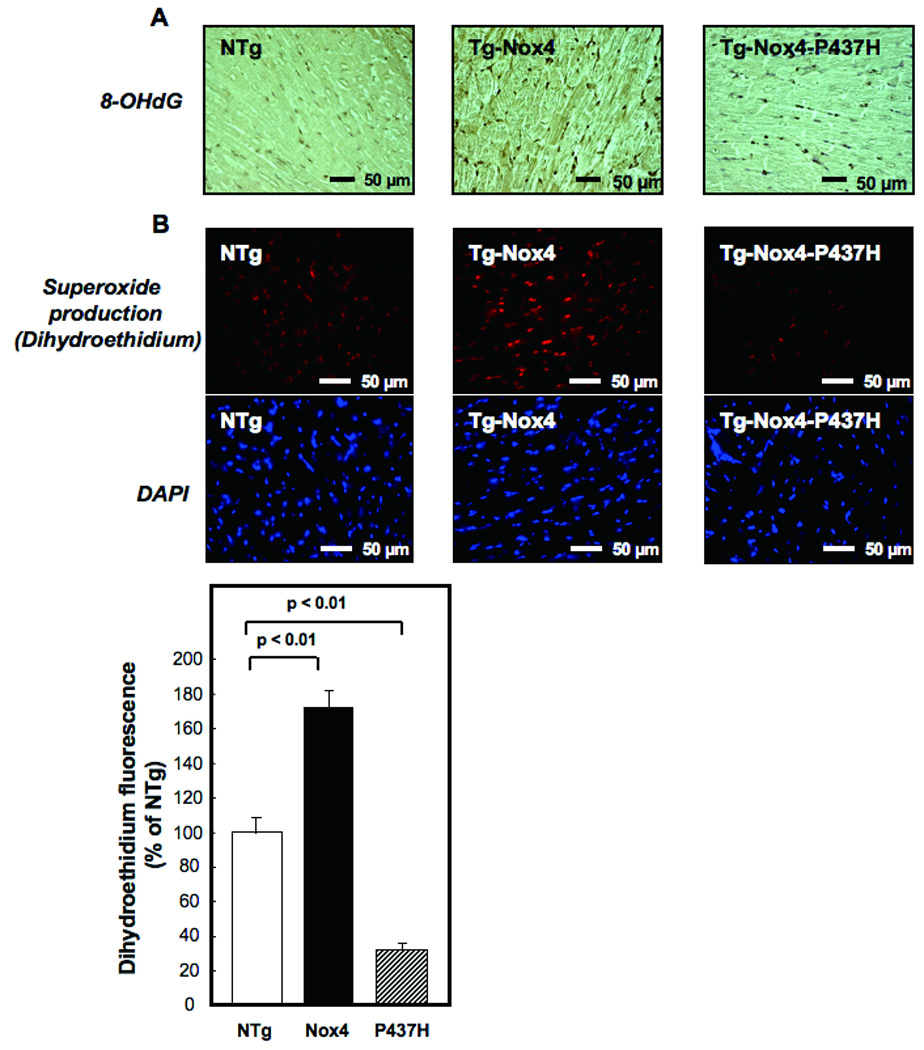
The extent of oxidative stress in the LV was determined by 8-hydroxyl-deoxyguanosine (8-OHdG) staining (A) and dihydroethidium (DHE) staining (B). In B, DAPI staining shows nuclei. LV sections were prepared from Tg-Nox4, Tg-Nox4-P437H and NTg mice.
Nox4 stimulates apoptosis but not hypertrophy in cultured cardiac myocytes
In order to evaluate the direct effect of Nox4 upon growth and death, we overexpressed Nox4 in cardiac myocytes, using adenovirus transduction (Online Fig. IVA). Cell size was similar in control myocytes and Nox4-overexpressing myocytes, regardless of the multiplicity of infection (Fig. 4A). Apoptotic cell death, as evaluated by TUNEL staining, was dose-dependently increased when Nox4 was overexpressed in cardiac myocytes (Fig. 4B). Nox4-induced increases in TUNEL positive myocytes were significantly attenuated in the presence of Bcl-xL, suggesting that the mitochondrial component of apoptosis may be stimulated by Nox4 (Fig. 4C). Overexpression of Nox4 induced release of cytochrome c into the cytosolic fraction (Fig. 4D). Taken together, these results show that Nox4 stimulates apoptosis in a cell autonomous fashion, but it does not induce hypertrophy at the single myocyte level. Thus the enlargement of cardiac myocytes seen in Tg-Nox4 mice after aging may be a compensatory response of the heart against LV dysfunction.
Figure 4.
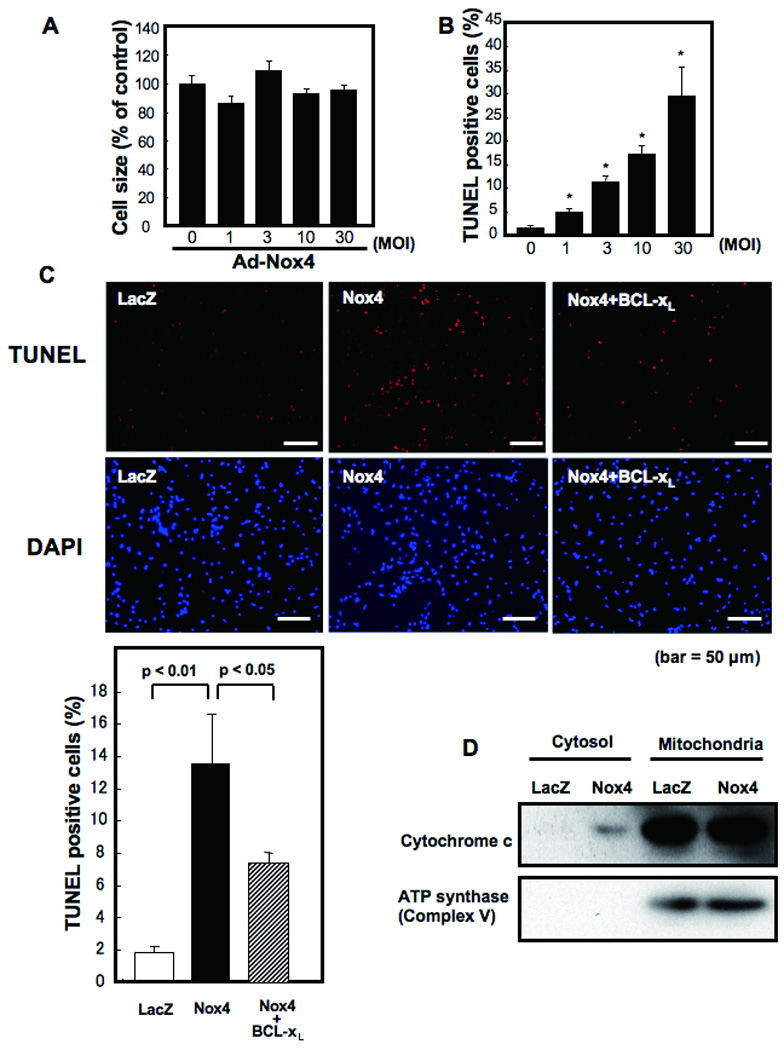
Cultured neonatal rat cardiac myocytes were transduced with Ad-Nox4 or Ad-LacZ at various multiplicities of infection (MOI) for 48 hours. Data are from 4–6 experiments. (A) Mean cell size of control cardiac myocytes is expressed as 100%. Changes were not significant. (B) The number of apoptotic cells was evaluated with TUNEL staining. *p<0.05 vs control (0 MOI). (C) Cultured cardiac myocytes were transduced with Ad-LacZ, Ad-Nox4, or Ad-Nox4 + Ad-Bcl-xL. Representative TUNEL and DAPI staining and the results of the quantitative analysis are shown. (D) Cultured cardiac myocytes were transduced with Ad-LacZ or Ad-Nox4. Cytosolic and mitochondrial fractions were isolated and expression of cytochrome c and F0F1 ATP synthase was evaluated with immunoblot analyses.
Nox4 is primarily localized in mitochondria
In order to elucidate the mechanism by which Nox4 stimulates cardiac dysfunction and increases in cell death during aging, we evaluated the intracellular localization of Nox4 in cardiac myocytes. Immunostaining of cardiac myocytes with the monoclonal antibody (3D2) and confocal microscopic analyses showed that endogenous Nox4 is localized in the perinuclear region, and co-localized with F1F0 ATP synthase in oxidative phosphorylation Complex V, a marker of mitochondria (Fig. 5A). When endogenous Nox4 was downregulated by transduction of adenovirus harboring shRNA-Nox4, the perinuclear staining with the anti-Nox4 antibody was markedly attenuated (Fig. 5A). Staining of overexpressed HA-Nox4 with anti-Nox4 antibody and with anti-HA antibody exhibited identical perinuclear staining patterns (Online Fig. VAB). These results are consistent with the notion that Nox4 is localized in perinuclear organelles, including mitochondria. Interestingly, staining of p22phox showed an almost identical staining pattern to that of Nox4 (Fig. 5B), suggesting that Nox4 is a major partner of p22phox in cardiac myocytes.
Figure 5.
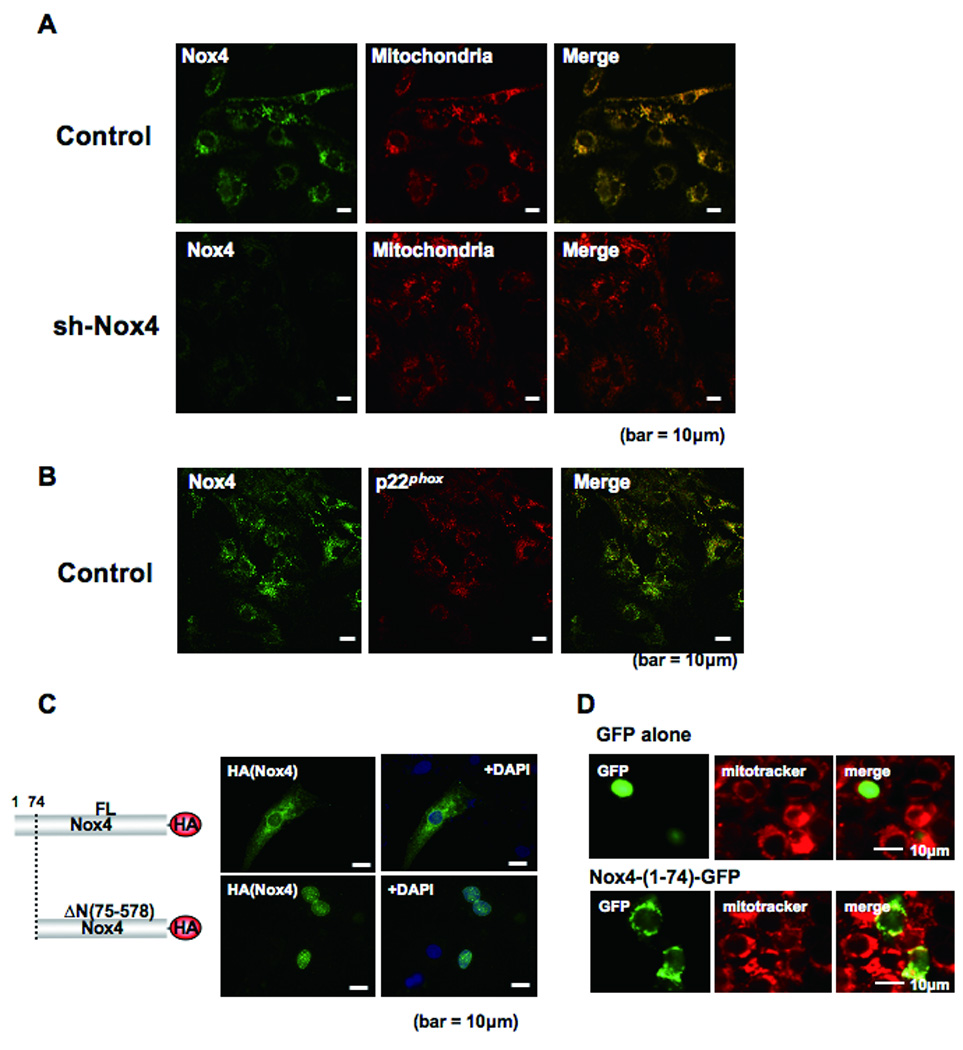
Subcellular localization of Nox4 in cardiac myocytes. A) Cultured cardiac myocytes were transduced with adenovirus harboring shRNA-scramble or shRNA-Nox4. Myocytes were co-stained with anti-Nox4 antibody and anti-F0F1 ATP synthase antibody, a marker of mitochondria. Merged images are shown on the right. B) Cultured cardiac myocytes were co-stained with anti-Nox4 and anti-p22phox antibodies. A merged image is shown on the right. C) Cultured cardiac myocytes were transduced with an expression plasmid harboring full length (FL) Nox4-HA or Nox4 lacking MLS (ΔN(75–578))-HA. Forty-eight hours after transfection, cells were stained with anti-HA antibody and DAPI. Truncation of the N-terminal region (1–74) causes disappearance of the perinuclear staining of Nox4 in cardiac myocytes. D) Cardiac myocytes were transduced with expression plasmids harboring either GFP alone or Nox4 (1–74)-GFP. Representative images of GFP, Mitotracker and merged images are shown.
Proteins localized in mitochondria have a mitochondrial localization signal (MLS) in their N-terminus. An amino acid sequence consistent with the MLS is found in the N-terminal region of Nox4. To further confirm the localization of Nox4, we made an N-terminally deleted Nox4 mutant (Nox4-ΔN: residues 75–578) and tested its localization. We found that Nox4-ΔN was no longer localized in the perinuclear region (Fig. 5C). On the other hand, an N-terminal fragment of Nox4 (1–74) tagged with GFP in its N-terminal region, but not GFP alone, was localized in the perinuclear region, as evidenced by co-staining with Mitotracker (Fig. 5D). All these findings support the notion that Nox4 is primarily localized in mitochondria.
Nox4 enhances O2− production in mitochondria
In order to obtain biochemical evidence of Nox4 in mitochondria, we prepared mitochondrial fractions from 12 month old NTg and Tg-Nox4 mouse hearts and examined them for protein expression of Nox4 and O2− production. The purity of the mitochondrial fraction was confirmed by the absence of BiP, a marker of endoplasmic reticulum (ER), and histone H3, a marker of the nuclear fraction (Fig. 6A). Immunoblot analyses showed that Nox4 is present in the mitochondrial fraction, although a lesser amount was also found in the microsomal fraction (Fig. 6A). In contrast, expression of endogenous Nox2 was observed primarily in the microsome fraction presumably at the plasma membrane and the level of Nox2 was not increased in Tg-Nox4 (Fig. 6A). The lucigenin chemiluminescent assay for O2− production indicated that NADH-dependent O2− production was easily detected in the mitochondrial fraction obtained from NTg mouse hearts, and it was significantly greater in Tg-Nox4 than in NTg. NADPH-dependent O2− production was also observed in both Tg-Nox4 and NTg, but at a lower level than the NADH-dependent O2− production (Fig. 6B). The NADH- and NADPH-dependent O2− production was also observed in the microsomal fraction, although at much lower levels than in the mitochondrial fraction. The NADH-dependent ROS production in the mitochondrial fractions was inhibited by rotenone, an inhibitor of Complex I, by ~ 50%, and further inhibited by diphenyleneiodonium, an inhibitor of NADPH oxidases, by another ~ 40% (Fig. 6C). The fluorescent intensity of Mitosox, a specific indicator of O2− production in the mitochondria, was also enhanced in myocytes overexpressing Nox4 (Fig. 6D). Thus, these results suggest that significant levels of Nox-dependent O2− producing activity in the heart exist in the mitochondrial fraction. Since NADH-dependent O2− production in the mitochondria is partially rotenone-dependent, O2− production by Nox4 may increase electron leakage from mitochondrial Complex I, thereby further stimulating O2− production in mitochondria.
Figure 6.
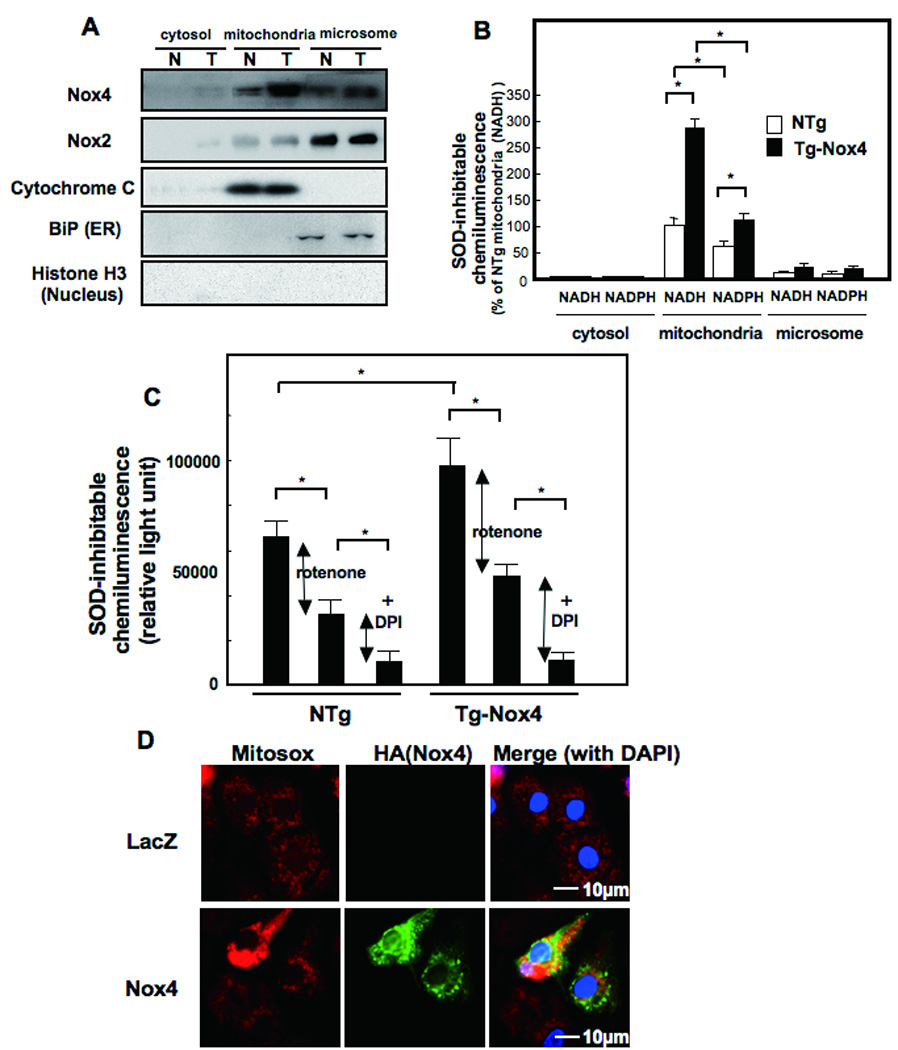
Localization of Nox4 in cardiac myocytes. (A and B) Cytosolic, mitochondrial membrane, and microsomal fractions were prepared from aging NTg and Tg-Nox4 mouse hearts. (A) Immunoblot analyses were conducted with anti-Nox4 monoclonal antibody (3D2) and anti-Nox2 polyclonal antibody. The purity of the mitochondrial fraction was confirmed by the lack of histone H3 or BiP staining. The purity of cytosolic fraction was confirmed by the lack of cytochrome c. (B, C) NADH- (B, C) and NADPH-dependent (B) O2− release was determined by the lucigenin method. The SOD inhibitable component of O2− release from each fraction was determined. *p<0.05. (C) NADH-dependent and SOD inhibitable O2− release from the mitochondrial membrane fraction consists of Rotenone-sensitive (shown by red arrow) and Rotenone-insensitive, DPI-sensitive (shown by blue arrow) components. The Rotenone-insensitive, DPI-sensitive components may include O2− release from Nox localized in the mitochondria *p<0.05. (D) Cultured neonatal rat cardiac myocytes were transduced with adenovirus harboring LacZ or Nox4. The extent of oxidative stress in the mitochondria was evaluated using MitoSOX™.
Upregulation of Nox4 induces mitochondrial damage in the heart
In order to evaluate whether increased production of O2− by Nox4 leads to mitochondrial dysfunction, the effect of Nox4 upon mitochondrial membrane potential was evaluated with TMRE and JC-1 staining in cardiac myocytes. Nox4 induced depolarization of the mitochondrial membrane potential (Fig. 7A), suggesting that mitochondrial function/integrity was attenuated in the presence of increased expression of Nox4. In order to evaluate whether apoptotic cell death induced by Nox4 overexpression is mediated through increased O2− in mitochondria, we expressed MnSOD, which is localized in mitochondria and dismutates O2−, in cardiac myocytes. MnSOD significantly attenuated Nox4-induced increases in O2−, as evaluated by DHE staining, in cardiac myocytes (Fig. 7B), and suppressed Nox4-induced increases in apoptosis, as evaluated by TUNEL assays (Fig. 7C), suggesting that mitochondrial O2− plays an essential role in mediating apoptosis in the presence of Nox4 overexpression in cardiac myocytes.
Figure 7.
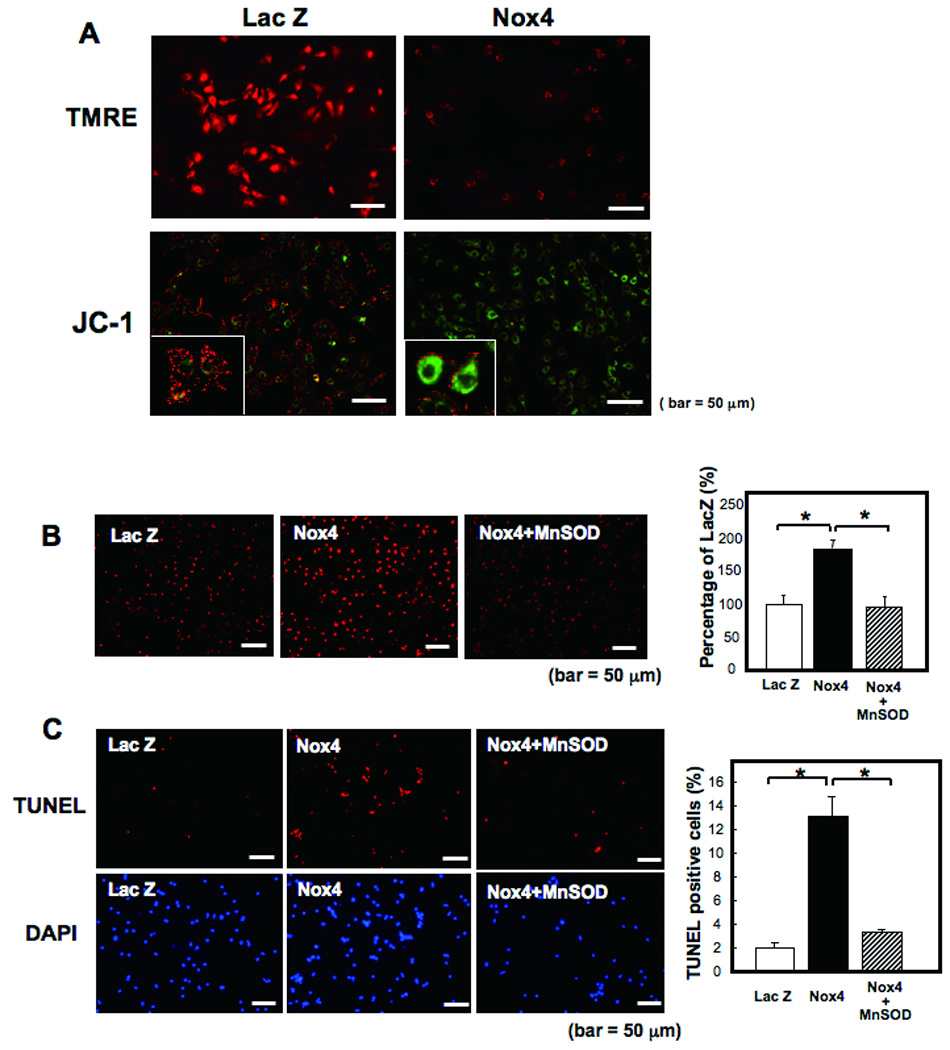
A) Cultured neonatal rat cardiac myocytes were transduced with adenovirus harboring Nox4 or Lac Z. After 48 hours, the cells were subjected to TMRE and JC-1 staining for mitochondrial membrane potential assessment. Note that red staining indicates polarized mitochondria in TMRE and JC-1 staining. Green staining indicates depolarized mitochondria in JC-1 staining. B,C) Cultured neonatal rat cardiac myocytes were transduced with adenovirus harboring Nox4 or Lac Z, or co-transduced with Ad-Nox4 and Ad-MnSOD. B) After 48 hours, superoxide production was evaluated with DHE staining. *p<0.05. C) The number of apoptotic cells were evaluated with TUNEL staining. *p<0.05.
We further investigated how increased expression of Nox4 leads to the impairment of mitochondrial function. Increased O2− production in the mitochondria may result in greater oxidation of mitochondrial proteins in Tg-Nox4 than in NTg. We tested this hypothesis using a proteomic approach. The results of ICAT analysis 15 demonstrated that the cysteine residues of many mitochondrial proteins are oxidized in Tg-Nox4 to a greater extent than in NTg. In particular, proteins involved in the TCA cycle, including aconitase-2 and citrate synthase, and the electron transport chain (ETC), including components of the NADH dehydrogenase complex, were significantly oxidized at cysteine residues (Fig. 8A and Online Fig. VI). In addition, components of the MPTP complex, including adenine nucleotide translocase type 1 (ANT1), were highly oxidized in Tg-Nox4 mouse hearts. Consistent with previous reports that oxidation attenuates the function of mitochondrial proteins 19, aconitase-2 and citrate synthase activities were significantly lower in Tg-Nox4 than in NTg (Fig. 8B). We confirmed that mitochondrial proteins, including aconitase-2 and ANT1, are similarly oxidized by either increased expression of Nox4 or clinically relevant stimuli such as TAC, as evaluated by iodoacetamide-biotin labeling experiments (Fig. 8C).
Figure 8.

Overexpression of Nox4 causes mitochondrial dysfunction. Purified mitochondrial fractions were prepared from aging Tg-Nox4 and NTg mouse hearts and subjected to ICAT proteomics or biochemical assays. A) The ICAT signal at cysteine 385 of aconitase, cysteine 126 of aconitase, cysteine 101 of citrate synthase, cysteine 206 of NADH dehydrogenase (51 kD subunit), cysteine 367 of NADH dehydrogenase (71 kD subunit), and cysteine 160 of adenine nucleotide translocase type 1 (ANT1) were significantly lower in Tg-Nox4 hearts than in NTg hearts, suggesting that these cysteines are oxidized. (B) The aconitase activity and the citrate synthase activity in the heart were compared among Tg-Nox4, Tg-Nox4-P437H and NTg mice. C) Twelve month old NTg and Tg-Nox4 mouse hearts were lysed in the presence of biotinylated iodoacetamide. Biotinylated proteins were pulled down on streptavidin beads and subjected to immunoblotting (Left panel). Three month old NTg mice were subjected to TAC or sham operation. The hearts were treated in the same way as above (Right panel). D) Evaluation of mitochondrial biogenesis. Twelve month old NTg or Tg-Nox4 mouse hearts were subjected to immunoblotting using antibodies raised against PGC-1α and TFAM (Left panel). Quantitative real-time PCR for mitochondrial DNA (Right panel). *p<0.05 vs NTg.
We also evaluated mitochondrial biogenesis as another mechanism through which Nox4 promotes mitochondrial dysfunction. Expression of genes involved in mitochondrial biogenesis in the heart, including PGC-1α and TFAM, was downregulated, and the mtDNA content was reduced in Tg-Nox4 compared to NTg, suggesting that mitochondrial biogenesis is attenuated in response to upregulation of Nox4 (Fig. 8D). Taken together, these results suggest that increased O2− production in the mitochondria caused by Nox4 upregulation induces oxidative mitochondrial damage, including oxidation of mitochondrial proteins, thereby leading to mitochondrial dysfunction and apoptotic cell death.
Discussion
Using a newly generated anti-Nox4 antibody and cardiac specific Nox4 transgenic mice, we here demonstrate three major properties of Nox4 in the heart. First, Nox4 is upregulated upon aging and hypertrophic stimulation, including pressure overload. Second, overexpression of Nox4 in the heart increases O2− production and induces cardiac dysfunction accompanied by increased fibrosis and apoptosis, but not obvious cardiac hypertrophy at the organ level. Third, Nox4 is localized primarily at mitochondria and oxidizes mitochondrial proteins involved in the TCA cycle and the electron transport chain, thereby leading to mitochondrial dysfunction.
The O2− producing activity of Nox4 is believed to be determined primarily at the level of protein expression 9. Our results, therefore, suggest that O2− production by Nox4 should be enhanced in cardiac myocytes subjected to hypertrophic stimuli and aging, which may contribute to increases in oxidative stress in hypertrophied hearts. Oxidative stress at middle age, as evaluated by DHE and 8-OHdG staining, was attenuated in Tg-Nox4-P437H hearts. These results suggest that Nox4 plays an important role in mediating increases in O2− and oxidative stress due to hypertrophic stimulation or aging in the heart. The molecular mechanism which mediates upregulation of Nox4 in response to hypertrophic stimuli remains to be elucidated.
An important question is whether upregulation of Nox4 stimulates cardiac hypertrophy. Overexpression of Nox4 in transgenic mice did not induce cardiac hypertrophy at baseline at young ages. On the other hand, although middle-aged Tg-Nox4 mice did not exhibit hypertrophy compared to NTg at the organ level, their myocyte size was greater than that of NTg. However, since cardiac function was attenuated in the middle-aged Tg-Nox4 hearts, the increase in cardiac myocyte size could be caused secondarily due to reduced LV function. The lack of cardiac hypertrophy at the organ level despite the presence of hypertrophy at the cell level may be due to myocyte loss. This notion is supported by the results of in vitro experiments, in which overexpression of Nox4 induces apoptosis, but not hypertrophy, in myocytes. Alternatively, Nox4 may not only stimulate apoptosis but also either gradually induce hypertrophy or enhance aging-induced hypertrophy in vivo. For example, if myocytes with Nox4 upregulation express antioxidants or cell survival factors sufficient for preventing apoptosis, such cells may eventually undergo hypertrophy. Nox4 induces hypertrophy in the kidney and mesangial cells 20, and proliferation in pulmonary vascular smooth muscle cells 21. Thus, the effect of Nox4 upon cell growth appears to be cell type-dependent.
Overexpression of Nox4 in Tg-Nox4 hearts significantly increased apoptosis in an age-dependent manner, which was accompanied by increased fibrosis and reduced LV function. Since overexpression of Nox4 significantly increased apoptosis in cultured cardiac myocytes, Nox4 must have cell-autonomous pro-apoptotic effects. Nox4 also induces differentiation of cardiac fibroblasts into myofibroblasts, thereby stimulating cardiac fibrosis 22. Enhanced expression of Nox4 due to aging increases apoptosis and fibrosis, which may contribute to the age-dependent decline in cardiac function. The reason why cardiac function is normal at baseline in young Tg-Nox4 remains to be elucidated. One possibility is that some other factors are necessary for Nox4 to exert its adverse effects on the heart. Although Nox4 is believed to produce O2− without cytosolic co-factors 23, the activity of Nox4 may be regulated by co-factors. Insulin induces Nox4 ROS generation within 5 min 24, a response which might be too fast to involve upregulation of Nox4, and Rac1 is required for Nox4 activation in mesangial cells 25. Another possibility is that factors suppressing Nox4-derived ROS may be decreased or inactivated with aging 26, 27.Our results suggest that catalase is upregulated in young Tg-Nox4 compared to NTg mice. Thus, the heart at young ages may have an ability to adapt to the increased Nox4 expression. Alternatively, the slowly progressive phenotype in the Tg-Nox4 heart could be secondary to other unknown functions of Nox4.
We expect that Nox4-P437H, which cannot use NADPH or NADH, forms a heterodimer with p22phox and disrupts interaction between Nox4 and p22phox , thereby inhibiting O2− production by endogenous Nox4 28. In theory, Nox4-P437H could inhibit O2− production by Nox2, another Nox isoform in cardiac myocytes, since Nox2 also forms a heterodimer with p22phox. We believe, however, that the effect of Nox4-P437H is primarily mediated through Nox4 because both endogenous Nox4 and Nox4-P437H are primarily localized in intracellular membranes, whereas Nox2 is localized in the plasma membrane.
Nox4 is localized at focal adhesions in vascular smooth muscle cells 29, in the nucleus in vascular endothelial cells 30, in the endoplasmic reticulum (ER) in human endothelial cells 28, and in mitochondria in mesangial cells 31. The diverse subcellular localization of Nox4 in previous reports could be due to differences in cell types and/or in the specificity of Nox4 antibodies. Our results, obtained with the newly generated anti-Nox4 specific antibody, suggest that Nox4 is localized in the peri-nuclear region, including in mitochondria, in cardiac myocytes. Immunoblots and biochemical analyses of subcellular fractions from mouse hearts (Fig. 6), as well as the presence of the functional MLS (Figs. 5CD), support the notion that Nox4 is localized primarily in the mitochondria and partially in the microsomes in cardiac myocytes.
Overexpression of Nox4 enhances O2− production in mitochondria at both the cellular and tissue levels (Fig. 6), suggesting that a similar degree of Nox4 upregulation in response to hypertrophic stimuli (Fig. 1C) could increase O2− production in mitochondria. O2− produced by Nox4 is rapidly dismutated to H2O2, which is freely diffusible in cells. Thus, in theory, increased production of ROS even outside mitochondria could lead to oxidative damage in mitochondrial proteins. However, increased oxidation of cysteine residues in many mitochondrial proteins in Tg-Nox4 was observed in young mice without obvious cardiac phenotype. Thus, Nox4 localized in the mitochondria could promote oxidation of mitochondrial proteins more efficiently.
In the cardiac mitochondrial fraction of Tg-Nox4, both NADH- and NADPH-dependent O2− production was significantly increased. Interestingly, a greater level of O2− was produced from NADH than from NADPH, consistent with the notion that Nox4 can use NADH efficiently as an electron donor 9, 17, 23. Importantly, both rotenone, an inhibitor of Complex I, and DPI, an inhibitor of NADPH oxidase, significantly inhibited the NADH-dependent ROS production in the mitochondrial fraction (Fig. 6C). Thus, we speculate that Nox4 increases O2− production in mitochondria, which triggers mitochondrial dysfunction, thereby leading to O2− production/leakage from mitochondria (Online Fig. VII). Nox4-induced increases in ROS may further stimulate “ROS-induced ROS release (RIRR)” 32, 33, where Complex I is an important source of ROS 32, and oxidation of the MPTP and depolarization of the mitochondrial membrane potential play an important role in enhancing ROS 33.
We have shown previously that oxidation of the MPTP complex occurs during pressure overload, and is suppressed drastically by overexpression of thioredoxin 1, an anti-oxidant, in mouse hearts 15. Consistently, critical redox-sensitive cysteine residues of NADH dehydrogenase flavoprotein I, a component of Complex I, ANT1, a key component of the MPTP complex, and aconitase-2, an established redox sensitive protein, are highly oxidized in Tg-Nox4. Since cysteine oxidation of these proteins also occurs by pressure overload, upregulation of Nox4 may play an important role in mediating cysteine oxidation of mitochondrial proteins during cardiac stress. Upregulation of Nox4 also negatively regulates mitochondrial biogenesis. Taken together, Nox4 strongly drives oxidative stress in mitochondria in the heart, either alone or by enhancing RIRR, which in turn enhances mitochondrial dysfunction and myocardial cell death, thereby leading to the development of cardiac dysfunction.
Novelty and Significance.
What is known?
The Nox2 and Nox4 isoforms of NAD(P)H oxidase, enzymes producing superoxide from molecular oxygen, are expressed in cardiac myocytes.
Nox2 mediates increases oxidative stress and cardiac hypertrophy in response to some forms of stress in the heart.
What new information does this article contribute?
Upregulation of Nox4 in response to hypertrophic stimuli induces cell death and cardiac dysfunction in the heart.
Nox4 is localized primarily in mitochondria and increases superoxide production in cardiac myocytes.
Upregulation of Nox4 induces oxidation and dysfunction of mitochondrial proteins.
Summary.
Essentially nothing is known regarding the isoform-specific function of Nox4 in the heart. We wished to elucidate whether increased expression of Nox4 increases oxidative stress, whether Nox4 affects growth and death of cardiac myocytes, and where Nox4 is localized in cardiac myocytes.
Using a newly generated Nox4-specific monoclonal antibody and cardiac specific Nox4 overexpressing mice, we demonstrate that increased expression of Nox4 in the heart induces apoptosis of cardiac myocytes and left ventricular dysfunction. Nox4 is localized primarily in mitochondria and produces superoxide in cardiac myocytes. Upregulation of Nox4 induces oxidation of mitochondrial proteins, which in turn induces mitochondrial dysfunction, including mPTP opening and cytochrome c release.
The findings that Nox4 is localized in mitochondria and that increased expression of Nox4 induces mitochondrial dysfunction are particularly important because pathological hypertrophy is accompanied by increases in oxidative stress in mitochondria, mitochondrial dysfunction, and consequent increases in cardiac myocyte apoptosis. Thus, our results suggest that Nox4 may be an important source of oxidative stress in the failing heart.
Targeted inhibition or deletion of Nox4 specifically may reduce oxidative stress and cell death, thereby improving heart function in patients with pathological cardiac hypertrophy.
Supplementary Material
Acknowledgments
The authors thank Daniela Zablocki for critical reading of the manuscript.
Sources of Funding
This work was supported in part by U.S. Public Health Service Grants HL 59139, HL67724, HL69020, HL91469, and AG27211, and Foundation Leducq Transatlantic Network of Excellence
Non-standard Abbreviations and Acronyms
- 8-OHdG
8-hydroxyl-deoxyguanosine
- Ad
adenovirus
- CSA
cross sectional area
- DHE
dihydroethidium
- DPI
diphenyleneiodonium
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- ETC
electron transport chain
- FAD
flavin adenine dinucleotide
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- ICAT
isotope-coded affinity tag
- LV
left ventricle
- MLS
mitochondrial localization signal
- MPTP
mitochondrial permeability transition pore
- NTg
non-transgenic
- RIRR
ROS-induced ROS release
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TAC
transverse aortic constriction
- Tg
transgenic
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WGA
wheat germ agglutinin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- 1.Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- 2.Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002;8:132–140. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- 3.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 4.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 5.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. Febs J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 7.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 9.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krause KH. Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol. 2007;42:256–262. doi: 10.1016/j.exger.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 13.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 14.Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, Colucci WS. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation. 2004;109:1168–1171. doi: 10.1161/01.CIR.0000117229.60628.2F. [DOI] [PubMed] [Google Scholar]
- 15.Fu C, Hu J, Ago T, Sadoshima J, Li H. Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res. 2008;7:3789–3802. doi: 10.1021/pr800233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem. 2004;279:51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- 17.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 18.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 19.Bulteau AL, Lundberg KC, Ikeda-Saito M, Isaya G, Szweda LI. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci U S A. 2005;102:5987–5991. doi: 10.1073/pnas.0501519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 21.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 22.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 23.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol Renal Physiol. 2003;285:F219–F229. doi: 10.1152/ajprenal.00414.2002. [DOI] [PubMed] [Google Scholar]
- 26.Ji LL, Dillon D, Wu E. Myocardial aging: antioxidant enzyme systems and related biochemical properties. Am J Physiol. 1991;261:R386–R392. doi: 10.1152/ajpregu.1991.261.2.R386. [DOI] [PubMed] [Google Scholar]
- 27.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–742. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 28.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 29.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 30.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, Sumimoto H. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells. 2005;10:1139–1151. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 31.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 33.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.