

Abstract
Based on the template of a recently introduced derivatization reagent for aldehydes, 4-(2-(trimethylammonio)ethoxy)benzeneaminium dibromide (4-APC), a new derivatization agent was designed with additional features for the analysis and screening of biomarkers of lipid peroxidation. The new derivatization reagent, 4-(2-((4-bromophenethyl)dimethylammonio)ethoxy)benzenaminium dibromide (4-APEBA) contains a bromophenethyl group to incorporate an isotopic signature to the derivatives and to add additional fragmentation identifiers, collectively enhancing the abilities for detection and screening of unknown aldehydes. Derivatization can be achieved under mild conditions (pH 5.7, 10 °C). By changing the secondary reagent (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide instead of sodium cyanoborohydride), 4-APEBA is also applicable to the selective derivatization of carboxylic acids. Synthesis of the new label, exploration of the derivatization conditions, characterization of the fragmentation of the aldehyde and carboxylic acid derivatives in MS/MS, and preliminary applications of the labeling strategy for the analysis of aldehydes in urine and plasma are described.
Figure.
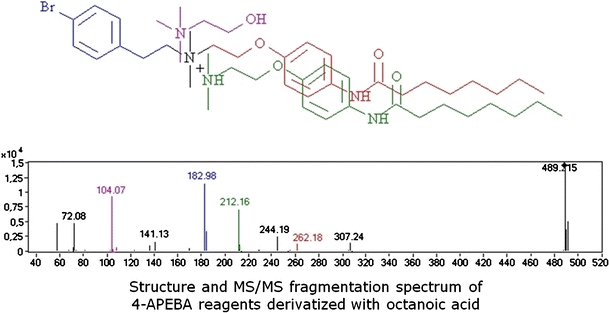
Structure and MS/MS fragmentation spectrum of 4-APEBA reagents derivatized with octanoic acid
Electronic supplementary material
The online version of this article (doi:10.1007/s00216-010-3575-1) contains supplementary material, which is available to authorized users.
Keywords: Derivatization, Aldehydes, Carboxylic acids, 4-APEBA, 4-APC, LC–MS/MS, Lipid peroxidation
Introduction
One of the incentives in the initial development of combined liquid chromatography–mass spectrometry (LC–MS) was to avoid the need for analyte derivatization [1], which is often needed in gas chromatography (GC) and may be considered as one of the limitations of GC–MS. In GC, derivatization of more polar compounds is obligatory to enable the GC analysis, whereas in GC–MS it may also improve ionization and/or fragmentation characteristics. However, because analyte ionization in LC–MS heavily relies on liquid-phase or gas-phase acid–base chemistry, analyte derivatization must be advantageous for analytes with poor properties in that respect. Early accounts on the derivatization of, for instance, prostaglandins [2] and steroids [3] for LC–MS show this. At present, analyte derivatization in LC–MS is frequently used in the analysis of compound classes like steroids [4, 5], sugars [6], and amino acids [7]. In the latter case, the derivatization is not only directed at improving the detectability but also at enhancing the chromatographic properties. In general, analyte derivatization may be performed for various reasons: increase the stability of the analyte, improve the separation from matrix components in sample pretreatment and/or chromatography, enhance the ionization efficiency, and/or alter the fragmentation characteristics in MS.
Two compound classes for which derivatization for LC–MS is highly desirable or even obligatory are aldehydes and carboxylic acids. Both are frequently analyzed as they are considered important biomarkers of oxidative stress and free radical damage. Lipid peroxidation (LPO), one of the main consequences of free radical damage, is implied in various diseases. Aldehydes and carboxylic acids may be formed as a result of LPO and, as such, both classes have been extensively studied as potential biomarkers of reactive oxidative species and/or radical damage [8–10]. Next to GC–MS [11] and various other methods [12, 13], LC–MS has become an important tool in the analysis of both aldehydes [14–17] and carboxylic acids [18, 19]. However, some problems are associated with this. Aldehydes can be measured directly in plasma but, when extracted from urine, they may suffer from degradation to aliphatic carboxylic acids [20]. Moreover, the volatility and intrinsically low response of aldehydes in LC–MS present analytical challenges. Carboxylic acids, on the other hand, are more stable but cannot be easily analyzed in positive-ion mode. In all these analytically challenging cases, the concept of derivatization can play an accelerating role.
From an organic–chemical perspective, derivatization involves an organic reaction of an analyte molecule with a suitable derivatization reagent. Thus, derivatization reagents contain a reactive functional group that (selectively) reacts with a complementary functional group in the analyte. For LC–MS, the derivatization strategy is typically directed at the incorporation of a group with a charge (cationic groups for positive-ion mode and strongly acidic functionality for negative-ion mode) or other groups that enhance ionization (secondary or tertiary amine for positive-ion mode or aromatic nitro groups in negative-ion mode). This combination of a chemical and analytical component can be found in a variety of agents that are available for aldehyde derivatization, such as the commercially available hydrazine-based Girard T and P reagents and dinitrophenylhydrazine [15, 21–23].
However, many of these off-the-shelf reagents still possess drawbacks, such as lack of selectivity or formation of unstable derivatives. More advanced derivatization agents, in which the chemical structure of the reagent has been fine-tuned to address such issues, have been reported [4, 17, 22–26], but mostly on a scattered basis. Recently, we reported [27, 28] a novel derivatization agent, 4-(2-(trimethylammonio)ethoxy)benzeneaminium dibromide (4-APC; Fig. 1a). 4-APC was designed for the selective determination of aldehydes in biological samples by positive-ion electrospray ionization–mass spectrometry (ESI–MS). Inspired by the interesting chemical and analytical properties of 4-APC, we decided to use it as a design template to address some additional stringent features of (oxidative) biomarker analysis. First, incorporation of an element with multiple stable isotopes would give a significant advantage as it allows the specific screening for unknown derivatives easily visible by their incorporated isotopic signature. Second, the signal-to-noise ratio should be as high as possible. Third, the fragmentation patterns of the adducts in tandem MS (MS/MS) should give several different types of information, thus enabling powerful MS acquisition strategies such as not only selected reaction monitoring (SRM) but also neutral loss and/or precursor-ion analysis modes. Fourth, an ideal scenario would involve the use of one derivatization agent for multiple analyte classes with reactivity being directed by a co-reagent.
Fig. 1.

Relevant reaction pathways. a Reaction of 4-APC and hexanal (blue) through a transient imine. b Reaction of 4-APEBA and hexanal (blue) through a transient imine. c Reaction of 4-APEBA with hexanoic acid (green) through a transient O-acyl-isourea. In reaction C, the R and R1 groups arbitrarily represent the two peripheral groups of EDC
In this publication, we describe the design, synthesis, and analytical properties of a novel derivatization reagent 4-(2-((4-bromophenethyl)dimethylammonio)ethoxy)benzenaminium dibromide (4-APEBA; 1 in Figure 1b) that addresses these four demands. Compared to the first-generation reagent 4-APC, 4-APEBA contains an extra bromophenethyl group which results in several benefits. The isotopic signature of 79Br and its 81Br isotope (100:98) provides confirmation of the presence of bromine in the derivatives. Furthermore, the total molecular mass of 4-APEBA derivatives is higher than that of 4-APC derivatives resulting in a higher signal-to-noise ratio in biological samples. The fragmentation of 4-APEBA derivatives displays an additional advantage in the form of consistent fragments and neutral losses. Last, it will be shown how a simple switch of co-reagent (EDC instead of NaBH3CN) turns 4-APEBA into a selective derivatization reagent for aliphatic carboxylic acids. Preliminary studies on aldehyde analysis in urine and plasma demonstrate its applicability in biological samples.
Experimental
Chemicals
Pentanal, hexanal, heptanal, octanal, nonanal, decanal, pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, nonanoic acid, sodium cyanoborohydride (NaBH3CN), ammonium acetate, 2,2′-azobis(2-amidopropane) dihydrochloride (AAPH), dichloromethane (DCM), NaOH, Na2SO4, KOH, ethyl acetate (EtOAc), methyl-tert-butyl ether (MTBE), aq. HBr solution, ethanol (EtOH), 2-(4-bromophenyl)ethanamine, formaline, formic acid (for synthesis), and human plasma were all purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). Methanol (MeOH), acetonitrile (MeCN), and formic acid (for analysis) were purchased from Biosolve (Valkenswaard, The Netherlands). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were purchased from Perbio Science (Etten-leur, The Netherlands). 4-APC dibromide was prepared according to the procedure reported before [28]. The urine was obtained from five human volunteers.
2-(4-Bromophenyl)-N,N-dimethylethanamine (3)
A solution of 2-(4-bromophenyl)ethanamine 2 (4.0 g, 20.1 mmol) in formaline (30% aq, 60 ml) and formic acid (50 ml) was heated to reflux for 5 h. The solution was concentrated by rotary evaporation. The solid residue was stirred with NaOH solution (10%, 50 ml) for 0.5 h. DCM was added; the whole was stirred vigorously for 5 min, and the layers were separated. The aqueous layer was extracted twice more with DCM. The combined organic layers were dried (Na2SO4) and concentrated. Some solid condense was observed in the rotary evaporator. The residue was mixed with water (50 ml) and sonicated for 5 min. DCM (50 ml) and solid KOH (approximately 10 g) were added; the whole was stirred vigorously for 5 min, and the layers were separated. The organic layer was dried (Na2SO4), concentrated, and dried. The title compound was obtained as a yellow oil (3.8 g, 16.7 mmol, 83%). This compound was pure enough for further manipulation. The synthesis of this compound by a different route has been disclosed, but no nuclear magnetic resonance (NMR) data have been reported [29]. 1H-NMR data are presented in the Electronic Supplementary Material.
N-(4-(2-Bromoethoxy)phenyl)acetamide (4)
Prepared according to Eggink et al. [28].
2-(4-Acetamidophenoxy)-N-(4-bromophenethyl)-N,N-dimethylethanaminium bromide (5)
Amine 3 (3.4 g, 14.9 mmol) and bromide 4 (3.8 g, 14.7 mmol) were mixed with MeCN (18 ml). The suspension was briefly warmed (approximately 10 s), resulting in a dark-brown clear solution. The solution was stirred in the dark for 4 days at room temperature. During this time, the product gradually precipitates. EtOAc (20 ml) was added dropwise to the suspension. The solid is filtered, washed with EtOAc (2×), and dried. The title compound is obtained as an off-white solid (5.15 g, 72%) with sufficient purity for further synthetic manipulations. An analytical sample can be obtained by recrystallization from a mixture of MeOH, MeCN, and MTBE to give beige crystals. M.p. = 194.4–195.2 °C. NMR data are presented in the Electronic Supplementary Material.
4-(2-((4-Bromophenethyl)dimethylammonio)ethoxy) benzenaminium dibromide (1, 4-APEBA)
Salt 5 (2.5 g, 5.17 mmol) was mixed with water (28 ml), giving a suspension. Then, aq. HBr solution (48%, 46 ml) was added, resulting in a clear solution. The mixture was heated to reflux for 45 min. The solvent was removed by rotary evaporation, and the residue was dried at 80 °C for 2 h. The solid residue was scraped off the walls of the flask and transferred to another flask. The solid was recrystallized from warm (not hot) EtOH/water. This afforded brown crystals (1.4 g, m.p. = 229.8–230.6 °C) which were shown to contain both 20% EtOH solvate and ∼2% of an impurity. The following second recrystallization effectively removed both these impurities. A total of 1.2 g of the material was mixed with H2O (2.0 ml). The suspension was warmed to ∼60 °C, and more H2O was added until complete dissolution (final volume ∼5 ml H2O). The mixture was left overnight in the dark at room temperature. The crystals were filtered, washed with ice-cold water (2.0 ml + 3.0 ml), and dried. This afforded beige needles with >98% purity (660 mg, extrapolated yield 29%). The product is stable when kept as a solid in the dark. Full chemical characterization and solution stability of 4-APEBA are presented in the Electronic Supplementary Material.
Sample pretreatment and derivatization
Aldehyde derivatization
For 4-APC derivatization reactions, 200 µl of 2.5 mg/ml 4-APC dibromide in 150 mM ammonium acetate buffer pH 5.7, 50 µl 0.75 mg/ml NaBH3CN in methanol, and 250 µl of the 500 nM aldehyde standards pentanal, hexanal, heptanal, octanal, nonanal, and decanal (in H2O) were taken. For the 4-APEBA derivatization reactions, 200 µl of 3 mg/ml 4-APEBA dibromide in 150 mM ammonium acetate buffer pH 5.7, 50 µl of 0.5 mg/ml NaBH3CN in methanol, and 250 µl of the aldehyde standards pentanal, hexanal, heptanal, octanal, nonanal, and decanal (in H2O) were taken. The derivatization reactions were carried out at 10 °C in the thermostated Agilent 1200 autosampler. After 3 h, the first sample was injected in the LC–MS/MS for analysis.
The urine/plasma samples were centrifuged at 13,600 rpm for 15 min at 10 °C before performing the derivatization reaction. The derivatization was carried out in the same way as the standard solutions. After 3 h, the first sample was injected in the LC–MS/MS for analysis.
Carboxylic acid derivatization
For the derivatization reactions, 200 µl of 3 mg/ml 4-APEBA dibromide in H2O, 50 µl of 100 mM NHS buffer pH 5.7, 50 µl of 290 mM EDC in H2O, and 200 µl of carboxylic acid standards pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, and nonanoic acid (1 mM in H2O) were used. In this case, the derivatization was carried out at 60 °C for 1 h in the thermostatic column oven in closed vials before injection.
In vitro oxidation of human plasma and analysis of formed aldehydes
Two hundred microliters of human plasma was mixed with 50 µl of AAPH solution (final concentration 1 mM), and the mixture was incubated at 37 °C for 4 h. An amount of 50 µl was taken from the solution and mixed with 50 µl of cold ACN, vortexed for 1 min, and centrifuged for 20 min at 13,600 rpm at 4 °C. For the derivatization, 50 µl of sample was mixed with 40 µl of 3 mg/ml 4-APEBA dibromide in 150 mM ammonium acetate buffer pH 5.7 and 50 µl of 0.5 mg/ml NaBH3CN in H2O. The control experiment was performed similarly with the exception that 50 µl of H2O was added instead of AAPH, without any incubation time.
HPLC
All high-performance liquid chromatography (HPLC) separations were performed on an Agilent 1200 HPLC system (Agilent Technologies, Amstelveen, The Netherlands) controlled by the Agilent Masshunter software. A Waters XTerra MS reversed-phase column (C18, 100 × 2.1 mm, 3 µm) at 45 °C and with a flow rate of 150 µl/min was used for the separation of the derivatized aldehydes, carboxylic acid standards, and urine and plasma samples. Samples were injected (10 µl) from a thermostatic autosampler kept at 10 °C. The gradient elution was programmed as follows: After injection, 100% mobile phase A (95% H2O + 5% MeOH + 0.1% formic acid) was maintained for 5 min and then solvent B (5% H2O + 95% MeOH + 0.1% formic acid) was increased from 5% to 90% in either 15 min (short gradient) or 25 min (long gradient) with a 5-min hold at 90% B. After this, the column was reconditioned for 10 min at 100% mobile phase A. The effluent from the LC column was directed to the mass spectrometer.
Mass spectrometry
LC–MS or LC–MS/MS were performed on an Agilent quadrupole time-of-flight (Q-TOF) hybrid mass spectrometer equipped with an Agilent 1200 LC system (Agilent Technologies, Amstelveen, The Netherlands). Positive-ion ESI–MS data were collected using capillary voltage of 3.5 kV, a fragmentor voltage of 120 V, skimmer at 65 V, and spectrum acquisition in the m/z range of 100–1,000. The source gas temperature was set to 350 °C with a drying gas flow of 7 l/min and a nebulization gas pressure of 15 psig. The collision energy was optimized for the best fragmentation patterns and was typically 60–80 V. Two internal calibrants with m/z 622.0290 and 922.0098, sprayed with a separate nebulizer, were employed to improve mass accuracy.
Results and discussion
Design and synthesis
As explained, for identification and characterization of known and unknown LPO biomarkers, we required a derivative of 4-APC which was further tailored with respect to four design demands. The incorporation of an element with multiple stable isotopes was envisaged through incorporation of a bromo group, which has two major isotopes separated by two atomic mass units (79 and 81) in a 100:98 ratio. Several sites of Br attachment were considered. Given that steric requirements and the pK a of the aniline moiety (vide infra) play crucial roles in the actual derivatization reaction [27, 30], bromo substitution on the 3- or 2-position of the oxy-aniline head was deemed suboptimal because of potentially compromised steric accessibility and/or a likely drop in the pK a (e.g., pK a ≈ 4 for 3-bromo-4-methoxyaniline [31]). A better alternative was to replace one of the N-methyl groups in 4-APC by a 4-bromophenyl-containing group, also because this would simultaneously address the second demand of increased mass and associated increase in signal-to-noise ratio. To maintain the highest chemical stability and optimal MS sensitivity, a C2 spacer between the charged group and the bromophenyl moiety, i.e., a bromophenethyl group, was preferred over a C0 or C1 spacer. Moreover, it was hoped that this phenethyl group would deliver an additional and reliable fragmentation site, thus aiding in the third demand.
All these new chemical features were deliberately installed remote from the aniline head. This ensures an anilinium pK a very similar to that of 4-APC (∼5.3) [27], which itself was deemed important for two reasons. First, the reaction with aldehydes will remain highly predictable. Second, the unique pK a also opens doors for the reaction of the candidate aniline with other carboxyl groups, such as carboxylic acid biomarkers. A typical derivatization cocktail for carboxylic acids will involve the use of EDC [32], which provides a transient O-acyl-isourea which is trapped by a nucleophile. In aqueous media of any relevant pH, aliphatic amines (pK a ∼ 10–11) are substantially more deactivated by protonation compared to anilines, which possess a lower pK a. This very difference bodes well for assistance in the fourth demand by enabling a relatively straightforward reaction of the aniline candidate with carboxylic acids in the presence of EDC.
Taken together, the design steps ultimately called for the previously unknown 4-APEBA (1 in Fig. 1b) as the target compound. The developed synthesis was similar to that of 4-APC [28] with the difference that, instead of NMe3, 2-(4-bromophenyl)-N,N-dimethylethanamine (3) was required. The synthesis consists of four steps only requiring recrystallizations as purifications (see Electronic Supplementary Material). Using this readily scalable protocol, more than a gram of crystalline, pure, and stable 4-APEBA was prepared.
Reactivity and analytical properties of 4-APEBA
Derivatization of aldehydes
First, we cross-checked 4-APEBA in the derivatization of aldehydes against first-generation reagent 4-APC. For the latter, we already showed that the derivatization of aliphatic aldehydes took less than 30 min to complete [27]. As shown in Fig. 2a, the derivatization of aldehydes with 4-APEBA at 10 °C and pH 5.7 proceeds at the same speed as with 4-APC. This reinforces our earlier notion that maintaining the exact same aniline head would lead to identical derivatization chemistry (Fig. 1a, b). The low reaction temperature is important because of the volatility of the aldehydes. Calibration curves, ranging from 0 to 200 nM pentanal and heptanal derivatized with either 4-APC or 4-APEBA, show similar linearity, slope, and intercept (see Electronic Supplementary Material Figure S10). Despite the fact that for 4-APEBA-derivatized aldehydes the ion intensity is split over the two Br isotope peaks, the response for the 4-APC derivatives of pentanal and heptanal in the extracted-ion chromatograms (with m/z 265 and 293, respectively) is the same as that for the 4-APEBA derivatives of pentanal and heptanal (with m/z 433 and 461, respectively). This indicates that an about twofold increased response is obtained with the 4-APEBA labeling compared to the 4-APC labeling. Thus, 4-APEBA is at least an equally effective and sensitive derivatization agent as 4-APC.
Fig. 2.

Reaction kinetics at 10 °C of 4-APEBA with: a aldehydes (trans-2-pentenal, pentanal, hexanal, heptanal, octanal, nonanal, decanal) in the presence of NaBH3CN. b Aliphatic acids (pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, and nonanoic acid) in the presence of EDC and NHS
Derivatization of carboxylic acids
Next, we explored the reactivity of 4-APEBA with carboxylic acids. Carboxylic acids are a special class of carbonyl biomarkers because deprotonation under (sub)neutral conditions leaves the carbonyl group unreactive as an electrophile. However, as extensively demonstrated by others before [33, 34], treatment of a carboxylic acid with reagents like EDC transforms the carboxylic acid to a transient O-acyl-isourea [32], which can be trapped by a NH2 nucleophile through its now highly electrophilic carbonyl. Figure 1c depicts this reaction sequence for 4-APEBA in comparison to the reaction with aldehydes (Fig. 1b). Collectively, the protocol involving EDC allows attachment of the NH2 group to a carboxylic acid. Under physiological conditions, this sequence is expected to be especially applicable to amines with a somewhat lower pK a, such as 4-APEBA (vide supra).
To test this hypothesis, we turned to a reported procedure which uses a cocktail of EDC and NHS [35, 36]. The use of NHS has been reported to increase the efficiency of EDC-mediated coupling reactions. Indeed, preliminary experiments with a cocktail of 4-APC, NHS, and EDC gave amide adducts with simple carboxylic acids (data not shown). When extended to 4-APEBA, we found that the short-chain aliphatic carboxylic acids are fully reacted within 300 min at 10 °C (Fig. 2b). The derivatization process can be accelerated by heating the reaction mixture to ∼60 °C, which is compatible with carboxylic acids because they are relatively nonvolatile. Figure S11, Electronic Supplementary Material, shows the extracted-ion chromatograms of 4-APEBA-derivatized pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, and nonanoic acid. The MS traces show the typical isotopic pattern for one bromine.
The stability of the reaction products has not yet been systematically investigated. In our current experience, no changes in peak areas were observed when samples standing in the autosampler were analyzed on three consecutive days.
MS/MS fragmentation of 4-APEBA aldehyde derivatives
Because the 4-APEBA derivatives of aldehydes and carboxylic acids contain a permanent positive charge, they can be readily analyzed using positive-ion ESI–MS. The M+ ion is observed for all compounds without fragmentation. Using collision-induced dissociation in MS/MS, two types of fragments are observed, that is: compound-specific fragments that still contain the (derivatized) aldehyde or carboxylic acid group and reactant-specific fragments resulting from bond cleavages with charge retention on the reactant side. The m/z values of the compound-specific fragments depend on the aldehyde or carboxylic acid analyzed, whereas the m/z values of the reactant-specific fragments are the same, irrespective of the compound analyzed.
In MS/MS, all tested 4-APEBA-derivatized aldehydes showed similar fragmentation. As an example, the spectrum of the nonanal derivative (C27H42BrN2O+ with m/z 489.213) is shown in Fig. 3a. The M+ precursor ion shows characteristic neutral losses of 227 and 255 amu, corresponding to the loss of 4-bromophenethyl dimethylamine (loss of C10H14BrN) and of both 4-bromophenethyl dimethylamine and ethene (loss of C12H18BrN). The m/z values of the resulting two fragments depend on the aldehyde derivatized, that is m/z 262 and 234 for the 4-APEBA-derivatized nonanal (cf. Fig. 3a). In terms of cleavage site and fragmentation pattern, these losses correspond to the losses of 59 and 87 amu in 4-APC-derivatized aldehydes [28]. Proposed structures for these fragments, involving a stable oxirane ring in the ion with m/z 262 and a protonated quinone imine in the ion with m/z 234, are given in the Electronic Supplementary Material (Figure S12A and S12B). In addition, four characteristic fragments are observed, the m/z values of which are independent of the aldehyde derivatized. The fragment ion with m/z 183 (C8H8Br+) corresponds to the 4-bromophenethyl cation (for structure, see Electronic Supplementary Material Figure S12C), whereas the other fragments are related to the quaternary ammonium group, that is the immonium ions with m/z 58 (C3H8N+) and m/z 72 (C4H10N+), and the quaternary ammonium ion with m/z 104 (C5H14NO+).
Fig. 3.

Typical MS/MS spectra of 4-APEBA-derivatized carbonyl compounds a 4-APEBA-derivatized nonanal, b 4-APEBA-derivatized octanoic acid
MS/MS fragmentation of 4-APEBA-derivatized carboxylic acid
The fragmentation of 4-APEBA-derivatized carboxylic acids in MS/MS is dominated by the cleavage between the 4-bromophenethyl group and the rest of the molecule, resulting in two complementary fragments, that is, an abundant fragment ion with m/z 183 due to the 4-bromophenethyl cation (C8H8Br+, see Electronic Supplementary Material Figure S13B) and an [M–182]+ ion, which is found with m/z 307 for 4-APEBA-derivatized octanoic acid (Fig. 3b).
Next to this structure-specific [M–182]+ fragment ion, another structure-specific fragment ion is observed due to the loss of 227 amu, which corresponds to the loss of 4-bromophenethyl dimethylamine (loss of C10H14BrN, also observed with 4-APEBA-derivatized aldehydes (see Electronic Supplementary Material Figure S13A)). Fragments related to the quaternary ammonium group are also observed for 4-APEBA-derivatized carboxylic acids (ions with m/z 58,72, and 104).
Features of 4-APEBA derivatization in biomarker detection and screening
After derivatization, both the aldehydes (pentanal, hexanal, heptanal, octanal, nonanal, and decanal) and the carboxylic acids (pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid, nonanoic acid) can be readily separated on a reversed-phase C18 column using a methanol/water gradient (containing 0.1% formic acid).
Based on the (fragmentation) characteristics of the 4-APEBA derivates, one may envisage various strategies to optimize detection of known aldehyde and carboxylic acid biomarkers, to confirm their identity, and/or to screen for unknown aldehyde or carboxylic acid biomarkers.
In single MS applications, the presence of the bromine in the 4-APEBA label provides an isotopic signature to all derivatized compounds of two isotope peaks of almost equal abundance with an m/z difference of two.
In MS/MS applications, this isotopic signature can still be retained if a wider than unit mass precursor-ion selection window is applied, as is currently often practiced in ion trap and Q-TOF instruments. In the current research, a Q-TOF instrument was applied. In targeted analysis with a Q-TOF instrument, MS1 may be set to select particular precursor ions in prespecified time segments of the chromatogram (time-scheduled product-ion MS/MS). Next to extracted-ion chromatograms for known m/z values of compound-specific fragments (Fig. 4a), the resulting data sets may be interrogated to provide either neutral loss chromatograms (fixed neutral loss of 227 amu for both aldehydes (Fig. 4b) and carboxylic acids or 182 amu for carboxylic acids) or extracted-ion chromatograms for the common (reactant-specific) fragments with m/z 183 and 58 (Fig. 4c).
Fig. 4.

LC–MS/MS traces of 4-APEBA derivatives of butanal, pentanal, hexanal, heptanal, octanal, nonanal, and decanal: a Combined XIC of compound-specific fragments, b extracted-ion chromatogram (EIC) of the fixed neutral loss of the mass 227 amu, c EIC of the reactant-specific fragment ion with m/z of 183. For the separation, the long gradient described in experimental was used
Although a Q-TOF instrument was used by us, targeted analysis could also readily be performed using a triple–quadrupole instrument, operated either in the SRM mode to detect compound-specific transitions of M+ to particular fragments, in the neutral loss scan mode (fixed neutral loss of 227 amu for aldehydes and carboxylic acids or 182 amu for carboxylic acids) or in the precursor-ion scan mode (with fixed (reactant-specific) product ions with m/z 183 and/or 58). The use of a triple–quadrupole instrument was not attempted in this project.
Biological applications
Analysis of aldehydes in urine
New mechanistic insights on LPO products continue to emerge, often because biomarkers of novel structure are found [20]. We believe that our reagents 4-APC [27, 28] and especially 4-APEBA can be very useful in the detection and screening of LPO biomarkers. Based on the use of derivatization with 4-APC, we have previously described the detection of elevated aldehyde concentrations in urine [27] as well as the formation of oxidized aldehydes like 4-HNE in plasma by the treatment with an radical inducer [28]. The 4-APEBA derivatization agent is designed to extend this work, as it shows important features for the detection of unknown biomarkers by LC–MS/MS.
As an example of the use of 4-APEBA derivatization in the analysis of known aldehydes, urine samples of five healthy volunteers were spiked with 20 nM (final concentration) of hexanal, heptanal, octanal, nonanal, and decanal prior to derivatization with 4-APEBA. The urine samples were analyzed using full-spectrum LC–MS. The five combined extracted chromatograms (XIC) of pentanal, hexanal, heptanal, octanal, nonanal, and decanal derivatives are shown in Fig. 5. The peaks are labeled with the observed peak areas (not corrected using the urinary creatinine value). Although pentanal was not in the spike, it was detected in two out of the five urine samples. Hexanal was observed with an elevated peak area in one of the samples as well, whereas heptanal, octanal, nonanal, and decanal showed quite similar peak areas between the five samples. This relatively simple experiment clearly demonstrates that sample-to-sample variation can be detected.
Fig. 5.

Combined extracted-ion chromatograms for five 4-APEBA-derivatized urine samples spiked with 20 nM of hexanal, heptanal, octanal, nonanal, and decanal. Individual peaks are labeled with the peak area. For the separation, the short gradient described in experimental was used
Based on the accurate m/z values from the Q-TOF mass spectrometer, the identity of the six derivatives was confirmed with a mass accuracy within 5 ppm using internal calibration, as demonstrated in Table 1.
Table 1.
Chemical formulas generated from extracted HR mass measurements
| Compounds | t R | Formula (M+) | Score | Mass | Calc mass | Abs diff (ppm) |
|---|---|---|---|---|---|---|
| Pentanal | 11.14 | C23H34BrN2O+ | 100 | 433.1860 | 433.18545 | 1.27 |
| Hexanal | 11.76 | C24H36BrN2O+ | 100 | 447.20222 | 447.2011 | 2.50 |
| Heptanal | 12.42 | C25H38BrN2O+ | 100 | 461.21465 | 461.21675 | 4.55 |
| Octanal | 13.10 | C26H40BrN2O+ | 100 | 475.23164 | 475.2324 | 1.59 |
| Nonanal | 13.79 | C27H42BrN2O+ | 100 | 489.2489 | 489.24805 | 1.74 |
| Decanal | 14.46 | C28H44BrN2O+ | 100 | 503.26547 | 503.2637 | 3.51 |
Screening and confirmation for unknown 4-APEBA derivatives
In order to demonstrate the potential of the 4-APEBA derivatization in the nontargeted screening of aldehyde biomarkers of LPO, we subjected human plasma to a radical inducer (AAPH) to force oxidative conditions. Human plasma prior to and after the AAPH treatment were derivatized with 4-APEBA/NaBH3CN. Subsequently, LC–MS and LC–MS/MS analysis was performed, and the data were collected.
By comparison of the MS data for the control and the AAPH-treated plasma, various new or elevated peaks appeared (data not shown). MS/MS experiments were performed to collect fragmentation data. Data for one of the elevated peaks are shown in Fig. 6. The extracted-ion chromatogram of m/z 477 shows two peaks, the first of which is about twice as high in the treated plasma compared to the control (Fig. 6a). Mass spectra of the two peaks show a doublet with m/z 477 and 479 (ratio 100:98) for the first peak and a doublet with m/z 475 and 477 (ratio 100:98) for the second peak (data not shown). This indicates that both peaks (most likely) are 4-APEBA derivatives. In the MS/MS spectrum, all the reactant-specific fragments with m/z 183, 58, 72, and 104 were present for both peaks. The MS/MS of the first elevated peak is shown in Fig. 6b. Based on the accurate mass data, chemical formulas were determined for both compounds in Fig. 6a (see Electronic Supplementary Material Table S1 for the generated formulas with the two samples). The formula of the second peak is consistent with an octanal derivative. The formula of the first peak indicates the presence of a C6 chain which, judging from the C/O ratio, is highly oxidized, e.g., a C6 peroxide. The AAPH radical inducer is known to generate peroxides [20]. This unknown compound is also present in the control plasma. Further identification of this unknown product was not attempted; the MS/MS spectrum does not provide clear information for further elucidation. The radical inducer neither increased nor decreased the abundance of the octanal in the plasma, which is somewhat in contrast to our earlier findings [28]. Nevertheless, this example shows how 4-APEBA derivatization can be used to screen biological samples for both known and unknown aldehydes and to detect elevated levels of known or unknown aldehydes. Further research in this direction is currently performed and results will be reported in due course.
Fig. 6.

Analysis of aldehyde biomarkers in plasma. a Comparison of extracted-ion chromatogram of m/z 477 for control (red) and AAPH-treated (blue) plasma, b MS/MS spectrum of the elevated peak at 17 min. The following gradient was used: 100% mobile phase A (95% H2O + 5% MeOH + 0.1% formic acid) was maintained for 5 min and then solvent B (5% H2O + 95% MeOH + 0.1% formic acid) was increased from 5% to 90% in 25 min (long gradient) with a 5-min hold at 90% B. After this, the column was reconditioned for 10 min at 100% mobile phase A
Conclusion
In the present publication, we demonstrate the design and synthesis of the novel derivatization agent 4-APEBA. Based on our first-generation reagent 4-APC, the design of 4-APEBA addressed four stringent issues in biomarker analysis. This led to the incorporation of a bromophenethyl unit. The reagent can be used on either aldehydes (using NaBH3CN as co-reagent) or carboxylic acids (using EDC as co-reagent). The new features of 4-APEBA are perfectly suited for screening and quantification purposes. The incorporated bromine provides a clearly detectable doublet in the mass spectra which significantly enhances peak recognition. Compared to the first-generation reagent 4-APC, 4-APEBA displays similar fragmentation patterns of the adducts. However, it provides a clear surplus value through the fixed neutral loss as well as one fixed charged (monobromo) fragment (with m/z 183) for all derivatives. This enables efficient screening for unknown biomarkers of LPO. Using 4-APEBA in conjunction with MS/MS and high-resolution MS (Q-TOF), we were able to detect potential highly oxidized aldehyde species in human plasma treated with AAPH. Future work from our labs will elaborate further on the design platform for derivatization reagents with the ultimate goal to identify these biomarkers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 499 kb)
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.McFadden WH. J Chromatogr Sci. 1980;18:97–115. [Google Scholar]
- 2.Voyksner RD, Bush ED, Brent D. Biomed Environ Mass Spectrom. 1987;14:523–531. doi: 10.1002/bms.1200140908. [DOI] [PubMed] [Google Scholar]
- 3.Lindberg C, Blomqvist A, Paulson J. Biol Mass Spectrom. 1992;21:525–533. doi: 10.1002/bms.1200211102. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths WJ, Liu S, Alvelius G, Sjovall J. Rapid Commun Mass Spectrom. 2003;17:924–935. doi: 10.1002/rcm.1002. [DOI] [PubMed] [Google Scholar]
- 5.Higashi T, Shimada K. Anal Bioanal Chem. 2004;378:875–882. doi: 10.1007/s00216-003-2252-z. [DOI] [PubMed] [Google Scholar]
- 6.Harvey DJ. J Am Soc Mass Spectrom. 2000;11:900–915. doi: 10.1016/S1044-0305(00)00156-2. [DOI] [PubMed] [Google Scholar]
- 7.Uutela P, Ketola RA, Piepponen P, Kostiainen R. Anal Chim Acta. 2009;633:223–231. doi: 10.1016/j.aca.2008.11.055. [DOI] [PubMed] [Google Scholar]
- 8.Guichardant M, Lagarde M. Eur J Lipid Sci Technol. 2009;111:75–82. doi: 10.1002/ejlt.200800069. [DOI] [Google Scholar]
- 9.Culbertson SM, Porter NA. Free Radic Res. 2000;33:705–718. doi: 10.1080/10715760000301231. [DOI] [PubMed] [Google Scholar]
- 10.Mariani E, Polidori MC, Cherubini A, Mecocci P. J Chromatogr B. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Spiteller G, Kern W, Spiteller P. J Chromatogr A. 1999;843:29–98. doi: 10.1016/S0021-9673(98)01078-4. [DOI] [Google Scholar]
- 12.Esterbauer H, Zollner H. Free Radic Biol Med. 1989;7:197–203. doi: 10.1016/0891-5849(89)90015-4. [DOI] [PubMed] [Google Scholar]
- 13.Wheatley RA. Trends Anal Chem. 2000;19:617–628. doi: 10.1016/S0165-9936(00)00010-8. [DOI] [Google Scholar]
- 14.Andreoli R, Manini P, Corradi M, Mutti A, Niessen WMA. Rapid Commun Mass Spectrom. 2003;17:637–645. doi: 10.1002/rcm.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolliker S, Oehme M, Dye C. Anal Chem. 1998;70:1979–1985. doi: 10.1021/ac9709458. [DOI] [PubMed] [Google Scholar]
- 16.Nagy K, Pollreisz F, Takats Z, Vekey K. Rapid Commun Mass Spectrom. 2004;18:2473–2478. doi: 10.1002/rcm.1648. [DOI] [PubMed] [Google Scholar]
- 17.Zurek G, Karst U. J Chromatogr A. 1999;864:191–197. doi: 10.1016/S0021-9673(99)01041-9. [DOI] [PubMed] [Google Scholar]
- 18.Yang WC, Adamec J, Regnier FE. Anal Chem. 2007;79:5150–5157. doi: 10.1021/ac070311t. [DOI] [PubMed] [Google Scholar]
- 19.Yang WC, Mirzaei H, Liu XP, Regnier FE. Anal Chem. 2006;78:4702–4708. doi: 10.1021/ac0600510. [DOI] [PubMed] [Google Scholar]
- 20.Schneider C, Porter NA, Brash AR. J Biol Chem. 2008;283:15539–15543. doi: 10.1074/jbc.R800001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stafiej A, Pyrzynska K, Ranz A, Lankmayr E. J Biochem Biophys Methods. 2006;69:15–24. doi: 10.1016/j.jbbm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Hong HH, Wang YS. Anal Chem. 2007;79:322–326. doi: 10.1021/ac061465w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson DW. Rapid Commun Mass Spectrom. 2007;21:2926–2932. doi: 10.1002/rcm.3175. [DOI] [PubMed] [Google Scholar]
- 24.Barry SJ, Carr RM, Lane SJ, Leavens WJ, Monte S, Waterhouse I. Rapid Commun Mass Spectrom. 2003;17:603–620. doi: 10.1002/rcm.957. [DOI] [PubMed] [Google Scholar]
- 25.Williams TI, Lovell MA, Lynn BC. Anal Chem. 2005;77:3383–3389. doi: 10.1021/ac048265+. [DOI] [PubMed] [Google Scholar]
- 26.Quirke JME, Van Berkel GJ. J Mass Spectrom. 2001;36:1294–1300. doi: 10.1002/jms.233. [DOI] [PubMed] [Google Scholar]
- 27.Eggink M, Wijtmans M, Ekkebus R, Lingeman H, de Esch IJ, Kool J, Niessen WMA, Irth H. Anal Chem. 2008;80:9042–9051. doi: 10.1021/ac801429w. [DOI] [PubMed] [Google Scholar]
- 28.Eggink M, Charret S, Wijtmans M, Lingeman H, Kool J, Niessen WMA, Irth H. J Chromatogr B. 2009;877:3937–3945. doi: 10.1016/j.jchromb.2009.09.043. [DOI] [PubMed] [Google Scholar]
- 29.Van der Auweraer M, De Schryver FC, Borsenberger PM, Fitzgerald JJ. J Phys Chem. 1993;97:8808–8811. doi: 10.1021/j100136a027. [DOI] [Google Scholar]
- 30.Dirksen A, Hackeng TM, Dawson PE. Angew Chem Intl Ed. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 31.Gillois M, Rumpf P (1954) Bull Soc Chim Fr 112
- 32.Nakajima N, Ikada Y. Bioconjug Chem. 1995;6:123–130. doi: 10.1021/bc00031a015. [DOI] [PubMed] [Google Scholar]
- 33.Ford QL, Burns JM, Ferry JL. J Chromatogr A. 2007;1145:241–245. doi: 10.1016/j.chroma.2007.01.096. [DOI] [PubMed] [Google Scholar]
- 34.Novak P, Kruppa GH. Eur J Mass Spectrom. 2008;14:355–365. doi: 10.1255/ejms.963. [DOI] [PubMed] [Google Scholar]
- 35.Grabarek Z, Gergely J. Anal Biochem. 1990;185:131–135. doi: 10.1016/0003-2697(90)90267-D. [DOI] [PubMed] [Google Scholar]
- 36.Staros JV, Wright RW, Swingle DM. Anal Biochem. 1986;156:220–222. doi: 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 499 kb)