

Abstract
The degree of H-bonding is thought to play an important role in defining collagen recognition sites or regions that contain disease causing collagen mutations. For collagen model peptides, structure determination by standard NMR approaches is limited due to their rod-like anisotropic shape and repeating sequence. We demonstrate that NMR 15N relaxation experiments and their dependence on rotational diffusion anisotropy can be used to obtain novel structural information about the orientation of the NH bonds relative to the protein backbone in these rod-like systems. 15N relaxation measurements on a triple helical peptide that models a collagen sequence just C-terminal to the unique collagenase cleavage site indicate that the angle between the NH bond vector and the diffusion tensor of the Gly residues needs to be readjusted. After placing the Gly amide protons out of the C′-N-Cα plane, the hydrogen bond angles and distances are re-calculated and are shown to be both closer to 180° and shorter. The data suggest that deviations of the Gly amide protons from its standard position arise from H-bonding effects and that these may impact on the hydrogen bond strengths in this collagen recognition region.
Knowledge of the structure and dynamics of the collagen triple helix is important for understanding its many interactions with receptors and other matrix molecules as well as the perturbations that are caused by collagen disease mutations.1 Collagen possesses a unique triple helical conformation, which consists of three supercoiled polyproline II-like helices with a repetitive Gly-X-Y sequence. The unique triple helical conformation is stabilized by inter-chain hydrogen bonding and an extensive hydration network as seen in the high resolution x-ray structures of collagen model peptides.2a Gly residues from the three chains are closely packed in the center of the triple helix and the backbone amide proton of Gly forms H-bonds with the carbonyl oxygen of the X-residue in the adjacent chain. The degree of the H-bonding may play an important role in recognition of collagen by other molecules and in determining the severity of collagen disease arising from Gly mutations; aspects of the frequency and strength of this hydrogen bonding have been studied by NMR, x-ray and computational approaches.1d,2 Herein we report 15N relaxation measurements on a collagen-like model peptide that reveal that the orientations of the Gly NH bonds relative to the protein backbone have an unanticipated geometry and suggest that the H-bonding may be responsible for this effect.
Standard NMR approaches of using NOEs and J-couplings to obtain high resolution structures are limited for collagen model peptides.1d Complications include their rod-like nature and repetitive sequences. The close packing of the three chains and the linear nature results in only a small number of short-range distances from NOE experiments. For globular proteins with intermediate anisotropy, there have been a few examples of the use of 15N relaxation rates for global structure refinement and for determination of inter-domain orientations.3 Here we demonstrate that we can use 15N relaxation measurements and their dependence on rotational diffusion anisotropy to obtain novel structural information about NH bonds in the very anisotropic collagen model peptide.
The triple helical peptide T3-785 (with sequence (POG)3ITGARGLAG(POG)4Y where O stands for Hydroxyproline) was designed to model an imino acid poor region occurring one triplet C-terminal to the unique collagenase cleavage site in type III collagen.4a The crystal structure of T3-785 shows that the peptide is a long straight rod2a and calculation of the relative ratio of the principal components of the inertia tensor from the crystal structure coordinates indicates that the peptide can be modeled as an axially symmetric rotor (Supporting Information, Inertia tensor).4b These rotational properties can be described by a cylinder model and prolate ellipsoid model.4c NMR hydrogen exchange data indicate that the Gly residues in the peptide have a rigid backbone and that Gly is H-bonded as indicated by the high protection factors.4a
In a protein with axially symmetric diffusion, 15N relaxation parameters R1 and R2 depend on the orientation of the N-H bond relative to the unique axis of the diffusion tensor (angle θ) (Supporting Information, Diffusion tensor, Figure S1). Using the experimental 15N R2/R1 ratios and the known structure, the diffusion tensor can be derived from the fitting program r2r1_diffusion and the angle θ which defines the orientation of the N-H bond can be obtained.4b Relaxation rates R1 and R2 were obtained at 500MHz for labeled residues in the central region of T3-785 including G15, L16, A17, and G18 and at the C-terminal end, G24 (Supporting Information, Table S1). The R2 values are almost identical for all the labeled residues, while the R1 values show small variations with G15 and G18 having the largest R1 values (R1=2.02 and R1=2.04, respectively) and A17 the smallest values (R1=1.79). The differences in R1 result in a range of R2/R1 ratios from 5.6 to 6.64.
The derivation of the diffusion tensor from R2/R1 requires using residues for which there is an absence of conformational exchange and large amplitude internal motion. All 12 labeled residues (four labeled residues per chain) showed no evidence of conformational exchange on the ms timescale as determined from R2Hahn-echo experiments and no evidence of large amplitude internal motions as seen from NOE values that are uniformly greater than 0.6. These results suggest no significant dynamics of the peptide in solution. Therefore all the labeled residues could in principle be included in the derivation of the diffusion tensor, but it was not possible to use the relaxation data for all four residues and obtain meaningful results.
Selection of which labeled residues to use in the derivation of the diffusion tensor is based on the following three criteria; an F-value analysis for selection of an isotropic vs. anisotropic model to describe the triple helix, the proper alignment of the unique axis of the diffusion tensor with the long symmetric axis of the peptide, and the comparison of the experimental D||/D⊥ and τc to the values calculated from the theoretical cylinder and prolate ellipsoid models. When the relaxation data for four labeled residues G15-L16-A17-G18 or for only two glycine residues G15 and G18 was used, an anisotropic model for the triple helix showed no improvement in the fit relative to an isotropic model, as indicated by high p-values (0.23 and 0.30, respectively), and the unique axis of the diffusion tensor was not parallel to the long symmetric axis of the peptide (Figure 1A). However, when using the six data points for L16-A17 only, a low p-value of 0.03 indicated that the anisotropic model was significantly better than the isotropic model, and the unique axis of the diffusion tensor was aligned in parallel to the long axis of the peptide (Figure 1A), both of which are expected for this very anisotropic system. From the experimental data, the ratio of the principal values of the diffusion tensor D||/D⊥ is 13.1, and the overall correlation time τc is 6.92ns. These values are comparable to those obtained from a cylinder model (D||/D⊥=12.3 and τc=6.98ns) and prolate ellipsoid model (D||/D⊥ =11.9), indicating that experimental values are consistent with those obtained from theoretical models (Supporting Information, Hydrodynamic models).4 These three criteria all indicate that using the relaxation data for L16-A17 only is the best selection for derivation of the diffusion tensor.
Figure 1.

(A) The relative position of triple helical peptide in the diffusion tensor frame obtained by fitting the relaxation data of G15 and G18 only (purple), L16 and A17 only (red), or all the labeled residues GLAG (blue). (B) Plots of R2/R1 vs the θ angle. The line shows the back-calculated R2/R1 based on the diffusion tensor and the colored dots (Gly in green, Leu in red, Ala in blue) show the experimental values; The figure assumes that θ′= θ.
R2/R1 values were back calculated for all the labeled residues G15-L16-A17-G18 given the diffusion tensor obtained above (Figure 1B) in order to compare the experimental and theoretical values and to understand the basis for why G15 and G18 needed to be eliminated from the diffusion tensor derivation. The back-calculated R2/R1 values for G15 and G18 deviate from the experimental values and are too large by approximately 12%. Possible reasons for these deviations could result either from the non-collinearity of the principal axis of the shielding tensor with the NH bond5a or from the uncertainty in the positions of the H-atoms relative to the backbone. Calculations suggest that non-collinearity of the principal axis of the shielding tensor with the NH bond does not account for the inability to fit the Gly residues (Supporting Information, CSA contribution).
The divergence of the experimental data from the theoretical data can be improved by altering the positions of the backbone amide protons of Gly residues by adjusting the angle θ to ~71° (Figure 1B). The X-ray structure does not contain information about protons, as their electron density is too weak to permit accurate determination. The amide protons are added to the X-ray structure with the program REDUCE with the assumption that the amide proton is positioned in the C′-N-Cα plane, while the NH vector is a little inclined away from the line that bisects the C′-N-Cα angle (~4 degrees) toward the N-Cα bond.5b In order to make the calculated R2/R1 values consistent with the experimental values, the angle θ for the G15 and G18 is adjusted by placing the amide proton out of the C′-N-Cα plane by 17.0+/−5.3 degrees. There is precedence for this as a 1Å structure of BPTI obtained by neutron diffraction and X-ray data have shown that some amide protons are out-of-plane by 0.4 +/− 4.8°.5c More recently, residual dipolar coupling (RDC) studies of the third lgG-binding domain of protein G (GB3) indicated that most NH vectors were located in the C′-N-Ca plane, while 38 of them were out of the plane with a maximum out-of-plane angle of 11.5 for K13. 6a, 7
It has been suggested that deviations of the amide proton from its standard position may arise from secondary structure differences or from H-bonding.6a After modification of the positions of the glycine amide protons according to the new θ angle, the hydrogen bond angles were re-calculated. The angles have improved relative to the ones without modification; on average, the N-H-O angle is closer to 180° and the H-O distances are shorter (Table 1). No clashes were caused by the repositioning of the amide protons as checked by the program MolProbity.6b With the modified θ angle for the Gly NH, the diffusion tensor can be obtained by using 500, 600 or 800MHz NMR relaxation data simultaneously for all the labeled residues without the need to consider the non-collinearity of the CSA.
Table 1.
Hydrogen bond information of the labeled Glycines before and after the modification of the amide protons.
| Before modification | After modification | |||
|---|---|---|---|---|
| H-bond | Angle N-H-O | Dist H-O | Angle N-H-O | Dist H-O |
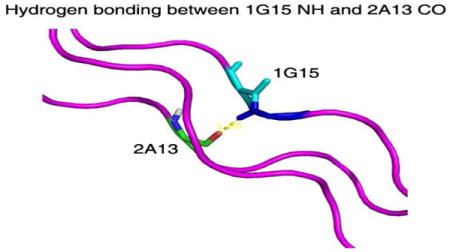
| 1G15 NH -2A13 C=O | 158 | 1.83 | 168 | 1.8 |
| 1G18 NH -2L16 C=O | 155 | 1.89 | 175 | 1.84 |
| 2G15 NH -3A13 C=O | 145 | 2.05 | 175 | 1.93 |
| 2G18 NH -3L16 C=O | 154 | 2.01 | 161 | 1.99 |
| 3G15 NH -1L16 C=O | 163 | 1.91 | 169 | 1.9 |
| 3G18 NH -1P19 C=O | 146 | 2.15 | 170 | 2.05 |
We propose that in peptide T3-785 the H-bonding rather than uniform PPII secondary structure may be responsible for the deviation of the Gly amide protons from their standard position. This is supported by the fact that only the H-bonded Gly residues require a modification of their θ angles while the X and Y residues remain in their standard positions. Although the re-positioning of the Gly NH vectors results in improved H-bond angles and lengths, the distortion of the Gly amide protons may impact on the hydrogen bond strengths in this collagen recognition region.
The use of 15N relaxation experiments to obtain long range orientational restraints extends the structural tools available to the triple helix system and may also be applied to nucleic acid systems that have similar anisotropic rod-like structural characteristics to the triple helix protein. The experiments provide information about local N-H vector orientations and distortions that can be related to H-bonding properties. This powerful new tool may be also be used to complement the short range NOEs and J-coupling values found in anisotropic systems for more complete structure determination.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM45302 and NSF grants DBI-0403062 and DBI-0320746. We thank David Case, Seho Kim and Barbara Brodsky for helpful discussions.
Footnotes
Supporting Information Available: Inertia tensor, Diffusion tensor, Hydrodynamic models, CSA contribution, Figure S1 and Table S1. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Myllyharju J, Kivirikko KI. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]; (b) Lauer-Fields JL, Juska D, Fields GB. Biopolymers. 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]; (c) Di Lullo GA, Sweeney SM, Korkko J, Ala-Kokko L, San Antonio JD. J Biol Chem. 2002;277:4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]; (d) Li Y, Brodsky B, Baum J. J Biol Chem. 2009;284:20660–20667. doi: 10.1074/jbc.M109.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Kramer RZ, Bella J, Mayville P, Brodsky B, Berman HM. Nat Struct Biol. 1999;6:454–457. doi: 10.1038/8259. [DOI] [PubMed] [Google Scholar]; (b) Nerenberg PS, Stultz CM. J Mol Biol. 2008;382:246–256. doi: 10.1016/j.jmb.2008.07.009. [DOI] [PubMed] [Google Scholar]; (c) Radmer RJ, Klein TE. Biochemistry. 2004;43:5314–5323. doi: 10.1021/bi035676w. [DOI] [PubMed] [Google Scholar]
- 3.(a) Tjandra N, Garrett DS, Gronenborn AM, Bax A, Clore GM. Nat Struct Biol. 1997;4:443–449. doi: 10.1038/nsb0697-443. [DOI] [PubMed] [Google Scholar]; (b) Fushman D, Xu R, Cowburn D. Biochemistry. 1999;38:10225–10230. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]; (c) Hashimoto Y, Smith SP, Pickford AR, Bocquier AA, Campbell ID, Werner JM. J Biomol NMR. 2000;17:203–214. doi: 10.1023/a:1008341609461. [DOI] [PubMed] [Google Scholar]; (d) Wu H, Blackledge M, Maciejewski MW, Mullen GP, King SM. Biochemistry. 2003;42:57–71. doi: 10.1021/bi026762j. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fan P, Li MH, Brodsky B, Baum J. Biochemistry. 1993;32:13299–13309. doi: 10.1021/bi00211a043. [DOI] [PubMed] [Google Scholar]; (b) Tjandra N, Fella SE, Pastor RW, Bax A. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]; (c) Hall JB, Fushman D. J Biomol NMR. 2003;27:261–275. doi: 10.1023/a:1025467918856. [DOI] [PubMed] [Google Scholar]
- 5.(a) Boyd J, Redfield C. J Am Chem Soc. 1998;120:9692–9693. [Google Scholar]; (b) Word JM, Lovell SC, Richardson JS, Richardson DC. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]; (c) Wlodawer A, Walter J, Huber R, Sjolin L. J Mol Biol. 1984;180:301–329. doi: 10.1016/s0022-2836(84)80006-6. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ulmer TS, Ramirez BE, Delaglio F, Bax A. J Am Chem Soc. 2003;125:9179–9191. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]; (b) Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.RDCs could potentially provide a complementary approach to obtaining orientational information about the N-H bond vectors in triple helical peptides. Attempts to find a suitable alignment medium (bicelles, gels and pf1 phage) have thus far been unsuccessful.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.