

Abstract
This multicenter, randomized, open-label phase III trial (planned enrollment: 700 patients) was conducted to test the hypothesis that single-agent sunitinib improves progression-free survival (PFS) compared with capecitabine as treatment for advanced breast cancer (ABC). Patients with HER2-negative ABC that recurred after anthracycline and taxane therapy were randomized (1:1) to sunitinib 37.5 mg/day or capecitabine 1,250 mg/m2 (1,000 mg/m2 in patients >65 years) BID on days 1–14 q3w. The independent data-monitoring committee (DMC) determined during the first interim analysis (238 patients randomized to sunitinib, 244 to capecitabine) that the trial be terminated due to futility in reaching the primary endpoint. No statistical evidence supported the hypothesis that sunitinib improved PFS compared with capecitabine (one-sided P = 0.999). The data indicated that PFS was shorter with sunitinib than capecitabine (median 2.8 vs. 4.2 months, respectively; HR, 1.47; 95% CI, 1.16–1.87; two-sided P = 0.002). Median overall survival (15.3 vs. 24.6 months; HR, 1.17; two-sided P = 0.350) and objective response rates (11 vs. 16%; odds ratio, 0.65; P = 0.109) were numerically inferior with sunitinib versus capecitabine. While no new or unexpected safety findings were reported, sunitinib treatment was associated with higher frequencies and greater severities of many common adverse events (AEs) compared with capecitabine, resulting in more temporary discontinuations due to AEs with sunitinib (66 vs. 51%). The relative dose intensity was lower with sunitinib than capecitabine (73 vs. 95%). Based on these efficacy and safety results, sunitinib should not be used as monotherapy for patients with ABC.
Keywords: Advanced breast cancer, Sunitinib malate, Capecitabine, Tyrosine kinase inhibitor
Introduction
Advanced breast cancer (ABC) remains an incurable disease, and new treatment options are urgently needed. For patients with HER2-positive disease, the introduction of anti-HER2 therapies has been associated with improved survival [1, 2]. Progress has been more limited for patients with HER2-negative disease, however. Currently available chemotherapeutic agents, while active, yield responses that are generally short-lived [3]. Capecitabine, an oral fluoropyrimidine carbamate, is an approved standard of care for ABC. In patients previously treated with anthracyclines/taxanes, single-agent capecitabine results in an objective response rate (ORR) of 26%, a median time to tumor progression (TTP) of 3.4 months, and a median overall survival (OS) of 8.4 months [4].
Angiogenesis is known to play a key role in tumor growth and the metastatic spread of breast cancer [5] consistent with the finding that expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor beta (PDGFR-β) are associated with a poorer prognosis [5, 6]. Inhibition of angiogenesis with the anti-VEGF monoclonal antibody bevacizumab has been shown to increase progression-free survival (PFS) in a number of studies when combined with chemotherapy [7–9], although single-agent bevacizumab is not known to result in improved outcomes in patients with ABC [10].
In contrast to single-targeted bevacizumab, sunitinib malate (SUTENT®) is an oral multitargeted tyrosine kinase inhibitor of VEGFRs and PDGFRs, as well as other receptor tyrosine kinases [11–13]. Sunitinib is currently approved multinationally for the treatment of metastatic renal cell carcinoma (mRCC), and gastrointestinal stromal tumor (GIST) after disease progression on, or intolerance to, imatinib treatment [13]. Importantly, single-agent sunitinib has demonstrated antitumor activity in heavily pre-treated patients with metastatic breast cancer (N = 64) in a phase II trial [14]. Patients received sunitinib 50 mg/day for 4 weeks followed by 2 weeks off-treatment (Schedule 4/2) in 6-week cycles. The ORR was 11%; stable disease (SD) for 6 months or more was 5%, leading to a clinical benefit rate of 16%; and median TTP was 10 weeks. More recently, continuous daily dosing (CDD) of sunitinib 37.5 mg has been found to provide a feasible and convenient alternative dosing schedule for patients with mRCC or GIST [15–17].
In 2006, a multicenter, randomized, open-label phase III trial (SUN 1107) was initiated to test the hypothesis that single-agent sunitinib 37.5 mg on the CDD schedule would be superior to capecitabine in prolonging PFS in patients with ABC that had recurred after anthracycline and taxane therapy (or single-agent taxane only if an anthracycline was not indicated). It was hypothesized that inhibition of multiple signal transduction pathways in ABC by a multitargeted kinase inhibitor would translate into a long-term efficacy benefit superior to that of a single-agent fluoropyrimidine. However, based on both efficacy and safety analyses early in the conduct of the study, the trial was terminated due to futility as determined by the independent data-monitoring committee (DMC). This article presents the final results of the trial.
Methods
Patients
Eligible patients were female, aged ≥18 years with measurable, HER2-negative ABC that had recurred after anthracycline and taxane therapies (or single-agent taxane treatment if an anthracycline was not indicated) in the neoadjuvant, adjuvant, and/or advanced disease setting. Neither more than two chemotherapy regimens in the advanced setting nor any prior capecitabine treatment were permitted. Prior hormonal therapy or immunotherapy in the adjuvant and/or advanced settings was permitted. An interval of ≥3 weeks was required between major surgery or systemic therapy (except hormone therapy) and initiation of study treatment. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0–2, adequate organ function [including left ventricular ejection fraction ≥50% measured by multigated acquisition (MUGA) scan or echocardiogram (ECHO)], and resolution of all the acute toxic effects of prior therapy or surgical procedures to National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0 (NCI CTCAE v3.0) grade ≤1 (except alopecia). Brain metastases and cardiovascular disease or uncontrolled hypertension were also key exclusion criteria.
The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable local regulatory requirements and laws and was approved by the institutional review board or independent ethics committee of each participating center. All patients provided written informed consent.
Study design and treatment
This was a multinational, multicenter, randomized, open-label, phase III clinical trial comparing the efficacy and safety of single-agent sunitinib versus capecitabine administered as first-, second- or third-line therapy in subjects with ABC. Patients were randomized 1:1 to treatment with sunitinib or capecitabine using an interactive voice randomization system. Stratification factors of randomization were visceral disease (yes/no), sensitivity to prior taxane treatment (progression </≥12 months after taxane therapy), hormone receptor status (positive/negative), and country (study site location).
Sunitinib was administered orally at 37.5 mg on the CDD schedule. Patients experiencing severe toxicity were permitted 1-week periods off treatment as needed; dose reduction to 25 mg was also permitted. Dose escalation to 50 mg was permitted in patients not experiencing a response based on Response Evaluation Criteria in Solid Tumors (RECIST) [18] and experiencing only grade ≤1 non-hematologic or grade ≤2 hematologic toxicity attributed to sunitinib within the first 9 weeks of treatment. Capecitabine was administered orally within 30 min after the end of a meal at 1,250 mg/m2 (1,000 mg/m2 in patients >65 years) twice daily on days 1–14 every 3 weeks. Dose reduction in response to toxicity was permitted according to the approved label for capecitabine [4]. Patients requiring >3 weeks’ dose interruption due to persistent toxicity attributable to either drug were considered for discontinuation from the study. Study treatment was planned to continue until disease progression, unacceptable toxicity, or consent withdrawal.
Assessments
The primary study endpoint was PFS, defined as the time from randomization to first documentation of objective tumor progression or death due to any cause, whichever occurred first. Key secondary endpoints were TTP, ORR, duration of response, OS, and safety. Tumors were imaged using computed tomography or magnetic resonance imaging at baseline and at 6-week intervals from randomization until documented disease progression or initiation of a subsequent anticancer therapy. Additional scans were performed whenever disease progression was suspected, to confirm an objective response [partial response (PR) or complete response (CR)] ≥4 weeks after initial documentation of response, and at withdrawal from the study (if >6 weeks since the last assessment). Tumor response (based on RECIST) was evaluated by the investigators. Safety was assessed by physical examination and analysis of adverse events (AEs), hematology, blood chemistry, vital signs, and cardiac function (using 12-lead electrocardiogram and two-dimensional ECHO or MUGA scanning) at regular intervals. Treatment-emergent AEs were graded using NCI CTCAE v3.0.
Statistical methods
The sample size was determined based on the assumption that median PFS for patients receiving single-agent capecitabine was 4.2 months [19]. With no historical data for sunitinib in this population and assuming that inhibition of multiple targets in breast cancer would translate into greater efficacy, it was hypothesized that median PFS with sunitinib would be 33% higher (5.6 months) and that PFS of both treatment arms would follow an exponential distribution. Additional assumptions were that interim analyses would be performed at 33 and 67% of the total number of PFS events, with an overall one-sided type I error of α = 0.025. In order to achieve 90% power to detect a statistical difference in PFS between the treatment groups and a hazard ratio (HR; sunitinib/capecitabine) of 0.75, a minimum of 525 PFS events and target enrollment of 700 patients were planned. Interim analyses were planned to allow early stopping of the study for futility and for efficacy at the time of the second interim analysis. Jennison and Turnbull’s Rho stopping boundary (ρ = 3.0) was used for the futility analysis, and the O’Brien-Fleming stopping boundary was used for evidence of efficacy [20].
The intent-to-treat (ITT) population comprised all patients who were randomized. All efficacy analyses, except for analyses of duration of response (which was assessed only in responders), were performed on the ITT population. The as-treated (AT) population included all patients who received at least one dose of study medication and was the primary population for evaluating treatment administration and safety. Descriptive statistics were used to summarize all baseline characteristics, treatment administration, compliance, efficacy endpoints, and safety parameters. Time-to-event endpoints were analyzed by treatment arm using Kaplan–Meier methods. Between-treatment comparisons for time-to-event endpoints were conducted using log-rank tests stratified by visceral disease, sensitivity to prior taxane treatment, and hormone receptor status. HRs and 95% confidence intervals (CIs) were estimated using Cox proportional hazards models stratified by the factors used for the stratified log-rank tests and by treatment. Overall relative dose intensities were determined by averaging the relative dose intensities for each cycle (cumulative doses for each 21-day cycle divided by the planned dose for a 21-day cycle).
Results
Study conduct, patients, and treatment administration
As part of ongoing efficacy and safety review by an independent DMC, the first pre-specified interim analysis was planned to be performed after 33% of the planned 525 events had occurred. The Rho stopping boundary for this futility analysis was a HR ≥1.04. The analysis took place in March 2009 when 224 PFS events had occurred; based on the available PFS data, the HR for PFS was determined to be 1.49 in favor of capecitabine. With the predefined stopping boundary having been crossed, the DMC recommended that study enrollment be discontinued.
Between November 2006 and April 2009 when enrollment was stopped, 482 patients had been entered into the trial at 119 centers worldwide (Europe, 39%; Asia–Pacific, 36%; South America, 15%; North America, 8%; and Africa, 1%). Data cut-off for the results presented here was July 10, 2009. The ITT population comprised all 482 patients (sunitinib, 238; capecitabine, 244). Four patients randomized to the capecitabine arm did not receive treatment. All remaining 478 patients received at least one dose of study medication and comprised the AT population. The treatment groups were well balanced for demographic, disease, and prior treatment characteristics (Table 1). A total of 36% of patients in the sunitinib arm and 33% in the capecitabine arm had triple-negative (hormone receptor- and HER2-negative) disease, and 73 and 72% of sunitinib and capecitabine patients, respectively, had visceral disease. Sixty-five percent of patients in both arms were considered resistant to prior taxane treatment, and 84% of patients in both arms had received at least one line of prior systemic treatment in the advanced setting.
Table 1.
Patient, disease, and prior treatment characteristics at baseline
| Patient characteristics | Sunitinib (N = 238) | Capecitabine (N = 244) |
|---|---|---|
| Age, years | ||
| Mean | 53 | 53 |
| Range | 25–80 | 23–80 |
| ECOG PS, n (%)a | ||
| 0 | 142 (60) | 126 (52) |
| 1 | 86 (36) | 110 (45) |
| 2 | 4 (2) | 5 (2) |
| Primary diagnosis, n (%) | ||
| Metastatic | 228 (96) | 233 (95) |
| Locally recurrent | 10 (4) | 11 (5) |
| Histologic classification, n (%) | ||
| Ductal | 202 (85) | 218 (89) |
| Lobular | 24 (10) | 17 (7) |
| Other | 11 (5) | 7 (3) |
| Location of disease, n (%) | ||
| Visceral | 173 (73) | 175 (72) |
| Non-visceral | 65 (27) | 67 (28) |
| Number of involved sites, n (%)a | ||
| 1 | 52 (22) | 64 (26) |
| ≥2 | 184 (77) | 178 (73) |
| Most commonly affected sites, n (%) | ||
| Liver | 115 (48) | 125 (51) |
| Bone | 114 (48) | 104 (43) |
| Lymph node | 107 (45) | 109 (45) |
| Lung | 101 (42) | 91 (37) |
| Receptor status, n (%)a | ||
| Hormone receptor-positive | 148 (62) | 160 (66) |
| Hormone receptor-negative | 87 (37) | 82 (34) |
| Triple-negativeb | 86 (36) | 81 (33) |
| Prior taxane treatment, n (%)a | ||
| Sensitive | 84 (35) | 83 (34) |
| Refractory | 154 (65) | 159 (65) |
| Prior lines of systemic therapy for advanced disease, n (%) | ||
| 0 | 38 (16) | 39 (16) |
| ≥1 | 200 (84) | 205 (84) |
ECOG PS: Eastern Cooperative Oncology Group performance status; triple-negative: hormone receptor- and HER2-negative
aData not reported (n), sunitinib and capecitabine, respectively: ECOG PS, 6 and 3; no. involved sites, 2 and 2; progesterone receptor status, 3 and 2; prior taxane treatment, 0 and 2
bHER2 status not evaluable for one sunitinib patient and missing for one capecitabine patient
Administration of study treatment is summarized in Table 2. At the time of data cut-off, patients had received study drug for a median of 61 days in both treatment arms. Ninety-seven percent of patients in the sunitinib arm and 80% of patients in the capecitabine arm discontinued the study, most commonly due to disease progression (63 and 57% of patients in the sunitinib and capecitabine arms; Fig. 1). Sunitinib dosing was interrupted in 52% of patients for a median of 10 days, while capecitabine treatment was interrupted in 46% of patients for a median of 7 days. The sunitinib dose was reduced in 28% of patients, while capecitabine was dose-reduced in 35% of patients.
Table 2.
Treatment administration
| Sunitinib (N = 238) | Capecitabine (N = 240) | |
|---|---|---|
| Median number of cycles started (range) | 4 (1–27) | 5 (1–40) |
| Median number of days on drug (range)a | 61 (1–485) | 61 (4–540) |
| Dosing interruptions,bn (%) | 124 (52) | 110 (46) |
| Median average duration (range), days | 10 (3–43) | 7 (3–35) |
| Dosing delays, n (%) | NA | 109 (45) |
| Median average duration (range), days | NA | 7 (3–33) |
| Dose reductions,cn (%) | 66 (28) | 84 (35) |
| Median relative dose intensity (range), % | 73 (5–114) | 95 (19–124) |
NA: not applicable (only dosing interruptions are possible on a CDD schedule as the study drug is administered every day)
aTotal number of dosing days from first to last day of treatment
bDefined as ≥3 consecutive days on which treatment was not given
cDefined as any dose ≥10% lower than the previous dose
Fig. 1.

Patient disposition (CONSORT flow diagram)
The resulting median overall relative dose intensities were 73% for sunitinib and 95% for capecitabine.
Efficacy
Efficacy results are summarized in Table 3. PFS, the primary study endpoint, when analyzed using a one-sided log-rank test, yielded a P value of 0.999 (P = 0.002 when analyzed using a two-sided test, indicating PFS inferiority for therapy with sunitinib alone). The median PFS was 2.8 months in the sunitinib arm and 4.2 months in the capecitabine arm (Table 3; Fig. 2; HR, 1.47; 95% CI, 1.16–1.87), with 63 and 60% of patients having died or had disease progression at data cut-off. Median TTP results were nearly identical to the PFS results (data not shown).
Table 3.
Efficacy results
| Variable | Sunitinib (N = 238) | Capecitabine (N = 244) | Hazard ratio | 95% CI | P |
|---|---|---|---|---|---|
| Progression-free survival | |||||
| Events, n (%) | 151 (63) | 147 (60) | |||
| Median, months | 2.8 | 4.2 | 1.47 | 1.16–1.87 | 0.999a 0.002b |
| 95% CI | 2.4–4.0 | 3.8–5.5 | |||
| Overall survival | |||||
| Events, n (%) | 78 (33) | 71 (29) | |||
| Median, months | 15.3 | 24.6 | 1.17 | 0.84–1.63 | 0.825a 0.350b |
| 95% CI | 11.4–25.3 | 12.6–26.0 | |||
| Objective response rate, % | 11 | 16 | 0.65c | 0.4–1.1 | 0.109d |
| 95% exact CI | 8–16 | 12–22 | |||
| Duration of response | |||||
| Events, n (%) | 27 (11) | 40 (16) | |||
| Median, months | 6.9 | 9.3 | 2.79 | 1.04–7.46 | NA |
| 95% CI | 3.1–8.5 | 5.5–9.7 | |||
| Clinical benefit rate, % | 19 | 27 | 0.65c | 0.4–1.0 | 0.045d |
| 95% exact CI | 15–25 | 22–33 | |||
Objective response rate: % complete responses and partial responses; NA: not applicable; clinical benefit rate: % complete responses, partial responses, and stable disease ≥6 months
aOne-sided stratified log-rank test
bTwo-sided stratified log-rank test
cOdds ratio
dPearson χ2 test
Fig. 2.
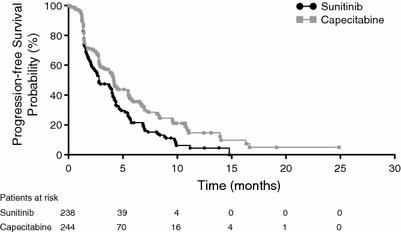
Kaplan–Meier estimate of progression-free survival
OS was shorter in the sunitinib arm than the capecitabine arm (15.3 vs. 24.6 months; HR, 1.17; 95% CI, 0.84–1.63; two-sided log-rank test P = 0.350; Table 3; Fig. 3). At data cut-off, 33% of sunitinib- and 29% of capecitabine-treated patients had died.
Fig. 3.
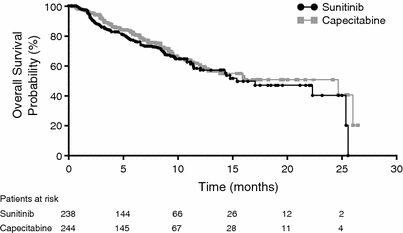
Kaplan–Meier estimate of overall survival
There were fewer confirmed responses in the sunitinib arm than in the capecitabine arm (1 CR and 26 PRs vs. 1 CR and 39 PRs, respectively). The corresponding ORRs were 11 and 16% (Pearson χ2 test P = 0.109; Table 3). Duration of response was also shorter with sunitinib (6.9 months; 95% CI, 3.1–8.5) than with capecitabine (9.3 months; 95% CI, 5.5–9.7). Likewise, with 19 and 26 patients having achieved SD for at least 6 months, the clinical benefit rate (percent objective responses plus SD ≥6 months) was lower with sunitinib (19%) than with capecitabine (27%; Pearson χ2 test P = 0.045).
Safety
Most patients in the study experienced treatment-related AEs, and frequencies were similar between the treatment arms overall (92 and 91% in the sunitinib and capecitabine arms; Table 4). The five most commonly reported treatment-related AEs on sunitinib were diarrhea (40%), hand–foot syndrome (32%), nausea (32%), fatigue (30%), and vomiting (28%); those on capecitabine were hand–foot syndrome (61%), diarrhea (34%), nausea (28%), fatigue (20%), and anorexia (16%). Treatment-related AEs that were reported at least 10% more frequently on sunitinib than on capecitabine were vomiting, dysgeusia, hypertension, thrombocytopenia, hypothyroidism, and decreased platelet count, while only hand–foot syndrome was reported at least 10% more frequently on capecitabine.
Table 4.
Treatment-related adverse eventsa occurring in at least 10% of patients in either treatment group
| Adverse event | n (%) | |||||
|---|---|---|---|---|---|---|
| Sunitinib (N = 238) | Capecitabine (N = 240) | |||||
| Any grade | Grade 3 | Grade 4 | Any grade | Grade 3 | Grade 4 | |
| Any adverse event | 218 (92) | 110 (46) | 17 (7) | 219 (91) | 71 (30) | 6 (3) |
| Diarrhea | 94 (40) | 11 (5) | 2 (1) | 81 (34) | 9 (4) | 2 (1) |
| Hand–foot syndrome | 77 (32) | 19 (8) | 0 | 146 (61) | 38 (16) | 0 |
| Nausea | 76 (32) | 2 (1) | 0 | 68 (28) | 1 (<1) | 0 |
| Fatigue | 71 (30) | 13 (6) | 2 (1) | 49 (20) | 3 (1) | 0 |
| Vomiting | 66 (28) | 5 (2) | 0 | 27 (11) | 3 (1) | 0 |
| Dysgeusia | 59 (25) | 0 | 0 | 11 (5) | 0 | 0 |
| Mucosal inflammation | 59 (25) | 7 (3) | 0 | 36 (15) | 2 (1) | 1 (<1) |
| Anorexia | 54 (23) | 2 (1) | 0 | 38 (16) | 1 (<1) | 0 |
| Hypertension | 46 (19) | 8 (3) | 1 (<1) | 1 (<1) | 0 | 0 |
| Asthenia | 41 (17) | 14 (6) | 1 (<1) | 28 (12) | 1 (<1) | 0 |
| Neutropenia | 41 (17) | 24 (10) | 2 (1) | 27 (11) | 8 (3) | 1 (<1) |
| Stomatitis | 38 (16) | 2 (1) | 0 | 20 (8) | 0 | 0 |
| Thrombocytopenia | 38 (16) | 15 (6) | 4 (2) | 6 (3) | 1 (<1) | 2 (1) |
| Dyspepsia | 29 (12) | 2 (1) | 0 | 10 (4) | 0 | 0 |
| Hypothyroidism | 29 (12) | 1 (<1) | 0 | 1 (<1) | 0 | 0 |
| Decreased platelet count | 28 (12) | 12 (5) | 3 (1) | 3 (1) | 0 | 0 |
| Headache | 27 (11) | 2 (1) | 0 | 9 (4) | 0 | 0 |
| Rash | 26 (11) | 0 | 0 | 17 (7) | 2 (1) | 0 |
aMaximum CTCAE grade
Treatment-related grade 3/4 AEs were reported more frequently in the sunitinib arm (53%) than in the capecitabine arm (32%). The three most common treatment-related grade 3/4 AEs were neutropenia (11%), hand–foot syndrome (8%), and thrombocytopenia (8%) in the sunitinib arm and hand–foot syndrome (16%), diarrhea (5%), and neutropenia (4%) in the capecitabine arm. Likewise, serious AEs (SAEs) considered related to treatment were more frequent on sunitinib (16%) than on capecitabine treatment (5%). In the sunitinib arm, the most frequent were thrombocytopenia (n = 5) and diarrhea (n = 4); in the capecitabine arm, diarrhea (n = 6) was most frequent. Seven patients died on study as a result of AEs related to treatment: five in the sunitinib arm (one each due to abnormal hepatic function, cerebral hemorrhage, congestive cardiac failure, hepatic failure, and pulmonary embolism) and two in the capecitabine arm (due to hypotension and septic shock in one patient and subdural hematoma in one patient).
Temporary dosing discontinuations/interruptions and dose reductions due to AEs were required by 66 and 15% of patients in the sunitinib arm and 51 and 23% of patients in the capecitabine arm. Hand–foot syndrome led to temporary discontinuations/interruptions or dose reductions in 9% of patients in the sunitinib arm and 31% of patients in the capecitabine arm. Treatment-related AEs led to discontinuation from the study in a higher proportion of patients in the sunitinib arm (12%) than the capecitabine arm (5%). The most common sunitinib-related AEs leading to discontinuation were neutropenia and thrombocytopenia in three patients each, while the most common capecitabine-related AE leading to discontinuation was neutropenia in four patients.
Discussion
The primary objective of this study was to demonstrate superior PFS with sunitinib versus capecitabine as monotherapy for patients with recurrent, HER2-negative, ABC. This objective was not met, and the data indicate that PFS was indeed shorter in the sunitinib arm when compared with the capecitabine arm (median 2.8 vs. 4.2 months, respectively; HR, 1.47; 95% CI, 1.16–1.87). The one-sided log-rank test P value of 0.999 demonstrated that there was no statistical evidence of sunitinib superiority; the two-sided test (P = 0.002) indicated PFS inferiority for therapy with sunitinib alone.
The differences in OS, a secondary endpoint in the study, were not statistically significant (two-sided log-rank test P = 0.350), and median OS was numerically shorter in the sunitinib arm than in the capecitabine arm (15.3 vs. 24.6 months), although the survival curves overlapped (Fig. 3). The OS results should be interpreted with caution, since the study was not powered for evaluation of OS and OS data were not mature [the majority of patients in both arms were in follow-up at data cut-off, which yielded a high censoring rate (>65%) in both arms].
Similarly, the ORR obtained in the sunitinib arm was not statistically different from that obtained in the capecitabine arm (11 vs. 16%; P = 0.109), and the duration of response was shorter with sunitinib than capecitabine (median 6.9 vs. 9.3 months, respectively). These results again suggest that treatment with sunitinib was inferior to capecitabine in patients with ABC at the doses and schedules used in this study.
Sunitinib dosed at 37.5 mg on a CDD schedule in this population of patients with recurrent ABC yielded a safety profile consistent with that observed previously with the approved dosing schedule of 50 mg/day on Schedule 4/2 in patients with RCC and GIST [13] as well as metastatic breast cancer [14]. Like previous reports, the most common AEs observed in this study included constitutional symptoms such as diarrhea, nausea, fatigue, and vomiting and skin toxicities such as hand–foot syndrome. No new or unexpected safety findings were reported.
However, sunitinib treatment was associated with higher frequencies and greater severities of many common AEs compared with capecitabine. Treatment-related grade 3/4 AEs were reported more frequently with sunitinib than capecitabine (53 vs. 32%, respectively). Discontinuations from study due to treatment-related AEs were also more common with sunitinib than capecitabine (12 vs. 5%).
As previously reported [13, 14], AEs associated with sunitinib treatment were manageable through dosing interruption, dose reduction, and/or standard medical therapies.
The higher frequencies and severities of AEs in the sunitinib arm led to more temporary discontinuations due to AEs in the sunitinib arm versus the capecitabine arm (66 vs. 51%). Median dosing interruptions were longer in the sunitinib arm (10 vs. 7 days), and the relative dose intensity of sunitinib treatment was lower than that of capecitabine treatment (73 vs. 95%).
Based on these results, single-agent sunitinib 37.5 mg on a CDD schedule is not recommended for treatment of patients with ABC. There are at least several potential reasons for the observed outcome of the study: the safety profile that led to the lower relative dose intensity achieved with sunitinib; the heavily pretreated, heterogeneous patient population studied; the lack of sufficient activity of sunitinib as monotherapy on the CDD schedule; and a lack of dependency of ABC on pathways inhibited by sunitinib. It is unknown to what extent each of these factors contributed to the lack of efficacy in the sunitinib arm.
In this study, the comparator was the cytotoxic agent capecitabine, which exhibits activity and acceptable tolerability in this patient population, even when dose reductions are required. In the face of more frequent and severe toxicity with sunitinib, less sunitinib was administered. Population pharmacokinetic analyses in other tumor types, which have shown that increased sunitinib exposure correlates with improved clinical outcomes, highlight the importance of maintaining sunitinib dosing [21].
The relatively broad population of patients with ABC studied across multiple lines of therapy remains a potential contributing factor. A post-hoc subset analysis failed to reveal any specific clinically defined sub-population that benefited more from sunitinib than from capecitabine treatment (data not shown). However, the possibility of specific biomarker-defined populations that benefited cannot be excluded. Unfortunately, such biomarkers are still the subject of preclinical and early-phase clinical research for antiangiogenic agents.
Sunitinib may simply have lower single-agent activity in ABC than cytotoxic chemotherapeutic agents such as capecitabine in this unselected patient population. Capecitabine exhibited similar antitumor activity compared to historical data: the median PFS of 4.2 months obtained with capecitabine in this study corresponded well to the 4.2 months reported in another study of patients with previously treated metastatic breast cancer [19]. Similarly, the ORR of 11% in the sunitinib arm of this study was the same as that observed in a phase II study of heavily pretreated metastatic breast cancer patients [14], although the phase II study was performed with the intermittent sunitinib dosing schedule (50 mg/day, Schedule 4/2).
Lower-than-expected single-agent sunitinib activity in this patient population is consistent with the results obtained with other single-agent angiogenesis inhibitors, such as bevacizumab and sorafenib, in metastatic breast cancer [10, 22], despite the fact that the spectrum of kinase inhibition of sunitinib is broader than other commercially available antiangiogenic agents. Numerous clinical studies of bevacizumab have been conducted in combination with chemotherapy, and consequently bevacizumab plus paclitaxel or docetaxel are approved for first-line use in patients with HER2-negative metastatic breast cancer [7–9]. Combination trials of chemotherapy with other targeted antiangiogenic agents, such as sorafenib, have demonstrated improved PFS over chemotherapy alone [23]; however, to date, no targeted agent has demonstrated robust benefit as a single agent. Therefore, emerging evidence suggests that antiangiogenic agents may be best used in combination with chemotherapy and/or inhibitors of other signaling pathways in ABC. Results are pending from a phase III study comparing sunitinib plus capecitabine versus capecitabine alone as second-line treatment for patients with ABC.
Acknowledgments
We thank the patients and their families, the investigators (who are listed in the Appendix to this article), and the staff at Pfizer Oncology who participated in this study, which was sponsored by Pfizer Inc. Medical writing support was provided by Wendy Sacks, PhD at ACUMED® (Tytherington, UK) and was funded by Pfizer Inc. Final approval of the manuscript rested with the authors alone.
Conflict of interest disclosures C. H. Barrios has received research support from and served in a consultancy role for Pfizer. X. Pivot has received research support from and served in a consultancy role for Glaxo Smith Kline Beecham, Roche, Novartis, Bristol Myers Squibb, and Sanofi-Aventis. N. Harbeck has received honoraria for consulting and lectures from Pfizer. M. J. Brickman, K. Zhang, and K. A. Kern are employees of Pfizer and hold Pfizer stock. M. Martin has served in a consultancy role for Pfizer. M.-C. Liu, S. C. Lee, L. Vanlemmens, J.-M. Ferrero, T. Tabei, H. Iwata, K. Aogi, and R. Lugo-Quintana have no disclosures to report.
Appendix
In addition to the authors, the following investigators participated in the SUN 1107 study.
Argentina: Centro Medico San Roque—J Zarba; Centro Oncológico de Rosario—L Fein; Clínica Viedma S.A.—R Kowalyszyn; Hospital Italiano Regional Sur—A Ferro; Instituto Médico Especializado—G Jankilevich; Instituto Oncológico de Cordoba—E Richardet; Saint Dennis Medical Group S.A.—M Rondinon.
Australia: Austin Health—S White; Mount Medical Centre—A Chan; Royal Adelaide Hospital—M Brown; Royal Brisbane & Women’s Hospital—N McCarthy; Royal Melbourne Hospital—R Deboer; St Vincent’s Hospital—R Ward.
Brazil: Centro de Oncologia InRad—HCFMUSP—R Marques; Fundacao Antonio Prudente—Hospital A.C. Camargo/Hospital do Cancer—S Sanches; Hospital Nossa Senhora de Conceicao—J Pedrini; Instituto de Oncologia do Parana—J Camargo; Instituto Nacional do Cancer—J Bines.
Bulgaria: Klinika po himioterapiya SHATOnc—C Timcheva; Statsionar Stara Zagora InterDistrDispOncDIU—P Chilingirov.
Canada: Hopital du Saint Sacrement—C Desbiens; London Regional Cancer Program—T Vandenberg; QEII Health Sciences Centre—D Rayson; Sunnybrook Health Sciences Centre—M Trudeau.
Colombia: Fundacion Centro de Investigacion Clinica CIC—A Avila; Hospital Universitario San Vicente de Paul—L Suarez Vasquez; Instituto Nacional de Cancerologia—R Tejada.
France: Centre de Radiotherapie du pays Basque—M Rotarski; Centre Eugene Marquis— P Kerbrat; Centre Jean Perrin—P Chollet; Centre Oscar Lambret—J Bonneterre; Clinique Hartmann—J-M Vannetzel.
Germany: early phase solutions gmbh—K Ruffert; Frauenaertze Pruener Gang—V Schulz; Gemeinschaftspraxis, Innere Medizin, Haematologie und Onkologie—A Jakob; Klinik fuer Internistische Onkologie—K Mross; Krankenhaus Nordwest—E Jaeger; Onkologische Schwerpunktpraxis—L Mueller; Rhoen Klinikum Meiningen—H Graf; Staedtisches Klinikum Magdeburg—E Kettner; Technische Universitaet Muenchen—J Ettl; Universitaets-Frauenklinik Tuebingen—J Huober, K Krauss; Universitaetsklinik Charite Berlin—A Korfel; Universitaetsklinik Mainz—M Schmidt; Universitaetsklinikum Schleswig-Holstein—D Fischer, M Friedrich.
Hong Kong: Queen Elizabeth Hospital—R Ngan; Queen Mary Hospital—R J Epstein, R Liang; Tuen Mun Hospital—Y Tung; UNIMED Medical Institute—L Chow.
India: Chhatrapati Shahuji Maharaj Medical University—M Bhatt; Christian Medical College—N Peters; S. K. Soni Hospital—A Maru; Vedanta Institute of Medical Sciences—C Desai.
Italy: Azienda Ospedaliera Arcispedale Santa Maria Nuova—C Boni; Istituto Nazionale per lo Studio e la Cura dei Tumori “Fondazione Pascale”—A De Matteis; Ospedale Niguarda Ca’Granda—S Siena; Universita’ degli Studi di Firenze—G Biti.
Japan: Kitakyushu Municipal Medical Center—K Anan; National Cancer Center Hospital—Y Fujiwara; National Hospital Organization, Kyushu Cancer Center—S Ohno; National Hospital Organization, Nagoya Medical Center—Y Sato; National Hospital Organization, Osaka National Hospital—N Masuda; Osaka University Hospital—T Taguchi; Saitama Cancer Center—K Inoue; St. Luke’s International Hospital—S Nakamura; Tokyo Metropolitan Komagome Hospital—K Kuroi, S Saji.
Korea (Republic of Korea): Inha University Hospital—M H Lee; National Cancer Center—J Ro; Pusan National University Hospital—J S Chung; Seoul National University Hospital—S-A Im, D-Y Oh; Yeungnam University Medical Center—K–H Lee.
Mexico: Centro de Estudios Mastologicos SA de CV—E Sanchez-Forgach; Centro Estatal de Atencion Oncologica—J Aguilar-Melchor; Hospital Privado San Jose de Ciudad—L Perez-Michel; ISSSTEP—E Tellez-Bernal; Torre Medica Cristobal Colon—J De la Cruz-Vargas.
Peru: Clinica Anglo Americana—A Alarcon; Instituto de Oncologia & Radioterapia de la Clinica Ricardo Palma—W Rodriguez.
Philippines: Cardinal Santos Medical Center—M L Tiambeng; National Kidney and Transplant Unit—B Tiangco; St Luke’s Medical Center—R Li; Veterans Memorial Medical Center—V Chan.
Singapore: National Cancer Centre—R Ng, P C Siang Ang.
South Africa: Sandton Oncology Centre—D Vorobiof.
Spain: Centro Oncologico MD Anderson Internacional Espana—R Colomer, L Gonzalez Cortijo; Complejo Hospitalario de Jaen—P Sanchez Rovira; Complejo Hospitalario Materno Insular—A Murias Rosales; Complexo Hospitalario Universitario á Coruña—S Antolin Novoa, L Calvo; Consorci Sanitari del Maresme—P Lianes Barragan; Corporacio Sanitaria Parc Tauli—M A Segui; Fundacion Hospital Alcorcon—C Jara Sanchez; Hospital Clinico San Carlos—J A Garcia-Saenz; Hospital de Basurto—P Martinez del Prado; Hospital Universitario Carlos Haya—F Carabantes Oncon; Hospital Universitario de Salamanca—C Rodriguez; Hospital Universitario Marques de Valdecilla—A De Juan; Hospital Universitario Reina Sofia—J De La Haba; Hospital Universitario Virgen de la Victoria—E Alba; Institut Catala de Oncologia de Gerona—S Del Barco Berron.
Taiwan: Chang Gung Memorial Hospital—S-C Chen; Changhua Christian Hospital—C-S Chang; Chung-Ho Memorial Hospital—M-F Hou; National Cheng Kung University—T-W Chang; National Taiwan University—C-S Huang; Taipei Veterans General Hospital—C-H Su.
Turkey: Hacettepe University School of Medicine—K Altundag; Istanbul University—A Aydiner.
United Kingdom: Guy’s Hospital—A Rigg; Nottingham City Hospital—S Chan; Velindre Hospital—P Barrett-Lee; Yeovil District Hospital—G Sparrow.
Footnotes
This article was submitted on behalf of the SUN 1107 investigators.
The details of investigators who participated in the SUN 1107 study are given in the Appendix.
References
- 1.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 2.Blackwell KL, Burstein HJ, Sledge GW et al (2009) Updated survival analysis of a randomized study of lapatinib alone or in combination with trastuzumab in women with HER2-positive metastatic breast cancer progressing on trastuzumab therapy. In: San Antonio Breast Cancer symposium (Abstract 61)
- 3.Cardoso F, Di Leo A, Lohrisch C, Bernard C, Ferreira F, Piccart MJ. Second and subsequent lines of chemotherapy for metastatic breast cancer: what did we learn in the last two decades? Ann Oncol. 2002;13(2):197–207. doi: 10.1093/annonc/mdf101. [DOI] [PubMed] [Google Scholar]
- 4.Roche Pharmaceuticals (2006) XELODA (capecitabine) prescribing information, Nutley, NJ
- 5.Banerjee S, Dowsett M, Ashworth A, Martin LA. Mechanisms of disease: angiogenesis and the management of breast cancer. Nat Clin Pract Oncol. 2007;4(9):536–550. doi: 10.1038/ncponc0905. [DOI] [PubMed] [Google Scholar]
- 6.Paulsson J, Sjöblom T, Micke P, et al. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol. 2009;175(1):334–341. doi: 10.2353/ajpath.2009.081030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 8.Miles DW, Chan A, Romieu G et al (2009) Final overall survival (OS) results from the randomised, double-blind, placebo-controlled, phase III AVADO study of bevacizumab (BV) plus docetaxel (D) compared with placebo (PL) plus D for the first-line treatment of locally recurrent (LR) or metastatic breast cancer (mBC). Presented at the 32nd annual San Antonio Breast Cancer symposium, San Antonio, TX, USA, December 9–13 (Oral presentation; abstract 41)
- 9.Robert NJ, Dieras V, Glaspy J et al (2009) RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab (B) for first-line treatment of HER2-negative locally recurrent or metastatic breast cancer (MBC). J Clin Oncol 27:15s (Abstr 1005) [DOI] [PubMed]
- 10.Cobleigh MA, Langmuir VK, Sledge GW, et al. A phase I/II dose-escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol. 2003;30(5 Suppl 16):117–124. doi: 10.1053/j.seminoncol.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. [PubMed] [Google Scholar]
- 12.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–478. [PubMed] [Google Scholar]
- 13.Pfizer Inc (2009) SUTENT (sunitinib) prescribing information, New York, NY
- 14.Burstein HJ, Elias AD, Rugo HS, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26(11):1810–1816. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 15.Escudier B, Roigas J, Gillessen S, et al. Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27(25):4068–4075. doi: 10.1200/JCO.2008.20.5476. [DOI] [PubMed] [Google Scholar]
- 16.Barrios CH, Hernandez-Barajas D, Brown MP et al (2009) Phase II trial of continuous once-daily dosing of sunitinib as first-line treatment in patients with metastatic renal cell carcinoma. Presented at the joint 15th congress of the European CanCer Organisation and 34th congress of the European Society for Medical Oncology, Berlin, Germany, September 20–24 (Poster presentation; Abstract P-7122)
- 17.George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–1968. doi: 10.1016/j.ejca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 19.Miller KD, Chap LI, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23(4):792–799. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 20.Jennison T, Turnbull BW. Group sequential methods with applications to clinical trials, Chapman & Hall/CRC Interdisciplinary Statistics Series. Boca Raton: CRC Press LLC; 1999. [Google Scholar]
- 21.Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ (2009) Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol Dec 5 (Epub ahead of print) [DOI] [PubMed]
- 22.Bianchi G, Loibl S, Zamagni C, et al. Phase II multicenter, uncontrolled trial of sorafenib in patients with metastatic breast cancer. Anticancer Drugs. 2009;20(7):616–624. doi: 10.1097/CAD.0b013e32832b2ea0. [DOI] [PubMed] [Google Scholar]
- 23.Baselga J, Grupo Español de Estudio Tratamiento y Otras Estrategias Experimentales en Tumores Sólidos, Roché H et al (2009) SOLTI-0701: A multinational double-blind, randomized phase 2b study evaluating the efficacy and safety of sorafenib compared to placebo when administered in combination with capecitabine in patients with locally advanced or metastatic breast cancer (BC). San Antonio Breast Cancer symposium (Abstract 45)