

Abstract
There is an urgent need for new drugs for the treatment of tropical parasitic diseases such as human African trypanosomiasis, which is caused by Trypanosoma brucei. The enzyme trypanothione reductase (TryR) is a potential drug target within these organisms. Herein we report the screening of a 62000 compound library against T. brucei TryR. Further work was undertaken to optimise potency and selectivity of two novel-compound series arising from the enzymatic and whole parasite screens and mammalian cell counterscreens. Both of these series, containing either a quinoline or pyrimidinopyrazine scaffold, yielded low micromolar inhibitors of the enzyme and growth of the parasite. The challenges of inhibiting TryR with druglike molecules is discussed.
Keywords: human African trypanosomiasis, pyrimidopyridazines, quinolines, trypanosoma brucei, trypanothione reductase
Introduction
Human African trypanosomiasis (HAT) is a serious health problem in sub-Saharan Africa, with an estimated 50000 new cases each year. HAT, a fatal disease unless treated, is caused by the protozoan parasites Trypanosoma brucei gambiense and T. b. rhodesiense, which initially reside within the blood stream and then subsequently penetrate into the central nervous system giving rise to the classical symptoms of HAT. The current drugs to treat HAT are inadequate due to poor efficacy, side effects and the requirement for parenteral administration, which is not appropriate for a rural African setting.1,2
Herein we report a target-based approach to the discovery of novel inhibitors of trypanothione reductase (TryR) as a potential therapy for HAT. TryR is an NADPH-dependent flavoprotein disulphide oxidoreductase unique to, and essential for growth of, trypanosomatid parasites, whose function is to convert trypanothione disulphide (N1,N8-bis(glutathionyl)spermidine disulphide, T[S]2; Figure 1) into the physiologically relevant reduced dithiol (T[SH]2).3 In these parasites, T[SH]2 serves as a substitute for many of the metabolic and antioxidant functions ascribed to glutathione (GSH) in mammalian cells.4 Mammalian glutathione reductase (GR) is the nearest homologue to TryR. However, the host and parasite enzymes have significant differences in their active site architecture resulting in a pronounced ability to discriminate between their respective disulphide substrates. These features make TryR an attractive target for selective drug design.5
Figure 1.
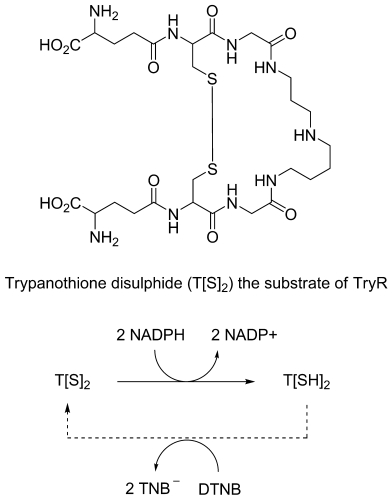
The structure of trypanothione and the principle of the DTNB-coupled assay for TryR.
Before carrying out a drug discovery programme TryR was assessed for appropriateness using the traffic light scoring system that we recently described.6 The process assesses a target in terms of: level of validation (genetic and chemical), assay feasibility, potential for toxicity and resistance, druggability, and level of structural information available allowing cross target comparisons and prioritisation for entry into hit discovery programmes. The results of this exercise are illustrated in Table 1. In brief, there was sufficient supporting information in the areas of genetic and chemical validation, assay feasibility and protein structure to progress the target into hit discovery. The main area for caution for this target was druggability since the active site is rather large and featureless.
Table 1.
Antiparasitic drug target evaluation for trypanothione reductase.
| Criteria | Status | Comments |
|---|---|---|
| Assay feasibility | green | Recombinant enzyme available in large quantities. 96 well assay in place |
| Genetic/chemical validation | green | Target essential in trypanosomes as evidenced by conditional knock-out.3 Some chemical validation from inhibitors such as trivalent antimonials and melaminophenyl arsenicals. |
| Druggability | amber | Several published low micromolar low molecular weight inhibitors. The active site of TryR is large and therefore the identification of potent low molecular weight inhibitors could be an issue. |
| Potential for resistance | amber | TryR is a single copy gene in T. brucei and no reported gene amplification in response to inhibitors has been reported. Reversal of competitive inhibitors by accumulation of T[S]2 is a potential liability |
| Potential for toxicity | green | Significant differences in substrate specificity and structure of TryR to corresponding human homologue, glutathione reductase. |
| Protein structural information | green | Crystal structures of recombinant T.cruzi TryR, alone and in combination with substrates and a covalently linked inhibitor |
A number of different TryR inhibitors have been reported in the literature. These can be broadly classified as tricyclics,7 polyamine analogues,5f,h,8 redox inhibitors,9 substrate analogues,10 and compounds identified through screening of a library of druglike compounds.11 To our knowledge, none of these series have progressed beyond the early discovery phase.
Results and Discussion
Hit discovery
In order to discover novel lead compounds against TryR, a high-throughput screen of an in-house designed diverse compound library12 (62000 compounds) was performed.
A nonenzymatically coupled colourimetric method for detecting TryR activity as developed by Fairlamb et al.13 was employed. In this assay, the activity of TryR is coupled to the reduction of DTNB (5,5′-dithiobis[2-nitrobenzoic acid]) by T(SH)2 to form the yellow thionitrobenzoate anion (TNB−) (see Figure 1). Unlike the direct assay, this method increases assay sensitivity, and allows the assay to proceed in a linear fashion for extended time periods with T[S]2 concentrations at or below Km.
The TryR screening assay was miniaturised to a 384-well plate format and optimised to the standard required to support a large-scale screening campaign. The assay was assessed for robustness in an automated environment yielding the following typical performance statistics: Z′=0.84±0.001; %CV (plate)=3.65±0.4; S:B=10±0.25; IC50 (clomipramine)=12.4±0.14 μm. Clomipramine was used as a standard throughout the screening process.
TryR was initially screened in single-point mode at 30 μm and the percentage inhibition (PI) value calculated for each compound (Figure 2). The distribution of hits deviated from normality due to a shoulder of activity between 20 and 50 PI, indicating a capacity of TryR to bind with a low affinity to a range of diverse structures. A statistical assessment of the screen relative to the error around the full signal controls (5× standard deviations) indicated that PI values ≥42% should be considered as highly significant. All compounds yielding PI ≥50% were therefore selected for re-testing in duplicate (30 μm). A total of 767 compounds were confirmed as hits giving a confirmation rate (primary to re-test screen) of 84% and an overall confirmed hit rate of 1.24%.
Figure 2.
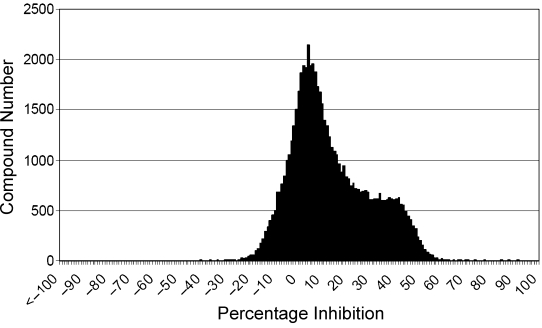
Single-point screen percentage inhibition (PI) distribution against TryR for all compounds returning PI values ≥50 (n=901).
A preliminary chemistry assessment of the confirmed hits identified a selection of 120 compounds for potency determinations. Each compound was subjected to a ten-point half-log titration in duplicate ranging from 30 μm to 1 nm. In all cases, residual material from the potency compound plates was subjected to LCMS analysis for structure and purity confirmation. Figure 3 illustrates the ability of this assay to report reliable potency values for test compounds, yielding a correlation coefficient (R2) value of 0.967 between replicate pIC50 determinations.
Figure 3.
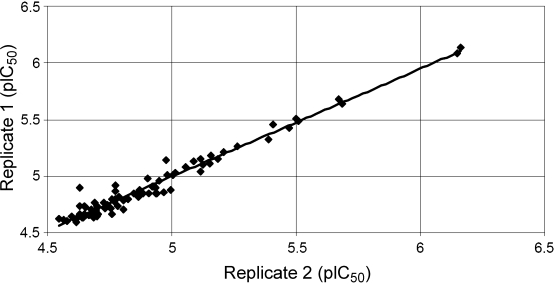
pIC50 correlation between two replicate assays.
Analysis of the potency data for the best hits revealed multiple putative hit series covering a range potencies from 0.7 to 30 μm. Key compounds were resynthesised or resupplied to further confirm their activity and identity. From the five series initially identified, two were deemed of sufficient interest to progress into formal hit validation. The aim of this phase was to rapidly assess the existence of defined structure–activity relationships (SAR), whilst securing a significant increase (>10-fold) in activity over the initial hit compound.
Hit series 1
Series 1 was based on a quinoline scaffold, which afforded a number of possible points of variation: the 5-methylfuran in the 2-position, the amide in the 4-position, and the 6-bromo substituent. To expand the SAR, we prepared or purchased 84 analogues. Table 2 provides data on the most potent and significant inhibitors.
Table 2.
Activity of series 1 (compounds 1–26) against TryR.
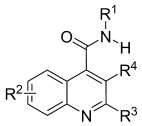 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R4 | TryR IC50 [μm] |
| 1 | CH2CH2NMe2 | 6-Br | 5′-Methyl-furan-2-yl | H | 1.7 |
| 2 | CH2CH2NHMe | 6-Br | 5′-Methyl-furan-2-yl | H | 1.1 |
| 3 | CH2CH2NMe3+ | 6-Br | 5′-Methyl-furan-2-yl | H | 1.2 |
| 4 | CH2CH2-N-morpholine | 6-Br | 5′-Methyl-furan-2-yl | H | 4.2 |
| 5 | 2-pyridyl methyl | 6-Br | 5′-Methyl-furan-2-yl | H | 4.7 |
| 6 | 3-pyridyl | 6-Br | 5′-Methyl-furan-2-yl | H | 7.2 |
| 7 | CH2CH2CH2-N-imidazole | 6-Br | 5′-Methyl-furan-2-yl | H | 5.1 |
| 8 | CH2CH2NMe2 | 6-Cl | 5′-Methyl-furan-2-yl | H | 3.1 |
| 9 | CH2CH2NMe2 | 7-Br | 5′-Methyl-furan-2-yl | H | 5.7 |
| 10 | CH2CH2NMe2 | 8-Br | 5′-Methyl-furan-2-yl | H | 5.8 |
| 11 | 2-pyridyl methyl | H | 5′-Methyl-furan-2-yl | H | 12.1 |
| 12 | CH2CH2NMe2 | H | 5′-Methyl-furan-2-yl | H | 14.4 |
| 13 | CH2CH2NMe2 | 6-Cl | 4′-Methylphenyl | CH3 | 26.5 |
| 14 | CH2CH2-N-morpholine | H | 5′-Methyl-furan-2-yl | H | 100 |
| 15 | CH2Ph | 6-Br | 5′-Methyl-furan-2-yl | H | >100 |
| 16 | CH2CH2 CH2-N-morpholine | 6-Cl | 5′-Methyl-furan-2-yl | H | 11.6 |
| 17 | CH2CH2NMe2 | 6-Br | 3′-Methoxyphenyl | H | >100 |
| 18 | CH2CH2NMe2 | 6-Br | Phenyl | H | >100 |
| 19 | CH2C6H4-p-SO2NH2 | 6-Br | 5′-Methyl-furan-2-yl | H | 8.4 |
| 20 | CH2CH2NMe2 | 6-Br | Furan-2-yl | H | 32.4 |
| 21 | CH2CH2NMe2 | 6-Br | Pyrid-4-yl | H | 96.3 |
| 22 | CH2CH2NMe2 | 6-Br | 5′-Methyl-thiophen-2-yl | H | >100 |
| 23 | -N-morpholine | 6-Br | 5′-Methyl-furan-2-yl | H | >100 |
| 24 | CH2-furan-2-yl | 6-Br | 5′-Methyl-furan-2-yl | H | >100 |
| 25 | CH2CH2Ph | 6-Br | 5′-Methyl-furan-2-yl | H | >100 |
| 26 | CH2CH2NMe2 | 6-F | 5′-Methyl-furan-2-yl | H | >100 |
The general synthetic route is shown in Scheme 1. In brief, the indoline-2,3-dione was reacted with an appropriate ketone to give the quinoline. The carboxylic acid could then be condensed with appropriate amines to form the required amides.
Scheme 1.
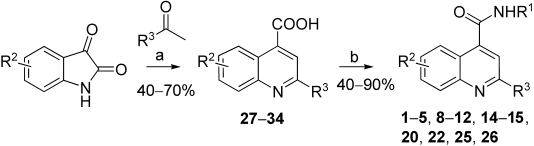
Synthetic route for hit series 1. Reagents and conditions: a) Ketone, KOH, EtOH/H2O, 110°C, 12 min, μW; b) R1NH2, CDI, CH2Cl2, RT, 18 h.
A summary of the SAR is given in Figure 4. Essentially, any variation of the 5-methylfuran at the 2-position led to a decrease in activity (1 and 17, 20 and 22; 8 and 13; 12 and 18); in the 4-position, amides with a basic substituent, such as dimethylaminoethyl were more potent than neutral aryl or alkylaryl amides (15, 23, 24, 25); and the 6-bromo group could be moved around the ring without too much effect on the activity (1, 9, 10). Although replacing the 6-bromo substituent with a chloro atom led to a small effect on activity (1 and 8, 16), replacement of the 6-bromo with hydrogen or fluorine led to loss in activity (1 and 12; 4 and 14, 1 and 26). Particularly with substituents on the 4-position, increases in MW and clogP led to small increases in potency, suggestive of a general surface contact between inhibitors and protein. There may be a similar effect with the 6-bromo group, as changes in location to the 7- or 8-position of the quinoline ring system, or even replacing it with a chlorine, led to only small effects on activity. It was difficult to deduce which changes, if any, would increase potency at these positions. It is possible that the 5-methylfuran at the 2-position is making a very specific hydrogen bonding and/or π-stacking interaction, which accounts for the requirement for this group at this position.
Figure 4.
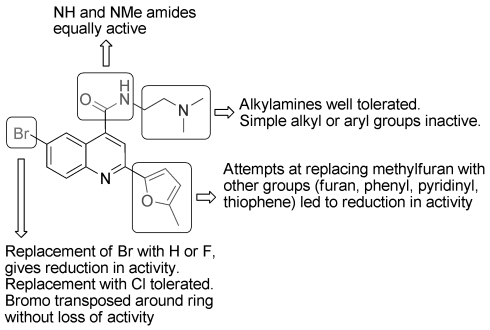
SAR for hit series 1.
Hit series 2
Series 2, containing the pyrimidopyridazines scaffold, gave five compounds with an inhibition of >62% in the initial screen. The general synthetic route is outlined in Scheme 2. The substituted 6-chlorouracil starting material was made by condensation of the appropriately substituted urea with malonic acid, followed by chlorination. The chloride was displaced with an appropriate hydrazine. The hydrazine intermediate (35) was then condensed with aldehyde and cyclisation was achieved by treatment with sodium nitrite followed by dehydration through microwave heating in DMF with molecular sieves to give 42.14 The free NH could be alkylated with various alkyl bromides to give the desired product. In total ∼30 compounds from this series were assayed. Table 3 provides data on the most potent and significant inhibitors. A summary of the SAR is given in Figure 5.
Scheme 2.
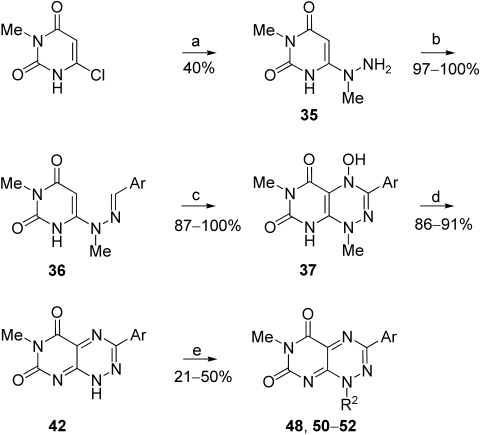
Synthetic Route for hit series 2. Reagents and conditions: a) H2NNHMe, EtOH, 80°C, 1 h; b) ArCOH, EtOH, 80°C, 2 h; c) NaNO2, AcOH, 0→20°C, 16 h; d) DMF, 90°C, 2 h; e) R2Br, Cs2CO3, 80°C, 3 h, DMF.
Table 3.
Activity of series 2 (compounds 31–44) against TryR.
 | ||||
|---|---|---|---|---|
| Compd | R1 | R2 | R3 | TryR IC50 [μm] |
| 39 | Me | Me | 4′-Methylphenyl | 8.6 |
| 40 | Me | CH2CH3 | Thiophen-2-yl | 12.8 |
| 41 | Me | CH2CH3 | 2′-Fluoro-5′-Bromophenyl | 10.2 |
| 42 | Me | H | 2′-Fluoro-5′-Bromophenyl | >100 |
| 43 | H | H | 2′-Fluoro-5′-Bromophenyl | 92 |
| 44 | Me | CH2CH3 | Pyridine-3-yl | >100 |
| 45 | Me | CH2CH3 | 4′-Bromothiophen-2-yl | 13.0 |
| 46 | Me | CH2CH3 | 4′-Trifluoromethylphenyl | 15.2 |
| 47 | Me | CH2CH3 | Phenyl | 11.3 |
| 48 | Me | CH2CH2NMe2 | 2′-Fluoro-5′-Bromophenyl | >100 |
| 49 | Me | CH2CH3 | CH=CHC6H5 | 2.6 |
| 50 | Me | CH(CH3)2 | 2′-Fluoro-5′-Bromophenyl | >50 |
| 51 | Me | (CH2)3CH3 | 2′-Fluoro-5′-Bromophenyl | >100 |
| 52 | Me | cyclopentyl | 2′-Fluoro-5′-Bromophenyl | >100 |
Figure 5.
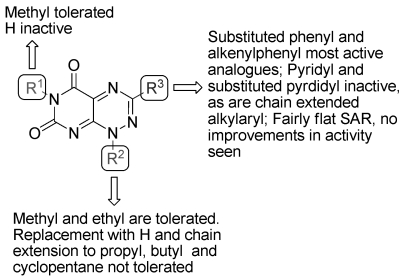
SAR for hit series 2.
Small alkyl groups were required in positions R1 (methyl) and R2 (methyl and ethyl) as replacement of R2 or both R1 and R2 with hydrogen led to a loss in activity (42 and 43). The methyl at R2 could be replaced by a small alkyl group; however, replacement with larger alkyl groups led to a decrease in activity. A number of different aromatic substituents were tolerated at R3, however, there was no clear SAR.
Selectivity and mode of action studies
The selectivity of these series with respect to nearest human homologue of TryR was assessed in glutathione reductase (GR) assays. Both TryR and GR assays were performed with substrate concentrations equal to Km allowing direct comparison of IC50 values for selectivity assessment. All of the compounds tested demonstrated significant selectivity for TryR over GR (Table 4). This is not surprising if we assume our binding model is correct as the binding site in GR is smaller, less hydrophobic and less negatively charged than that of TryR. Hence, if our compounds are binding by a general hydrophobic interaction and interaction with negatively charged residues in the active site, they are unlikely to interact with GR.
Table 4.
Selectivity of hit compounds and mode of inhibition.[a]
| Compd | Mode of inhibiton | Inhibitor constants [μm] | IC50 [μm] | EC50 [μm] | |||
|---|---|---|---|---|---|---|---|
| Ki | Ki′ | TryR | hGR[b] | Parasite[c] | MRC-5[c] | ||
| 1 | linear mixed | 1.6±0.2 | 3.2±0.3 | 1.7 | >100 | 10.3 | >50 |
| 2 | NT[d] | – | – | 1.1 | NT | 6.0 | 18.7 |
| 3 | NT | – | – | 1.2 | NT | >50 | >50 |
| 4 | linear mixed | 3.0±0.2 | 4.3±0.3 | 4.2 | 74.2 | 25.6 | >50 |
| 5 | linear mixed | 2.0±0.1 | 3.0±0.2 | 4.7 | 33.4 | 18.7 | 31.1 |
| 6 | NT | – | – | 7.2 | NT | 6.9 | NT |
| 7 | NT | – | – | 5.1 | NT | 16.5 | NT |
| 10 | NT | – | – | 5.8 | NT | 19.2 | NT |
| 12 | NT | – | – | 14.4 | NT | 32.1 | 43.7 |
| 19 | NT | – | – | 8.4 | NT | 14.5 | NT |
| 39 | linear uncompetitive | 4.6±0.3 (T[S]2) 29.4±1.7 (NADPH) | – | 8.6 | >100 | 1.22 | 1.62 |
| 40 | linear uncompetitive | 3.0±0.2 (T[S]2) 21.5±1.6 (NADPH) | – | 12.8 | 10.0 | 1.43 | 3.2 |
| 41 | NT | – | – | 10.2 | NT | 8.1 | NT |
| 45 | NT | – | – | 13.0 | NT | 3.4 | 12.9 |
[a] MOI: Compounds 1–19 are from hit series 1 and 39–45 from hit series 2. [b] hGR: human glutathione reductase. [c] Concentration giving 50% growth inhibition after 73 h exposure. [d] NT: not tested.
Mode of inhibition studies were carried out on key representatives of series 1 and 2. The data indicated that neither compound series acted as simple linear competitive inhibitors with respect to T[S]2. The mixed inhibition of compounds in series 1 indicates the compounds can either bind to the free enzyme or to the enzyme–substrate complex, implying that the compounds are not binding in an orientation that excludes binding of T[S]2. For series 2, the binding appears to be linear and uncompetitive, with compounds binding only to the enzyme–substrate complex. In enzymes such as TryR that obey a bi bi ping pong reaction mechanism, such an inhibition pattern can arise due to an inhibitor competing for the second independent substrate-binding site, NADPH. However, a similar uncompetitive inhibition pattern was observed when the concentration of the second substrate, NADPH, was varied in the presence of a fixed concentration of T[S]2. This finding indicates that compounds from series 2 bind at a site distinct from both substrate-binding sites. Since compounds of series2 have some structural similarity to the FAD co-factor of TryR, we propose that disruption of the orientation or displacement of FAD could account for the uncompetitive inhibition pattern. In theory, both series present significant advantages over competitive inhibitors where build up of substrate trypanothione during inhibition in vivo could reduce the efficacy of the inhibitor. Indeed, substrate build-up in the case of uncompetitive inhibition may even drive binding of the inhibitor.
The most potent compounds were also assessed for their ability to affect T. brucei and MRC-5 (prototypical mammalian cell line) proliferation in vitro (Table 4). Series 1 compounds showed weak inhibition of parasite growth. Whilst it is expected that cellular activity is likely to be lower than enzyme activity due to factors such as high intracellular substrate concentration, there was no clear correlation between enzyme inhibition and effect on trypanosomes. In the case of series 2, the cellular activity was more potent than would be predicted by the enzyme assay suggesting that these compounds are either selectively concentrated by the parasites or are acting off-target. However, the latter seems more likely, given the lack of selectivity apparent between the trypanosome and MRC-5 read-outs.
Conclusions
We have reported the identification of two novel compound series active against TryR in vitro from a high-throughput screening campaign. Both hit series were low molecular weight compounds with lead-like properties suitable for a medicinal chemistry optimisation programme. They are structurally very different to other TryR inhibitors reported in the literature and constitute novel chemical lead structures against TryR.
SAR studies were undertaken for both series. For series 1, there was some discernable SAR. Unfortunately, we were unable to significantly increase the potency of the compounds against the enzyme to a level likely to have therapeutic significance. The TryR active site contains both hydrophobic and acidic regions (for interaction with the spermidine moiety); it is likely that what we are observing is hydrophobic interactions between the hydrophobic regions of our inhibitors and the active site and electrostatic interactions between the positive charge on the inhibitors and the negatively charged region of the active site. In order to get a significant increase in potency, it will be necessary to build in some additional specific interactions between the inhibitor and the enzyme. This process would be greatly aided by a co-crystal structure of an inhibitor bound in the active site. Some cellular activity was observed, which implies that the compounds are able to penetrate into cells. However we believe that to get a significant correlation between enzyme and cellular activity will require enzyme inhibitors that are significantly more potent.
Data from the gene knockout studies indicated that it would be necessary to cause >90% loss in activity of TryR for cell death to occur.3 Therefore, either a very potent inhibitor (low nanomolar) of the enzyme is required or some kind of irreversible inhibition. Furthermore, the TryR active site is large and relatively solvent exposed, which may make the discovery of low molecular weight, highly potent, competitive inhibitors problematic, especially in the absence of structural information from inhibitors bound into the active site.
In summary, we have identified two novel series of TryR inhibitors from a HTS screen and have carried out SAR studies. Further work is needed to increase the potency of these compounds to a level where the activity is sufficient to achieve significant cellular activity. This would be greatly facilitated by structural information.
Experimental Section
Biology
Enzyme-based assays
Primary screening assay for TryR
Recombinant TryR from T. brucei was initially screened for inhibitors in single-point mode. Each test compound (500 nL) was transferred into 384-well clear polystyrene plates at an appropriate stock concentration. Clomipramine and DMSO were added to the appropriate control wells. Then 37.5 μL of reaction mix (3 nm TryR, 6 μm T[S]2, 50 μm DTNB, 40 mm Hepes, 1 mm EDTA, pH 7.4) was added to each well and the reaction initiated by addition of 150 μm NADPH (4 μL). Low signal controls received no NADPH. Plates were incubated for 35 min at room temperature and absorbance measured at 405±8 nm using the Envision platereader (Perkin–Elmer).
Initial single-point screening of compounds was conducted at 30 μm; subsequent ten-point screening was conducted between 30 μm and 1 nm or between 100 μm and 3 nm depending on stock concentrations. The final DMSO level was 1% in all cases.
Mode of inhibition studies
An assay mixture consisting of TryR, NADPH and DTNB was made up in 40 mm HEPES; 1 mm EDTA, pH 7.4. Assay mixture (180 μL) containing three different concentrations of test compound was added to three rows of a microtitre plate, a fourth row contained only the assay mixture. The test compound concentration ranged up to ∼1×IC50 value. T[S]2 was serially diluted across a fifth row of the plate to produce a 12-point range from 500 μm to 5.8 μm. The assay was initiated by transferring 20 μL of the T[S]2 row to each of the assay rows. The final 200 μL assay contained 150 μm NADPH; 50 μm DTNB and 20 mUmL−1 U TryR.
Reaction rates were measured from the linear increase in absorbance at 412 nm using a Molecular Devices Thermomax plate reader. Individual data sets were plotted as 1/v versus 1/[S] using Grafit 5.0 (Erithacus software) and inspected for diagnostic inhibition patterns. Data sets were then globally fitted by nonlinear regression to the appropriate equation for competitive, mixed or uncompetitive inhibition.
Cell-based assays
Measurement of the ability of the compounds to inhibit trypanosome (bloodstream form T. brucei brucei single marker) and human (MRC5, human lung fibroblast cells) cell growth was performed using a modification of the cell viability assay previously described by Raz et al.15
Trypanosome growth assay
Cell culture plates were stamped with 1μL of an appropriate stock concentration of test compound, followed by addition of 200 μL trypanosome culture (HMI9-T,16 10% FCS) to each well at a density of 2000 cells per well (except column 12 receiving media only).
MRC-5 growth assay
MRC-5 cells (2000 cells per well) were seeded in 200 μL DMEM, 10% FCS and allowed to adhere overnight. One microlitre of test compound was added to each well on the day of assay initiation.
Trypanosome and MRC-5 plates were incubated at 37°C in an atmosphere of 5% CO2 for 69 h, prior to the addition of 20 μL rezasurin (final concentration 50 μm). After a further 4 h, fluorescence was measured (excitation 528 nm; emission 590 nm) using a BioTek flx800 plate reader.
For cell assays, ten-point screening was conducted between 15 μm and 0.5 nm or between 50 μm and 2 nm, depending on stock concentrations. The final DMSO level was 0.5% in all cases.
Chemistry
1H NMR were recorded on either a Bruker Avance DPX 500 or on a Bruker Avance 300 spectrometer. Chemical shifts (δ) are expressed in ppm. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (br), or combination thereof. Low resolution electrospray (ES) mass spectra were recorded on a Bruker MicroTof mass spectrometer, run in positive ion mode, using either CH3OH, CH3OH/H2O (95:5) or H2O/CH3CN (1:1) + 0.1% formic acid as the mobile phase. High resolution electrospray measurements were performed on a Bruker MicroTof mass spectrometer. LC-MS analysis was performed using an Agilent HPLC 1100 with a diode array detector in parallel with a Bruker MicroTof mass spectrometer. A Phenomenex Gemini C18 column 110 A 50×3.0 mm, 5 μm particle size was used. The mobile phase was H2O/CH3CN + 0.1% HCOOH (80:20→5:95 over 3.5 min, 5:95 for 1.5 min; flow rate 0.5 mLmin−1).
Thin layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates using UV light and/or KMnO4 for visualisation. Column chromatography was performed using RediSep® 4 or 12 g silica prepacked columns with the specified eluent system.
Compounds 6, 7, 13, 16, 17, 19, 21, 23 and 24 were purchased from ChemBridge and compounds 18, 39, 40, 41, 44, 45, 46, 47 and 49 were purchased from ASINEX and submitted to the screening assay. They were checked for purity by LCMS and were found to be >90% pure.
All reactions were carried out under dry and inert conditions unless otherwise stated.
6-Bromo-N-(2-(dimethylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (1): Compound 27 (200 mg, 0.6 mm) and carbonyldiimidazole (CDI; 108 mg, 0.66 μm) were stirred in CH2Cl2 (15 mL) at 35°C for 20 min. N,N-dimethylethane-1,2-diamine (58 mg, 0.66 mm) was added, and the solution stirred at RT for 18 h. The solution was diluted with CH2Cl2 (30 mL) and washed with aq NaHCO3 (2×20 mL), H2O (1×30 mL) and brine (2×20 mL). The organic layer was dried (MgSO4), filtered and concentrated to afford a yellow/brown solid. The crude product was purified by flash column chromatography (6%, CH3OH/CH2Cl2) to give 1 as pale yellow solid (65 mg, 27%): 1H NMR (500 MHz, [D6]DMSO): δ=2.28 (6H, br s, 2CH3), 2.44 (3H, s, CH3), 2.51 (2H, m, CH2), 3.46 (2H, q, J=6.3 Hz, CH2), 6.39 (1H, m, ArH), 7.38 (1H, d, J=3.2 Hz, ArH), 7.91 (1H, d, J=2.2 Hz, ArH), 7.92 (1H, m, ArH), 7.96 (1H, m, ArH), 8.4 (1H, d, J=2.2 Hz, ArH), 8.85 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 402 (100) [M+H]+ (79Br), 404 (100) [M+H]+ (81Br); tR=5.0 min.
6-Bromo-N-(2-(methylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (2): Prepared as for 1 from 27 (150 mg, 0.45 mm) and CDI (82 mg, 0.5 mm) in CH2Cl2 (15 mL). The mixture was stirred at 35°C for 20 min, then treated with N-methylethane-1,2-diamine (37 mg, 0.5 mm) and stirred at RT for 18 h. The resultant solid product was collected by filtration and further purified by trituration with hot Et2O (3×10 mL) to give 2 as a pale yellow solid (37 mg, 21%): 1H NMR (500 MHz, [D6]DMSO): δ=2.29 (3H, s, CH3), 2.40 (3H, s, CH3), 2.72 (2H, m, CH2), 3.41 (2H, m, CH2), 6.39 (1H, br s, ArH), 7.37 (1H, s, ArH), 7.89 (1H, dd J=9.0 and 2.2 Hz, ArH), 7.96 (2H, m, 2ArH), 8.34 (1H, d, J=2.1 Hz, ArH), 8.88 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 388 (100) [M+H]+ (79Br), 390 (100) [M+H]+ (81Br); tR=4.2 min.
6-Bromo-N,N,N-trimethyl-2-(2-(5-methylfuran-2-yl)quinoline-4-carboxamido)ethanaminium iodide (3): CH3I (31 mg, 0.22 mm) was added to a solution of 1 (60 mg, 0.15 mm) in dry acetone (5 mL). The solution was stirred at RT for 48 h then concentrated. A solid was precipitated from the resulting gum on the addition of acetone (15 mL), which was isolated by filtration and washed with acetone (3×10 mL) to afford 3 as a beige solid (50 mg, 61%): 1H NMR (500 MHz, [D6]DMSO): δ=2.41 (3H, s, 2CH3), 3.19 (9H, s, 3CH3), 3.6 (2H, m, CH2), 3.79 (2H, m, CH2), 6.39 (1H, br s, ArH), 7.36 (1H, m, ArH), 7.94 (1H, dd, J=9.0, 2.2 Hz, ArH), 7.98 (2H, m, 2ArH), 8.34 (1H, d, J=2.0 Hz, ArH), 9.25 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 418 (100) [M+H]+ (79Br), 420 (100) [M+H]+ (81Br); tR=4.4 min.
6-Bromo-2-(5-methylfuran-2-yl)-N-(2-morpholinoethyl)quinoline-4-carboxamide (4): Prepared as for 1 from 27 (150 mg, 0.45 mm), CH2Cl2 (15 mL), CDI (82 mg, 0.5 mm) and 2-morpholinoethanamine (65 mg, 0.5 mm). The reaction was stirred for 18 h then concentrated. The crude solid was recrystallised from refluxing EtOAc to give 4 as a white crystalline solid (95 mg, 48%): 1H NMR (500 MHz, [D6]DMSO): δ=2.38 (3H, s, CH3), 2.48 (2H, m, CH2), 2.49 (4H, m, 2CH2), 4.49 (2H, m, CH2), 4.66 (4H, m, 2CH2), 6.39 (1H, m, ArH), 7.38 (1H, d, J=3.2 Hz, ArH), 7.91 (1H, d, J=2.2 Hz, ArH), 7.92 (1H, m, ArH), 7.96 (1H, m, ArH), 8.32 (1H, d, J=2.0 Hz, ArH), 8.86 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 444 (100) [M+H]+ (79Br), 446 (100) [M+H]+ (81Br); tR=4.5 min.
6-Bromo-2-(5-methylfuran-2-yl)-N-(pyridin-2-ylmethyl)quinoline-4-carboxamide (5): Prepared as for 1 from 27 (150 mg, 0.45 mm), CDI (82 mg, 0.5 mm) and pyrin-2-ylmethanamine (55 mg, 0.5 mm). The reaction was stirred for 18 h then concentrated. The crude solid was purified by trituration with hot Et2O (3×10 mL) to give 5 as pale yellow solid (70 mg, 37%): 1H NMR (500 MHz, [D6]DMSO): δ=2.39 (3H, s, CH3), 4.79 (2H, m, CH2), 6.39 (1H, br s, ArH), 7.35 (1H, dd, J=6.7, 5.0 Hz, ArH), 7.41 (1H, d, J=3.3 Hz, ArH), 7.49 (1H, d, J=7.8 Hz, ArH), 7.84 (1H, dt, J=7.7, 1.8 Hz, ArH), 7.92 (1H, dd, J=9.0, 2.2 Hz, ArH), 7.99 (1H, d, J=9.0 Hz, ArH), 8.03 (1H, s, ArH), 8.52 (1H, s, ArH), 8.61 (1H, m, ArH), 9.51 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 422 (100) [M+H]+ (79Br), 424 (100) [M+H]+ (81Br); tR=4.7 min.
6-Chloro-N-(2-(dimethylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (8): Compound 29 (468 mg, 1.63 mm) and CDI (260 mg, 1.8 mm) were stirred in CH2Cl2 (15 mL) for 10 min at RT. N,N-Dimethylethane-1,2-diamine (158 mg, 1.8 mm) was added, and the resulting solution was stirred at RT for 18 h. The reaction was diluted with CH2Cl2 (20 mL) and washed with aq NaHCO3 (2×20 mL), H2O (2×30 mL) and brine (1×50 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (2%, CH3OH/CH2Cl2) gave 8 as a white solid (200 mg, 34%): 1H NMR (500 MHz, [D6]DMSO): δ=2.26 (6H, s, 2CH3), 2.41 (3H, s, CH3), 2.48 (2H, m, CH2), 3.47 (2H, m, CH2), 6.39 (1H, br s, ArH), 7.34 (1H, br s, ArH), 7.77 (1H, m, ArH), 7.90 (1H, br s, ArH), 7.96 (1H, dd, J=9.0, 2.2 Hz, ArH), 8.1 (1H, br s, ArH), 8.84 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 358 [M+H]+; tR=5.0 min.
7-Bromo-N-(2-(dimethylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (9): Prepared as for 1 from 31 (265 mg, 0.8 mm), CH2Cl2 (15 mL), CDI (144 mg, 0.88 mm) and N,N-dimethylethane-1,2-diamine (77 mg, 0.66 mm). The reaction was stirred at RT for 18 h. Purification by flash column chromatography (3%, CH3OH/CH2Cl2) gave 9 as a white solid (85 mg, 38%): 1H NMR (300 MHz, CDCl3): δ=2.3 (6H, s, 2CH3), 2.45 (3H, s, CH3), 2.68 (2H, t, J=5.8 Hz, CH2), 3.70 (2H, q, J=5.4 Hz, CH2), 6.21 (1H, m, ArH), 7.35 (2H, m, ArH), 7.92 (1H, s, ArH), 8.05 (1H, m, ArH), 8.16 ppm (1H, m, ArH); LCMS (ES+): m/z (%): 402 (100) [M+H]+ (79Br), 404 (100) [M+H]+ (81Br); tR=5.1 min.
8-Bromo-N-(2-(dimethylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (10): Prepared as for 1 from 32 (200 mg, 0.6 mm), CH2Cl2 (10 mL), CDI (108 mg, 0.66 mm) and N,N-dimethylethane-1,2-diamine (58 mg, 0.66 mm). The reaction was stirred for 18 h at RT. Purification by flash column chromatography (3%, CH3OH/CH2Cl2) gave 10 as a white solid (60 mg, 25%): 1H NMR (500 MHz, [D6]DMSO): δ=2.24 (6H, s, 2CH3), 2.43 (3H, s, CH3), 2.8 (2H, m, CH2), 3.46 (2H, m, CH2), 6.40 (1H, br s, ArH), 7.38 (1H, d, J=3.3 Hz, ArH), 7.47 (1H, m, ArH), 7.91 (1H, s, ArH), 8.12 (1H, dd, J=8.4, 1.2 Hz, ArH), 8.17 (1H, dd, J=7.5, 1.2 Hz, ArH), 8.84 ppm (1H, br t, NH); LCMS (ES+): m/z (%): 402 (100) [M+H]+ (79Br), 404 (100) [M+H]+ (81Br); tR=5.2 min.
2-(5-Methylfuran-2-yl)-N-(pyridin-2-ylmethyl)quinoline-4-carboxamide (11): Compound 34 (100 mg, 0.4 mm) and CDI (72 mg, 0.44 mm) were dissolved in CH2Cl2 (4 mL). The solution was stirred for 10 min at RT, then pyrin-2-ylmethanamine (48 mg, 0.44 mm) was added. The reaction was stirred at RT for 48 h and worked up as previously described (see details for 8). The product was recrystallised from from Et2O to give 11 as a white solid (80 mg, 58%): 1H NMR (500 MHz, [D6]DMSO): δ=2.44 (3H, br s, CH3), 4.68 (2H, m, CH2), 6.39 (1H, m, ArH), 7.31 (1H, m, ArH), 7.35 (1H, m, ArH), 7.48 (1H, d, J=7.8 Hz, ArH), 7.6 (1H, ddd, J=8.2, 6.9, 1.2 Hz, ArH), 7.80 (1H, ddd, J=8.4, 6.9, 1.4 Hz, ArH), 7.85 (1H, dt, J=7.7, 1.8 Hz, ArH), 7.97 (1H, s, ArH), 8.05 (1H, d, J=8.0 Hz, ArH), 8.16 (1H, m, ArH), 8.58 (1H, ddd, J=4.8, 1.6 and 0.8 Hz, ArH), 9.47 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 344 [M+H]+; tR=4.7 min.
N-(2-(Dimethylamino)ethyl)-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (12): Prepared as for 11 from 34 (100 mg, 0.4 mm), 8 mL CH2Cl2, CDI (72 mg, 0.44 mm) and N,N-dimethylaminoethylenediamine (39 mg, 0.44 mm). The reaction was stirred for 18 h at RT. Column chromatography (5%, CH3OH/CH2Cl2) gave 12 as a white solid (30 mg, 24%): 1H NMR (500 MHz, [D6]DMSO): δ=2.22 (6H, s, 2CH3), 2.41 (3H, s, CH3), 2.48 (2H, m, CH2), 3.46 (2H, m, CH2), 6.39 (1H, m, ArH), 7.32 (1H, d, J=3.3 Hz, ArH), 7.59 (1H, ddd, J=8.2, 6.9, 1.2 Hz, ArH), 7.78 (1H, ddd, J=8.4, 6.9, 1.4 Hz, ArH), 7.86 (1H, s, ArH), 8.03 (1H, d, J=8.4 Hz, ArH), 8.11 (1H, m, ArH), 8.8 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 324 [M+H]+; tR=4.3 min.
2-(5-Methylfuran-2-yl)-N-(2-morpholinoethyl)quinoline-4-carboxamide (14): Prepared as for 11 from 34 (150 mg, 0.6 mm), 10 mL CH2Cl2, CDI (108 mg, 0.66 mm) and 2-morpholinoethanamine (86 mg, 0.66 mm). The reaction was stirred for 18 h at RT, concentrated and purified by triturating the crude solid with hot Et2O to give 14 (50 mg, 23%): 1H NMR (500 MHz, [D6]DMSO): δ=2.41 (3H, s, CH3), 2.48 (4H, m, 2CH2), 2.52 (2H, m, CH2), 3.49 (2H, q, J=6.5 Hz, CH2), 3.64 (4H, t, J=4.4 Hz, 2CH2), 6.39 (1H, m, ArH), 7.34 (1H, d, J=3.3 Hz, ArH), 7.59 (1H, ddd, J=8.2, 6.9, 1.2 Hz, ArH), 7.79 (1H, ddd, J=8.4, 6.9, 1.4 Hz, ArH), 7.85 (1H, s, ArH), 8.05 (1H, d, J=8.4 Hz, ArH), 8.18 (1H, m, ArH), 8.79 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 366 [M+H]+; tR=4.1 min.
N-Benzyl-6-bromo-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (15): Prepared as for 1 from 27 (150 mg, 0.45 mm), CH2Cl2 (10 mL), CDI (82 mg, 0.5 mm) and benzylamine (54 mg, 0.5 mm). The reaction was stirred for 18 h at RT. Flash column chromatography (2%, CH3OH/CH2Cl2) gave 15 as a beige solid (16 mg, 8%): 1H NMR (500 MHz, CDCl3): δ=2.36 (3H, s, CH3), 4.70 (2H, d, J=5.7 Hz, CH2), 6.09 (1H, m, ArH), 6.59 (1H, br s, NH), 6.95 (1H, m, ArH), 7.31 (1H, m, ArH), 7.39 (4H, m, 4ArH), 7.58 (1H, s, ArH), 7.66 (1H, dd, J=9.0, 2.1 Hz, ArH), 7.74 (1H, d, J=9.0 Hz, ArH), 8.2 ppm (1H, d, J=2.1 Hz, ArH); LCMS (ES+): m/z (%): 421 (100) [M+H]+ (79Br), 423 (100) [M+H]+ (81Br); tR=5.7 min.
6-Bromo-N-(2-(dimethylamino)ethyl)-2-(furan-2-yl)quinoline-4-carboxamide (20): Prepared as for 1 from 33 (200 mg, 0.63 mm), CH2Cl2 (15 mL), CDI (115 mg, 0.7 mm) and N,N-dimethylethane-1,2-diamine (62 mg, 0.7 mm). The reaction was stirred at RT for 18 h. Flash column chromatography (6%, CH3OH/CH2Cl2) gave 20 as a pale yellow solid (65 mg, 27%): 1H NMR (500 MHz, [D6]DMSO): δ=2.29 (6H, br s, 2CH3), 2.51 (2H, m, CH2), 3.48 (2H, q, J=6.5 Hz, CH2), 6.78 (1H, m, ArH), 7.47 (1H, m, ArH), 7.95 (1H, dd, J=9.0, 2.2 Hz, ArH), 7.99 (3H, m, 3ArH), 8.44 (1H, d, J=2.2 Hz, ArH), 8.9 ppm (1H, br s, NH); LCMS (ES+): m/z (%): 388 (100) [M+H]+ (79Br), 390 (100) [M+H]+ (81Br); tR=4.9 min.
6-Bromo-N-(2-(dimethylamino)ethyl)-2-(5-methylthiophen-2-yl)quinoline-4-carboxamide (22): Prepared as for 11 from 30 (209 mg, 0.6 mm), CH2Cl2 (15 mL), CDI (108 mg, 0.66 mm) and N,N-dimethylethane-1,2-diamine (58 mg, 0.66 mm). The reaction was stirred for 18 h at RT. Flash column chromatography (3%, CH3OH/CH2Cl2) gave 22 as a beige solid (73 mg, 29%): 1H NMR (500 MHz, [D6]DMSO): δ=2.28 (6H, br s, 2CH3), 2.44 (3H, s, CH3), 2.60 (2H, m, CH2), 3.50 (2H, q, J=6.3 Hz, CH2), 6.92 (1H, br s, ArH), 7.76 (1H, m, ArH), 7.89 (2H, m, 2ArH), 8.11 (1H, s, ArH), 8.40 (1H, d, J=2.1 Hz, ArH), 8.9 ppm (1H, m, NH); LCMS (ES+): m/z (%): 419 (100) [M+H]+ (79Br), 421 (100) [M+H]+ (81Br); tR=5.1 min.
6-bromo-2-(5-methylfuran-2-yl)-N-phenethylquinoline-4-carboxamide (25): Prepared as for 1 from 27 (200 mg, 0.6 mm), CH2Cl2 (10 mL), CDI (108 mg, 0.66 mm) and phenethylamine (84 μL, 0.66 mm). The reaction was stirred for 18 h at RT. Purification by flash column chromatography (3%, CH3OH/CH2Cl2) gave 25 as a cream solid (58 mg, 28%): 1H NMR (500 MHz, CDCl3): δ=2.43 (3H, s, CH3), 4.70 (2H, d, J=5.7 Hz, CH2), 5.23 (2H, m, CH2), 6.11 (1H, m, ArH), 6.58 (1H, m, ArH), 6.97 (1H, d, J=3.3 Hz, ArH), 7.31 (1H, m, ArH), 7.61 (3H, m, 3ArH), 7.61 (1H, s, ArH), 7.66 (1H, dd, J=9.0, 2.1 Hz, ArH), 7.74 (1H, d, J=9.0 Hz, ArH), 8.21 ppm (1H, d, J=2.1 Hz, ArH); LCMS (ES+): m/z (%): 434 (100) [M+H]+ (79Br), 436 (100) [M+H]+ (81Br); tR=5.2 min.
N-(2-(dimethylamino)ethyl)-6-fluoro-2-(5-methylfuran-2-yl)quinoline-4-carboxamide (26): Prepared as for 1 from 28 (150 mg, 0.55 mm), CH2Cl2 (10 mL), CDI (90 mg, 0.61 mm) and N,N-dimethylethane-1,2-diamine (54 mg, 0.61 mm). The reaction was stirred for 18 h at RT. Purification by flash column chromatography (3%, CH3OH/CH2Cl2) gave 26 as a white solid (50 mg, 24%): 1H NMR (500 MHz, [D6]DMSO): δ=2.29 (6H, br s, 2CH3), 2.46 (3H, s, CH3), 2.53 (2H, m, CH2), 3.46 (2H, q, J=6.5 Hz, CH2), 6.37 (1H, dd, J=3.3, 1.0 Hz, ArH), 7.34 (1H, d, J=3.3 Hz, ArH), 7.72 (1H, m, ArH), 7.92 (1H, br s, ArH), 7.95 (1H, dd, J=10.4, 2.9 Hz, ArH), 8.1 (1H, dd, J=9.3, 5.6 Hz, ArH), 8.85 ppm (1H, m, NH); LCMS (ES+): m/z (%): 341 [M+H]+; tR=4.0 min.
6-Bromo-2-(5-methylfuran-2-yl)quinoline-4-carboxylic acid (27): A solution of 5-bromoindoline-2,3-dione (1.25 g, 5 mm) in EtOH/H2O (20 mL, 1:1) in a 30 mL microwave reactor tube was treated with 1-(5-methylfuran-2-yl)ethanone (0.62 g, 5 mm). This tube was transferred to the microwave reactor and, following a 1 min pre-stirring, was heated to 110°C for 13 min. The reaction mixture was acidified to pH 3 using AcOH, diluted with H2O (60 mL) and extracted with EtOAc (2×80 mL). The organic layer was washed with H2O (2×80 mL) and brine (80 mL), dried (MgSO4), filtered and concentrated to give a yellow/brown solid. The crude product was redissolved in a minimum amount of hot EtOAc (∼40 mL) and the pure product was allowed to crystallise out yielding 27 (500 mg, 30%): RF=0.1 (10%, CH3OH/CH2Cl2); 1H NMR (500 MHz, [D6]DMSO): δ=2.42 (3H, br s, CH3), 6.40 (1H, dd, J=3.3, 0.9 Hz, ArH), 7.39 (1H, d, J=3.3 Hz, ArH), 7.83 (1H, dd, J=9.0, 2.4 Hz, ArH), 8.07 (1H, d, J=9.0 Hz, ArH), 8.31 (1H, s, ArH), 8.77 (1H, d, J=2.4 Hz, ArH), 14.16 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 332 (100) [M+H]+ (79Br), 334 (100) [M+H]+ (81Br); tR=0.5–0.6 min.
6-Fluoro-2-(5-methylfuran-2-yl)quinoline-4-carboxylic acid (28): Prepared as for 27 from 5-fluoroindoline-2,3-dione (0.94 g, 5.51 mm), 1-(5-methylfuran-2-yl)ethanone (0.65 mL, 6.06 mm) and KOH (2.8 g, 50 mm) in EtOH/H2O (20 mL, 1:1) to give 28 as a pale cream solid (600 mg, 40%): 1H NMR (500 MHz, [D6]DMSO): δ=2.45 (3H, br s, CH3), 6.40 (1H, m, ArH), 7.40 (1H, d, J=3.3 Hz, ArH), 7.94 (1H, dd, J=9.0, 2.2 Hz, ArH), 8.00 (1H, d, J=8.9 Hz, ArH), 8.31 (1H, s, ArH), 8.92 (1H, d, J=2.1 Hz, ArH), 14.10 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 271 [M+H]+; tR=0.5–0.6 min.
6-Chloro-2-(5-methylfuran-2-yl)quinoline-4-carboxylic acid (29): Prepared as for 27 from 5-chloroindoline-2,3-dione (1 g, 5.51 mm), 1-(5-methylfuran-2-yl)ethanone (0.65 mL, 6.06 mm) and KOH (2.8 g, 50 mm) in EtOH/H2O (20 mL, 1:1) to give 29 as a pale cream solid (600 mg, 38%): 1H NMR and LCMS confirm purity at >95%; 1H NMR (500 MHz, [D6]DMSO): δ=2.42 (3H, br s, CH3), 6.40 (1H, br d, J=3.2 Hz, ArH), 7.39 (1H, d, J=3.3 Hz, ArH), 7.94 (1H, dd, J=2.2, 9.0 Hz, ArH), 8.00 (1H, d, J=8.9 Hz, ArH), 8.3 (1H, s, ArH), 8.92 (1H, m, ArH), 14.15 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 288 [M+H]+; tR=0.5–0.6 min.
6-Bromo-2-(5-methylthiophen-2-yl)quinoline-4-carboxylic acid (30): Prepared as for 27 from 2-acetyl-5-methylthiophene (0.77 g, 5.5 mm) to give carboxylic acid 30 as a yellow solid (1.08 g, 62%): 1H NMR and LCMS confirm purity at >95%; 1H NMR (500 MHz, [D6]DMSO): δ=2.47 (3H, br s, CH3), 6.92 (1H, m, ArH), 7.92 (3H, m, 3ArH), 8.42 (1H, s, ArH), 8.88 (1H, br s, ArH), 14.15 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 348 (100) [M+H]+ (79Br), 350 (100) [M+H]+ (81Br); tR=0.5–0.6 min.
7-Bromo-2-(5-methylfuran-2-yl)quinoline-4-carboxylic acid (31): Prepared as for 27 from 6-bromoindoline-2,3-dione (1 g, 3.3 mm). The crude product was triturated with hot Et2O to isolate purified carboxylic acid 31 as a pale brown solid (350 mg, 36%): 1H NMR and LCMS confirm purity at >95%; 1H NMR (500 MHz, [D6]DMSO): δ=2.42 (3H, s, CH3), 6.40 (1H, m, ArH), 7.42 (1H, m, ArH), 7.50 (1H, m, ArH), 8.18 (1H, s, ArH), 8.22 (1H, s, ArH), 8.54 (1H, m, ArH), 14.2 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 332 (100) [M+H]+ (79Br), 334 (100) [M+H]+ (81Br); tR=0.5–0.6 min.
8-Bromo-2-(5-methylfuran-2-yl)quinoline-4-carboxylic acid (32): Prepared as for 27 from 7-bromoindoline-2,3-dione (1 g, 3.3 mm). The crude product was triturated with hot Et2O to isolate purified carboxylic acid 32 as a pale brown solid (400 mg, 41%): 1H NMR and LCMS confirm purity at >95%; 1H NMR (500 MHz, [D6]DMSO): δ=2.42 (3H, s, CH3), 6.42 (1H, m, ArH), 7.39 (1H, d, J=3.3 Hz, ArH), 7.53 (1H, dd, J=8.4, 7.6 Hz, ArH), 8.2 (1H, dd, J=7.5, 1.1 Hz, ArH), 8.28 (1H, s, ArH), 8.62 (1H, dd, J=8.5, 1.1 Hz, ArH), 14.2 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 332 (100) [M+H]+ (79Br), 334 (100) [M+H]+ (81Br); tR=0.5–0.6 min.
6-Bromo-2-(furan-2-yl)quinoline-4-carboxylic acid (33): 5-Bromoindoline-2,3-dione (1.25 g, 5 mm) and KOH flakes (2.8 g, 50 mm) were added to a 30 mL microwave reactor tube containing EtOH/H2O (20 mL, 1:1). The tube was transferred to the microwave reactor and, following a 1 min prestirring, was heated to 110°C for 3 min. The tube was then removed from the reactor and 2-acetylfuran (0.6 g, 5.5 mm) was added. Heating in the microwave was recommenced (110°C for 12 min) to achieve complete reaction. The reaction mixture was then acidified to pH 3 using AcOH, diluted with H2O (60 mL) and extracted with EtOAc (2×80 mL). The organic layer was washed with H2O (2×80 mL) and brine (80 mL), dried (MgSO4), filtered and concentrated to give a pale yellow solid. The crude solid was heated to reflux in EtOAc (20 mL) and the remaining insoluble cream solid was filtered off to give 33 (900 mg, 60%): Product purity was determined by 1H NMR and LCMS as >98%; RF=0.3 (10%, CH3OH/CH2Cl2); 1H NMR (500 MHz, [D6]DMSO): δ=6.77 (1H, m, ArH), 7.50 (1H, d, J=3.4 Hz, ArH), 7.97 (1H, dd, J=9.0, 2.1 Hz, ArH), 8.01 (2H, m, 2ArH), 8.38 (1H, s, ArH), 8.91 (1H, m, ArH), 14.17 ppm (1H, br s, COOH); LCMS (ES+): m/z (%): 318 [M+H]+; tR=0.5–0.6 min.
2-(5-Methylfuran-2-yl)quinoline-4-carboxylic acid (34): Prepared as for 23 from indoline-2,3-dione (1 g, 6.79 mm), KOH flakes (3.8 g, 50 mm) and 2-acetyl-5-methylfuran (0.79 mL, 6.79 mm). The crude solid was redissolved in refluxing EtOAc (30 mL) and the purified product was precipitated out and filtered off, washing with cold EtOAc (20 mL) to give compound 34 (500 mg, 31%): Product purity was determined by 1H NMR and LCMS as >98%; RF=0.15 (10%, CH3OH/CH2Cl2); 1H NMR (500 MHz, [D6]DMSO): δ=2.42 (3H, br s, CH3), 6.38 (1H, m, ArH), 7.36 (1H, d, J=3.3 Hz, ArH), 7.62 (1H, ddd, J=8.2, 6.9, 1.2 Hz, ArH), 7.79 (1H, ddd, J=8.4, 6.9, 1.4 Hz, ArH), 7.87 (1H, s, ArH), 8.02 (1H, d, J=8.4 Hz, ArH), 8.11 ppm (1H, m, ArH); LCMS (ES+): m/z (%): 254 [M+H]+; tR=0.5–0.6 min.
1-Methyl-6-(1-methylhydrazinyl)pyrimidine-2,4-(1H,3H)dione (35): 6-Chloro-3-methyluracil (2.0 g, 12.5 mmol) and methyl-hydrazine (0.861 mL, 16.2 mmol) were refluxed in EtOH (20 mL) for 1.5 h. The reaction was cooled to RT and concentrated in vacuo to afford pyrimidine 35 as a pale yellow solid (1.96 g, 100%): 1H NMR (500 MHz, [D6]DMSO): δ=2.67 (1H, br s, NH), 3.09 (3H, s, CH3), 3.11 (3H, s, CH3), 3.41 (2H, br s, NH2), 4.23 ppm (1H, s, CH); LCMS (ES+): m/z (%): 288 [M+H]+ (100); tR=2.7–2.9 min.
6-(2-(5-Bromo-2-fluorobenzylidene)-1-methylhydrazinyl)-1-methylpyrimidine-2,4(1H,3H)dione (36): Compound 35 (1.0 g, 4.07 mmol) and 5-bromo-2-fluoro-benzaldehyde (0.959 mL, 4.07 mmol) were refluxed in EtOH (40 mL) for 2 h. The reaction was cooled to RT, filtered and washed with Et2O to afford benzylidene 36 as a cream solid (1.61 g, 100%): 1H NMR (500 MHz, [D6]DMSO): δ=3.13 (2H, s, CH3), 3.39 (3H, s, CH3), 5.31 (1H, d, J=2.3 Hz, CH), 7.28 (1H, dd, J=10.4, 8.8 Hz, ArH), 7.60 (1H, ddd, J=8.8, 4.6, 2.6 Hz, ArH), 7.88 (1H, s, NH), 8.74 (1H, dd, J=6.6, 2.6 Hz, ArH), 11.05 ppm (1H, d, J=2.3 Hz, imine CH); LCMS (ES+): m/z (%): 381 (100) [M+H]+ (79Br), 383 (100) [M+H]+ (81Br); tR=3.9 min.
3-(4-Bromo-2-fluorophenyl)-4-hydroxy-8-methyl-6,8-dihydropyrimido[5,4-e,1,2,4]triazine-5,7(1H,4H)dione (37): Benzylidene 36 (0.443 g, 1.22 mmol) was dissolved in AcOH/H20 (3.45/0.17 mL) and the solution was cooled to 0°C. NaNO2 (0.126 g, 1.83 mmol) was added portionwise and the reaction was warmed to RT over 16 h. Et2O (10 mL) was added and the reaction was stirred at RT for 1 h. The resultant yellow precipitate was filtered and washed with Et2O to afford 37 (0.460 g, 99%): 1H NMR (500 MHz, [D6]DMSO): δ=3.25 (3H, s, CH3), 7.39 (1H, dd, J=10.6, 8.8 Hz, ArH), 7.72 (1H, ddd, J=8.8, 4.1, 2.7 Hz, ArH), 8.11 (1H, dd, J=6.8, 2.7 Hz, ArH), 12.30 ppm (1H, br s, OH).
3-(5-Bromo-2-fluorophenyl)-6-methylpyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (42): Compound 37 (0.430 g, 1.12 mmol) was heated in anhyd DMF (5 mL) at 90°C for 2 h. The reaction was cooled to RT and iced H2O was added. The resultant brown precipitate was filtered and dried in vacuo to afford pyrimido triazine 42 (0.357 g, 91%): 1H NMR (500 MHz, [D6]DMSO): δ=3.30 (3H, s, CH3), 7.48 (1H, dd, J=10.4, 8.9 Hz, ArH), 7.86 (1H, ddd, J=8.9, 4.1, 2.7 Hz, ArH), 8.15 (1H, dd, J=6.6, 2.7 Hz, ArH) and 13.00 ppm (br s, 1H, NH); LCMS (ES+): m/z (%): 351 (100) [M+H]+ (79Br), 353 (100) [M+H]+ (81Br); tR=3.9 min.
3-(5-Bromo-2-fluorophenyl)pyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (43): 3-(5-Bromo-2-fluorophenyl)-4-hydroxy-1-methyl-6,8-dihydropyrimido[5,4-e][1,2,4]triazine-5,7(1H,4H)-dione (0.430 g, 1.12 mmol) was heated in anhyd DMF (5 mL) at 90°C for 3 h in a sealed tube with 4 Å molecular sieves. The reaction was cooled to RT and iced H2O was added. The resultant brown precipitate was filtered and dried in vacuo to afford pyrimido triazine 43 (48 mg, 77%): 1H NMR (500 MHz, [D6]DMSO): δ=7.29 (1H, dd, J=5.0, 3.7 Hz, ArH), 7.86 (1H, ddd, J=5.0, 1.2 Hz, ArH), 8.01 (1H, dd, J=3.7, 1.2 Hz, ArH), 11.97 (1H, br s, NH), 12.49 ppm (1H, br s, NH).
3-(5-Bromo-2-fluorophenyl)-1-((2-dimethylamino)ethyl)-6-methylpyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (48): Compound 42 (50 mg, 0.14 mmol), Cs2CO3 (97 mg, 0.30 mmol), 2-dimethylaminoethylchloride (20 mg, 0.14 mmol) and anhyd DMF (2 mL) were heated at 80°C for 3 h. The reaction mixture was then filtered and diluted with EtOAc, the organic layer was washed with H2O (3×10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude was purified by column chromatography (CH2Cl2/MeOH/NH3, 95:5:0.1) to give pyrdimido triazine 48 as a yellow solid (5 mg, 9%): 1H NMR (500 MHz, CDCl3): δ=2.11 (6H, br s, 2CH3), 2.80 (2H, br s, CH2), 3.48 (3H, s, CH3), 4.13 (2H, t, J=6.2 Hz, CH2), 7.04 (1H, dd, J=10.3, 8.8 Hz, ArH), 7.49 (1H, ddd, J=8.8, 4.2, 2.6 Hz, ArH,), 8.13 ppm (1H, dd, J=6.6, 2.6 Hz, ArH); LCMS (ES+): m/z (%): 395 (100) [M+H]+ (79Br), 397 (100) [M+H]+ (81Br); tR=0.9–1.1 min.
3-(5-Bromo-2-fluorophenyl)-1-isopropyl-6-methylpyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (50): Compound 42 (50 mg, 0.14 mmol), Cs2CO3 (97 mg, 0.30 mmol), isopropylbromide (0.015 mL, 0.14 mmol) and anhyd DMF (2 mL) were heated at 150°C for 10 min in a Biotage microwave reactor. The reaction mixture was filtered and the filtrate was concentrated in vacuo. The crude was purified by column chromatography (CH2Cl2/MeOH/NH3, 95:5:0.1) to give pyrdimido triazine 50 as a yellow solid (5 mg, 9%): 1H NMR (500 MHz, CDCl3): δ=8.35 (1H, dd, J=6.6, 2.6 Hz, ArH), 7.65 (1H, ddd, J=8.8, 4.2, 2.6 Hz, ArH), 7.18 (1H, dd, J=10.3, 8.8 Hz, ArH,), 5.90 (1H, sep, J=7.0 Hz, CH), 3.57(3H, s, CH3), 1.72 ppm (6H, d, J=7.0 Hz, isopropy-CH3); LCMS (ES+): m/z (%): 399 (100) [M+H]+ (79Br), 401 (100) [M+H]+ (81Br); tR=4.8 min.
3-(5-Bromo-2-fluorophenyl)-1-butyl-6-methylpyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (51): Compound 42 (50 mg, 0.14 mmol), Cs2CO3 (69 mg, 0.21 mmol), n-butylbromide (0.015 mL, 0.14 mmol) and anhyd DMF (2 mL) were heated at 80°C for 6 h. The reaction mixture was filtered and the filtrate was concentrated in vacuo. The crude was purified by column chromatography (hexane/Et2O, 9:1) to give pyrdimido triazine 51 as a yellow solid (5 mg, 9%): 1H NMR (500 MHz, CDCl3): δ=8.33 (1H, dd, J=6.6, 2.6 Hz, ArH), 7.53 (1H, ddd, J=8.7, 4.2, 2.6 Hz, ArH), 7.16 (1H, dd, J=10.2, 8.8 Hz, ArH,), 4.53 (2H, t, J=7.7 Hz, CH2), 3.56 (3H, s, CH3), 1.81–1.78 (2H, m, CH2), 1.50–1.43 (2H, m, CH2) and 1.00–0.97 ppm (3H, m, CH3).
3-(5-Bromo-2-fluorophenyl)-1-cyclopentyl-6-methylpyrimido[5,4-e,1,2,4]triazine-5,7(1H,6H)dione (52): Compound 42 (50 mg, 0.14 mmol), K2CO3 (40 mg, 0.29 mmol), cyclopentylbromide (0.020 mL, 0.14 mmol) and anhyd DMF (2 mL) were heated at 150°C for 10 min in a Biotage microwave reactor. The reaction mixture was filtered and the filtrate concentrated in vacuo. The crude was purified by column chromatography (CH2Cl2/MeOH/NH3 95:5:0.1) to give pyrdimido triazine 52 as a yellow solid (5 mg, 9%): 1H NMR (500 MHz, CDCl3): δ=8.19 (1H, dd, J=6.7, 2.6 Hz, ArH), 7.54 (1H, ddd, J=8.8, 4.2, 2.6 Hz, ArH), 7.11 (1H, dd, J=10.4, 8.8 Hz, ArH,), 4.96 (1H, m, cyclopentyl CH), 3.51 (3H, s, CH3), 2.36–2.31 (2H, m, cyclopentyl CH2), 2.09–2.03 (4H, m, cyclopentyl CH2) and 1.73–1.65 ppm (2H, m, cyclopentyl CH2).
Acknowledgments
We would like to acknowledge the Wellcome Trust (WT 077705, WT 079838 and WT 083481) for funding and Irene Hallyburton and Bhavya Rao for technical support.
Supplemental material
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.200900262.
References
- 1.Stuart K, Brun R, Croft SL, Fairlamb AH, Gurtler RE, McKerrow J, Reed S, Tarleton R. J. Clin. Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett MP, Boykin DW, Brun R, Tidwell R. Br. J. Pharmacol. 2007;152:1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Mol. Microbiol. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 4a.Augustyns K, Amssoms K, Yamani A, Rajan PK, Haemers A. Current Pharmaceutical Design. 2001;7:1117–1141. doi: 10.2174/1381612013397564. [DOI] [PubMed] [Google Scholar]
- b.Fairlamb AH, Cerami A. Annu. Rev. Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- c.Hunter WN, Bailey S, Habash J, Harrop SJ, Helliwell JR, Aboagye-Kwarteng T, Smith K, Fairlamb AH. J. Mol. Biol. 1992;227:322–333. doi: 10.1016/0022-2836(92)90701-k. [DOI] [PubMed] [Google Scholar]
- 5a.Zhang Y, Bond CS, Bailey S, Cunningham ML, Fairlamb AH, Hunter WN. Protein Sci. 1996;5:52–61. doi: 10.1002/pro.5560050107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Werbovetz KA. Curr. Med. Chem. 2000;7:835–860. doi: 10.2174/0929867003374615. [DOI] [PubMed] [Google Scholar]
- c.Stump B, Eberle C, Kaiser M, Brun R, Krauth-Siegel RL, Diederich F. Org. Biomol. Chem. 2008;6:3935–3947. doi: 10.1039/b806371k. [DOI] [PubMed] [Google Scholar]
- d.Holloway GA, Baell JB, Fairlamb AH, Novello PM, Parisot JP, Richardson J, Watson KG, Street IP. Bioorg. Med. Chem. Lett. 2007;17:1422–1427. doi: 10.1016/j.bmcl.2006.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- e.Martyn DC, Jones DC, Fairlamb AH, Clardy J. Bioorg. Med. Chem. Lett. 2007;17:1280–1283. doi: 10.1016/j.bmcl.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- f.Hamilton CJ, Saravanamuthu A, Poupat C, Fairlamb AH, Eggleston IM. Bioorg. Med. Chem. 2006;14:2266–2278. doi: 10.1016/j.bmc.2005.11.004. [DOI] [PubMed] [Google Scholar]
- g.Parveen S, Khan MOF, Austin SE, Croft SL, Yardley V, Rock P, Douglas KT. J. Med. Chem. 2005;48:8087–8097. doi: 10.1021/jm050819t. [DOI] [PubMed] [Google Scholar]
- h.Dixon MJ, Maurer RI, Biggi C, Oyarzabal J, Essex JW, Bradley M. Bioorg. Med. Chem. 2005;13:4513–4526. doi: 10.1016/j.bmc.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 6.Frearson JA, Wyatt PG, Gilbert IH, Fairlamb AH. Trends Parasitol. 2007;23:589–595. doi: 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Khan MOF, Austin SE, Chan C, Yin H, Marks D, Vaghjiani SN, Kendrick H, Yardley V, Croft SL, Douglas KT. J. Med. Chem. 2000;43:3148–3156. doi: 10.1021/jm000156+. [DOI] [PubMed] [Google Scholar]
- b.Parveen S, Khan MOF, Austin SE, Croft SL, Yardley V, Rock P, Douglas KT. J. Med. Chem. 2005;48:8087–8097. doi: 10.1021/jm050819t. [DOI] [PubMed] [Google Scholar]
- c.Chan C, Yin H, Garforth J, McKie JH, Jaouhari R, Speers P, Douglas KT, Rock PJ, Yardley V, Croft SL, Fairlamb AH. J. Med. Chem. 1998;41:148–156. doi: 10.1021/jm960814j. [DOI] [PubMed] [Google Scholar]
- d.Bonse S, Santelli-Rouvier C, Barbe J, Krauth-Siegel RL. J. Med. Chem. 1999;42:5448–5454. doi: 10.1021/jm990386s. [DOI] [PubMed] [Google Scholar]
- e.Chibale K, Haupt H, Kendrick H, Yardley V, Saravanamuthu A, Fairlamb AH, Croft SL. Bioorg. Med. Chem. Lett. 2001;11:2655–2657. doi: 10.1016/s0960-894x(01)00528-5. [DOI] [PubMed] [Google Scholar]
- f.Benson TJ, McKie JH, Garforth J, Borges A, Fairlamb AH, Douglas KT. Biochem. J. 1992;286:9–11. doi: 10.1042/bj2860009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- g.Chibale K, Visser M, Yardley V, Croft SL, Fairlamb AH. Bioorg. Med. Chem. Lett. 2000;10:1147–1150. doi: 10.1016/s0960-894x(00)00154-2. [DOI] [PubMed] [Google Scholar]
- h.Chibale K, Visser M, vanSchalkwyk D, Smith PJ, Saravanamuthu A, Fairlamb AH. Tetrahedron. 2003;59:2289–2296. [Google Scholar]
- 8a.Li Z, Fennie MW, Ganem B, Hancock MT, Iija MK, Rattendi D, Bacchi CJ, O’Sullivan MC. Bioorg. Med. Chem. Lett. 2001;11:251–254. doi: 10.1016/s0960-894x(00)00643-0. [DOI] [PubMed] [Google Scholar]
- b.Ponasik JA, Strickland C, Faerman C, Savvides S, Karplus PA, Ganem B. Biochem. J. 1995;311:371–375. doi: 10.1042/bj3110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- c.Hamilton CJ, Saravanamuthu A, Fairlamb AH, Eggleston IM. Bioorg. Med. Chem. 2003;11:3683–3693. doi: 10.1016/s0968-0896(03)00344-4. [DOI] [PubMed] [Google Scholar]
- d.Chitkul B, Bradley M. Bioorg. Med. Chem. Lett. 2000;10:2367–2369. doi: 10.1016/s0960-894x(00)00471-6. [DOI] [PubMed] [Google Scholar]
- e.Orain D, Bradley M. Tetrahedron Lett. 2001;42:515–518. [Google Scholar]
- f.DeLuca S, Ulhaq S, Dixon MJ, Essex J, Bradley M. Tetrahedron Lett. 2003;44:3195–3197. [Google Scholar]
- 9a.Salmon-Chemin L, Lemaire A, DeFreitas S, Deprez B, Sergheraert C, Davioud-Charvet E. Bioorg. Med. Chem. Lett. 2000;10:631–635. doi: 10.1016/s0960-894x(00)00056-1. [DOI] [PubMed] [Google Scholar]
- b.Cuellar MA, Salas C, Cortes MJ, Morello A, Maya JD, Preite MD. Bioorg. Med. Chem. 2003;11:2489–2497. doi: 10.1016/s0968-0896(03)00193-7. [DOI] [PubMed] [Google Scholar]
- 10.Garrard EA, Borman EC, Cook BN, Pike EJ, Alberg DG. Org. Letts. 2000;2:3639–3642. doi: 10.1021/ol0065423. [DOI] [PubMed] [Google Scholar]
- 11a.Richardson JL, Nett IR, Jones DC, Abdille MH, Gilbert IH, Fairlamb AH. ChemMedChem. 2009;4:1333–1340. doi: 10.1002/cmdc.200900097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Patterson S, Jones DC, Shanks EJ, Frearson JA, Gilbert IH, Wyatt PG, Fairlamb AH. ChemMedChem. 2009;4:1341–1353. doi: 10.1002/cmdc.200900098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. ChemMedChem. 2008;3:435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamilton CJ, Saravanamuthu A, Eggleston IM, Fairlamb AH. Biochem J. 2003;369:529–537. doi: 10.1042/BJ20021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacrampe JFA, Connors RW, Ho CY, Richardson A, Freyne EJE, Buijnsters PJJ, Bakker AC. PCT Int. Appl. WO 2004007499.
- 15.Räz B, Iten M, Grether-Bühler Y, Kaminsky R, Brun R. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 16.Greig N, Wyllie S, Patterson S, Fairlamb AH. FEBS J. 2009;276:376–386. doi: 10.1111/j.1742-4658.2008.06788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.