

Summary
RNA interference (RNAi) is a form of posttranscriptional gene silencing mediated by microRNA (miRNA) and small interfering RNA (siRNA). In Drosophila melanogaster, the RNase III enzymes Dicer-1 and Dicer-2 generate miRNA and siRNA, respectively. We describe the methods for the expression, purification, and analysis of recombinant Dicer-1 and Dicer-2 enzymes. Our studies demonstrate that Dicer-1 and Dicer-2 display different substrate specificities and ATP requirements.
Keywords: RNA interference, Dicer-1, Dicer-2, baculovirus, dsRNA, siRNA, pre-miRNA, miRNA
1. Introduction
RNA interference (RNAi) is a form of posttranscriptional gene silencing mediated by 21- to 25-nucleotide microRNA (miRNA) and small interfering RNA (siRNA). These tiny regulatory RNAs control many important biological processes, such as development, antiviral defense, heterochromatin formation, and maintenance of genomic stability (1–4). It follows that misregulation of miRNAs has been linked to human diseases, such as cancer (5,6). Moreover, siRNAs or miRNAs have been widely used as a powerful gene-silencing tool for functional genomic analyses in multiple model organisms, including humans (1,3).
In principle, miRNAs and siRNAs can be viewed as two parallel branches of the RNAi pathway, which consists of initiation and effector steps. In the initiation step, siRNAs are derived from mostly exogenous long dsRNAs, whereas miRNAs originate from endogenous (∼ 60–70 nt) stem-loop precursor miRNAs (pre-miRNAs). Both siRNAs and miRNAs are produced as 21- to 25-nt duplexes with 2-nt 3′-overhangs and 5′-phosphate and 3′-hydroxyl termini. While siRNAs are perfectly complementary, miRNAs frequently contain mismatches, bulges, or G:U wobble base pairs. In the effector step, nascent siRNA and miRNA duplexes are assembled into the respective RNA-induced silencing complexes that are simply referred to as siRISC and miRISC. In RISCs, single-stranded siRNA or miRNA functions as the guide RNA to direct sequence-specific cleavage and/or translational repression of complementary mRNA (1,2,7,8).
Both siRNAs and miRNAs are produced by Dicers, a family of large (∼ 200 kDa) multidomain RNaseIII enzymes that exist in most eukaryotic organisms (9). A typical Dicer contains a putative RNA helicase domain, a DUF (domain of unknown function) 283 domain, and a PAZ domain at the amino (N)-terminus as well as two RNase III domains and a dsRNA-binding domain at the carboxyl (C)-terminus. The tandem RNase III domains of Dicer form an intramolecular dimer to make a pair of cuts on dsRNA, creating a characteristic 2-nt 3′-overhang (10,11). In most organisms, such as C. elegans and humans, a single Dicer generates both siRNAs and miRNAs. However, two Dicer enzymes, Dicer-1 and Dicer-2, have been identified in the Drosophila genome (10,12,13). Both genetic and biochemical studies have implicated that Dicer-1 and Dicer-2 are involved in the biogenesis of miRNAs and siRNAs, respectively (14–18).
Dicer enzymes do not function alone. We have previously purified the siRNA-generating enzyme from Drosophila S2 cells and found that it consisted of Dicer-2 and R2D2 (15). R2D2 contains two dsRNA-binding domains (R2) and forms a heterodimeric complex with Dicer-2 (D2). Although R2D2 does not regulate siRNA production, R2D2 and Dicer-2 coordinately bind siRNA and facilitate its incorporation into the effector siRISC complex (15,19). An R2D2-like protein, Loquacious (Loqs, also known as R3D1), has recently been identified as a cofactor for Dicer-1 in the miRNA pathway. The loqs gene encodes at least two alternatively spliced proteins, Loqs-L (long) and Loqs-S (short). Both Loqs isoforms contain three putative dsRNA-binding domains. Loqs-L forms a stable complex with Dicer-1 and greatly enhances its miRNA-generating activity. It remains uncertain if the Dicer-1/Loqs complex facilitates miRNA loading onto the miRISC complex (16–18).
Therefore, the Dicer-1/Loqs-L and Dicer-2/R2D2 complexes function in parallel to generate miRNAs and siRNAs in Drosophila cells. The functional differences of the two Dicer complexes can be explained by either that Dicer-1 and Dicer-2 possess distinct biochemical activities or that Loqs-L and R2D2 help define the functional specificities for Dicer-1 and Dicer-2 (16). Here we describe the detailed protocols for the expression, purification, and analysis of recombinant Dicer-1 and Dicer-2 enzymes. Our reconstitution studies indicate that Dicer-1 and Dicer-2 are biochemically distinct enzymes with different substrate specificities and ATP requirements.
2. Materials
2.1. For Expression of Dicer
The BAC-to-BAC® Baculovirus Expression System (Invitrogen, Carlsbad, CA, Cat# 10359-016), which includes the pFastBac™ 1 vector, E. coli DH10 Bac competent cells, Cellfectin transfection reagent, and an instruction manual.
Recombinant Dicer bacmid DNA, prepared from 2 mL of bacteria culture, and resuspended in 40 μL of sterile TE.
Complete IPL-41 medium: 1 L of incomplete IPL-41 medium (Invitrogen Cat# 11405-081) supplemented with 100 mL of fetal bovine serum (Gemini Bio-Products, West Sacramento, CA, Cat# 100-106, heat-inactivated at 55 °C for 30 min), 10 mL of Pluronic®-F68 (Invitrogen, Cat# 24040-032), 10 mL of 100X Antibiotic-antimycotic (Invitrogen, Cat# 15240-062), 20 mL of 50X Tryptose Phosphate Broth (130 mg/mL, powder is ordered from Sigma, St. Louis, MO, Cat# T9157), and 20 mL of 50X Yeastolate Ultrafiltrate (Invitrogen, Cat# 18200-048).
Buffers and reagents for SDS-Polyacrylamide gel electrophoresis (PAGE): 4X SDS sample buffer (0.25 M Tris-HCl pH 6.8, 8% SDS, 30% glycerol, 0.02% bromophenol blue, and 10% β-mercaptoethanol); 10X running buffer (250 mM Tris, 1.92 M glycine, and 1% SDS; do not adjust pH).
Buffers and reagents for Western blot: 10X transfer buffer (312.5 mM Tris, 2.4 M glycine); rabbit anti-His antibody (Bethyl, Montgomery, TX, Cat#A190-114A); HRP-conjugated anti-rabbit IgG antibody (Sigma, Cat# A6154); ECL reagents (PerkinElmer, Waltham, MA, Cat# NEL102).
2.2. For Purification of Dicer
Preparation of cell lysates: Buffer A [10 mM KOAc, 10 mM HEPES, pH 7.4, 2 mM Mg(OAc)2, 5 mM β-mercaptoethanol]; Buffer B (buffer A plus 1 M NaCl); Protease inhibitors, including Pefabloc SC (Roche, Indianapolis, IN, Cat# 1-585-916), Leupeptin (Roche, Cat# 1-034-626), and Pepstatin (Roche, Indianapolis, IN, Cat# 1-359-053). Store 1,000X stocks (1 mg/mL Pefabloc SC, 5 mg/mL Leupeptin, and 0.7 mg/mL Pepstain) at −20 °C in small aliquots.
Nickel purification: Ni-NTA Agarose beads (Qiagen, Valencia, CA, Cat# 30230); 50% slurry in ethanol, store at 4 °C; Imidazole (Sigma), 2.5 M of stock dissolved in buffer A; Econo-Pac® disposable chromatography column (Bio-Rad, Hercules, CA, Cat# 732-1010).
Ion-exchange chromatography: 1 mL of SP-Sepharose column (GE Healthcare, Piscataway, NJ, Cat# 17-1151-01) and 1 mL of Q-Sepharose column (GE Healthcare, Piscataway, NJ, Cat# 17-1153-01).
Recombinant protein stocking: Glycerol (Sigma) and liquid nitrogen.
Buffers and reagents for SDS-PAGE and Coomassie blue staining: 4X SDS sample loading buffer and 10X running buffer (see Section 2.1). Coomassie blue staining solution (Bio-Rad, Cat# 161-0436) and destaining solution (10% methanol and 10% acetic acid in water).
2.3. For Assay of Dicer
Templates for in vitro transcription of dsRNA: The sense and antisense (250 bp) templates were generated from the firefly luciferase gene by PCR using primers 5′-GCGTAATACGACTCACTATAGATGCACATATCGAGGTGGA-3′ and 5′-GAACACCACGGTAGGCTGC-3′ (sense template); 5′-GATGCACAT-ATCGAGGTGGA-3′ and 5′-GCTAATACGACTCACTATAGAACACCACGG-TAGGCTGC-3′ (antisense template). The two long primers contain T7 polymerase promoter sequence.
Synthetic RNA oligonucleotides: 61 nt pre-let7, 5′-UGAGGUAGUAGGUUGUA-UAGUAGUAAUUACACAUCAUACUAUACAAUGUGCUAGCUUUCUU-3′. 21-nt RNA marker, 5′-CGUACGCGGAAUACUUCGATT-3′. Both are custom-ordered from Dharmacon, Chicago, IL.
Isotopes: [α-32P] GTP (40 mCi/mL, Cat # 32010XT01) and [γ-32P]ATP (10 mCi/mL, Cat # 38101X) from MP Biomedicals, Solon, OH.
Reagents for radiolabeling: Riboprobe® System-T7 Kit (Promega, Madison, WI, Cat# P1440) contains T7 polymerase, 5X T7 transcription buffer, 100 mM DTT, 10 mM ATP, UTP, CTP, and GTP, nuclease-free water, RNase inhibitor, and RNase-free DNase I. Polynucleotide kinase (PNK) and 10X PNK buffer (NEB, Ipswich, MA, Cat# M0201S).
Buffers for the assays: Buffer A (see Section 2.2), buffer X (500 mM ammonium acetate, 5 mM EDTA, 0.5% SDS); 10X dsRNA processing buffer [500 mM K-acetate, 150 mM HEPES (pH7.4); 18 mM magnesium acetate, 25 mM DTT]; and 10X pre-miRNA processing buffer [1 M K-acetate, 150 mM HEPES (pH 7.4), 100 mM magnesium acetate, and 25 mM DTT].
Other reagents for the assays: 10 mM of ATP (Ambion, Austin, TX, Cat# 8110G), SUPERase•In™ (Ambion, Cat# 2696), and phenol/chloroform (5:1, pH 4.5, Ambion, Cat# 9722), glycogen (5 mg/mL, Ambion), 3 M and 0.3 M sodium acetate (pH5.2), diethyl pyrocarbonate (DEPC)-treated water [add 1 mL DEPC (Simga) in 1 L deionized water, stir overnight at room temperature, then autoclave for 30 min to inactivate DEPC].
Buffers and reagents for denaturing polyacrylamide gel electrophoresis: Formamide load dye (Ambion), 16% denaturing polyacrylamide gel (freshly made with 5 mL 16% Urea gel mix, 40 μL 10% ammonium persulfate, and 4 μL TEMED), and 10X TBE running buffer (Ambion). For 16% urea gel mix, we made a solution containing 7 M Urea, 16% Acrylamide/Bis (from 40% Acrylamide/Bis 19:1 solution, Bio-Rad, Cat# 161-0144), and 1X TBE (from 10X TBE running buffer) and stored at 4 °C.
3. Methods
3.1. Expression of Dicer
Both recombinant Dicer-1 and Dicer-2 enzymes are expressed in insect cells using the BAC-to-BAC® Baculovirus Expression System (15,16). Thus, for simplicity, we will refer to both Dicer-1 and Dicer-2 as “Dicer” when not specified in the sections below. The cDNAs of Dicer-1 and Dicer-2 are cloned by reverse transcription from total RNA of Drosophila S2 cells using the RLM-RACE kit from Ambion (Cat# 1700). To simplify purification of recombinant Dicers, polyhistidine (His)-tags were added by polymerase chain reaction (PCR) to the N- and C-termini of Dicers. We found that the addition of double His-tags greatly enhanced the expression levels of Dicer viruses. After verified by sequencing, the double His-tagged Dicer cDNAs were subcloned into the pFastBac™ 1 vector. The pFastBac-Dicer plasmids were transformed into E. coli DH10Bac competent cells to generate recombinant bacmids. The recombinant Dicer bacmids were isolated from positive bacteria transformants and verified by PCR that they contained the full-length Dicer cDNA (Fig. 1). The protocols for this section are described in detail in the manufacturer's instruction manual and will not be repeated here.
Fig. 1.
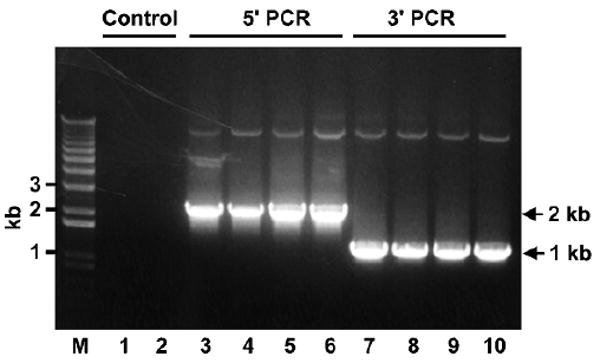
Recombinant Dicer-1 bacmids contain full-length Dicer-1 cDNA. The 5′ and 3′ PCR reactions were performed without DNA template (lane 1 and 2) or with 1 μL of recombinant Dicer-1 bacmids prepared from four positive transformants (lanes 3–10). The 5′ PCR reactions use a 5′ bacmid (forward) primer (5′-GTTTTCCCAGTCACGAC-3′) and a 5′ Dicer-1 (reverse) primer (5′-CCGTCCAGCAATGATCAAAG-3′). The 3′ PCR reactions use a 3′ Dicer-1 (forward) primer (5′-TTCCACAAGTTCTTCCGGCA-3′) and a 3′ bacmid (reverse) primer (5′-CAGGAAACAGCTATGAC-3′). The predicted size of 5′ and 3′ PCR products are ∼ 2 kb and 1 kb.
3.1.1. Culture of Sf21 Insect Cell
We have had good results with Sf21 cells from Invitrogen (Cat# 10497-013), but the reader is welcome to try insect cells from other sources. Every 3 months, thaw 1 vial of low-passage frozen Sf21 cells in a 25 °C water bath. Transfer the cells to a 15-mL tube. Add 10 mL (prewarmed to 25 °C) of complete IPL-41 medium and mix well by pipetting up and down.
Spin at 1,000 rpm for 5 min. Remove the supernatant and resuspend the cell pellet with 5 mL of completed IPL-41 medium. Count the cell density with a hemocytometer under a microscope. Transfer the cells into a 250-mL tissue culture flask (Corning, NY). Add complete IPL-41 medium to dilute the cell density to 1 × 106 cells/mL.
Incubate the suspension culture in a 27 °C shaker at 125 rpm. The doubling time of Sf21 cells is ∼ 24 h, and the optimal cell density should be kept between 0.5–5 × 106 cells/mL. Healthy Sf21 cells are round in shape and uniform in size.
It is best to freeze large numbers of small aliquots of low-passage Sf21 cells at the beginning. Spin 250 mL of mid-log-phase Sf21 cell culture (∼ 4 × 106 cells/mL) in a sterile centrifuge bottle at 1,000 rpm for 5 min. Remove the supernatant, and gently resuspend the cell pellet with 50 mL of complete IPL-41 medium containing 10% DMSO. Aliquot to freezing vials at 2 × 107 cells/mL/vial.
Place the vials into a room-temperature Cryo 1 °C freezing container (Nalgene, Rochester, NY, Cat# 5100-0001) and put the container into a −80 °C freezer. The Nalgene container allows the temperature to drop at a rate of 1 °C per min. After 24 h, thaw one vial for testing and move the remaining frozen vials into a −190 °C liquid nitrogen freezer for permanent storage (see Notes 4.1).
3.1.2. Generation of Recombinant Dicer Virus (Passage 1 and 2)
Seed total 2 × 106 Sf21 cells in 4 mL of complete IPL-41 medium in a T-25 (cm2)-flask (BD Bioscience, San Jose, CA, Cat# 353109). Incubate at 27 °C to allow cells to attach for at least 15 min. We often perform two transfections for each bacmid for backup.
Solution A: Dilute 15 μL of bacmid DNA into 200 μL of IPL-41 medium. Solution B: Dilute 12 μL of Cellfectin Reagent (inverting the tube 5 to 10 times before removing) into 200 μL of IPL-41 medium. Add solution B dropwise into solution A, mix gently by inverting the tube 8–10 times, and incubate at room temperature for 30–45 min.
Wash the attached Sf21 cells once with 2 mL of incomplete IPL-41 medium (no supplements). Transfer 1.6 mL of incomplete IPL-41 medium and then add 400 μL of lipid-DNA complexes (A and B) dropwise into the T-25 flask. Gently rock the flask several times to mix the content. Incubate at 27 °C for 4–5 h.
Remove the transfection mixture, add 5 mL of complete IPL-41 medium, and incubate at 27 °C for 8–10 days. Check the cells under a microscope for contamination or cell lysis. Since Dicers are large proteins, it takes 8–10 days for more complete cell lysis in order to obtain high-titer viruses.
When 50–60% of the cells are lysed or floating, blow the cells off and transfer to a sterile 15-mL tube. Spin at 1,000 rpm for 5 min and transfer the supernatant into a fresh 15-mL tube. This is called the passage 1 (P1) virus.
Transfer the entire P1 virus into a 250-mL flask containing 45 mL of Sf21 cells (2 × 106 cells/mL). Incubate in a 27 °C shaker (125 rpm) for 5–7 days. Check the cells under a microscope. When more than 90% cells are lysed, harvest the cells by spinning and transfer the supernatant to a 50-mL tube. This is called the passage 2 (P2) virus.
Both P1 and P2 viruses can be stored at 4 °C wrapped with aluminum foil for protection from light. For long-term storage, it is best to store the virus in 7% DMSO at −80 °C.
3.1.3. Test the Expression Level of Dicer Virus
Plate 300 μL (2 × 106 cells/mL) of Sf21 cells in a 24-well dish. Add 3 μL of P2 Dicer virus in each well. Save one well for no virus control. Gently mix by rocking the dish several times. Incubate at 27 °C for 40–44 h.
Remove the medium by aspirating. Place the dish on ice. Add 400 μL of 2% SDS/water to each well. Sit on ice for 1 min.
Resuspend the cell lysates and transfer to 1.5-mL microcentrifuge tubes.
Vortex vigorously for 30 sec to break the viscous genomic DNA. Transfer 45 μL of each sample to a new microcentrifuge tube containing 15 μL of 4X SDS sample buffer. Boil the samples for 5 min. Spin at 14,000 rpm for 2 min to precipitate insoluble material. Load 20 μL of the top layer on a 6% SDS-PAGE gel and run until the front dye migrates to the bottom of the gel.
Transfer the SDS-PAGE gel to a nitrocellulose membrane for 2 h at 400 mA. Perform Western blot using rabbit anti-His (primary, 1:5,000) antibody and HRP-conjugated anti-rabbit IgG (secondary, 1:10,000) antibody (Fig. 2).
Fig. 2.
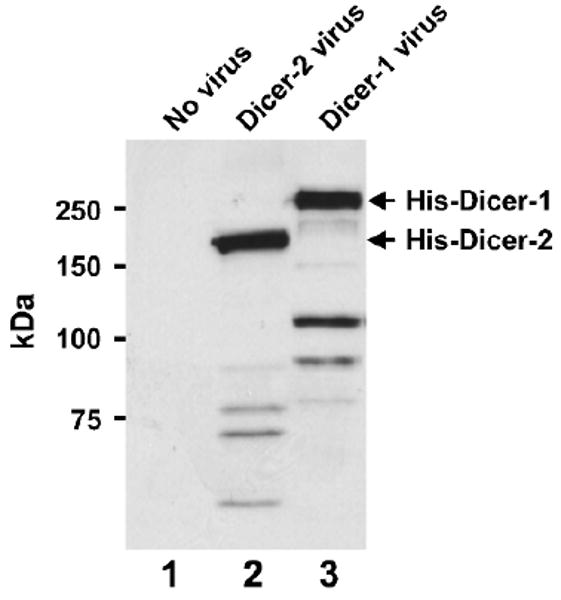
Expression test of P2 baculovirus. Western blot was performed with anti-His antibodies using lysates prepared from Sf21 cells that are mock treated (lane 1) or infected with P2 viruses of Dicer-2 (lane 2) or Dicer-1 (lane 3).
3.1.4. Amplification of Dicer Virus (P3, P4) and Large-Scale Protein Production
Grow 600 mL of Sf21 cells to 2.5 × 106 cells/mL in a 2-L flask. Add 25 mL of Dicer P2 virus and incubate in a 27 °C shaker (125 rpm) for 5–7 days.
Check the cells under a microscope. When more than 90% cells are broken, distribute the culture to multiple 50-mL sterile tubes and spin at 2,000 rpm for 5 min. The supernatant is called the P3 virus, which can be amplified to the P4 virus in the same manner. Store the P3 and P4 viruses at 4 °C wrapped in aluminum foil.
For large-scale protein production, grow 2 × 600 mL of Sf21 cells in 2-L flasks to a cell density of 2.5 × 106 cells/mL. Add 25 mL of the P3 Dicer virus into each of two flasks. Incubate in a 27 °C shaker (125 rpm) for 42–48 h.
Spin down the cells in 1-L centrifuge bottles at 2,500 rpm for 20 min at 4 °C. Remove the supernatant and resuspend each pellet with 20 mL of PBS. Transfer the cell suspension into 50-mL tubes and spin again at 4,000 rpm for 15 min at 4 °C. Remove the supernatant. The cell pellets can be immediately used to make lysate for purifying recombinant Dicer proteins or can be stored at −80 °C for future use.
3.2. Purification of Dicer
Both Dicer-1 and Dicer-2 recombinant proteins are highly purified by nickel affinity purification followed by SP- and Q-Sepharose chromatography (Fig. 3). Nickel affinity purification is based on the double His-tags of recombinant Dicers, which is the same for Dicer-1 and Dicer-2. However, the column behaviors of Dicer-1 and Dicer-2 are different for the SP- and Q-Sepharose chromatography. On the SP-Sepharose column, Dicer-1 flows through, whereas Dicer-2 binds and is eluted at 180–220 mM of sodium chloride. On the Q-Sepharose column, both Dicer-1 and Dicer-2 bind but are respectively eluted at 350–380 mM and 260–300 mM sodium chloride. All purification steps are performed at 4 °C on an ACTA FPLC machine purchased from Amersham. After the multistep purification, we can typically get ∼ 0.6–1.0-mg recombinant Dicer proteins with more than 90% purity from 1.2 L of insect cell culture (Fig. 3).
Fig. 3.
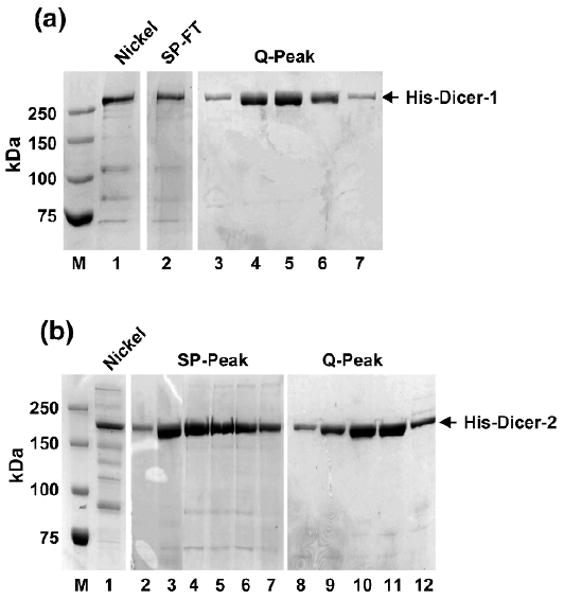
Purification of recombinant Dicer-1 and Dicer-2. The His-tagged Dicer-1 and Dicer-2 proteins are both purified by Ni+2-affinity purification followed by SP-Sepharose and Q-Sepharose chromatography. (a) Lane 1: 250 mM imidazole elution, also as input of SP column; lane 2: SP flowthrough, also as input of Q column; lanes 3–7: Dicer-1 peak fractions of Q column. (b) Lane 1: 250 mM imidazole elution, also as input of SP column; lane 2: Dicer-2 peak fractions of SP column; also combined as input of Q column; lane 3: Dicer-2 peak fractions of Q column.
3.2.1. Preparation of Cell Lysate and Nickel Purification
Quick-thaw the cell pellet by placing the frozen tube in a beaker filled with room-temperature water. Resuspend the cell pellet thoroughly (from 1.2 L Dicer virus-infected culture, 15–20 mL pellet volume) in three times pellet volume of buffer A freshly supplemented with protease inhibitors and 20 mM of imidazole (see Notes 4.2). Let the cells swell in hypotonic buffer A by staying on ice for 20 min.
Pour the cell suspension into a 100-mL glass douncer and make 40 strokes to break the cells completely. Transfer the whole-cell lysates into 50-mL centrifuge tubes and spin at 20,000 × g at 4 °C for 30 min. The supernatant is called S20.
Wash 2-mL nickel beads (4 mL 50% slurry) three times with 10 mL of buffer A containing 20 mM of imidazole in a 15-mL conical tube. Each time, resuspend the beads thoroughly, spin at 1,000 rpm for 3 min, and remove the supernatant.
Transfer S20 of step 2 to a 50-mL conical tube. Use S20 to resuspend the prewashed nickel beads and transfer back to the 50-mL conical tube. Rotate at 4 °C for 4 h to allow recombinant Dicer proteins to bind with the nickel beads.
Pour the lysate/beads mixture into a 25-mL Econo-Pac® disposable chromatography column. Break the bottom plastic cap, let the beads settle down by gravity, and collect the flowthrough.
Wash the beads sequentially with 200 mL of buffer B containing 20 mM of imidazole, 50 mL of buffer A containing 20 mM of imidazole, and 10 mL of buffer A containing 50 mM of imidazole. Elute with 10 mL of buffer A (freshly supplemented with protease inhibitors and 250 mM imidazole). Nickel elution can be filtered and used for the next purification steps or stored at −80 °C with 10% glycerol for future use.
Take 15 μL of the elution and mix with 5 μL of the 4X SDS sample buffer. Boil for 5 min and load onto a 6% SDS-PAGE to check the quantity and quality of recombinant Dicer proteins by Coomassie Blue staining.
3.2.2. Ion-Exchange Chromatography for Dicer-1 Purification
Filter the Dicer-1 nickel elution with a 0.2-μm syringe filter. Wash the SP- and Q-Sepharose columns and the super-loop of the FPLC system with buffer A.
Inject the sample into the super-loop and load it onto the 1-mL SP-Sepharose column. Collect the flowthrough that contains recombinant Dicer-1 proteins.
Reinject the SP flowthrough back into the super-loop and load it onto the 1-mL Q-Sepharose column. Perform a 20-mL 0–45% buffer B gradient elution followed by a 5-mL 100% buffer B step wash (fraction volume = 1 mL). The peak of Dicer-1 is at 34–38% buffer B elution.
Take 15 μL of SP flowthrough and Q peak elution of Dicer-1, mix with 5 μL of 4X SDS sample loading buffer, boil for 5 min, and load onto a 6% SDS-PAGE followed by Coomassie Blue staining. Compare the nickel elution with SP flowthrough and Q peak fractions to see the increasing purity of recombinant Dicer-1.
Take 1 μL of Q peak fractions to measure the concentration of purified recombinant Dicer-1 proteins. They typically fall between 0.3 and 0.5 mg/mL. Add 10% glycerol, mix well, and store at −80 °C in small aliquots.
3.2.3. Ion-Exchange Chromatography for Dicer-2 Purification
Filter the Dicer-2 nickel elution with a 0.2-μm syringe filter. Wash the SP- and Q-Sepharose columns and the super-loop with buffer A.
Inject the sample into the super-loop and load onto the 1-mL SP-Sepharose column. Perform a 15-mL 0–30% gradient buffer B elution followed by a 5-mL 100% buffer B step wash (fraction volume = 1 mL). The peak of Dicer-2 is at 18–22% buffer B elution.
Combine the SP peak fractions, and dilute with two volumes of buffer A to reduce the salt concentration. Reinject it into the FPLC system and load onto the 1-mL Q-Sepharose column. Perform a 20-mL 0–40% gradient buffer B elution followed by a 5-mL 100% buffer B step wash (fraction volume = 1 mL). The peak of Dicer-2 is at 26–30% buffer B elution.
Take 15 μL of the SP and Q peak fractions, mix with 5 μL of 4 × SDS sample loading buffer, boil for 5 min, and load onto a 6% SDS-PAGE followed by Coomassie Blue staining. Compare the nickel elution with SP and Q peak fractions to see the increasing purity of recombinant Dicer-2.
Take 1 μL of the Q peak fractions to measure the concentration of purified recombinant Dicer-2. They typically fall between 0.3 and 0.5 mg/mL. Add 10% glycerol, mix well, and store at −80 °C in small aliquots.
3.3. Assay of Dicer
Genetic studies have suggested that Dicer-1 and Dicer-2 are respectively involved in miRNA and siRNA production (14). By biochemical fractionation, it has been shown that the Dicer-1/Loqs-L complex processes pre-miRNA into mature miRNA, whereas the Dicer-2/R2D2 complex cleaves long dsRNA into siRNA (15–18). The functional specificities of the two Dicer complexes can be explained by the fact that Dicer-1 and Dicer-2 possess different biochemical activities or that Loqs-L and R2D2 help define the functional specificities for their respective Dicer partners. Here we compare the abilities of purified recombinant Dicer-1 and Dicer-2 enzymes to process radiolabeled pre-miRNA or long dsRNA in the absence or presence of ATP (see Notes 4.3). Our studies indicate that, despite sharing extensive sequence homology, Drosophila Dicer-1 and Dicer-2 enzymes display different substrate specificities and ATP requirements. While Dicer-1 prefers pre-miRNA as its ideal substrate, Dicer-2 is a much better enzyme for processing dsRNA (Fig. 4). Furthermore, Dicer-1 produces miRNA or siRNA independent of ATP, whereas Dicer-2 absolutely requires ATP hydrolysis for efficient siRNA production (Fig. 5).
Fig. 4.
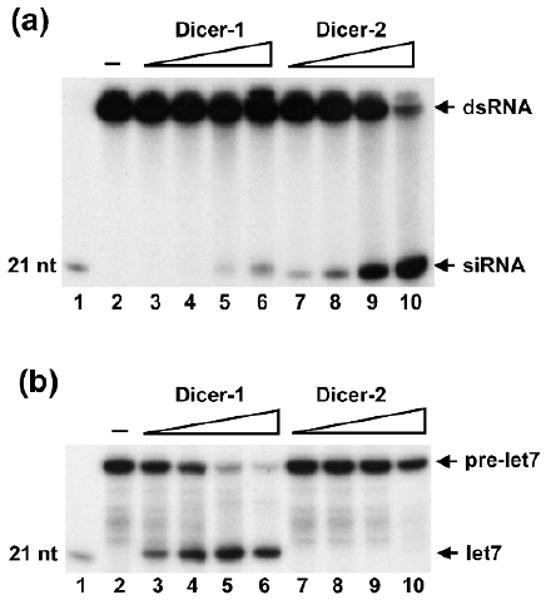
Dicer-1 and Dicer-2 display different substrate specificities. (a) The dsRNA-processing assays were performed with 0.03, 0.1, 0.3, 1 nmol of recombinant Dicer-1 (lanes 3–6) and Dicer-2 (lanes 7–10) enzymes. Dicer-2 is a much better enzyme than Dicer-1 in cleaving dsRNA into siRNA. (b) The pre-miRNA processing assays were performed with 0.03, 0.1, 0.3, 1 nmol of recombinant Dicer-1 (lanes 3–6) and Dicer-2 (lanes 7–10) enzymes. While Dicer-1 efficiently processes pre-miRNA into 21 nt mature miRNA (lanes 3–6), Dicer-2 cannot cleave pre-miRNA at these concentrations (lanes 7–10). Lane 1: radiolabeled 21 nt marker, lane 2: buffer control.
Fig. 5.
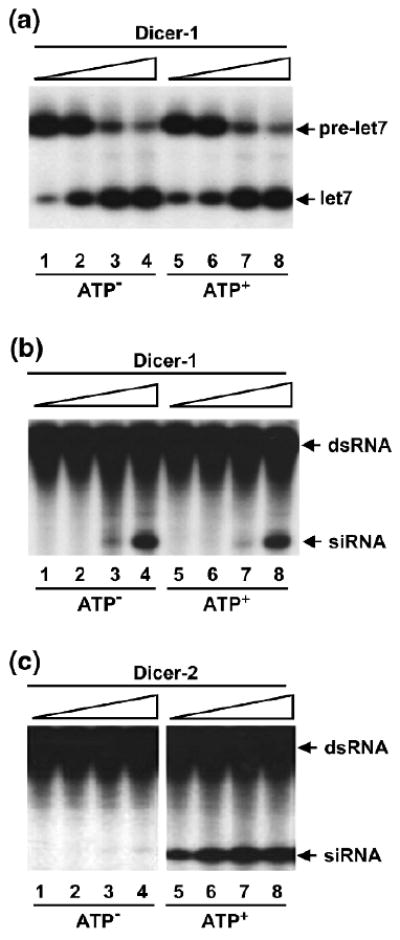
Dicer-1 and Dicer-2 have different ATP requirements. (a) Dicer-1 processes pre-miRNA into miRNA in an ATP-independent manner. The pre-miRNA-processing assays were performed with 0.03, 0.1, 0.3, 1 nmol recombinant Dicer-1 in the absence (lanes 1–4) or presence (lanes 5–8) of ATP. (b) Dicer-1 cleaves dsRNA into siRNA in an ATP-independent manner. The dsRNA-processing assays were performed with 0.03, 0.1, 0.3, 1 nmol recombinant Dicer-1 in the absence (lanes 1–4) or presence (lanes 5–8) of ATP. (c) Dicer-2 requires ATP for efficient siRNA production. The dsRNA-processing assays were performed with 1.25, 2.5, 5, 10 pmol recombinant Dicer-2 in the absence (lanes 1–4) or presence (lanes 5–8) of ATP.
3.3.1. Preparation of Radiolabeled dsRNA
Set up a 20-μL in vitro transcription reaction using the Riboprobe®-T7 Kit: 4 μL of 5 × T7 transcription buffer, 2 μL of 100 mM DTT, 1 μL of RNase inhibitor, 4 μL of NTP mix (10 μL ATP/UTP/CTP + 2 μL GTP + 8 μL water), 1.5 μL of sense template DNA (250 ng/μL), 1.5 μL of antisense template DNA (250 ng/μL), 5 μL [α-32P] of GTP, and 1 μL of T7 polymerase. Mix well and incubate at 37 °C for 1.5 h.
Add 1 μL of RNase-free DNase I to the reaction tube, mix well, and incubate at 37 °C for 15 min.
Stop the reaction with addition of 115 μL of nuclease-free water and 15 μL of 3 M sodium acetate (pH 5.2). Incubate at 90 °C for 1 min.
Add 150 μL of acid phenol/chloroform at room temperature. Close the cap tightly and vortex vigorously for 30 sec. Spin at 13,000 rpm for 5 min at room temperature. Transfer the aqueous phase to a new microcentrifuge tube and repeat this step. The two phenol/chloroform extractions will facilitate annealing of dsRNA substrates.
Transfer the aqueous phase to a new microcentrifuge tube. Add 3.5 μL of 5 mg/mL glycogen and 300 μL of −20 °C 100% ethanol. Invert the tube several times to mix well. Incubate at −20 °C for 1 h to overnight.
Spin at 13,000 rpm for 5 min at 4 °C. Remove the supernatant and wash the pellet with 0.5 mL of 70% ethanol. Remove all supernatant, air-dry the pellet for 5–10 min, and resuspend in 100 μL of nuclease-free water.
Count 1 μL with a scintillation counter and dilute the probe to 1 × 105 cpm/μL. Store the probe in aliquots at −20 °C, which will be good for 2–3 weeks.
3.3.2. Preparation of Radiolabeled Pre-miRNA
Set up a 20-μL 5′ end-labeling reaction in a microcentrifuge tube with the following: 2 μL of 10X PNK buffer, 8 μL of synthetic pre-let7 RNA (2 μM), 8 μL of [γ-32P] ATP, 2 μL of PNK. Mix well and incubate at 37 °C for 2 h.
Add 20 μL of formamide loading dye to stop the reaction. Incubate at 95 °C for 5 min. Load onto a 5-well 6% denaturing polyacrylamide gel.
Run at 300 V for 15 min. Expose to an autoradiography film for 1 min to detect the position of radiolabeled pre-miRNA. Cut the pre-miRNA containing band from the gel and place it into a special microcentrifuge tube.
Add 500 μL of buffer X into the tube. Crush the gel slice with an accompanying pestle (20 strokes). Rotate at room temperature for 1 h.
Spin the tube at 13,000 rpm for 2 min. Transfer the aqueous phase to a Wizard SV minicolumn (included in Wizard Plus SV Minipreps DNA Purification System, Promega, Cat# A1460), place the minicolumn in a microcentrifuge tube, and spin at 13,000 rpm for 2 min.
Transfer the aqueous phase (∼450 μL) to a new microcentrifuge tube. Add 3.5 μL of glycogen and 1 mL of −20 °C ethanol. Invert the tube several times to mix well. Incubate at −20 °C for 1 h to overnight.
Spin at 13,000 rpm for 5 min at 4 °C. Remove the supernatant and wash the pellet with 0.5 mL of 70% ethanol. Remove all the supernatant, air-dry the pellet for 5–10 min, and dissolve it in 50 μL of nuclease-free water.
Count 1 μL with scintillation counter and dilute the probe to 4 × 104 cpm/μL. Store in aliquots at −20 °C.
3.3.3. DsRNA-Processing Assays of Dicer-1 and Dicer-2
Make 90 μL of the master mix for 10 reactions with the following: 10 μL of 10X dsRNA processing buffer, 10 μL of 10 mM ATP, 2 μL of SUPERase•In™, 10 μL of dsRNA probe (1 × 105 cpm/μL), and 58 μL of nuclease-free water.
Distribute 9 μL of the master mix to each microcentrifuge tube, and add 1 μL of buffer A or recombinant Dicer-1 or Dicer-2 proteins of various concentrations. Incubate the reaction tubes at 30 °C for 30 min.
Stop the reactions by the addition of 200 μL of 0.3 M sodium acetate (pH 5.2) and extract with 200 μL of phenol/chloroform. Transfer the aqueous phase to new microcentrifuge tubes containing 3.5 μL of glycogen. Add 0.5 mL of −20 °C 100% ethanol to each tube. Invert the tube several times. Incubate at −20 °C for 30 min.
Spin at 13,000 rpm for 5 min at 4 °C. Remove all the supernatant, air-dry the pellet for 5–10 min, and resuspend the pellet in 5 μL of formamide loading dye. Boil for 5 min and load onto a 16% denaturing polyacrylamide gel. Use a radiolabeled synthetic 21-mer RNA oligonucleotide as the size marker.
Run at 300 V for 40 min. Expose to an autoradiography film at −80 °C for 1 to 2 h. Develop the film and compare the dsRNA processing activities of Dicer-1 and Dicer-2.
3.3.4. Pre-miRNA-Processing Assays of Dicer-1 and Dicer-2
Make 90 μL of the master mixture for 10 reactions with the following: 10 μL of 10X pre-miRNA processing buffer, 10 μL of 10 mM ATP, 5 μL of SUPERase•In™, 10 μL of pre-miRNA probe (4 × 104 cpm/μL), and 55 μL of nuclease-free water. For the rest of the procedure, follow steps 2–5 as above.
4. Notes
4.1. Notes on Expression
1. Frozen stocks of bacmids and viruses should be in small aliquots (each for one use). Repetitive freeze-and-thaw cycles will destroy their structures and activities.
2. Amplification of P2 virus to the P3 and P4 viruses is essential to get large amounts of higher-titer viruses. However, we found that the expression levels of Dicer viruses would decrease with more than four passages.
3. For protein productions, do not let the cells grow more than 48 h after virus infection. Cells will begin to lyse after 48 h, resulting in more degradation products of recombinant Dicer proteins.
4.2. Notes on Purification
4. For nickel affinity purification, beta-mercaptoethanol is used instead of DTT because DTT will destroy the nickel beads. The amount of nickel beads to use is based on the expression levels of Dicer proteins.
5. It is critical to store recombinant Dicer in 10% glycerol at −80 °C. Freezing without glycerol will kill the enzymes. Fast freezing in liquid nitrogen is recommended.
4.3. Notes on Assay
6. When performing radioactive experiments, always take care to avoid contaminating the environment, and protect yourself and people around you from exposure to the radioactive materials.
7. To minimize RNase contamination, all solutions and buffers used in these experiments should be made with DEPC-treated water. Always wear gloves, and use RNase-free tips and reagents designated to the RNA bench.
8. When preparing radiolabeled dsRNA or pre-miRNA substrates, it is best to verify the quality of the radiolabeled probes by running 1 μL of probe on a denaturing PAGE prior to performing the actual assays. The pre-miRNA substrate needs to be PAGE gel-purified before use. We use a mixture of sense and antisense DNA templates (each contains a single T7 promoter) to generate blunt-ended dsRNA, which has been shown to be the ideal substrate for Dicer enzymes (15,16).
9. For assays of ATP requirements of Dicer-1 and Dicer-2, it is desirable to check if purified Dicer-1 and Dicer-2 proteins have residual ATP contamination. Alternatively, simply perform one set of regular reactions at 4 °C (ATP won't be hydrolyzed at this temperature) and compare the activities between 4 °C and 30 °C.
Acknowledgments
We thank Dr. Zain Paroo for critical reading of the manuscript and Feng Jiang for technical assistance. Q. L. is a W. A. “Tex” Moncrief Jr. Scholar in Medical Research and a Damon Runyon Scholar supported by the Damon Runyon Cancer Research Foundation (DRS-43). The work is also supported by a Welch grant (I-1608).
References
- 1.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr Opin Struct Biol. 2005;15:331–341. doi: 10.1016/j.sbi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Sontheimer EJ, Carthew RW. Silence from within: Endogenous siRNAs and miRNAs. Cell. 2005;122:9–12. doi: 10.1016/j.cell.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 5.Esquela-Kerscher A, Slack FJ. Oncomirs—MicroRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 6.Plasterk RH. MicroRNAs in animal development. Cell. 2006;124:877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 7.Tomari Y, Zamore PD. Perspective: Machines for RNAi. Genes Dev. 2005;19:517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 8.Tang G. siRNA and MiRNA: An insight into RISC. Trends Biochem Sci. 2005;30:106–114. doi: 10.1016/j.tibs.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Nat Struct Mol Biol. 2004;11:214–218. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Kolb FA, Jashiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial Rnase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 11.MacRae IJ, Zhou K, Li F, et al. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 13.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YS, Nakahara K, Pham JW, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the RNAi/miRNA silence pathway. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, Rand TA, Kalidas S, et al. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science. 2003;301:1921–1925. doi: 10.1126/science.1088710. [DOI] [PubMed] [Google Scholar]
- 16.Jiang F, Ye X, Liu X, Fincher L, McKearin D, Liu Q. Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev. 2005;19:1674–1679. doi: 10.1101/gad.1334005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito K, Ishizuka A, Siomi H, Siomi MC. Processing of pre-microRNAs by the Dicer-1-Loquacious complex in Drosophila cells. PLoS Biol. 2005;3:e235. doi: 10.1371/journal.pbio.0030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forstemann K, Tomari Y, Du T, et al. Normal microRNA maturation and germ-line stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS Biol. 2005;3:e236. doi: 10.1371/journal.pbio.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomari Y, Matranga C, Haley B, Martinez N, Zamore PD. A protein sensor for siRNA asymmetry. Science. 2004;306:1378–1380. doi: 10.1126/science.1102755. [DOI] [PubMed] [Google Scholar]