

Abstract
AIM
To study the steady-state plasma and intracellular pharmacokinetics of raltegravir, etravirine, darunavir and ritonavir in heavily pre-treated patients.
METHODS
Patients on a salvage regimen containing raltegravir, etravirine, darunavir and ritonavir were eligible for inclusion. During a 12 h dosing interval plasma and peripheral blood mononuclear cells were collected. Drug concentrations were measured using a validated LC-MS/MS assay and pharmacokinetic analysis was performed using non-linear mixed effect modelling.
RESULTS
Irregular absorption was observed with raltegravir and darunavir, which may be caused by enterohepatic cycling. Relative bioavailability of ritonavir was low, when compared with other ritonavir regimens. Raltegravir plasma pharmacokinetics showed wide interpatient variability, while intracellular raltegravir concentrations could not be detected (<0.001 mg l−1 in cell lysate). The intracellular to plasma ratios for etravirine, darunavir and ritonavir were 12.9, 1.32 and 7.72, respectively, and the relative standard error of these estimates were 16.3%, 12.3% and 13.0%.
CONCLUSIONS
The observed distinct intracellular accumulation indicated that these drugs have different affinity for the cellular compartment. The relatively high intracellular accumulation of etravirine may explain its efficacy and its previously described absence of PK–PD relationships in the therapeutic concentration range, when compared with other non-nucleoside reverse transcriptase inhibitors. Lastly, the intracellular concentrations of ritonavir seem sufficient for inhibition of viral replication in the cellular compartment in PI-naive patients, but not in patients with HIV harbouring PI resistance.
Keywords: darunavir, etravirine, intracellular, pharmacokinetics, raltegravir, ritonavir
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The combination of raltegravir, etravirine and ritonavir boosted darunavir is a potent antiretroviral regimen for patients who have been heavily pre-treated for HIV-infection. All these agents have to exert their action intracellularly. However, only little is known about the cellular pharmacology of these agents.
WHAT THIS STUDY ADDS
We investigated the steady-state plasma and cellular pharmacokinetics of raltegravir, etravirine, darunavir and ritonavir and the observed distinct intracellular accumulation ratios indicated that these antiretroviral drugs have different affinity for the cellular compartment.
Introduction
Since the recent approval of raltegravir, darunavir and etravirine by the European Medicines Evaluation Agency a new potent antiretroviral salvage regimen has become available for patients infected with the human immunodeficiency virus (HIV) who are resistant against all previous regimens.
Raltegravir is the first drug in the class of integrase inhibitors and inhibits viral replication by preventing integration of the viral DNA into host DNA. It is dosed 400 mg twice daily with or without food [1]. Darunavir is a drug from the class of the protease inhibitors (PIs). It shows activity against different strains of HIV-1 harbouring PI resistance [2]. Darunavir is dosed 600 mg with 100 mg ritonavir twice daily with food or 800 mg with 100 mg ritonavir once daily. Darunavir is mainly metabolized by the cytochrome P450 3A4 isoenzyme. The PI ritonavir at a dose of 100 mg is co-administered to increase the bioavailability of darunavir by means of CYP3A inhibition in the intestines and liver [3]. Lastly, etravirine is a new drug in the class of non-nucleoside reverse transcriptase inhibitors (NNRTIs). Etravirine is still active against strains of HIV-1 that show resistance against other NNRTIs. Etravirine is dosed 200 mg twice daily and should be taken with food.
The pharmacokinetic interactions between raltegravir, etravirine and darunavir/ritonavir are considered to be minimal or clinically irrelevant: ritonavir slightly decreases raltegravir exposure by 16%, darunavir/ritonavir decreases etravirine exposure by 37% and there is only a minimal pharmacokinetic interaction between etravirine and raltegravir, although the latter has been recently debated [4–7].
Thus far only little is known about the plasma, and particularly the cellular pharmacokinetics of these drugs in heavily pre-treated patients. The viral integrase, protease and reverse transcriptase are all expressed within the infected cell. Therefore, raltegravir, darunavir and etravirine all have to accumulate intracellularly to exert their action. No in vivo data, however, are available on the cellular accumulation of these drugs. We therefore investigated the steady-state pharmacokinetics of raltegravir, darunavir and etravirine in plasma and peripheral blood mononuclear cells (PBMCs) in heavily pre-treated patients on a regimen containing these agents.
Methods
Study design
Ambulatory patients were recruited from the outpatient clinic of the Slotervaart Hospital, Amsterdam, the Netherlands. Patients of 18 years and older with confirmed HIV-infection were eligible for the study if they had used raltegravir, darunavir or etravirine for at least 2 weeks prior to inclusion and were willing to give written informed consent and to participate in and comply with the study. Patients were excluded if they were pregnant or breast feeding during the study. Approval was obtained from the institutional review board of the Slotervaart Hospital.
The pharmacokinetic profile of all drugs was assessed during a 12 h dosing interval. After an overnight fast, patients were admitted to the hospital. All medication was ingested with a light breakfast. Concomitant medication was ingested according to the corresponding regulations regarding food intake.
Blood (8 ml) was collected in Vacutainer® cell preparation tubes (CPT) from Becton Dickinson Vacutainer® systems (Franklin Lakes, NJ, USA) just before and at approximately 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8 and 12 h after drug intake. Within 2 h after collection, the tubes were centrifuged at 1500 g for 30 min at ambient temperature to separate the PBMCs. After centrifugation, the cells were resuspended in the plasma fraction by gently inverting the unopened tubes five times. The cell-suspension in plasma was then transferred to a 15 ml conical centrifuge tube and subsequently centrifuged at 600 g for 10 min at 4°C. Thereafter the plasma layer was transferred to a cryotube and stored at −20°C for further analysis. The remaining cell pellet was washed with 14 ml of ice-cold phosphate buffered saline (PBS) containing 0.1% BSA (w : v). Thereafter, the cell pellet was again resuspended in 14 ml of PBS containing 0.1% BSA (w : v) and a 200 µl aliquot was taken for counting lymphocytes and monocytes using a differential counter, model Cell-Dyn 4000 (Abbott Laboratories, Abbott Park, IL, USA). The remaining cell suspension was centrifuged at 600 g for 10 min at 4°C. The supernatant was aspirated and the cell pellet was stored at −20°C for further analysis.
Bioanalysis
The concentrations of raltegravir, darunavir and etravirine were determined simultaneously from plasma and cells using validated assays by means of high performance liquid chromatography coupled with tandem mass spectrometry using dibenzepine, D5-saquinavir and 13C6-efavirenz as internal standards. Briefly, plasma sample pre-treatment consisted of protein precipitation with a mixture of methanol and acetonitrile (1:1 v : v) using 50 µl plasma. Drugs were extracted from cell pellets using methanol in water (1:1 v : v). Chromatographic separation was performed on a Phenomenex Gemini C18 column of 150 × 2.0 mm (Torrence, Ca, USA) with a quick stepwise gradient using an acetate buffer (pH 5) and methanol, at a flow rate of 0.25 ml min−1 in an analytical run time of only 10 min. The accuracies and precisions were 100 ± 12% and less than 15%, respectively, across all concentrations in all matrices. The assays were validated according to FDA guidelines as previously described [8–12]. The concentration ranges in plasma covered the concentrations that can be encountered in routine clinical practice and the validated limit of quantitation of raltegravir, darunavir and ritonavir in PBMC lysate was 1 ng ml−1 and 5 ng ml−1 for etravirine.
Pharmacokinetic analysis
Plasma concentration vs. time data were analysed using the non-linear mixed effect modelling program NONMEM 6.2 (ICON development solutions, Ellicott city, MD, USA). The first-order conditional estimation method with the interaction option was used. The adequacy of the tested models was evaluated using statistical and graphical methods. The minimal value of the objective function (OFV, equal to minus twice the likelihood) provided by NONMEM was used as goodness of fit characteristic to discriminate between hierarchical models using the log likelihood ratio test. A P value of 0.05, representing a decrease in OFV of 3.84 was considered statistically significant (chi-square distribution, degrees of freedom = 1). The covariance option in NONMEM was used to calculate estimate precisions, expressed as relative standard error (RSE). XPose and Perl speaks NONMEM were used for graphical and statistical model diagnostics [13, 14]. The program Piraña was used for run deployment and analysis and as an interface to NONMEM, PsN and our computer cluster [15].
For raltegravir, darunavir and etravirine the plasma pharmacokinetics were described using one depot and one central compartment, with first order absorption and elimination. Inter-individual and residual variability was estimated with an exponential error model and correlation between individual random effects of apparent volume of distribution (V/F) and apparent oral clearance (CL/F) was estimated.
To describe the gradual and variable onset of oral drug absorption, a chain of transition compartments between the depot and central compartment was tested, as previously described by Kappelhoff et al. [16]. The optimal number of transition compartments was tested for each drug and the mean absorption time (MAT) was calculated using the formula MAT = (n+ 1)/ktr in which n represents the number of transition compartments and ktr represents the transition rate constant. The basic pharmacokinetic model as used for raltegravir, darunavir and etravirine is schematically depicted in Figure 1A.
Figure 1.
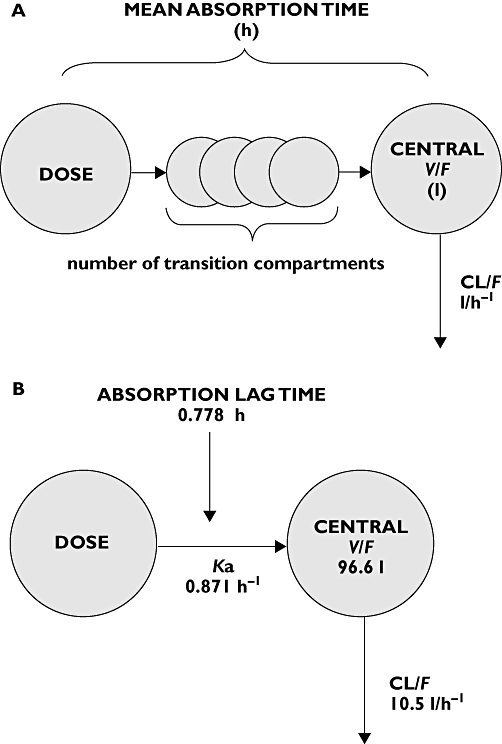
A) Basic pharmacokinetic model for raltegravir, darunavir and etravirine. B) Basic pharmacokinetic model for ritonavir
For description of ritonavir plasma pharmacokinetics, a previously developed and validated model for ritonavir was used: a one-compartment model with first order absorption and elimination with an absorption lag time. Inter-individual and inter-occasion variability were estimated with an exponential error model. The residual error was described using both an additive and proportional component [17]. The population pharmacokinetic model for ritonavir is schematically depicted in Figure 1B. Our data were added to the existing ritonavir dataset and the co-administration of darunavir on CL/F, V/F, relative bioavailability (F) and absorption lag-time of ritonavir was tested.
The intracellular drug concentration was calculated from a measured volume of 0.4 pl for each PBMC [18]. The intracellular accumulation ratio for all agents was estimated with NONMEM, with the formula:
whereby the observed intracellular concentrations (CPBMC,obs) were predicted from the predicted plasma concentrations (Cplasma,pred) and the interindividual (eps(x)) and residual error were described with an exponential model.
Results
Patients
Eleven HIV-infected patients, all male, participated in this study. All patients were heavily pre-treated with antiretroviral agents from all classes thus far available (see Table 1). Ten patients were infected with HIV-1 and one patient was infected with HIV-2. Seven patients used the combination raltegravir, etravirine and darunavir/ritonavir. One patient used the combination of raltegravir, etravirine, darunavir/ritonavir and tenofovir, one patient used the combination of raltegravir, etravirine and lamivudine, one patient used the combination of raltegravir, darunavir/ritonavir and efavirenz and the patient with HIV-2 infection used the combination of raltegravir and darunavir/ritonavir. Baseline characteristics of the patients are shown in Table 1.
Table 1.
Patient characteristics at day of pharmacokinetic sampling
| Parameter | Median | Range |
|---|---|---|
| Age (years) | 56 | 45–59 |
| Plasma HIV-1 RNA (copies ml−1) | <40 | <40–58 |
| CD4+ lymphocytes (cells µl−1) | 210 | 81–270 |
| CD8+ lymphocytes (cells µl−1) | 1170 | 510–2120 |
| ASAT (U l−1) | 24 | 11–40 |
| ALAT (U l−1) | 39 | 16–83 |
| AP (U l−1) | 98 | 78–131 |
| GGT (U l−1) | 53 | 41–107 |
| Total bilirubin (µmol l−1) | 12 | 8–14 |
| Serum creatinine (µmol l−1) | 82 | 46–98 |
| Number of different antiretroviral drugs in antiretroviral treatment history | ||
| Nucleoside/nucleotide reverse transcriptase inhibitors | 6 | 5–7 |
| Protease inhibitors | 3 | 2–6 |
| Non-nucleoside reverse transcriptase inhibitors | 1 | 0–1 |
ALAT, alanine aminotransferase; AP, alkaline phosphatase; ASAT, aspartate aminotransferase; GGT, gamma-glutamyltransferase.
Pharmacokinetic analysis
Plasma
In three out of 11 patients the last blood collection (12 h after intake), could not be performed due to obstruction of the intravenous cannula used for collection. All observed pharmacokinetics are shown in Table 2. All estimated pharmacokinetic parameters are given in Table 3A, B. Observed inter-individual variability in raltegravir plasma pharmacokinetics was high. Maximum raltegravir concentrations were observed between 1 and 5 h after intake. Pre-dose and maximum concentrations ranged from 0.00631 to 0.175 mg l−1 and from 0.232 to 2.85 mg l−1, respectively. In three out of 11 patients a secondary increase in plasma concentrations of raltegravir was observed occurring 2–6 h after intake. For pharmacokinetic analysis, data were log transformed and two transition compartments were used for description of the absorption process.
Table 2.
Observed pharmacokinetics in plasma and PBMCs
| Raltegravir | Etravirine | Darunavir | Ritonavir | ||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Plasma | Plasma | PBMCs | Plasma | PBMCs | Plasma | PBMCs |
| n | 11 | 9 | 10 | 10 | |||
| Cpre-dose (mg l−1) | 0.0690 ± 0.0507 | 0.235 ± 0.092 | 4.53 ± 6.01 | 1.89 ± 1.27 | 1.45 ± 1.11 | 0.124 ± 0.0741 | 0.976 ± 0.485 |
| Cmax (mg l−1) | 0.709 ± 0.760 | 0.444 ± 0.135 | 9.40 ± 6.47 | 4.47 ± 1.82 | 9.73 ± 5.37 | 0.350 ± 0.213 | 2.55 ± 1.22 |
| tmax (h) | 3.08 ± 2.47 | 2.89 ± 0.735 | 2.64 ± 0.907 | 2.23 ± 0.934 | 2.26 ± 1.31 | 3.42 ± 1.66 | 2.74 ± 2.29 |
Table 3.
| A)Estimated pharmacokinetic parameters of raltegravir, etravirine and darunavir | |||
|---|---|---|---|
| Raltegravir (RSE)* | Etravirine (RSE)* | Darunavir (RSE)* | |
| Number of transition compartments | 2 | 4 | 2 |
| Mean absorption time (h) | 1.49 (27.9%) | 1.77 (14.8%) | 0.965 (22.3%) |
| CL/F (l h−1) | 191 (16.0%) | 55.3 (9.11%) | 21.3 (13.9%) |
| V/F (l) | 820 (25.5%) | 781 (13.6%) | 220 (14.5%) |
| Accumulation ratio | – | 12.9 (16.3%) | 1.32 (12.3%) |
| IIV MAT (%) | 75.0 (59.5%) | 36.9 (14.8%) | 62.8 (22.3%) |
| IIV CL/F (%) | 49.7 (59.5%) | 26.3 (23.2%) | 43.0 (31.1%) |
| Correlation IIV CL/F and IIV V/F | 0.273 (47.6%) | 0.0697 (45.2%) | 0.133 (37.5%) |
| IIV V/F (%) | 72.1 (35.6%) | 30.3 (60.3%) | 40.5 (38.2%) |
| IIV accumulation ratio (%) | – | 55.1 (52.4%) | 35.4 (52.7%) |
| Exponential residual error plasma observations (%) | 68.1 | 13.2 | 19.4 |
| Exponential residual error intracellular observations (%) | – | 53.3 | 75.2 |
| B)Estimated pharmacokinetic parameters of ritonavir | |||
| Ritonavir (RSE)* | |||
| Absorption lag time (h) | 0.778 | ||
| Ka (h−1) | 0.871 | ||
| CL/F (l h−1) | 10.5 | ||
| V/F (l) | 96.6 | ||
| Relative bioavailability (%) | 20.3 (20.1%) | ||
| Accumulation ratio | 7.72 (13.0%) | ||
| IIV Ka (%) | 160 (20.9%) | ||
| IIV CL/F (%) | 37.8 (20.8%) | ||
| IIV V/F (%) | 48.5 (36.2%) | ||
| Correlation IIV V/F and IIV Ka | 0.426 (29.3%) | ||
| IIV accumulation ratio (%) | 40.7 (77.5%) | ||
| Proportional error plasma observations (%) | 19.5 (20.7%) | ||
| Additive error plasma observations (mg l−1) | 0.0236 (53.8%) | ||
| IIV additive error plasma observations (%) | 105 (106%) | ||
| Exponential error intracellular observations (%) | 37.7 (14.9%) | ||
RSE, Relative Standard Error of estimate.
Plasma pharmacokinetics of all other agents were less variable. For etravirine, a rapid absorption with a slow onset was observed, followed by a slow elimination. Maximum concentrations were reached 1.5–4 h after intake. Pre-dose and maximum concentrations ranged from 0.135 to 0.430 and from 0.305 to 0.647 mg l−1, respectively. For description of the oral absorption four transition compartments were used.
Darunavir absorption was fast with a variable onset. Maximum concentrations were reached 0.5–3.75 h after intake. Darunavir pre-dose and maximum concentrations ranged from 0.682 to 5.07 mg l−1 and 2.27 to 7.99 mg l−1, respectively. In the darunavir plasma concentrations vs. time curves, in five out of 10 patients a small secondary peak was observed, approximately 1–2 h after the first peak. Two transition compartments were used for the description of the oral absorption.
Lastly, ritonavir absorption, like darunavir, was fast but with a variable onset. Maximum concentrations were reached 1.5–6 h after intake. Ritonavir pre-dose and maximum concentrations ranged from 0.0307 to 0.253 mg l−1 and from 0.0888 to 0.694 mg l−1. CL/F and V/F of ritonavir were significantly increased compared with the reference population described earlier [17], indicating that the relative bioavailability of ritonavir in our study subjects was significantly decreased. The relative bioavailability of ritonavir was estimated to be 20.3% (P < 0.0001) compared with the reference population.
Intracellular
A median of 4.83 milion PBMCs (interquartile range 3.05–8.13 million) was isolated at each timepoint from 8 ml of blood.
Only trace quantities (below the limit of quantitation of 0.001 mg l−1 in lysate) of raltegravir were detected in the PBMC lysate. The intracellular accumulation ratio of 12.9 (16.3% RSE estimate precision) of etravirine was high. Although visual inspection of the data suggested that etravirine was eliminated more slowly from the cellular compartment than from the plasma compartment, separate estimation of cellular pharmacokinetics did not improve the model. The observed intracellular darunavir concentrations were of the same order of magnitude as darunavir plasma concentrations and the accumulation ratio was estimated to be 1.32 (12.3% RSE estimate precision). In contrast, ritonavir concentrations were low in plasma, but high in cells with an estimated accumulation ratio of 7.72 (14.5% RSE estimate precision).
All observed pharmacokinetic parameters are shown in Table 2. All estimated pharmacokinetic parameters of raltegravir, etravirine and darunavir are given in Table 3A. Table 3B shows the estimated pharmacokinetic parameters of ritonavir. Figure 2 depicts the measured pharmacokinetic data in plasma and PBMCs, respectively, together with the median population prediction (bold black line) and the 90% confidence intervals (grey area) of the pharmacokinetic models. As observed from the 90% prediction intervals and median curves, the models adequately captured the observed pharmacokinetic data of all agents.
Figure 2.
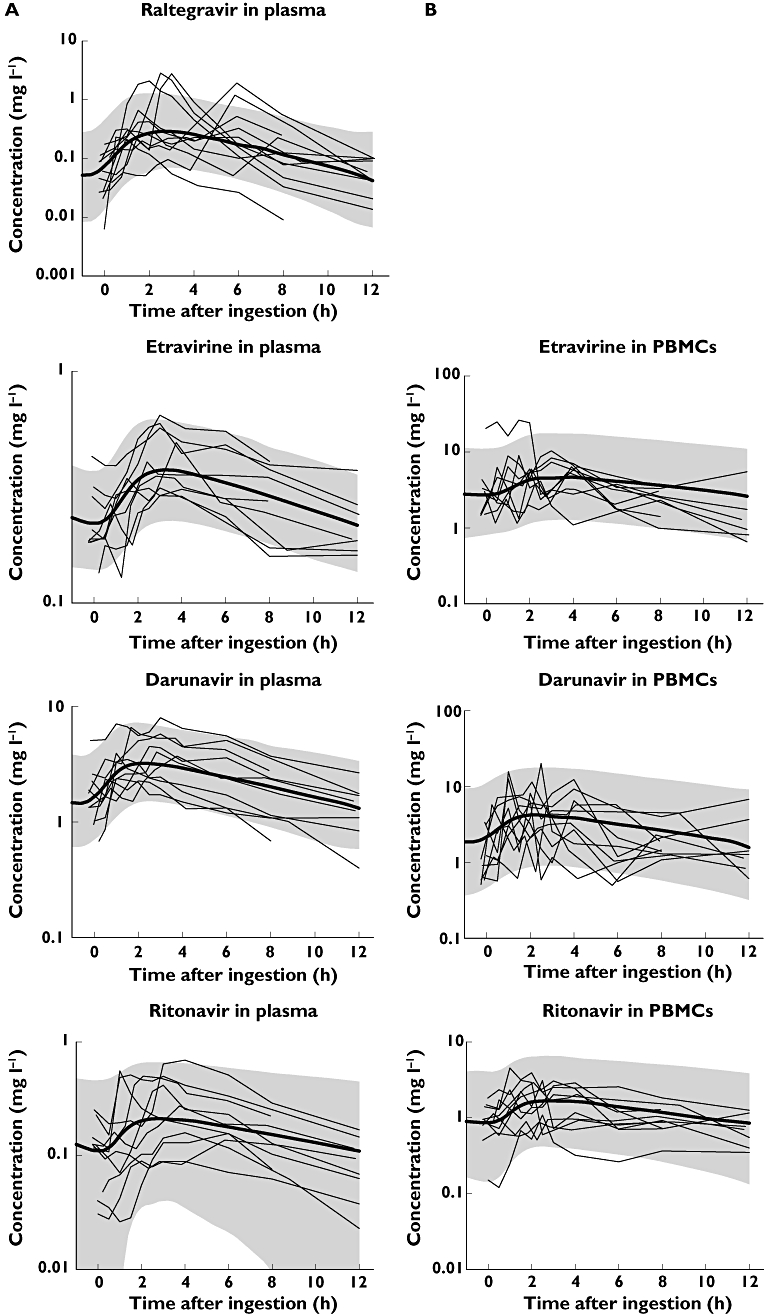
A) pharmacokinetics in plasma. B) pharmacokinetics in PBMCs
Discussion
We report the steady-state pharmacokinetics of raltegravir 400 mg twice daily, etravirine 200 mg twice daily and darunavir/ritonavir 600/100 mg twice daily in plasma and PBMCs in heavily pre-treated individuals from an outpatient cohort of HIV-infected patients.
Observed raltegravir concentrations in plasma were highly variable, consistent with previous studies investigating raltegravir exposure [5, 19]. At a single time point, concentrations could vary more than 50-fold from one individual to another. Secondary increases of plasma concentrations and variable absorption both contributed to this variability. In humans, raltegravir is metabolized to its metabolite raltegravir-glucuronide. This metabolite is mainly excreted in the bile. However, in a biotransformation study of raltegravir, Kassahun and coworkers found that only raltegravir, but not raltegravir-glucuronide could be detected in faeces, indicating that raltegravir-glucuronide is hydrolyzed to raltegravir in the gastro-intestinal tract [20, 21]. It is therefore not unlikely that raltegravir undergoes enterohepatic cycling, explaining the observed secondary increase in plasma concentrations in some individuals. Due to the high interpatient and residual variability in raltegravir pharmacokinetics, standard errors of parameters were relatively high. Larger pharmacokinetic studies are required to characterize fully the pharmacokinetics of raltegravir.
Notably, only trace quantities of raltegravir were observed in PBMCs, indicating a low, but unmeasurable, accumulation ratio. Further optimization of the intracellular raltegravir assay (e.g. a more sensitive detector) and collection of a larger sample volume for isolation of PBMCs, may be useful to attempt to quantify intracellular raltegravir concentrations.
Etravirine pharmacokinetics in plasma were less variable and were in the range of expected values when co-administered with darunavir [6]. Although a large inter- and intra-individual variability of intracellular etravirine accumulation was observed, intracellular concentrations were high, in contrast with previous studies showing accumulation ratios of 1.3 and 0.005 for the NNRTIs efavirenz and nevirapine, respectively [22, 23].
Observed darunavir plasma concentrations were lower than previously described, but all predose concentrations were above the protein-corrected EC50 for protease inhibitor resistant viruses of 0.55 mg l−1[3]. The observed lower exposure to darunavir could not be explained by co-administered drug or food intake, as no interacting drugs were used and darunavir was ingested with food in all patients. Population pharmacokinetic analyses of darunavir from outpatient cohorts are warranted and may identify covariates for darunavir pharmacokinetics explaining these differences. The secondary peaks in the plasma concentration vs. time curves have been reported previously with the PI lopinavir [24]. As with raltegravir, this phenomenon may be explained by enterohepatic cycling or by absorption taking place in more than one place in the gastro-intestinal tract.
Observed intracellular concentrations were comparable with plasma concentrations of darunavir and the intracellular accumulation ratio of 1.32 was comparable with the accumulation ratio we have previously found for the protease inhibitor lopinavir [24]. The intracellular accumulation of darunavir was lower than previously found for the PIs nelfinavir and saquinavir, but higher than the intracellular accumulation of indinavir [18, 25–27].
The observed ritonavir plasma concentrations were low and variable. The low observed concentrations were explained by the low relative oral bioavailability of ritonavir of 20.3%. It has been previously demonstrated that plasma pharmacokinetics of ritonavir are influenced by concomitantly administered antiretroviral drugs. The observed low relative bioavailability of ritonavir in our study may be explained by saturation of ritonavir first pass metabolism at higher doses or saturation of this pathway by concomitantly given drugs in the comparator dataset [17], which was not observed in our dataset. The intracellular accumulation of ritonavir was high, with an accumulation ratio of 7.72. This accumulation ratio was comparable with the accumulation ratio of ritonavir in PBMCs when co-administered with lopinavir, but higher than observed in combination with saquinavir [18, 24, 26].
Although measured concentrations in the PBMC lysate cannot discriminate between intracellular or membrane-bound concentrations, the observed distinct affinities of raltegravir, etravirine, darunavir and ritonavir for the PBMCs indicate that it is a separate compartment and that measured concentrations may reflect intracellular concentrations. Intracellular accumulation of antiretroviral drugs may depend on the presence of other agents, as previously seen with lopinavir, saquinavir and nelfinavir [26, 28, 29]. Studies investigating the intracellular accumulation of raltegravir, etravirine, darunavir and rironavir in patients taking different combinations therefore seem warranted. At present, there is only little known about the cellular pharmacology of raltegravir. Although plasma concentrations of raltegravir and etravirine in plasma are approximately of the same order of magnitude, their intracellular accumulation is not. This may imply that cellular pharmacology of raltegravir is different from other antiretroviral drugs like PIs and NNRTIs.
In special cases there may be a place for therapeutic drug monitoring of antiretroviral treatment, for example, in special populations lacking thorough pharmacokinetic studies or with unknown interactions. The observed pharmacokinetic variability of raltegravir in plasma and the fact that raltegravir plasma trough concentrations do not seem to be related to effectiveness [19], render pharmacokinetic monitoring of raltegravir therapy at a single time point useless. Proper strategies for monitoring plasma pharmacokinetics of raltegravir need therefore to be established. Darunavir trough concentrations in combination with baseline resistance have been shown to predict virological success on a darunavir-containing regimen [30]. As shown in our study, plasma concentrations may be lower than previously shown in pharmacokinetic studies and that pre-dose concentrations may be near the protein-corrected EC50 for protease inhibitor resistant viruses of 0.55 mg l−1[3], indicating that in special cases pharmacokinetic monitoring of treatment may be useful.
In conclusion, we have investigated plasma and intracellular pharmacokinetics of raltegravir, etravirine, darunavir and ritonavir. Irregular absorption was observed with raltegravir and darunavir, which may be caused by enterohepatic cycling. Relative bioavailability of ritonavir was low when compared with other ritonavir containing regimens. Intracellular raltegravir could not be detected. The intracellular to plasma ratios for etravirine, darunavir and ritonavir were 12.9, 1.32 and 7.72, respectively. The observed distinct intracellular accumulation indicated that these drugs have different affinities for the cellular compartment.
Lastly, population pharmacokinetic studies in large outpatient cohorts may further help to elucidate covariates influencing plasma and intracellular pharmacokinetics of etravirine, darunavir/ritonavir and raltegravir.
Competing interests
None declared.
This work was financially supported by Merck Sharp & Dohme BV, Haarlem, the Netherlands.
REFERENCES
- 1.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez PO, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51:5843–55. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 2.Dierynck I, De WM, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81:13845–51. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rittweger M, Arasteh K. Clinical pharmacokinetics of darunavir. Clin Pharmacokinet. 2007;46:739–56. doi: 10.2165/00003088-200746090-00002. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MS, Kakuda TN, Hanley W, Miller J, Kost JT, Stoltz R, Wenning LA, Stone JA, Hoetelmans RM, Wagner JA, Iwamoto M. Minimal pharmacokinetic interaction between the human immunodeficiency virus nonnucleoside reverse transcriptase inhibitor etravirine and the integrase inhibitor raltegravir in healthy subjects. Antimicrob Agents Chemother. 2008;52:4228–32. doi: 10.1128/AAC.00487-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwamoto M, Wenning LA, Petry AS, Laethem M, De SM, Kost JT, Breidinger SA, Mangin EC, Azrolan N, Greenberg HE, Haazen W, Stone JA, Gottesdiener KM, Wagner JA. Minimal effects of ritonavir and efavirenz on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 2008;52:4338–43. doi: 10.1128/AAC.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scholler-Gyure M, Kakuda TN, Sekar V, Woodfall B, De SG, Lefebvre E, Peeters M, Hoetelmans RM. Pharmacokinetics of darunavir/ritonavir and TMC125 alone and co-administered in HIV-negative volunteers. Antivir Ther. 2007;12:789–96. [PubMed] [Google Scholar]
- 7.Menard A, Solas C, Mokthari S, Bregigeon S, Drogoul MP, Tamalet C, Lacarelle B, Martin IP. Etravirine-raltegravir, a marked interaction in HIV-1-infected patients: about four cases. AIDS. 2009;23:869–71. doi: 10.1097/QAD.0b013e328329915f. [DOI] [PubMed] [Google Scholar]
- 8.Ter Heine R, Alderden-Los CG, Rosing H, Hillebrand MJ, van Gorp EC, Huitema AD, Beijnen JH. Fast and simultaneous determination of darunavir and eleven other antiretroviral drugs for therapeutic drug monitoring: method development and validation for the determination of all currently approved HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors in human plasma by liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:2505–14. doi: 10.1002/rcm.3119. [DOI] [PubMed] [Google Scholar]
- 9.Ter Heine R, Rosing H, van Gorp EC, Mulder JW, van der Steeg WA, Beijnen JH, Huitema AD. Quantification of protease inhibitors and non-nucleoside reverse transcriptase inhibitors in dried blood spots by liquid chromatography-triple quadrupole mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;867:205–12. doi: 10.1016/j.jchromb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Ter Heine R, Hillebrand MJ, Rosing H, van Gorp EC, Mulder JW, Beijnen JH, Huitema AD. Quantification of the HIV-integrase inhibitor raltegravir and detection of its main metabolite in human plasma, dried blood spots and peripheral blood mononuclear cell lysate by means of high-performance liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2009;49:451–8. doi: 10.1016/j.jpba.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Ter Heine R, Rosing H, van Gorp EC, Mulder JW, Beijnen JH, Huitema AD. Quantification of etravirine (TMC125) in plasma, dried blood spots and peripheral blood mononuclear cell lysate by liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2009;49:393–400. doi: 10.1016/j.jpba.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 12.Ter Heine R, Davids M, Rosing H, van Gorp EC, Mulder JW, van der Heide YT, Beijnen JH, Huitema AD. Quantification of HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors in peripheral blood mononuclear cell lysate using liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:575–80. doi: 10.1016/j.jchromb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson EN, Karlsson MO. Xpose – an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 14.Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Keizer RJ, Zandvliet AS, Huitema AD. A simple infrastructure and graphical user interface (GUI) for distributed NONMEM analysis on standard network environments – abstract 1237. Population Approach Group in Europe (PAGE) meeting 17. 29-1-2008.
- 16.Kappelhoff BS, Huitema AD, Yalvac Z, Prins JM, Mulder JW, Meenhorst PL, Beijnen JH. Population pharmacokinetics of efavirenz in an unselected cohort of HIV-1-infected individuals. Clin Pharmacokinet. 2005;44:849–61. doi: 10.2165/00003088-200544080-00006. [DOI] [PubMed] [Google Scholar]
- 17.Kappelhoff BS, Huitema AD, Crommentuyn KM, Mulder JW, Meenhorst PL, van Gorp EC, Mairuhu AT, Beijnen JH. Development and validation of a population pharmacokinetic model for ritonavir used as a booster or as an antiviral agent in HIV-1-infected patients. Br J Clin Pharmacol. 2005;59:174–82. doi: 10.1111/j.1365-2125.2004.02241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khoo SH, Hoggard PG, Williams I, Meaden ER, Newton P, Wilkins EG, Smith A, Tjia JF, Lloyd J, Jones K, Beeching N, Carey P, Peters B, Back DJ. Intracellular accumulation of human immunodeficiency virus protease inhibitors. Antimicrob Agents Chemother. 2002;46:3228–35. doi: 10.1128/AAC.46.10.3228-3235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller M, Danovich R, Fransen S, Gupta S, Huang W, Nguyen B, Parkin N, Petropoulos C, Teppler H, Witmer M, Zhao J, Hazuda D. Analysis of resistance to the HIV-1 integrase inhibitor raltegravir: results from BENCHMRK-1 and -2. 48th Annual ICAAC/IDSA. 28-10-0008.
- 20.Kassahun K, McIntosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, Azrolan N, Iwamoto M, Wagner JA, Wenning LA. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos. 2007;35:1657–63. doi: 10.1124/dmd.107.016196. [DOI] [PubMed] [Google Scholar]
- 21.Monteagudo E, Pesci S, Taliani M, Fiore F, Petrocchi A, Nizi E, Rowley M, Laufer R, Summa V. Studies of metabolism and disposition of potent human immunodeficiency virus (HIV) integrase inhibitors using 19F-NMR spectroscopy. Xenobiotica. 2007;37:1000–12. doi: 10.1080/00498250701652323. [DOI] [PubMed] [Google Scholar]
- 22.Almond LM, Edirisinghe D, Dalton M, Bonington A, Back DJ, Khoo SH. Intracellular and plasma pharmacokinetics of nevirapine in human immunodeficiency virus-infected individuals. Clin Pharmacol Ther. 2005;78:132–42. doi: 10.1016/j.clpt.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Almond LM, Hoggard PG, Edirisinghe D, Khoo SH, Back DJ. Intracellular and plasma pharmacokinetics of efavirenz in HIV-infected individuals. J Antimicrob Chemother. 2005;56:738–44. doi: 10.1093/jac/dki308. [DOI] [PubMed] [Google Scholar]
- 24.Crommentuyn KM, Mulder JW, Mairuhu AT, van Gorp EC, Meenhorst PL, Huitema AD, Beijnen JH. The plasma and intracellular steady-state pharmacokinetics of lopinavir/ritonavir in HIV-1-infected patients. Antivir Ther. 2004;9:779–85. [PubMed] [Google Scholar]
- 25.Ford J, Cornforth D, Hoggard PG, Cuthbertson Z, Meaden ER, Williams I, Johnson M, Daniels E, Hsyu P, Back DJ, Khoo SH. Intracellular and plasma pharmacokinetics of nelfinavir and M8 in HIV-infected patients: relationship with P-glycoprotein expression. Antivir Ther. 2004;9:77–84. [PubMed] [Google Scholar]
- 26.Ford J, Boffito M, Wildfire A, Hill A, Back D, Khoo S, Nelson M, Moyle G, Gazzard B, Pozniak A. Intracellular and plasma pharmacokinetics of saquinavir-ritonavir, administered at 1,600/100 milligrams once daily in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2004;48:2388–93. doi: 10.1128/AAC.48.7.2388-2393.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hennessy M, Clarke S, Spiers JP, Mulcahy F, Kelleher D, Meadon E, Maher B, Bergin C, Khoo S, Tjia J, Hoggard P, Back D, Barry M. Intracellular indinavir pharmacokinetics in HIV-infected patients: comparison with plasma pharmacokinetics. Antivir Ther. 2003;8:191–8. [PubMed] [Google Scholar]
- 28.Chandler B, Detsika M, Owen A, Evans S, Hartkoorn RC, Cane PA, Back DJ, Khoo SH. Effect of transporter modulation on the emergence of nelfinavir resistance in vitro. Antivir Ther. 2007;12:831–4. [PubMed] [Google Scholar]
- 29.Janneh O, Jones E, Chandler B, Owen A, Khoo SH. Inhibition of P-glycoprotein and multidrug resistance-associated proteins modulates the intracellular concentration of lopinavir in cultured CD4 T cells and primary human lymphocytes. J Antimicrob Chemother. 2007;60:987–93. doi: 10.1093/jac/dkm353. [DOI] [PubMed] [Google Scholar]
- 30.Molto J, Santos JR, Perez-Alvarez N, Cedeno S, Miranda C, Khoo S, Else L, Llibre JM, Valle M, Clotet B. Darunavir inhibitory quotient predicts the 48-week virological response to darunavir-based salvage therapy in human immunodeficiency virus-infected protease inhibitor-experienced patients. Antimicrob Agents Chemother. 2008;52:3928–32. doi: 10.1128/AAC.00520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]