

Abstract
AIMS
To i) investigate the pharmacokinetics of total and unbound plasma melphalan using a population approach, ii) identify clinical factors that affect melphalan disposition and iii) evaluate the role of melphalan exposure in melphalan-related toxicity and disease response.
METHODS
Population pharmacokinetic modelling (using NONMEM) was performed with total and unbound concentration–time data from 100 patients (36–73 years) who had received a median 192 mg m−2 melphalan dose. Model derived estimates of total and unbound melphalan exposure (AUC) in patients with serious melphalan toxicity and those who had a good disease response (≥90% decrease in paraprotein concentrations) were compared using the Mann-Whitney test.
RESULTS
A two compartment model generated population mean estimates for total and unbound melphalan clearance (CL) of 27.8 and 128 l h−1, respectively. Estimated creatinine clearance, fat free mass and haematocrit were important determinants of total and unbound CL, reducing the inter-individual variability in total CL from 34% to 27% and in unbound CL from 42% to 30%. Total AUC (range 4.9–24.4 mg l−1 h) and unbound AUC (range 1.0–6.5 mg l−1 h) were significantly higher in patients who had oral mucositis (≥grade 3) and long hospital admissions (P < 0.01). Patients who responded well had significantly higher unbound AUC (median 3.2 vs. 2.8 mg l−1 h, P < 0.05) when assessed from diagnosis to post-melphalan and higher total AUC (median 21.3 vs. 13.4 mg l−1 h, P= 0.06), when assessed from pre- to post-melphalan.
CONCLUSIONS
Creatinine clearance, fat free mass and haematocrit influence total and unbound melphalan plasma clearance. Melphalan exposure is related to melphalan toxicity while the association with efficacy shows promising trends that will be studied further.
Keywords: melphalan, myeloma, optimal dosing, population pharmacokinetics, transplantation
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
There has been one previous population pharmacokinetic analysis of total melphalan given as a short infusion in 84 adults (mixed diagnoses) and creatinine clearance and body size were found to be important determinants of total melphalan clearance. Dose and exposure to total melphalan were found to correlate with the development of mucositis.
WHAT THIS STUDY ADDS
This is the largest population pharmacokinetic study on melphalan conducted to date. It is the first conducted in a uniform patient population (patients with multiple myeloma) and the first in which both total and unbound melphalan pharmacokinetics are examined. Factors found to be important determinants of total and unbound plasma clearance of melphalan were creatinine clearance, fat free mass and haematocrit. Haematocrit has not previously been identified as an influential covariate in any previous study. The importance of total and unbound melphalan exposure on transplant outcome was demonstrated by preliminary pharmacodynamic results showing significant associations with melphalan-related toxicity. A preliminary analysis of the association with disease response showed promising trends, but will be examined in more detail with longer follow-up of the whole cohort.
Introduction
High dose melphalan is one of the most active agents in the treatment of multiple myeloma, with several clinical trials demonstrating its superiority to conventional chemotherapy in terms of the complete (CR) and very good partial response (VGPR) rates, event-free survival (EFS) and overall survival (OS) [1, 2]. Even in an era where biological agents such as lenalidomide and bortezomib are incorporated into frontline therapy for myeloma, consolidation of the initial therapy with high dose melphalan remains standard. However the toxicity of high dose melphalan is profound: prolonged cytopenias occur in all patients, necessitating rescue with autologous stem cell transplantation (ASCT). Gastrointestinal toxicity, including anorexia, mucositis, nausea, vomiting and diarrhoea, is also very common [3–5]. Both severity and duration of myelosuppression are dose-dependent [5] and the gastrointestinal toxicity is dose-limiting. Conversely, insufficient dose intensity can lead to suboptimal response, as previously observed in patients with amyloidosis [6]. In this study, the partial response rate to melphalan (≥50% reduction in the serum or urine M protein) was significantly higher in the group receiving standard high doses compared with the group who had intermediate, risk-adjusted doses (75% vs. 53%, P < 0.01).
Melphalan is eliminated by both renal excretion and spontaneous chemical degradation to its mono- and di-hydroxy metabolites [7, 8]. The latter pathway has been shown to be a relatively minor contributor (<5%) [9] because plasma protein binding retards the hydrolysis rate of melphalan [9]. In water and in urine, however, melphalan undergoes rapid chemical decomposition [8]. This has made it difficult to study the 24 h urinary excretion of melphalan and has led to some confusion about the role of renal excretion in melphalan elimination. Highly variable estimates of the fraction of melphalan that is renally excreted have been obtained, ranging from 3% to 93% in nine adults (mean ± SD 34 ± 33%), even after attempts to freeze the urine specimens rapidly, suggesting that there may be decomposition in the bladder [7]. However, the fact that greater than 60% of the dose was recovered in the urine obtained from three patients in the study by Reece et al. [7] suggests that renal excretion is likely to be the major elimination pathway for melphalan.
In patients with multiple myeloma the standard melphalan dose for patients undergoing ASCT is 200 mg m−2. Dose modifications have been recommended in patients with impaired renal function [3, 10], while obese patients often receive a dose based on adjusted ideal body weight or capped at a body surface area of 2 m2. The optimal dose, that produces a complete disease response with acceptable toxicity, is unknown. In order to ensure that every patient is administered the optimal dose it is necessary to have a comprehensive understanding of i) the pharmacokinetics of melphalan and the factors that affect disposition and ii) inter-patient variability in drug exposure and its association with toxicity and efficacy in uniform disease populations.
While there has been one previous population pharmacokinetic study on melphalan in adults [11], there have been no previous studies in which unbound melphalan was examined and none conducted on a uniform disease population. The aims of this study were to i) investigate the pharmacokinetics of total and unbound plasma melphalan in a large population of patients with multiple myeloma undergoing high dose therapy, ii) identify clinical factors that may affect the disposition of the drug, iii) develop limited sampling strategies that will aid in the pharmacokinetic monitoring of melphalan and iv) examine the role of exposure to total and unbound melphalan in melphalan-related toxicity and disease response.
Methods
This study was a prospective, multi-centre, observational investigation of the pharmacokinetics of melphalan in patients who underwent ASCT as part of their treatment for multiple myeloma. This study was registered with the Australian Clinical Trials Registry (Registration number: ACTRN0126000231549). The Ethics Committees at each of the six participating hospitals approved the study and all the participants provided written informed consent.
Clinical and biochemical determinations
The Vitros Fusion 5.1 analyser (Ortho Clinical Diagnostics Australia, Mulgrave, VIC, Australia) enzymatic assay was used to measure plasma creatinine concentrations in samples taken on the day of melphalan pharmacokinetic analysis. Such enzymatic assay methods for plasma creatinine have been standardized with the international reference method of isotope dilution mass spectrometry [12]. The Vitros 5.1 analyser was also used to determine pre-ASCT total protein, albumin and transferrin concentrations, while serum electrophoresis was used to determine pre-ASCT paraprotein concentrations. The haematocrit value was recorded either on the day of melphalan administration (preferably) or, if this value was not available, on the closest day prior.
Creatinine clearance (CLcr) was estimated from plasma creatinine concentration, age and total body weight (TBW) using the Cockcroft & Gault equation [13] given as follows:
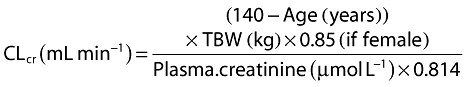 |
The Cockroft & Gault formula, applied using the patient's actual weight and a creatinine assay that is aligned with the isotope dilution mass spectrometry method, has been shown to provide a good estimate of isotopic glomerular filtration rate in 167 Australian patients with body mass index (BMI) values ranging from 15 to 51 kg m−2[14]. CLcr was normalized to a standard weight of 70 kg by dividing by total body weight and multiplying by 70.
BMI and body surface area (BSA) were calculated using published equations [15, 16].
Fat free mass (FFM, kg) was determined using the equations of Janmahasatian et al. [17]:
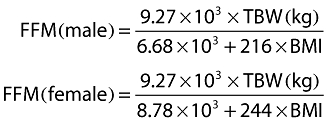 |
Drug administration and blood sampling
Melphalan (Alkeran®, GlaxoSmithKline Australia Pty Ltd, Boronia Victoria, Australia) was administered as an intravenous infusion over a median of 35 min (range 15–95 min). Blood sampling for melphalan concentration measurements occurred either from a catheter that had been inserted in the arm (78 patients) or from the second lumen of a double lumen central line (the other lumen was used for drug administration). To avoid contamination, after flushing the cannula, 5 ml of blood was withdrawn prior to taking each sample. Blood collection times for 63 initial patients were at the end of the infusion, then at 5, 10, 20, 30, 40 and 50 min, then 1, 2, 3, 4 and 8 h after the end of the melphalan infusion. In 37 subsequent patients blood sampling (five or six) occurred at times within the optimal sampling windows shown in Table 1, which were identified using d-optimality, implemented by the POPT software (http://www.winpopt.com). Plasma was prepared by centrifugation at 1200 g for 10 min at 4°C (Beckman CS-15R, Beckman Instruments, CA, USA). Samples were stored at −40°C until analysis.
Table 1.
Optimal sampling times and windows to assess melphalan population pharmacokinetics following intravenous infusion
| Sample number | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Optimal sampling time (h) | 0.083 | 0.66 | 0.66 | 1.19 | 2.81 |
| Optimal sampling window (h) | 0.08–0.15 | 0.46–0.73 | 0.46–0.73 | 1.08–1.36 | 2.41–3.33 |
Two windows are identical, corresponding to two identical sampling times. Two samples should be taken in this window (at different times).
Melphalan assay
Total and unbound melphalan concentrations were measured in plasma samples using our previously published high performance liquid chromatography assay [18]. Samples were prepared using methanol precipitation (total melphalan) and ultrafiltration (unbound melphalan) [18]. Total melphalan concentrations were measured in all samples, while unbound melphalan concentrations were measured in five or six samples per patient (timed according to the optimal design schedule, Table 1). The total melphalan assay was linear to at least 40 µg ml−1 and had excellent inter-day precision (<9% for 2.5–40 µg ml−1 melphalan), accuracy (<3% deviation from nominal concentration) and recovery (91–110% for 0.5–40 µg ml−1). The unbound melphalan assay was linear to at least 2.5 µg ml−1 and also had excellent inter-day precision (<11% for 0.7–2.5 µg ml−1 melphalan) and recovery (89–93% for 0.25–2.5 µg ml−1 melphalan). Detection limits were 0.1 µg ml−1 and 0.05 µg ml−1 for the total and unbound melphalan assays, respectively. No compounds interfered with the melphalan assay.
Population pharmacokinetic analysis
Population pharmacokinetic modelling of both total (n= 1057) and unbound (n= 691) melphalan concentrations was performed with NONMEM 6, version 2 (Globomax LL, Hanover, MD, USA) that had been installed on a Pentium D personal computer running Windows XP and Compaq Visual Fortran Compiler (version 6.6, Compaq Computer Corporation, Houston, Texas, USA). The program Wings for NONMEM version 613 (developed by Dr Nicholas Holford, Auckland University; http://wfn.sorceforge.net) was used as a front-end processor. Graphical output from the NONMEM analyses were obtained using CrossGraphs version 2.3 (PPD Development, Cambridge MA, USA) and Microsoft Excel (Microsoft corporation, Troy NY, USA). The first order conditional estimation method (FOCE) that took into account the η-ε interaction was used throughout the model building and evaluation procedures. Population pharmacokinetic models for total and unbound melphalan were developed separately in a series of steps: i) base model development, ii) covariate model development and iii) covariate model evaluation.
Base model development (Step 1)
Base models were developed that did not include covariate effects. The structural and statistical models used to fit the total and unbound melphalan concentration vs. time data were derived from our previous analysis of melphalan in children [19]. A two compartment model with first order elimination from the central compartment was used, parameterized with use of clearance (CL), volume of distribution of the central compartment (V1), inter-compartmental clearance (Q) and volume of distribution of the peripheral compartment (V2). Inter-patient variability was described using an exponential random effects model, defined as:
where θi represents the pharmacokinetic parameter for the ith individual, 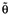 is the typical value of pharmacokinetic parameter in the population (e.g. population mean) and ηi quantifies the deviation of θi from
is the typical value of pharmacokinetic parameter in the population (e.g. population mean) and ηi quantifies the deviation of θi from 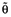 with a distribution (0, ω2). Intra-patient variability was described by a combined additive and proportional error model, given by:
with a distribution (0, ω2). Intra-patient variability was described by a combined additive and proportional error model, given by:
where Ŷ are the predicted and Y the measured concentrations in the ith individual at the jth sampling time and where ε1 (proportional component) and ε2 (additive component) are random effects quantifying the residual errors, both with a distribution (0, σ2). Residual errors (ε) represent the differences between the model predictions and the data and include intra-patient variability, assay error and model misspecification error.
Covariate model development (Step 2)
Covariates screened for their possible influence on total and unbound melphalan pharmacokinetic parameters included TBW (kg), BSA (m2), FFM (kg), age (years), CLcr (ml min−1 70 kg−1), CLcr (ml min−1), sex, albumin concentration (g l−1), total protein concentration (g l−1) and haematocrit (HCT, %). The covariates were implemented in the model using two different approaches:
In addition, the effects of the size covariates, as well as albumin and total protein concentrations were evaluated on both CL and V1 simultaneously. Each of the covariates, except TBW, was centred to the median value in the population (shown in Table 2). TBW was centred to 70 kg. The influence of TBW and FFM on CL and V1 was assessed with the use of an allometric scaling function [20], in which the exponent was fixed to 0.75 for CL and 1.0 for V1:
Table 2.
Characteristics of the 100 patients (59 male, 41 female) with multiple myeloma
| Characteristic | Median | Range |
|---|---|---|
| Melphalan dose (mg) | 368 | 150–450 |
| Melphalan dose (mg m−2) | 192 | 115–216 |
| Age (years) | 57 | 36–73 |
| Weight (kg) | 78 | 42–132 |
| Height (cm) | 168 | 147–185 |
| Body surface area (m2) | 1.9 | 1.3–2.6 |
| Body mass index (kg m−2) | 27.6 | 19.2–40.9 |
| Fat free mass (kg) | 53.3 | 34.4–80.5 |
| CLcr (ml min−1) | 97 | 29–234 |
| CLcr (ml min−1 70 kg−1) | 88 | 26–205 |
| Haematocrit (%) | 34 | 20–45 |
| Albumin (g l−1) | 37 | 14–49 |
| Total bilirubin (µmol l−1) | 3 | 2–37 |
| Total protein (g l−1) | 71 | 39–117 |
| C-reactive protein (mg l−1) | 6 | 0–74 |
The influence of individual covariates on pharmacokinetic parameters was first examined by plotting the empirical Bayesian estimates of the pharmacokinetic parameters generated from the base model against each covariate. Covariates identified as potentially influential were tested for inclusion in the population pharmacokinetic models by adding these individually into the base population pharmacokinetic model and noting the changes in the objective function value (OBV). A decrease in the objective function by more than 6.63 corresponds to a significance level of P < 0.01 (d.f. = 1) using the likelihood ratio test. Covariates found to reduce significantly the objective function value when tested in the initial screening procedure were cumulatively added to the population pharmacokinetic model using parameterizations that reflected the physiology of the processes involved. Since total clearance is the sum of the independent clearances for all the different pathways of elimination (including renal clearance, hepatic clearance and other methods of elimination) an additive model (CL = CLrenal+ CLhepatic+ CLother) best reflects the physiology of the processes. A number of evaluation criteria were then used to select the most appropriate covariate model including i) a low value for the objective function (OFV), ii) low estimates for sigma, iii) low estimates of inter-subject variability in the pharmacokinetic parameters, iv) good agreement between model-predicted and observed melphalan concentrations and v) good model performance as assessed by a visual predictive check, comparing observed concentration vs. time data and the 90% confidence interval generated using 500 simulated concentration–time data sets.
Covariate model evaluation: (Step 3)
A bootstrap procedure was used to assess the accuracy and robustness of the covariate models. This was performed in an automated fashion using the bootstrap option in the Wings for NONMEM software. The results from 1000 successful runs were obtained (including minimization successful and minimization terminated due to rounding errors [21]). The mean and 95% confidence intervals were calculated for all population pharmacokinetic parameters, as well as the % difference between the bootstrap mean and the estimate derived from the original dataset.
Model-derived pharmacokinetic parameters and other variables
A number of additional pharmacokinetic parameters for total and unbound melphalan were derived from the posthoc estimates of the primary pharmacokinetic parameters including CL and V1 normalized to weight and surface area, the rate constants (k10, k12, k21), as well as the distributional half-life (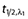 ) and the elimination half-life (
) and the elimination half-life (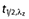 ). Total and unbound AUC were determined by dividing the dose (mg) by the individual posterior Bayesian estimates of total and unbound CL, respectively. Fraction unbound (fu) was determined for each of the six samples collected from each patient by dividing the measured unbound melphalan concentration by the total melphalan concentration. Linearity in melphalan protein binding was then examined by using one-way anova to test for significant differences in fu for the six specifically-timed samples collected from each patient. Overall fraction unbound for each patient was then determined by dividing the unbound AUC by the total AUC.
). Total and unbound AUC were determined by dividing the dose (mg) by the individual posterior Bayesian estimates of total and unbound CL, respectively. Fraction unbound (fu) was determined for each of the six samples collected from each patient by dividing the measured unbound melphalan concentration by the total melphalan concentration. Linearity in melphalan protein binding was then examined by using one-way anova to test for significant differences in fu for the six specifically-timed samples collected from each patient. Overall fraction unbound for each patient was then determined by dividing the unbound AUC by the total AUC.
Investigating the effects of paraprotein and transferrin concentrations and myeloma type on total and unbound melphalan clearance
The dataset was incomplete with respect to pre-ASCT paraprotein (n= 77) and transferrin concentrations (n= 67), so these covariates could not be considered for inclusion in the population pharmacokinetic models and were therefore tested for significant associations with total and unbound melphalan clearance using the correlation coefficient of determination. The Mann-Whitney test was used to compare total and unbound melphalan clearance in patients with IgA and IgG myeloma.
Investigating the effect of total and unbound melphalan exposure on toxicity post-transplant and disease response
Gastrointestinal toxicity, including clinical oral mucositis, functional oral mucositis, colitis, nausea, vomiting and diarrhoea, was monitored daily from 2 days prior to stem cell re-infusion (day 0), then up to day 14 and 28 (in the event of ongoing gastrointestinal toxicity). Grade of toxicity was assigned on a daily basis using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3) [22], with the patient's overall grade being the maximum level achieved during the period of monitoring. Duration of hospital admission was calculated as the number of days from date of melphalan administration to date of hospital discharge following admission for management of post transplant complications. In those patients who had melphalan and ASCT on an outpatient basis, this time period included the days between melphalan administration and hospital admission. The Mann-Whitney test was then used to test for significant differences in total and unbound AUC between patients who had ≥ grade 3 toxicity or long hospital admissions (≥21 days, the 75th percentile) and those who had toxicity grades of 0–2 or shorter hospital admissions (<21 days). The exception was the toxicity of vomiting: total and unbound melphalan AUCs were compared between patients who had ≥ grade 2 vomiting and those who had grade 0–1 vomiting as there were only six patients who had ≥ grade 3 vomiting.
In patients with multiple myeloma, serum monoclonal paraprotein concentrations were monitored from diagnosis and throughout treatment to follow response to treatment. Paraprotein concentrations were recorded at diagnosis, immediately prior to melphalan, then at 6 weeks post melphalan. Data were not available for all patients due to i) the test not being performed at the correct time, ii) the presence of overlying bands on electrophoresis preventing the accurate quantitation of the patient's paraprotein or iii) the data were missing. Disease response criteria conformed to those previously established for multiple myeloma [23]. Overall disease response was based on the % change in paraprotein concentrations from diagnosis to post melphalan and was classified as complete response (CR) (100% decrease), very good partial response (VGPR) (≥90% decrease), partial response (PR) (50–89% decrease), minimal response (MR) (25–49% decrease), or no change (increase–24% decrease). Melphalan-related disease response was based on the percentage (%) change in paraprotein concentrations from pre- to post-melphalan (classifications were as above for overall disease response) and was assessed in patients whose maximum response to prior treatment was a VGPR or less. The Mann-Whitney test was then used to test for significant differences in total and unbound AUC between patients who achieved a CR or VGPR and the remainder.
Results
Patient characteristics
The characteristics of the 100 participants (59 male, 41 female) are summarized in Table 2. Myeloma type, as classified by paraprotein type, was IgG (58 patients), IgA (21 patients), light chain only (7) and non-secretory (1 patient). Data on paraprotein type were missing for 13 patients.
Population pharmacokinetics
The population pharmacokinetic parameters derived from the base models for total and unbound melphalan are shown in Table 3. In the covariate screen, the potential covariates that were identified for potential inclusion in the population pharmacokinetic models for total and unbound melphalan included CLcr (ml min−1 70 kg−1), CLcr (ml min−1), the body size covariates (TBW, BSA, FFM) and haematocrit (Table 4). Patients with low values for CLcr, haematocrit and FFM tended to have low total and unbound clearance of melphalan (P < 0.01) as shown in Figures 1 and 2.
Table 3.
Population pharmacokinetic parameters for total and unbound melphalan using the base model
| Total melphalan | Unbound melphalan | |||
|---|---|---|---|---|
| Parameter | Population mean | Interindividual variability (%CV) | Population mean | Interindividual variability (%CV) |
| CL (l h−1) | 27.8 | 33.6 | 128 | 41.7 |
| V1 (l) | 13.1 | 59.5 | 60.1 | 57.2 |
| Q (l h−1) | 31.3 | 42.4 | 160 | 45.8 |
| V2 (l) | 15.1 | 34.4 | 72 | 33.6 |
| Random residual variability | ||||
| σ1 (SD) | 0.072 | 0.042 | ||
| σ2 (SD) | 0.082 | 0.029 | ||
CL, clearance; %CV, coefficient of variation; Q, intercompartmental clearance; SD, standard deviation; V1, volume of distribution into the central compartment; V2, volume of distribution into the peripheral compartment.
Table 4.
Covariate screen: Objective function changes after adding individual covariates into the base population pharmacokinetic models for total and unbound melphalan
| Model for total melphalan | Model for unbound melphalan | ||||
|---|---|---|---|---|---|
| Covariate | Covariate equations | ΔOBV | P value* | ΔOBV | P value* |
| CLcr (ml min−1 /70 kg−1) | CL =θ1+θ2× (CLcr/88) V1 =θ3 | −34 | <0.01 | −26 | <0.01 |
| CLcr (ml min−1) | CL =θ1+θ2× CLcr/97 V1 =θ3 | −37 | <0.01 | −28 | <0.01 |
| HCT | CL =θ1+θ2× (HCT/34) V1 =θ3 | −8 | <0.01 | −12 | <0.01 |
| TBW | CL =θ1+θ2× (TBW/70)0.75V1 =θ3× (TBW/70) | −4 | <0.05 | −15 | <0.01 |
| TBW | CL =θ1× (TBW/70)0.75V1 =θ2× (TBW/70) | +3 | NS | −11 | <0.01 |
| FFM | CL =θ1+θ2× (FFM/53)0.75V1 =θ3× (FFM/53) | −12 | <0.01 | −12 | <0.01 |
| FFM | CL =θ1× (FFM/53)0.75V1 =θ2× (FFM/53) | −3 | NS | −4 | <0.05 |
| BSA | CL =θ1+θ2× (BSA/1.9) V1 =θ3× (BSA/1.9) | −6 | <0.05 | −10 | <0.01 |
| BSA | CL =θ1× (BSA/1.9) V1 =θ2× (BSA/1.9) | −3 | NS | −6 | <0.05 |
| Age | CL =θ1+θ2× (Age/57) V1 =θ3 | +9 | NS | +5 | NS |
| Sex | CL =θ1+θ2× (1−sex) V1 =θ3 | −1 | NS | 0 | NS |
| ALB | CL =θ1× (ALB/37) V1 =θ2× (ALB/37) | +28 | NS | +50 | NS |
| TPR | CL =θ1× (TPR/71) V1 =θ2× (TPR/71) | −1 | NS | +9 | NS |
Significance in the change in Objective function value (ΔOBV) was assessed using the likelihood ratio test, ALB, albumin; BSA, body surface area; CLcr, estimated creatinine clearance; FFM, fat free mass; HCT, haematocrit; NS, Not significant; TBW, total body weight; TPR, total protein.
Figure 1.
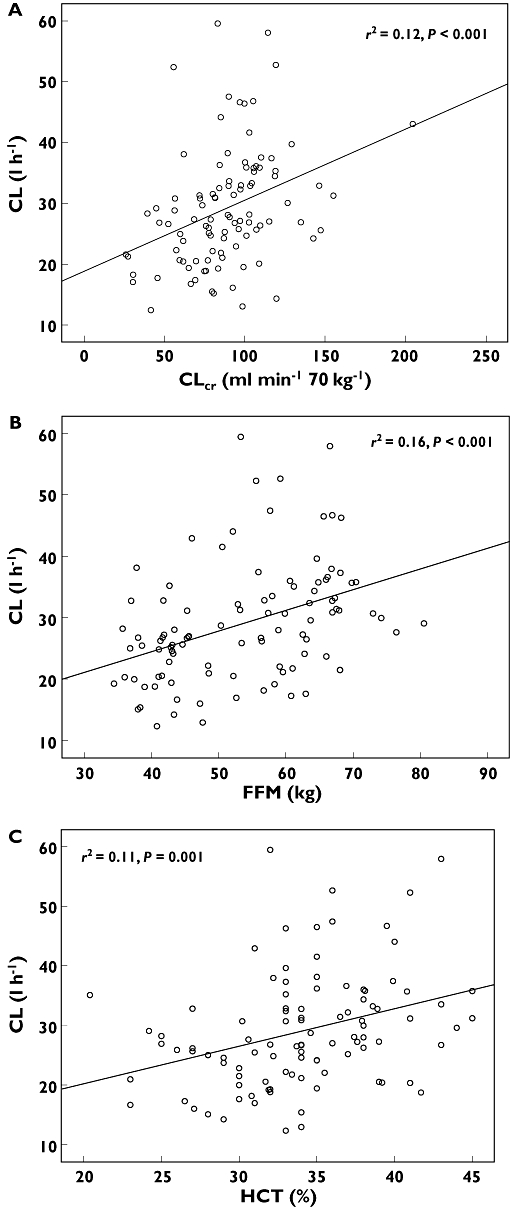
Scatterplots showing the associations between total melphalan plasma clearance and the covariates of A) estimated creatinine clearance (CLcr), B) fat free mass (FFM) and C) haematocrit (HCT)
Figure 2.
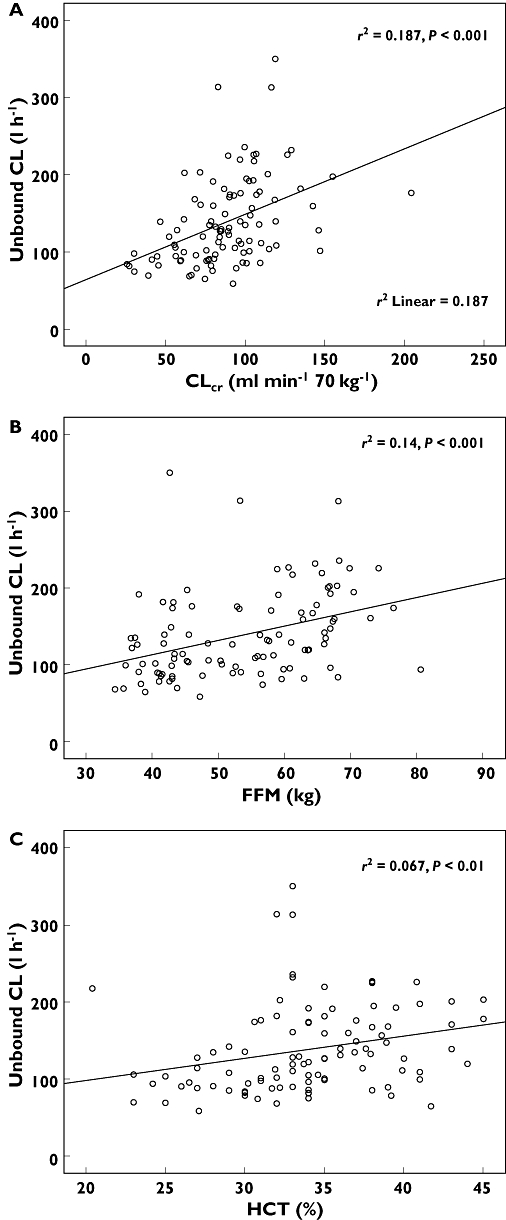
Scatterplots showing the associations between unbound melphalan plasma clearance and the covariates of A) estimated creatinine clearance (CLcr), B) fat free mass (FFM) and C) haematocrit (HCT)
After individually testing each potentially influential covariate, CLcr was found to be the most influential, individually reducing the objective function value by more than 30 units for total melphalan, and by more than 26 units for unbound melphalan. Therefore, clearance of total and unbound melphalan was divided into non-renal (CLNR) and renal (CLR) components, as follows: CL = CLNR+ CLR. Haematocrit and body size (FFM) were added to CLNR. Of the body size covariates tested, FFM was selected for inclusion in the models because it reduced the OBV by an amount that was significant at the P < 0.01 level for both total and unbound melphalan when incorporated into the model for CL in the format of TVCL =θCL+θCOV× Covariate (Table 4). FFM was also associated with decreases in the OBV value for both total and unbound melphalan when incorporated into the model for CL using the physiologically superior format of TVCL =θCL× Covariate (Table 4). The final structural models for CL and V1 of total melphalan incorporated CLcr (ml min−1 70 kg−1) and had the format: CL = CLNR+ CLR, where
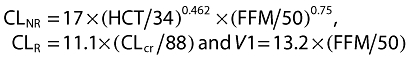 |
The final structural model for CL and V1 of unbound melphalan incorporated CLcr (ml min−1 70 kg−1) and had the format: CL = CLNR+ CLR, where
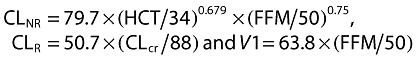 |
The population pharmacokinetic parameter estimates generated using the final covariate models for total and unbound melphalan are shown in Table 5. Renal clearance of total and unbound melphalan could be estimated from the population pharmacokinetic parameters shown in Table 5 and was found to be approximately 40%. Addition of the covariates into the population pharmacokinetic models substantially reduced the inter-individual variability in CL and V1. Inter-individual variability in clearance of total melphalan was reduced by 25% from the base model value of 34% while that of unbound melphalan was reduced by 29% from the base model value of 42%. Inter-individual variability in V1 for total melphalan was reduced by 13% from the base model value of 60%, while that of unbound melphalan was reduced by 42% from the base model value of 57%. There was generally good agreement between observed and population predicted total and unbound melphalan concentrations (Figure 3). Figure 4A, B shows the visual predictive checks for the covariate models for total and unbound melphalan, respectively, and demonstrates good model performance. Using the covariate models the population pharmacokinetic parameter estimates generated using 1000 replicate data sets in the bootstrap analyses were comparable with those generated using the original data set (Table 5), indicating that the accuracy and stability of the models for total and unbound melphalan were acceptable. Mean values for all fixed and random effect parameters were within ±1.5% for total melphalan and within ±7% for unbound melphalan.
Table 5.
Population pharmacokinetic parameter estimates for total and unbound melphalan in 100 patients with multiple myeloma using separately developed Covariate Models that incorporated CLcr with units ml min−1 70 kg−1
| Total melphalan | Unbound melphalan | |||
|---|---|---|---|---|
| Parameter | Mean | Bootstrap mean (%diff, 95% CI) | Mean | Bootstrap mean (%diff, 95% CI) |
| Fixed effects | ||||
| CLNR (l h−1) | ||||
| θ1 | 17 | 17 (0%, 13.5–21.3) | 79.7 | 80.3 (0.8%, 64.8, 97.3) |
| θ2 | 0.462 | 0.463 (0.2%, 0.060, 0.954) | 0.679 | 0.682 (0.4%, 0.284, 1.070) |
| CLR (l h−1) | 11.1 | 11.2 (0.9%, 6.8, 14.6) | 50.7 | 49.8 (−1.8%, 34.8, 66.1) |
| V1 (l) | 13.2 | 13.3 (0.8%, 11.0, 15.6) | 63.8 | 65.0 (1.9%, 50.6, 78.1) |
| Q (l h−1) | 30.6 | 30.5 (−0.3%, 26.5, 34.2) | 152 | 146.5 (−3.6%, 123.0, 171.0) |
| V2 (l) | 15 | 15 (0%, 13.8, 16.2) | 71.6 | 70.3 (−1.8%, 62.7, 78.4) |
| Interindividual variability | ||||
| ωCL (CV%) | 26.7 | 26.7 (0%, 21.7, 31.8) | 29.8 | 29.8 (0%, 23.0, 37.1) |
| ωV1 (CV%) | 57.9 | 57.9 (0%, 38.0, 75.9) | 38.7 | 39.8 (2.8%, 17.6, 65.6) |
| ωQ (CV%) | 41.1 | 41.5 (1%, 27.3, 55.9) | 49.6 | 46.3 (−6.7%, 26.9, 60.5) |
| ωV2 (CV%) | 34.5 | 34.1 (−1.2%, 25.9, 42.9) | 35.4 | 37.6 (6.2%, 26.8, 49.2) |
| Random residual variability | ||||
| σ1 (SD) | 0.072 | 0.072 (0%, 0.060, 0.083) | 0.138 | 0.131 (−5.1%, 0.104, 0.155) |
| σ2 (SD) | 0.082 | 0.081 (−1.2%, 0.060, 0.107) | 0.027 | 0.028 (3.7%, 0.003, 0.051) |
| OBV | −876 | −1535 | ||
| Structural models: | ||||
| CL = CLNR+ CLR, where CLNR=θ1× (HCT/34)θ2× (FFM/50)0.75 and CLR=θ3× (CLcr/88), V1 =θ4× (FFM/50), Q =θ5, V2 =θ6 | ||||
95% CI = lower and upper limits of the 95% confidence interval for population pharmacokinetic parameters obtained with 1000 bootstrap runs. %diff = (bootstrap mean – Covariate model mean)/Covariate model mean × 100, OBV = Objective function value. CL, clearance; CLcr, estimated creatinine clearance (ml min−1 70 kg−1); %CV, coefficient of variation; FFM, fat free mass (kg); HCT, haematocrit (%); Q, Intercompartmental clearance; SD, standard deviation; V1, Volume of distribution into the central compartment; V2, Volume of distribution into the peripheral compartment; WT, weight.
Figure 3.
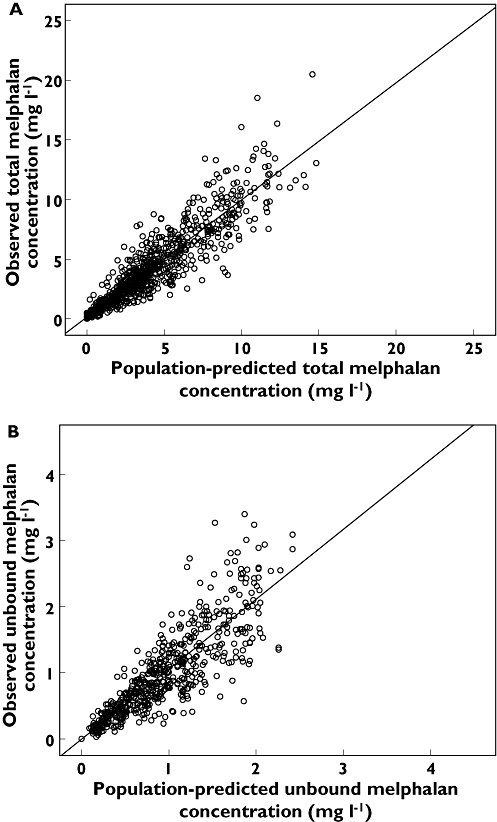
Scatterplot of observed and population-predicted concentrations for A) the Covariate Model for total melphalan and B) the Covariate Model for unbound melphalan
Figure 4.
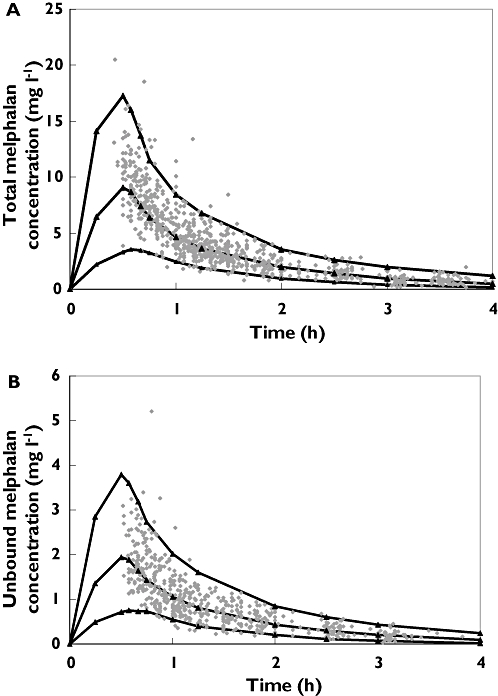
Visual predictive check of A) total and B) unbound melphalan concentration vs. time curves, comparing observed data (solid data points) with the 5th, 50th and 95th percentiles of simulated data (n= 500) generated using the final Covariate Population Pharmacokinetic models (solid lines)
We also compared the population pharmacokinetic results of our final covariate model for clearance that had the structure of CL = CLNR+ CLR and incorporated CLcr with units of ml min−1 70 kg−1 (Table 5) with the results obtained when CLcr was incorporated with units of ml min−1 (Table 6). We found that while the population mean estimates were quite similar, the models for total and unbound melphalan that incorporated CLcr (ml min−1) tended to be less stable, deviating from the mean values obtained from 1000 bootstrap runs by up to 11% in the model for total melphalan and by up to 48% in the model for unbound melphalan.
Table 6.
Population pharmacokinetic parameter estimates for total and unbound melphalan in 100 patients with multiple myeloma using separately developed Covariate Models that incorporated CLcr with units ml min−1
| Total melphalan | Unbound melphalan | |||
|---|---|---|---|---|
| Parameter | Mean | Bootstrap mean (%diff, 95% CI) | Mean | Bootstrap mean (%diff, 95% CI) |
| Fixed effects | ||||
| CLNR (l h−1) | ||||
| θ1 | 17.5 | 17.7 (1.1%, 14.1, 22.6) | 81.1 | 81.3 (0.2%, 64.7, 100) |
| θ2 | 0.402 | 0.358 (−10.9%, −0.073, 0.930) | 0.587 | 0.615 (4.8%, 0.216, 1.070) |
| CLR (l h−1) | 10.4 | 10.3 (−1.0%, 5.6, 13.5) | 49.7 | 49.1 (−1.2%, 49.3, 64.6) |
| V1 (l) | 13.2 | 13.2 (0%, 10.9, 15.8) | 69.3 | 68.5 (−1.2%, 50.6, 78.1) |
| Q (l h−1) | 30.2 | 30.3 (0.3%, 26.4, 34.4) | 144 | 142.8 (−0.8%, 118.0. 169.0) |
| V2 (l) | 14.8 | 14.9 (0.7%, 13.7, 16.2) | 69.7 | 69 (−1.0%, 61.2, 76.9) |
| Interindividual variability | ||||
| ωCL (CV%) | 26.8 | 26.8 (0%, 21.9, 31.5) | 27.8 | 28.5 (2.5%, 21.6, 36.6) |
| ωV1 (CV%) | 59.2 | 62.6 (5.7%, 43.4, 79.8) | 26.4 | 39.1 (48.1%, 12.9, 68.0) |
| ωQ (CV%) | 40.2 | 41.1 (2.2%, 26.6, 55.1) | 45.4 | 46.1 (1.5%, 26.6, 61.6) |
| ωV2 (CV%) | 34.5 | 33.7 (−2.6%, 30.6, 42.9) | 41.0 | 37.2 (−9.3%, 25.3, 49.9) |
| Random residual variability | ||||
| σ1 (SD) | 0.071 | 0.071 (0%, 0.060, 0.082) | 0.135 | 0.131 (−3.0%, 0.104, 0.155) |
| σ2 (SD) | 0.082 | 0.080 (−2.4%, 0.059, 0.106) | 0.029 | 0.029 (0%, 0.004, 0.053) |
| OBV | −868 | −1525 | ||
| Structural models: | ||||
| CL = CLNR+ CLR, where CLNR=θ1× (HCT/34)θ2× (FFM/50)0.75 and CLR=θ3× (CLcr/97), V1 =θ4× (FFM/50), Q =θ5, V2 =θ6 | ||||
95% CI = lower and upper limits of the 95% confidence interval for population pharmacokinetic parameters obtained with 1000 bootstrap runs. %diff = (bootstrap mean – Covariate model mean)/Covariate model mean × 100, OBV = objective function value. CL, clearance; CLcr, estimated creatinine clearance (ml min−1); %CV, coefficient of variation; FFM, fat free mass (kg); HCT, haematocrit (%); Q, Intercompartmental clearance; SD, standard deviation; V1, Volume of distribution into the central compartment; V2, Volume of distribution into the peripheral compartment; WT, weight.
Model-derived pharmacokinetic parameters and other variables
The derived pharmacokinetic parameters for total and unbound melphalan are shown in Table 7. There were no significant differences in the mean values for the elimination and distributional rate constants and half-lives that were derived from the population pharmacokinetic models for total and unbound melphalan using a Wald test. One way anova indicated no significant difference in fu in each of the specifically timed samples collected from each patient (Table 8), suggesting that melphalan protein binding is linear over the range of melphalan concentrations measured in this study.
Table 7.
Model-derived pharmacokinetic parameter and other variables for total and unbound melphalan in 100 patients with multiple myeloma. Data are presented as median (interquartile range)
| Pharmacokinetic parameter/variable | Total melphalan | Unbound melphalan |
|---|---|---|
| CL (l h−1 kg−1) | 0.35 (0.28–0.43) | 1.59 (1.27–2.14) |
| CL (l h−1 m−2) | 14.4 (11.9–17.2) | 65.0 (51.3–88.0) |
| V1 (l kg−1) | 0.18 (0.14–0.21) | 0.79 (0.62–0.99) |
| V1 (l m−2) | 7.3 (5.6–9.0) | 32.2 (26.0–40.7) |
| k10 (h−1) | 1.95 (1.58–2.65) | 2.0 (1.8–2.4) |
| k12 (h−1) | 2.2 (1.8–2.9) | 2.4 (1.9–3.2) |
| k21 (h−1) | 2.0 (1.8–2.4) | 2.1 (2.0–2.3) |
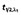 (h) (h) |
0.13 (0.10–0.15) | 0.12 (0.10–0.13) |
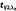 (h) (h) |
0.97 (0.86–1.06) | 0.92 (0.84–1.05) |
| AUC (mg l−1 h) | 12.8 (10.8–15.1) | 2.80 (2.08–3.37) |
| Fraction unbound | 0.21 (0.17–0.27) |
Table 8.
Total melphalan concentrations and fu in each of the specifically-timed plasma samples collected from each patient. Data are mean ± standard deviation (SD)
| Sample number | Time after infusion start (h) | Total melphalan concentration (µg ml−1) | fu |
|---|---|---|---|
| 1 | 0.78 ± 0.27 | 7.85 ± 2.56 | 0.24 ± 0.08 |
| 2 | 0.90 ± 0.27 | 6.10 ± 1.93 | 0.24 ± 0.08 |
| 3 | 1.10 ± 0.27 | 4.62 ± 1.56 | 0.23 ± 0.09 |
| 4 | 1.26 ± 0.27 | 3.88 ± 1.37 | 0.24 ± 0.09 |
| 5 | 1.79 ± 0.29 | 2.45 ± 0.94 | 0.24 ± 0.09 |
| 6 | 2.87 ± 0.38 | 1.16 ± 0.60 | 0.24 ± 0.10 |
| Significance* | NS DF = 5585, F = 0.23 |
Significance tested using one-way anova, Bonferroni test for multiple comparisons. NS, Not significant.
The effects of paraprotein and transferrin concentrations and myeloma type on total and unbound melphalan clearance
Pre-ASCT paraprotein concentration was not significantly associated with total or unbound melphalan clearance using the correlation coefficient of determination (r2= 0.017, r2= 0.001, respectively), and neither was transferrin concentration (r2= 0.003, r2= 0.002, respectively). Patients with IgA myeloma did not have significantly altered pharmacokinetic parameters for total or unbound melphalan compared with patients with IgG myeloma.
Influence of total and unbound melphalan exposure on toxicity post-transplant and disease response
The pharmacodynamics of high dose melphalan in patients with multiple myeloma are summarized in Table 9. Unbound AUC, which ranged from 1.0 to 6.49 mg l−1 h, was significantly higher for patients who had severe (grade 3 or 4) clinical or functional oral mucositis (P < 0.001) or nausea (P < 0.05) and those whose duration of hospital admission was ≥21 days (the 75th percentile, P < 0.001) using the Mann-Whitney test. Total AUC, which ranged from 4.9 to 24.4 mg l−1 h, was significantly higher in patients who had severe clinical oral mucositis (P < 0.05), severe functional oral mucositis (P < 0.01) and in those whose duration of hospital admission was ≥21 days (P < 0.01). Patients who achieved an overall disease response of CR or VGPR had significantly (P < 0.05) higher unbound AUC than the remainder (median 3.2 vs. 2.8 mg l−1 h). The patients who achieved a melphalan-related disease response of CR and VGPR had higher total AUC than the remainder (median 21.3 vs. 13.4 mg l−1 h), but the result was not significant (P= 0.062).
Table 9.
Pharmacodynamics of high dose melphalan in patients with multiple myeloma. Data are median (lower and upper limits of 95% confidence interval)
| Pharmacodynamic endpoint | AUC (mg l−1 h)Median (95%CI†, n) | Unbound AUC (mg l−1 h)Median (95%CI†, n) |
|---|---|---|
| Mucositis oral (clinical) | ||
| Grade 3–4 | 16.9 (8.2, 24.1, n= 13) | 4.4 (2.6, 5.5, n= 13) |
| Grade 0–2 | 13.4 (7.4, 25.4, n= 78) | 2.8 (1.4, 5.3, n= 78) |
| Significance* | P < 0.05 | P < 0.001 |
| Mucositis oral (functional) | ||
| Grade 3–4 | 16.9 (8.2, 24.1, n= 21) | 3.9 (1.7, 5.3, n= 21) |
| Grade 0–2 | 13.4 (7.3, 25.7, n= 72) | 2.8 (1.3, 5.7, n= 72) |
| Significance* | P < 0.01 | P < 0.001 |
| Nausea | ||
| Grade 3 | 14.3 (11.8, 24.6, n= 14) | 3.7 (1.7, 6.8, n= 14) |
| Grade 0–2 | 13.9 (7.4, 25.3, n= 79) | 2.9 (1.4, 5.3, n= 79) |
| Significance* | NS | P= 0.06 |
| Vomiting | ||
| Grade 2–3 | 14.7 (7.1, 25.3, n= 24) | 3.8 (2.0, 6.8, n= 24) |
| Grade 0–1 | 13.9 (7.4, 25.9, n= 67) | 2.7 (1.3, 5.3, n= 67) |
| Significance* | NS | P < 0.05 |
| Diarrhoea | ||
| Grade 3 | 14.4 (8.4, 24.1, n= 19) | 2.5 (1.0, 5.3, n= 19) |
| Grade 0–2 | 14.0 (7.3, 25.7, n= 73) | 3.1 (1.5, 5.7, n= 73) |
| Significance* | NS | NS |
| Days of hospital admission | ||
| ≥21 days (75th percentile) | 16.3 (7.1, 24.6, n= 24) | 3.9 (1.7, 6.8, n= 24) |
| <21 days | 13.3 (7.4, 26.0, n= 67) | 2.9 (1.3, 5.3, n= 67) |
| Significance* | P < 0.01 | P < 0.01 |
| Overall disease response | ||
| Complete, very good partial | 13.8 (7.4, 23.5, n= 23) | 3.2 (1.7, 5.1, n= 23) |
| Partial, minimal, no change | 14.0 (7.7, 25.3, n= 50) | 2.8 (1.1, 6.4, n= 50) |
| Significance* | NS | P < 0.05 |
| Melphalan-related disease response | ||
| Complete, very good partial | 21.3 (12.3, 27.5, n= 7) | 3.8 (1.7, 4.4, n= 7) |
| Partial, minimal, no change | 13.4 (7.4, 24.9, n= 60) | 2.8 (1.2, 6.1, n= 60) |
| Significance* | P= 0.062 | NS |
Mann-Whitney test.
Lower and upper limits of the 95% confidence interval, n= number of observations per group. NS, not significant.
Discussion
This is the largest published study on melphalan pharmacokinetics in patients with multiple myeloma. Our two compartment population pharmacokinetic model generated population mean estimates for total melphalan CL, V1, Q and V2 of 27.8 l h−1, 13.1 l, 31.3 l h−1 and 15.1 l, respectively, and these were similar to the previous adult results (84 patients, mixed diagnoses) of 33.06 l h−1, 18.26 l, 25.8 l h−1 and 15.1 l, respectively [11]. There have been no previous population pharmacokinetic studies on unbound melphalan. Our approach of modelling total and unbound melphalan concentrations separately is simple and straight forward and allows the development of accurate and stable population pharmacokinetic models for both total and unbound melphalan. The fact that there were no significant differences in the mean values for the elimination and distributional rate constants and half-lives provides added confidence in the models, as this is to be expected in a pharmacologically-linked linear system. This study is ongoing and the dataset is incomplete, especially with respect to the pre-transplant protein concentrations, including paraprotein and transferrin. Once all data have been collected, a more complex model that combines both the total and unbound concentration data will allow the determination of fraction unbound as a pharmacokinetic parameter, including population variability and covariate influences, and a more comprehensive assessment of the linearity of melphalan protein binding.
After testing a wide variety of patient characteristics and clinical factors, we found that estimated creatinine clearance, fat free mass and haematocrit were important determinants of melphalan total and unbound clearance, while fat free mass was also an important determinant of total and unbound melphalan volume of distribution into the central compartment. Inclusion of these factors significantly improved the population pharmacokinetic models based on the likelihood ratio test and substantially decreased the inter-individual variability in total and unbound clearance.
As renal excretion is an important elimination pathway for melphalan [7, 8], an effect of creatinine clearance on melphalan total and unbound clearance can be expected. In our study, low creatinine clearance was associated with low clearance of total and unbound melphalan (shown in Figures 1, 2), which would consequently lead to increased exposure. This finding is consistent with previous studies showing increased melphalan toxicity [3, 24] and improved survival with reduced doses [3] in patients who have impaired renal function. According to our model, total and unbound renal clearance of melphalan was approximately 40%, which is consistent with a 24 h urinary excretion of 34 ± 33% [8]. Renal function has previously been identified as an important determinant of melphalan clearance in population pharmacokinetic studies [11, 19]. Our final model incorporated CLcr with units of ml min−1 70 kg−1. We also tested a model that incorporated CLcr with units of ml min−1 (which is more familiar to clinicians), but this model was not as stable when evaluated using bootstrapping. It is possible that normalizing to body weight (or body surface area) improves model stability by having a centring effect.
The increasing prevalence of obesity [25] has drawn attention to the absence of high quality pharmacokinetic data for many chemotherapeutic agents, including melphalan, in this population of patients. Our study population had a broad weight range (42–132 kg) with 37% having body mass index values of greater than 30 mg kg−2 and therefore defined as obese, by the World Health Organization [26]. In our population pharmacokinetic modelling we found that of all the alternative body size descriptors, fat free mass best described both melphalan total and unbound clearance. Fat free mass has been proposed as the best size descriptor for use in pharmacokinetic studies and dose adjustments in the obese [27, 28]. Body size has previously been found to be an important predictor of total melphalan clearance in adults [11], but total body weight was used in that model.
The disease of multiple myeloma can be associated with anaemia, reduced haemoglobin and, consequently, low haematocrit. In this population of patients haematocrit values ranged from 23% to 44% and 35% of patients had haematocrit values less than 33%. Melphalan (37%) has been recovered from the red cell fraction of human whole blood [29], while in rats it has been demonstrated that binding (covalent) is primarily to proteins in red cell membranes [30]. Low haematocrit means reduced red blood cell count and, consequently, lower binding of melphalan to red blood cells and a higher non-red blood cell fraction. This could lead to higher plasma and ultrafiltrate concentrations and lower clearance values, as have been observed in this study (Figures 1, 2). Haematocrit has not been previously identified as a predictor of melphalan pharmacokinetic parameters in any other studies.
We examined whether the concentrations of specific proteins contributed to the large variability in total or unbound melphalan clearance but did not detect any significant associations with the pre-transplant levels of paraprotein, total protein, albumin or transferrin. A highly variable unbound melphalan fraction that was not associated with total protein or albumin concentrations has been previously observed [29], even though in vitro melphalan binds to albumin (60%) and α1-glycoprotein (20%) [8]. In multiple myeloma paraprotein concentrations are used to monitor response to therapy. Pre-melphalan paraprotein concentrations can vary widely, depending on response to previous treatment. Our finding (in a large population) that paraprotein concentrations do not influence total or unbound melphalan pharmacokinetics confirms results from previous smaller studies [8, 29].
We investigated the association between total and unbound melphalan exposure and toxicity post-melphalan and we found that patients who had severe (grade 3) gastrointestinal toxicity or a long hospital admission had significantly higher exposure to total and unbound melphalan. Unbound AUC was a more sensitive predictor of toxicity than total AUC since a greater level of significance was demonstrated for a greater number of toxicity endpoints. This is to be expected because use of unbound AUC eliminates the (perhaps substantial) population variability in protein binding. High total AUC has previously been observed to be associated with the occurrence of grade 1 or 2 gastrointestinal toxicity following 100 mg m−2 melphalan in children [31] and the development of mucositis in adults [11].
We also investigated the association between total and unbound exposure to melphalan and disease response. We observed a weak (P < 0.05) association between unbound melphalan AUC and overall disease response. A significant association between total melphalan AUC and melphalan- related disease response could not be demonstrated (P= 0.062), but this may reflect the fact that the study was insufficiently powered (at this point) to demonstrate an effect due to the small numbers of patients (n= 7) in the group who had achieved a CR or VGPR to melphalan. Additionally, this simple analysis does not take into account other factors, such as post ASCT therapy, that may also impact on disease response. These preliminary results are very promising and further longitudinal response data may enable us to characterize better the association between total and unbound melphalan exposure and efficacy in multiple myeloma.
In conclusion, population pharmacokinetic modelling of total and unbound melphalan shows that estimated creatinine clearance, fat free mass and haematocrit are important determinants of total and unbound melphalan clearance. Preliminary pharmacodynamic analyses demonstrate that higher drug exposure is associated with both increased toxicity and efficacy, with unbound exposure being a more sensitive predictor of toxicity and efficacy than total exposure. These results provide the promise of a melphalan dosing algorithm in myeloma that maximizes therapeutic efficacy and reduces toxicity.
Competing interests
A.J. McL. has received research funding from GlaxoSmithKline to support a postgraduate research scholarship for a student under his supervision.
Christa E. Nath is supported by the Leukaemia Research Support Fund of The Children's Hospital Westmead and by NHMRC Project Grant 396702. Lihua Zeng is also supported by NH MRC Project Grant 396702. We would like to thank the patients for taking part in the study, the nursing staff in the haematology units for their care of the patients, including taking blood samples for measurement of melphalan concentrations and the data managers for recording the clinical data.
REFERENCES
- 1.Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, Casassus P, Maisonneuve H, Facon T, Ifrah N, Payen C, Bataille R. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335:91–7. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 2.Lenhoff S, Hjorth M, Holmberg E, Turesson I, Weston J, Nielsen JL, Wisloff F, Brinch L, Carlson K, Carlsson M, Dahl IM, Gimsing P, Hippe E, Johnsen H, Lamvik J, Lofvenberg E, Nesthus I, Rodjer S. Impact on survival of high dose therapy with autologous stem cell support in patients younger than 60 years with newly diagnosed multiple myeloma: a population-based study. Nordic myeloma Study Group. Blood. 2000;95:7–11. [PubMed] [Google Scholar]
- 3.Cornwell GG, III, Pajak TF, McIntyre OR, Kochwa S, Dosik H. Influence of renal failure on myelosuppressive effects of melphalan: cancer and leukemia group B experience. Cancer Treat Rep. 1982;66:475–81. [PubMed] [Google Scholar]
- 4.Moreau P, Kergueris M-F, Milpied N, Le Tortorec SL, Mahe B, Bulabois C-E, Rapp M-J, Larousse C, Bataille R, Harousseau J-L. A pilot study of 220 mg/m2 melphalan followed by autologous stem cell transplantation in patients with advanced haematological malignancies: pharmacokinetics and toxicity. Br J Haematol. 1996;95:527–30. doi: 10.1046/j.1365-2141.1996.d01-1932.x. [DOI] [PubMed] [Google Scholar]
- 5.Sarosy G, Leyland-Jones B, Soochan P, Cheson BD. The systemic administration of intravenous melphalan. J Clin Oncol. 1988;6:1768–82. doi: 10.1200/JCO.1988.6.11.1768. [DOI] [PubMed] [Google Scholar]
- 6.Gertz MA, Lacy MQ, Dispenzieri A, Ansell A, Elliot SM, Gastineau DA, Inwards DJ, Micallef INM, Porrata LF, Tefferi A, Litzow MR. Risk-adjusted manipulation of melphalan dose before stem cell transplantation in patients with amyloidosis is associated with a lower response rate. Bone Marrow Transplant. 2004;34:1025–31. doi: 10.1038/sj.bmt.1704691. [DOI] [PubMed] [Google Scholar]
- 7.Reece PA, Hill HS, Green RM, Morris RG, Dale BM, Kotasek D, Sage RE. Renal clearance and protein binding of melphalan in patients with cancer. Cancer Chemother Pharmacol. 1988;22:348–52. doi: 10.1007/BF00254244. [DOI] [PubMed] [Google Scholar]
- 8.Gera S, Musch E, Osterheld HK, Loos U. Relevance of the hydrolysis and protein binding of melphalan to the treatment of multiple myeloma. Cancer Chemother Pharmacol. 1989;23:76–80. doi: 10.1007/BF00273521. [DOI] [PubMed] [Google Scholar]
- 9.Chang SY, Alberts DS, Farquhar D, Melnick LR, Walson PD, Salmon SE. Hydrolysis and protein binding of melphalan. J Pharm Sci. 1978;67:682–4. doi: 10.1002/jps.2600670530. [DOI] [PubMed] [Google Scholar]
- 10.Badros A, Barlogie B, Siegel E, Roberts J, Langmaid C, Zangari M, Desikan R, Shaver MJ, Fassas A, McConnell S, Muwalla F, Barri Y, Anaissie E, Munshi N, Tricot G. Results of autologous stem cell transplant in multiple myeloma patients with renal failure. Br J Haematol. 2001;114:822–9. doi: 10.1046/j.1365-2141.2001.03033.x. [DOI] [PubMed] [Google Scholar]
- 11.Kühne A, Sezer O, Heider U, Meineke I, Muhlke S, Niere W, Overbeck T, Hohloch K, Trümper L, Brockmöller J, Kaiser R. Population pharmacokinetics of melphalan and glutathione S-transferase polymorphisms in relation to side effects. Clin Pharmacol Ther. 2008;83:749–57. doi: 10.1038/sj.clpt.6100336. [DOI] [PubMed] [Google Scholar]
- 12.Junge W, Wilke B, Halabi A, Klein G. Determination of reference intervals for serum creatinine, creatinine excretion and creatinine clearance with an enzymatic and a modified Jaffe method. Clin Chim Acta. 2004;344:137–48. doi: 10.1016/j.cccn.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 14.Jones GRD, Imam SK. Validation of the revised MDRD formula and the original Cockroft and Gault formula for estimation of the glomerular filtration rate using Australian data. Pathology. 2009;41:379–82. doi: 10.1080/00313020902884980. [DOI] [PubMed] [Google Scholar]
- 15.Keys A, Fidanza F, Karvonen M, Kimura N, Taylor H. Indices of relative weight and obesity. J Chronic Dis. 1972;25:329–43. doi: 10.1016/0021-9681(72)90027-6. [DOI] [PubMed] [Google Scholar]
- 16.Mosteller RD. Simplified calculation of body surface area. N Engl J Med. 1987;317:1098. doi: 10.1056/NEJM198710223171717. [DOI] [PubMed] [Google Scholar]
- 17.Janmahasatian S, Duffull S, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean body weight. Clin Pharmacokinet. 2005;44:1051–65. doi: 10.2165/00003088-200544100-00004. [DOI] [PubMed] [Google Scholar]
- 18.Nath CE, Zeng L, Eslick A, Trotman J, Earl J. An isocratic UV HPLC assay for analysis of total and free melphalan concentrations in human plasma. Acta Chromatogr. 2008;20:383–98. [Google Scholar]
- 19.Nath CE, Shaw PJ, Montgomery K, Earl JW. Population pharmacokinetics of melphalan in paediatric blood or marrow transplant recipients. Br J Clin Pharmacol. 2007;64:151–64. doi: 10.1111/j.1365-2125.2007.02862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–32. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 21.Gastonguay MR, El-Tahtawy A. Effect of NONMEM minimization status and number of replicates on bootstrap parameter distributions for population pharmacokinetic models: a case study. Clin Pharmacol Ther. 2005;77:2. doi: 10.1016/j.clpt.2004.11.010. [Google Scholar]
- 22.National Cancer Institute, USA. Common terminology Criteria for Adverse Events v 3.0 (CTCAE) Publish date: 12 December 2003. Available at http://ctep.cancer.gov (last accessed 31 March 2003.
- 23.Bladē J, Samson D, Reece D, Apperley J, Björkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S, Vesole D. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high dose therapy and haemopoietic stem cell transplantation. Br J Haematol. 1998;102:1115–23. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 24.Carlson K. Melphalan 200 mg/m2 with blood stem cell support as first line myeloma therapy: impact of glomerular filtration rate on engraftment, transplantation-related toxicity and survival. Bone Marrow Transplant. 2005;35:985–90. doi: 10.1038/sj.bmt.1704948. [DOI] [PubMed] [Google Scholar]
- 25.Bjorntorp P. Obesity. Lancet. 1997;350:423–6. doi: 10.1016/S0140-6736(97)04503-0. [DOI] [PubMed] [Google Scholar]
- 26.World Health Organization. Report of a WHO Consultant on Obesity: Preventing and managing the global epidemic. Geneva: WHO; 1998. [PubMed] [Google Scholar]
- 27.Green B, Duffull SB. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br J Clin Pharmacol. 2004;58:119–33. doi: 10.1111/j.1365-2125.2004.02157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han PY, Duffull SB, Kirkpatrick CMJ, Green B. Dosing in obesity: a simple solution to a big problem. Clin Pharmacol Ther. 2007;82:505–8. doi: 10.1038/sj.clpt.6100381. [DOI] [PubMed] [Google Scholar]
- 29.Grieg NH, Sweeney DJ, Rapoport SI. Melphalan concentration dependent plasma protein binding in healthy humans and rats. Eur J Clin Pharmacol. 1987;32:179–85. doi: 10.1007/BF00542192. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed AE, Hsu TF, El-Azhary RA, Moawad H, Costanzi J. Macromolecular interactions of 14C-ring melphalan in blood. Biochem Pharmacol. 1982;31:1615–9. doi: 10.1016/0006-2952(82)90389-6. [DOI] [PubMed] [Google Scholar]
- 31.Vassal G, Tranchand B, Valteau-Couanet D, Mahē C, Couanet D, Schoeppfer C, Grill J, Kalifa C, Hill C, Ardiet C, Hartmann O. Pharmacodynamics of tandem high-dose melphalan with peripheral blood stem cell transplantation in children with neuroblastoma and medulloblastoma. Bone Marrow Transplant. 2001;27:471–7. doi: 10.1038/sj.bmt.1702806. [DOI] [PubMed] [Google Scholar]