

Abstract
AIM
Gabapentin enacarbil, a transported prodrug of gabapentin, provides sustained, dose-proportional exposure to gabapentin. Unlike gabapentin, the prodrug is absorbed throughout the intestinal tract by high-capacity nutrient transporters, including mono-carboxylate transporter-1 (MCT-1). Once absorbed, gabapentin enacarbil is rapidly hydrolyzed to gabapentin, which is subsequently excreted by renal elimination via organic cation transporters (OCT2). To examine the potential for drug–drug interactions at these two transporters, the pharmacokinetics of gabapentin enacarbil were evaluated in healthy adults after administration alone or in combination with either naproxen (an MCT-1 substrate) or cimetidine (an OCT2 substrate).
METHODS
Subjects (n= 12 in each study) received doses of study drug until steady state was achieved; 1200 mg gabapentin enacarbil each day, followed by either naproxen (500 mg twice daily) or cimetidine (400 mg four times daily) followed by the combination.
RESULTS
When gabapentin enacarbil was co-administered with naproxen, gabapentin Css,max increased by, on average, 8% and AUC by, on average, 13%. When gabapentin enacarbil was co-administered with cimetidine, gabapentin AUCss increased by 24% and renal clearance of gabapentin decreased. Co-administration with gabapentin enacarbil did not affect naproxen or cimetidine exposure. Gabapentin enacarbil was generally well tolerated.
CONCLUSIONS
No gabapentin enacarbil dose adjustment is needed with co-administration of naproxen or cimetidine.
Keywords: cimetidine, drug–drug interactions, gabapentin enacarbil, naproxen, XP13512/GSK1838262
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Gabapentin enacarbil is a transported prodrug of gabapentin that provides sustained, dose-proportional exposure to gabapentin by taking advantage of high-capacity transport pathways expressed throughout the intestinal tract. This prodrug has shown efficacy in multiple clinical trials for the treatment of moderate-to-severe primary restless legs syndrome and could potentially represent the first non-dopaminergic treatment for this important disease.
WHAT THIS STUDY ADDS
Unlike gabapentin, gabapentin enacarbil is actively absorbed from the intestine by multiple pathways, including the monocarboxylate transporter type-1 transporter (MCT-1). Although drug interactions of gabapentin have been reported in the literature, the distinctly different absorption pathway of gabapentin enacarbil requires a separate evaluation of the potential for interaction with other substrates of MCT-1. To achieve this, the pharmacokinetics of gabapentin enacarbil were examined in healthy adults when administered alone or in combination with naproxen, a known MCT-1 substrate.
After absorption, gabapentin enacarbil is completely hydrolyzed to gabapentin, and the released gabapentin is excreted by renal elimination. Gabapentin is a substrate for the organic cation transporter type-2 (OCT2) present in the kidney. To examine the potential for an elimination-site drug interaction resulting from administration of the prodrug, the pharmacokinetics of gabapentin enacarbil were examined in healthy adults when administered alone or in combination with cimetidine, a known substrate of OCT2.
Introduction
Gabapentin enacarbil ([1-({[({1-[(2-methylpropanoyl)oxy]ethyl}oxy)carbonyl] amino}methyl) cyclohexyl] acetic acid) is a transported prodrug of gabapentin that overcomes the pharmacokinetic limitations of gabapentin [1, 2]. Gabapentin enacarbil is stable in gastrointestinal contents and is actively absorbed after oral dosing by high-capacity nutrient transporters present throughout the intestinal tract. Administration of gabapentin enacarbil achieves efficient oral absorption and conversion to gabapentin, and provides dose proportional systemic gabapentin exposure up to 6000 mg [3]. The prodrug is formulated as an extended release tablet that provides sustained exposure to gabapentin and allows for a decreased dosing frequency compared with oral gabapentin [3]. Gabapentin enacarbil is currently under investigation for the treatment of moderate-to-severe primary restless legs syndrome (RLS), prophylaxis of migraine headache, and the treatment of neuropathic pain, and has demonstrated efficacy in the treatment of subjects with moderate-to-severe primary RLS [4].
In vitro studies have shown that gabapentin enacarbil is a substrate for multiple high-capacity nutrient transport pathways, including the monocarboxylate transporter type 1 (MCT-1) and the sodium-dependent multivitamin transporter (SMVT), which are abundant throughout the intestinal tract [1]. Following absorption by these pathways, the prodrug is rapidly converted to gabapentin by non-specific carboxylesterases primarily in enterocytes and to a lesser extent in the liver. Previous studies of oral gabapentin have demonstrated a pharmacokinetic drug interaction with naproxen that was not considered clinically significant; specifically, co-administration of naproxen led to a 12–15% increase in gabapentin exposure [5].
Although drug interactions with gabapentin have been reported in the literature, the distinctly different absorption pathway of gabapentin enacarbil requires a separate evaluation of the potential for interaction with other substrates of MCT-1. To achieve this, the pharmacokinetics of gabapentin enacarbil were examined in healthy adults when administered alone or in combination with naproxen, an MCT-1 substrate [6, 7]. MCT-1 appears to be the primary monocarboxylate transporter localized on the apical surface of cells lining the intestinal tract and is abundant in both the small and the large intestine [8]. This transporter is responsible for the absorption of various short-chain fatty acids and transports butyrate derived from bacterial fermentation. Several drugs (e.g. ibuprofen, ketoprofen, naproxen, pravastatin) have been shown to be substrates for MCT-1 [6, 9, 10] and published data support the use of naproxen as a suitable control substrate for MCT-1 [6, 7].
Gabapentin enacarbil is efficiently converted to gabapentin during absorption, prior to reaching the systemic circulation [3]. Gabapentin is primarily excreted renally, as unchanged drug. The renal excretion of gabapentin involves a component of active secretion via an organic cation transporter (OCT2) in the kidney [11]. Other substrates for OCT2 include histamine, and guanidine derivatives such as creatinine and cimetidine [12]. Cimetidine is an established histamine H2-receptor antagonist treatment that is widely used to reduce stomach acid secretion. It has been demonstrated that there was a small but insignificant decrease in oral clearance of gabapentin (14%) with a corresponding decrease in creatinine clearance when gabapentin was dosed with cimetidine. Although cimetidine altered the renal excretion of gabapentin and creatinine, the small decrease in excretion was not considered clinically relevant [5].
Because gabapentin enacarbil is a substrate for multiple high-capacity nutrient transport pathways (MCT-1), we evaluated the potential for a pharmacokinetic drug–drug interaction of gabapentin enacarbil with naproxen when dosed concomitantly at therapeutic doses in healthy subjects. In addition, as gabapentin released after the oral absorption of gabapentin enacarbil is a substrate for the OCT2 transporter, we evaluated the potential for a drug interaction with cimetidine when dosed concomitantly at therapeutic doses in healthy subjects.
Methods
Study design
The drug–drug interaction studies of gabapentin enacarbil and naproxen, and gabapentin enacarbil and cimetidine (XenoPort, Inc., Santa Clara, CA, USA) were open-label, three-period studies. They were conducted in accordance with the Declaration of Helsinki (1964) and approved by an Institutional Review Board (Aspire Independent Board, LLC, La Mesa, CA, USA, 91941 for gabapentin enacarbil and naproxen; and IntegReview, LTD Austin, Texas 78704 for gabapentin enacarbil and cimetidine). All subjects provided written informed consent prior to any study participation. The studies were conducted at single sites in the United States in April and May 2007 (gabapentin enacarbil and naproxen) and June and July 2007 (gabapentin enacarbil and cimetidine).
Subjects
In both studies, healthy adults 18–55 years of age with creatinine clearance values of ≥80 ml min−1 and body mass index of 18–30 kg m−2 were eligible for inclusion. Women of potential childbearing age were required to be sexually inactive for 14 days prior to screening and for the duration of the study, or to use acceptable birth control methods. Subjects with a history or presence of significant cardiovascular, pulmonary, hepatic, renal, haematologic, gastrointestinal, endocrine, immunological, dermatological, neurological or psychiatric disease, a history of seizures other than febrile seizures as a child, a history of alcoholism or drug abuse within the past 2 years or any report of acute illness or febrile event that had not resolved within 72 h of dosing were excluded from the study. Subjects were also excluded if they had an abnormal diet, given a recent blood or plasma donation, taken any medication other than study medication in the 14 days prior to dosing, used nicotine-containing products within 6 months prior to the study, recently consumed alcohol, recently engaged in strenuous exercise, had previous exposure to gabapentin enacarbil or participated in another clinical trial within 30 days. Subjects with hypersensitivity to gabapentin or related compounds, aspartame, naproxen or non-steroidal anti-inflammatory drugs (NSAIDS) or cimetidine; or who had elevated liver function tests, a creatine kinase value of greater than the upper limit of normal (234 IU l−1 for females and 397 IU l−1 for males), or haemoglobin <12.0 g dl−1 were also excluded.
Dosing regimen
In each study, subjects were dosed until they reached steady state; this equated to 5 days for gabapentin enacarbil and naproxen and 4 days for gabapentin enacarbil and cimetidine.
For the gabapentin enacarbil and naproxen study, in period 1, subjects received gabapentin enacarbil 1200 mg (two 600 mg extended-release tablets) once daily in the morning (Figure 1A). In period 2, subjects received naproxen 500 mg (two 250 mg Naprosyn® tablets; Roche, Basel, Switzerland) twice daily in the morning and evening. In period 3, subjects received gabapentin enacarbil 1200 mg once daily in the morning and naproxen 500 mg twice daily in the morning and evening.
Figure 1.
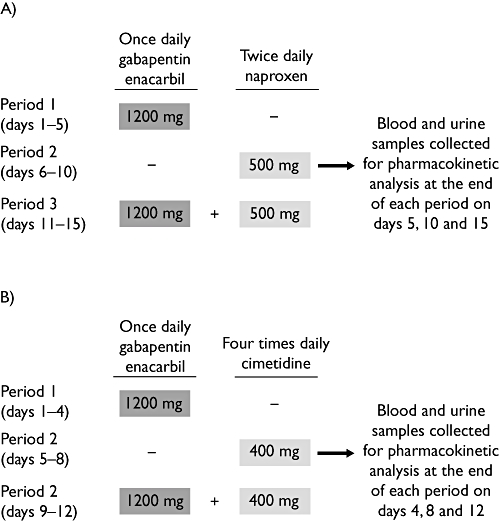
Design of (A) gabapentin enacarbil and naproxen study and (B) gabapentin enacarbil and cimetidine study
For the gabapentin enacarbil and cimetidine study, in period 1, subjects received gabapentin enacarbil 1200 mg (two 600 mg extended-release tablets) once daily in the morning (Figure 1B). In period 2, subjects received cimetidine (Tagamet® tablets; GlaxoSmithKline, Brentford, UK) 400 mg four times daily. In period 3, subjects received gabapentin enacarbil 1200 mg once daily in the morning and cimetidine 400 mg four times daily.
Subjects were given a standard meal (30% calories from fat) 30 min prior to dosing in both studies, except for the last daily dose of cimetidine, which was administered without a meal.
Sample collections
Pharmacokinetic samples were taken on the last day of each period of the gabapentin enacarbil and naproxen study (Figure 1A). On each of these days, blood samples (6 ml) were collected in tubes containing ethylene diamine tetra-acetic acid (EDTA) at pre-dose (0), 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 18 and 24 h after the morning dose. Two 1 ml aliquots were immediately quenched with methanol. The remaining blood sample was separated by centrifugation at 4°C within 15 min of collection and two 0.7 ml aliquots of plasma were retained. Blood and plasma samples were frozen at −80°C until analysis. In addition, a complete urine collection was obtained at 0–4 h, 4–8 h, 8–12 h and 12–24 h intervals after the morning dose on days 5, 10 and 15. Two 1.5 ml aliquots were stored at −20°C until transported for analysis.
Blood samples for pharmacokinetic analysis were taken on the last day of dosing in each period of the gabapentin enacarbil and cimetidine study (Figure 1B), prior to morning dosing and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 13, 14, 15, 16, 18 and 24 h after the morning dose. Venous blood samples (6 ml) were collected by venepuncture or cannulation into EDTA-containing Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ, USA) and processed immediately. The tubes were inverted several times and the blood separated by centrifugation at 4°C for 15 min. The resultant plasma samples were aliquoted and stored at −20°C or below until analysis. In addition, complete urine collections were obtained at 0–3 h, 3–6 h, 6–12 h and 12–24 h intervals after the morning dose on days 4, 8 and 12. Aliquots were stored at −20°C until analysis.
Sample analysis
Samples were analysed at CEDRA Corporation (Austin, TX, USA) for the determination of cimetidine or naproxen concentrations or at XenoPort Inc. (Santa Clara, CA, USA) for the determination of gabapentin and gabapentin enacarbil concentrations. All samples were analysed by validated high-pressure liquid chromatography coupled with a tandem mass spectrometer (LC-MS/MS) method.
For the analysis of gabapentin and gabapentin enacarbil, quenched blood, plasma or urine samples were injected on a Phenomenex Hydro-RP column (4 µm; 50 × 4.6 mm) (Phenomenex, Torrance, CA, USA) operated at 30°C. The mobile phases were (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. The gradient was set at time 0 min, 95%A; at 0.5 min, 95%A; at 1.8 min, 5%A; at 3 min, 5%A; at 3.5 min, 2%A; at 4.1 min, 95%A until 6 min. The flow rate was 1200 µl min−1 and the detection was by Sciex API 2000 (Applied Biosystems, Foster City, CA, USA) using positive ion multiple-reaction monitoring (MRM) mode with ion transitions (m/z) of 172.1/137.0 for gabapentin, 200/154 for L-4-chlorophenylalanine (internal standard for gabapentin), 330.1/197.9 for gabapentin enacarbil, and 403.4/172.2 for Leu-gabapentin phenylacetamide (internal standard for gabapentin enacarbil). The bioanalytical method was validated for gabapentin enacarbil over the concentration range 10–2500 ng ml−1 with quality control (QC) samples at 30, 200 and 1900 ng ml−1. The method for gabapentin in blood was validated over the concentration range 50–12 500 ng ml−1 with the QC samples at 150, 1000 and 9500 ng ml−1. The method for gabapentin in plasma was validated over the concentration range 80–10 000 ng ml−1 with the QC samples at 240, 1600 and 7500 ng ml−1. The method for gabapentin in urine was validated over the concentration range 50–12 500 ng ml−1 with the QC samples at 150, 1000 and 9400 ng ml−1.
Precision (% coefficient of variation) and accuracy (deviation from theoretical values) of the blood method were ≤6.8% and ≤101%, respectively, for gabapentin enacarbil and ≤10% and ≤102% for gabapentin, respectively. Precision and accuracy were ≤5.8% and ≤105%, respectively, for analysis of gabapentin in plasma and in urine.
For the analysis of naproxen in plasma, samples were extracted with an organic solvent (liquid–liquid extraction). Following centrifugation, the supernatant was transferred and evaporated. For the analysis of naproxen in urine, samples were diluted and extracted by solid-phase extraction. An aliquot of the reconstituted plasma or urine extract was injected onto a Sciex API 4000 LC-MS/MS equipped with an HPLC column. The MRM transitions (m/z) were 229/185 for naproxen and 232/188 for naproxen-D3 (internal standard). The analysis was validated over the concentration range 0.5–50 µg ml−1 for human plasma and urine with QC samples at 1.50, 20 and 40 µg ml−1. Precision and accuracy of the method were ≤3.6% and ≤105%, respectively, for plasma and ≤5.2% and ≤102%, respectively, for urine.
For the analysis of cimetidine, plasma or urine samples were extracted by solid-phase extraction. An aliquot of the reconstituted plasma or urine extract was injected onto a Sciex API 4000 LC-MS/MS equipped with an HPLC column. The MRM transitions (m/z) were 253/159 for cimetidine and 256/162 for cimetidine-D3 (internal standard). The bioanalysis method was validated over the concentration range 5–1000 ng ml−1 for human plasma and urine with QC samples at 15, 200 and 800 ng ml−1. Precision and accuracy of the method were ≤4.3% and ≤99%, respectively, for plasma, and ≤15% and ≤99%, respectively, for urine.
Pharmacokinetic assessments
For the gabapentin enacarbil and naproxen study, concentration data for gabapentin and gabapentin enacarbil in blood and urine, and for naproxen in plasma and urine were analysed. For the gabapentin enacarbil and cimetidine study, concentration data for gabapentin and cimetidine in plasma and urine were analysed. All pharmacokinetic data were analysed using non-compartmental methods using WinNonlin software (Pharsight Corporation, CA, USA). All concentration values below the limit of quantification were treated as zero for the pharmacokinetic analysis and the statistical calculation. Actual time points were used for the calculation of pharmacokinetic parameters. All concentration data and pharmacokinetic parameters were plotted using SigmaPlot (Systat Software Inc., CA, USA).
The maximum concentration at steady state (Css,max) and time to Css,max (tmax) were obtained by observation. The apparent elimination half-life (t1/2) was obtained by linear regression of three or more log-transformed data points in the terminal phase. The area under the concentration vs. time curve at steady state (AUCss) was obtained by the linear trapezoidal method using concentration data over the dosing interval (τ), which was 24 h for gabapentin and gabapentin enacarbil, 12 h for naproxen, and 6 h for cimetidine. Clearance at steady state (CLss/F) was calculated from gabapentin dose divided by AUCss. As gabapentin is excreted exclusively in urine without significant metabolism, the urinary recovery of gabapentin after oral dosing may be considered equivalent to the oral bioavailability.
Tolerability assessments
Tolerability was assessed using treatment-emergent adverse events (TEAEs), clinical laboratory evaluations (haematology, serum chemistry and urinalysis), physical examination, vital signs and electrocardiograms. Electrocardiograms and clinical laboratory evaluations were performed at screening and follow-up. Vital signs were measured at screening, follow-up and on days 1, 5, 10 and 15 of the gabapentin enacarbil and naproxen study and on days 1, 4, 5, 8, 9 and 12 of the gabapentin enacarbil and cimetidine study.
Statistical analysis
A sample size of 12 healthy male and female subjects, typical for studies of this nature, was chosen to estimate the effect of concomitant drug administration, to an adequate precision, on the pharmacokinetics of gabapentin enacarbil and naproxen and cimetidine. Mixed-effects analysis of variance (anova) was conducted to test for differences in AUCss and Css,max. Differences in gabapentin exposure between gabapentin enacarbil co-administered with naproxen or cimetidine and gabapentin enacarbil dosed alone were assessed. Differences in naproxen or cimetidine exposure between gabapentin enacarbil co-administered with naproxen or cimetidine and naproxen or cimetidine dosed alone were also evaluated. A pair-wise comparison was performed using log-transformed data with the subject variable included in the anova model. The resulting 95% confidence intervals for the estimated treatment ratios of AUCss and Css,max were compared with limits associated with bioequivalence, 80% and 125%.
Results
Subjects
Twelve healthy subjects (eight men and four women) participated in the gabapentin enacarbil and naproxen study. Mean age was 31 years (range 18–53 years), mean weight was 72 kg (range 58–100 kg) and mean body mass index was 24.2 kg m−2 (range 18.8–28.9 kg m−2). Ten subjects were Caucasian and two were African American. Ten subjects completed the study, one male subject discontinued treatment because of an adverse event and another male subject was withdrawn by the investigator.
Eleven men and one woman were enrolled in the gabapentin enacarbil and cimetidine study. Ten subjects were Caucasian and two were African American; mean age was 24.6 years (range 20–43 years). The mean weight of the subjects was 74.7 kg (range, 55.0–86.5 kg), and the mean body mass index was 24.2 kg m−2 (range 20.3–27.6 kg m−2). All subjects completed the study.
Pharmacokinetics following co-administration of gabapentin enacarbil and naproxen
Gabapentin
Following co-administration of gabapentin enacarbil and naproxen, no clinically relevant change in gabapentin exposure was observed compared with gabapentin enacarbil dosed alone (Figure 2). After once daily oral dosing of 1200 mg gabapentin enacarbil, the mean Css,max of gabapentin in blood was 6.10 µg ml−1 and the mean tmax was 5.20 h. When gabapentin enacarbil was co-administered with naproxen the Css,max of gabapentin in blood was 6.52 µg ml−1 and the mean tmax was 5.80 h (Table 1). Statistical analysis showed that gabapentin exposure increased slightly when gabapentin enacarbil was co-administered with naproxen. The Css,max of gabapentin increased by, on average, 8%. The estimated geometric mean ratio of gabapentin enacarbil and naproxen to gabapentin enacarbil (1.08) was close to 1 (95% confidence interval 0.98, 1.18). Similarly, AUCss increased by, on average, 13%; the estimated ratio was 1.13 (95% confidence interval 1.07, 1.20). The mean bioavailability of gabapentin enacarbil as gabapentin was 72% with gabapentin enacarbil alone vs. 76% when co-administered with naproxen (Table 1).
Figure 2.
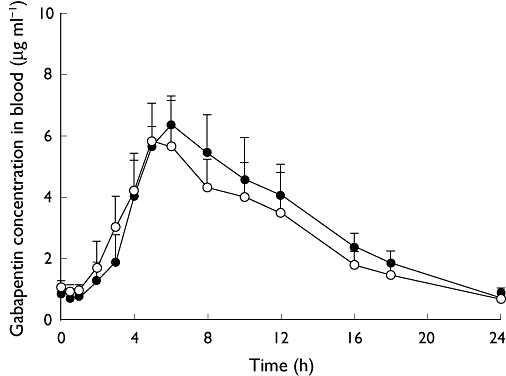
Mean (SD) concentrations of gabapentin in blood at steady state after dosing of gabapentin enacarbil alone and concomitantly with naproxen. n= 10 subjects per treatment. One subject (subject 007) was excluded from mean and SD calculations due to early termination during period 3. SD, standard deviation. Gabapentin enacarbil (–○–); Gabapentin enacarbil + naproxen (–•–)
Table 1.
Mean (SD) pharmacokinetic parameters for gabapentin at steady state after dosing of gabapentin enacarbil alone and concomitantly with naproxen
| Css,max | Css,min | tmax | t1/2 | AUCss | CLss/F | Vd/F | CLr | F | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | n | (µg ml−1) | (µg ml−1) | (h) | (h) | (µg ml−1 h) | (l h−1) | (l) | (l h−1) | (%) |
| 1200 mg gabapentin enacarbil once daily | 10 | 6.10 | 0.667 | 5.20 | 5.64 | 63.8 | 10.1 | 83.9 | 7.09 | 72.0 |
| (1.29) | (0.133) | (1.14) | (1.08) | (11.7) | (1.8) | (28.8) | (0.94) | (13.2) | ||
| 1200 mg gabapentin enacarbil once daily with 500 mg naproxen twice daily | 10 | 6.52 | 0.707 | 5.80 | 5.73 | 71.7 | 8.87 | 74.5 | 6.71 | 76.1 |
| (0.92) | (0.274) | (1.03) | (1.02) | (10.1) | (1.27) | (22.1) | (0.92) | (8.9) |
AUCss, area under the concentration–time curve at steady state; CLr, renal clearance; CLss/F, apparent clearance after oral dosing at steady state; Css,max, maximum concentration at steady state; Css,min, minimum concentration at steady state; F, percent of gabapentin dose recovered in urine in 24 h post-dose; SD, standard deviation; tmax, time to Css,max; t1/2, half-life; Vd/F, apparent volume of distribution.
Gabapentin enacarbil
Following oral administration of gabapentin enacarbil extended-release tablets, gabapentin enacarbil was rapidly absorbed and converted to gabapentin (Table 1). Exposure to gabapentin enacarbil was low and variable. There was no effect on gabapentin enacarbil exposure when co-administered with naproxen. After once daily oral dosing of 1200 mg gabapentin enacarbil alone, the mean Css,max of gabapentin enacarbil in blood was 0.045 µg ml−1 and the mean tmax was 4.30 h. Similarly, when gabapentin enacarbil was co-administered with naproxen, the mean Css,max of gabapentin enacarbil in blood was 0.039 µg ml−1 and the mean tmax was 4.31 h.
Naproxen
After twice daily dosing of 500 mg naproxen, the mean Cmax of naproxen in plasma was 99.2 µg ml−1 and the mean tmax was 3.92 h. When gabapentin enacarbil was co-administered with naproxen the mean Cmax of naproxen in plasma was 97.8 µg ml−1 and the mean tmax was 4.81 h (Table 2). Following co-administration of gabapentin enacarbil and naproxen, there was no clinically relevant change in naproxen exposure, compared with naproxen dosed alone (Table 2). Statistical analysis showed that for both Css,max and AUCss, the estimated means for the ratio of gabapentin enacarbil and naproxen to naproxen alone were close to the value of 1 (0.99 and 0.98, respectively). In addition, the associated 95% confidence intervals for both Css,max and AUCss were 0.94, 1.04 and 0.93, 1.04, respectively.
Table 2.
Mean (SD) pharmacokinetic parameters for naproxen at steady state after dosing of naproxen alone and concomitantly with gabapentin enacarbil
| Css,max | Css,min | tmax | t1/2 | AUCss | CLss/F | Vd/F | CLr | R | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | n | (µg ml−1) | (µg ml−1) | (h) | (h) | (µg ml−1 h) | (l h−1) | (l) | (l h−1) | (%) |
| 500 mg naproxen twice daily | 10 | 99.2 | 56.7 | 3.92 | 11.7 | 874 | 0.579 | 9.67 | 0.259 | 44.6 |
| (9.5) | (8.9) | (0.88) | (5.0) | (107) | (0.068) | (3.72) | (0.052) | (6.8) | ||
| 1200 mg gabapentin enacarbil once daily with 500 mg naproxen twice daily | 10 | 97.8 | 53.2 | 4.81 | 10.0 | 860 | 0.589 | 8.51 | 0.256 | 43.7 |
| (9.5) | (9.8) | (1.40) | (3.9) | (112) | (0.071) | (3.55) | (0.029) | (4.2) |
AUCss, area under the concentration–time curve at steady state; CLr, renal clearance; CLss/F, apparent clearance after oral dosing at steady state; Css,max, maximum concentration at steady state; Css,min, minimum concentration at steady state; R, percent of naproxen dose recovered in urine in 12 h post-dose; SD, standard deviation; tmax, time to Css,max; t1/2, half-life; Vd/F, apparent volume of distribution.
Pharmacokinetics following co-administration of gabapentin enacarbil and cimetidine
Gabapentin
Gabapentin exposure increased slightly when gabapentin enacarbil was co-administered with cimetidine compared with gabapentin enacarbil dosed alone (Figure 3). After once daily dosing of gabapentin enacarbil 1200 mg for 4 days, the mean Css,max of gabapentin in plasma was 7.05 µg ml−1 and the mean tmax was 5.61 h (Table 3). When gabapentin enacarbil and cimetidine were dosed concomitantly, the mean Css,max of gabapentin increased to 7.44 µg ml−1 and the mean tmax was 5.53 h. Similarly, when gabapentin enacarbil was dosed alone the mean gabapentin AUCss was 70.8 µg ml−1 h, compared with 87.6 µg ml−1 h when gabapentin enacarbil was co-administered with cimetidine.
Figure 3.
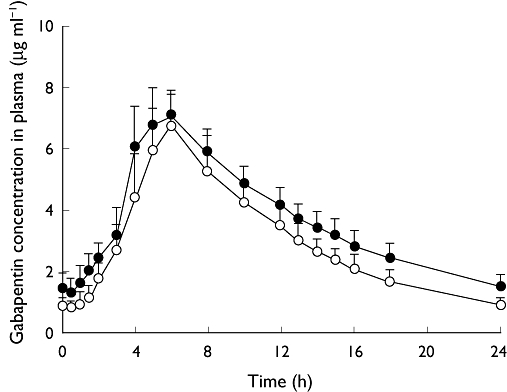
Mean (SD) concentrations of gabapentin in plasma at steady state after dosing of gabapentin enacarbil alone and concomitantly with cimetidine. SD, standard deviation. Gabapentin enacarbil (–○–); Gabapentin enacarbil + cimetidine (–•–)
Table 3.
Mean (SD) pharmacokinetic parameters for gabapentin in plasma and urine at steady state after dosing of gabapentin enacarbil alone and concomitantly with cimetidine
| Css,max | Css,min | tmax | t1/2 | AUCss | CLss/F | Vd/F | CLr | F | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | n | (µg ml−1) | (µg ml−1) | (h) | (h) | (µg ml−1 h) | (l h−1) | (l) | (l h−1) | (%) |
| 1200 mg gabapentin enacarbil once daily | 12 | 7.05 | 0.819 | 5.61 | 6.32 | 70.8 | 9.00 | 82.0 | 6.02 | 67.1 |
| (1.20) | (0.229) | (0.54) | (1.14) | (10.0) | (1.40) | (17.8) | (1.31) | (13.2) | ||
| 1200 mg gabapentin enacarbil once daily with 400 mg cimetidine four times daily | 12 | 7.44 | 1.30 | 5.53 | 8.12 | 87.6 | 7.20 | 83.7 | 4.95 | 68.5 |
| (0.94) | (0.52) | (0.70) | (1.57) | (9.1) | (0.72) | (14.5) | (0.68) | (5.6) |
AUCss, area under the concentration–time curve at steady state; CLr, renal clearance; CLss/F, apparent clearance after oral dosing at steady state; Css,max, maximum concentration at steady state; Css,min, minimum concentration at steady state; F, percentage of gabapentin dose recovered in urine in 24 h post-dose; SD, standard deviation; tmax, time to Css,max; t1/2, half-life; Vd/F, apparent volume of distribution.
The estimated geometric mean ratio for the Css,max of gabapentin from gabapentin enacarbil dosed with cimetidine compared with gabapentin enacarbil dosed alone was close to 1 (1.06) and the 95% confidence interval was 0.97, 1.17. The mean gabapentin AUCss increased by, on average, 24% when gabapentin enacarbil was co-administered with cimetidine compared with gabapentin enacarbil dosed alone. The estimated geometric mean ratio for the AUCss of gabapentin from gabapentin enacarbil dosed with cimetidine compared with gabapentin enacarbil dosed alone was 1.24 and the 95% confidence interval was 1.16, 1.34.
Apparent oral clearance and renal clearance of gabapentin decreased when gabapentin enacarbil was co-administered with cimetidine compared with gabapentin enacarbil dosed alone. CLss/F for gabapentin was 9.00 l h−1 for gabapentin enacarbil alone and 7.20 l h−1 with co-administration of cimetidine. These results are consistent with a mean decrease in CLss/F of approximately 20%.
Cimetidine
Following co-administration of gabapentin enacarbil and cimetidine, there was no clinically relevant change in cimetidine exposure compared with cimetidine dosed alone (Table 4). The Css,max and AUCss of cimetidine were similar when cimetidine was dosed alone or concomitantly with gabapentin enacarbil. After four-times-daily dosing of cimetidine 400 mg, the mean Css,max of cimetidine in plasma was 2.34 µg ml−1, the mean tmax was 2.42 h and the mean AUCss was 8.45 µg ml−1 h. When cimetidine was co-administered with gabapentin enacarbil, the mean Css,max of cimetidine was 2.26 µg ml−1, the mean tmax was 2.42 h and the mean cimetidine AUCss value was 8.36 µg ml−1 h (Table 4).
Table 4.
Mean (SD) pharmacokinetic parameters for cimetidine in plasma and urine at steady state after dosing of cimetidine alone and concomitantly with gabapentin enacarbil
| Css,max | Css,min | tmax | t1/2 | AUCss | CLss/F | Vd/F | CLr | R | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | n | (µg ml−1) | (µg ml−1) | (h) | (h) | (µg ml−1 h) | (l h−1) | (l) | (l h−1) | (%) |
| 400 mg cimetidine four times daily | 12 | 2.34 | 0.650 | 2.42 | 2.38 | 8.45 | 47.9 | 165 | 19.9 | 41.7 |
| (0.36) | (0.122) | (0.67) | (0.34) | (0.95) | (5.6) | (32) | (4.7) | (9.4) | ||
| 1200 mg gabapentin enacarbil once daily with 400 mg cimetidine four times daily | 12 | 2.26 | 0.641 | 2.42 | 2.74 | 8.36 | 48.5 | 194 | 17.1 | 35.5 |
| (0.49) | (0.149) | (0.76) | (0.79) | (1.03) | (6.0) | (69) | (4.1) | (9.2) |
AUCss, area under the concentration–time curve at steady state; CLr, renal clearance; CLss/F, apparent clearance after oral dosing at steady state; Css,max, maximum concentration at steady state; Css,min, minimum concentration at steady state; R, percentage of cimetidine dose recovered in urine in 6 h post-dose; SD, standard deviation; tmax, time to Css,max; t1/2, half-life; Vd/F, apparent volume of distribution.
The estimated geometric mean ratio for the Css,max of cimetidine in combination with gabapentin enacarbil compared with cimetidine dosed alone was close to 1 (0.96) and the 95% confidence interval was 0.89, 1.03. For cimetidine AUCss, the corresponding estimated geometric mean ratio value was 0.99 and the 95% confidence interval was 0.94, 1.04. The amount of cimetidine excreted in urine at 6 h post-dosing was evaluated. When administered alone, the mean percentage of dose recovery in urine for cimetidine was 42% compared with 36% when co-administered with gabapentin enacarbil.
Tolerability
TEAEs occurring in two or more subjects in any treatment group are listed in Tables 5 and 6. The most frequent TEAEs reported during the gabapentin enacarbil and naproxen study were fatigue with gabapentin enacarbil when dosed alone, dyspepsia with naproxen when dosed alone, and constipation with co-administered gabapentin enacarbil and naproxen. The frequency of TEAEs did not increase when gabapentin enacarbil was administered with naproxen compared with gabapentin enacarbil dosed alone (Table 5). All TEAEs resolved and were judged as mild in intensity, apart from three AEs of moderate intensity reported in two subjects. One subject withdrew from the study due to moderate hypersensitivity (rash) when receiving gabapentin enacarbil and concomitant naproxen on the first day of period 3; the event resolved after discontinuation of study medications and anti-histamine treatment. Another subject withdrew based on investigator judgment on day 2 of period 1 after receiving two doses of gabapentin enacarbil. This subject showed irrational behaviour (including agitation and paranoia, which were reported as AEs) during the study. It was learned that the subject had an undisclosed history of behavioural issues and was withdrawn from the study on investigator judgement. No severe or serious TEAEs were reported.
Table 5.
Treatment-emergent adverse events in two or more subjects in any treatment group: gabapentin enacarbil dosed alone or concomitantly with naproxen
| Treatment group | |||
|---|---|---|---|
| Gabapentin enacarbil | Naproxen | Gabapentin enacarbil with naproxen | |
| n= 12 | n= 11 | n= 11 | |
| Treatment-emergent adverse event | n (%) | n (%) | n (%) |
| Any | 11 (92) | 4 (36) | 7 (64) |
| Abdominal pain | 1 (8) | 0 | 2 (18) |
| Constipation | 0 | 1 (9) | 3 (27) |
| Dyspepsia | 0 | 2 (18) | 0 |
| Euphoric mood | 3 (25) | 0 | 2 (18) |
| Fatigue | 4 (33) | 0 | 1 (9) |
| Feeling drunk | 2 (17) | 0 | 0 |
| Feeling of relaxation | 3 (25) | 1 (9) | 0 |
| Headache | 3 (25) | 0 | 0 |
Table 6.
Treatment-emergent adverse events in two or more subjects in any treatment group: gabapentin enacarbil dosed alone or concomitantly with cimetidine
| Treatment group | |||
|---|---|---|---|
| Gabapentin enacarbil | Cimetidine | Gabapentin enacarbil with cimetidine | |
| n= 12 | n= 12 | n= 12 | |
| Treatment-emergent adverse event | n (%) | n (%) | n (%) |
| Any event | 11 (92) | 3 (25) | 11 (92) |
| Somnolence | 6 (50) | 0 | 6 (50) |
| Dizziness | 5 (42) | 0 | 6 (50) |
| Disorientation | 2 (17) | 0 | 3 (25) |
| Abnormal co-ordination | 2 (17) | 0 | 2 (17) |
| Lethargy | 2 (17) | 0 | 2 (17) |
| Thirst | 2 (17) | 0 | 2 (17) |
| Dysgeusia | 1 (8) | 0 | 2 (17) |
| Headache | 0 | 1 (8) | 2 (17) |
No serious TEAEs were reported during the gabapentin enacarbil and cimetidine study and all reported TEAEs were judged by the investigator as mild in intensity. No subjects withdrew from the study due to TEAEs. The nature or frequency of these TEAEs did not change whether gabapentin enacarbil was administered alone or with cimetidine.
For both studies, vital signs did not show any consistent pattern or trends to indicate drug-related effects and all electrocardiograms taken throughout the study were within normal limits. Laboratory results outside of the normal range were infrequent and none was considered clinically relevant by the investigator.
Discussion
No clinically relevant change in gabapentin exposure was observed when gabapentin enacarbil was co-administered with naproxen compared with gabapentin enacarbil dosed alone. Although there were slight increases in the Css,max and AUCss of gabapentin (8% and 13%, respectively), the 95% confidence intervals for the estimated treatment ratios were within the limits associated with bioequivalence. Similarly, there was no effect on gabapentin enacarbil or naproxen exposure when gabapentin enacarbil and naproxen were dosed concomitantly at clinically relevant doses. Therefore, dose adjustment is not necessary with co-administration of the two drugs.
Gabapentin enacarbil is targeted to high-capacity nutrient transporters, such as MCT-1, which are expressed in all regions of the intestinal tract. The pathway for absorption of gabapentin enacarbil is not saturated at clinically useful doses [3]. Thus, due to the high capacity of the MCT-1 transporter, there was no saturation of absorption of either gabapentin enacarbil or naproxen, a substrate of the MCT-1 transporter. In the literature, co-administration of naproxen (250 mg) and gabapentin (125 mg) increased gabapentin absorption by 12–15% and was not considered clinically significant [5, 13]. In the present study, co-administration of naproxen (500 mg) and gabapentin enacarbil (1200 mg) also increased gabapentin exposure within a comparable range; however, there is a distinctly different absorption pathway of gabapentin enacarbil compared with gabapentin. Given the high capacity of the MCT-1 pathway, coupled with its distribution along the length of the gastrointestinal tract, it is unlikely that oral administration of gabapentin enacarbil will interfere with other MCT-1 substrates at the site of absorption.
No clinically relevant drug–drug interaction was identified between gabapentin as delivered by gabapentin enacarbil and cimetidine after oral co-administration in healthy volunteers. Although there was a 24% increase in the AUCss of gabapentin and a corresponding 20% decrease in CLss/F for gabapentin enacarbil co-administered with cimetidine compared with gabapentin enacarbil dosed alone, this was not considered clinically relevant. This change in gabapentin exposure is in the expected range for subjects treated with gabapentin enacarbil [3]. Similarly, there was no effect on cimetidine exposure when gabapentin enacarbil and cimetidine were dosed concomitantly. Again, these results were consistent with published data for concomitant treatment with gabapentin and cimetidine [5]. When cimetidine 300 mg was dosed four times daily, the mean apparent oral clearance of gabapentin was reduced by 14% and creatinine clearance was reduced by 10%. Although cimetidine altered the renal excretion of gabapentin and creatinine, the small decrease in excretion was not considered clinically relevant [5].
Gabapentin is a weak acid that undergoes renal excretion and is excreted primarily in unaltered form [14]. The renal clearance of gabapentin is slightly higher than the glomerular filtration rate, suggesting some involvement of active secretion [5]. Gabapentin is believed to be a substrate of the renal transporter OCT2. The human organic transporter OCT2 is a multi-specific transporter of organic cations and is primarily responsible for uptake of organic cations across the basolateral membrane of renal tubular epithelial cells [15–17]. This transporter appears to interact with many organic cation drugs as well as dietary supplements [18]. OCT2 substrates include amantadine, cimetidine and memantine. Inhibitors of OCT2 include desipramine, phenoxy-benzamine and quinine [19]. There are no known reports of clinically relevant drug–drug interactions between gabapentin and these inhibitors. In addition, the organic cation transporter-1 (OCTN1) has also been shown to be involved in the renal clearance of gabapentin [20]. This transporter also appears to be inhibited by various organic cations, including cimetidine [21]. Thus, cimetidine was considered a suitable compound for evaluating the drug–drug interaction with gabapentin enacarbil.
The pKa for the carboxylate moiety of gabapentin enacarbil is 5.0 [1]. Antacids would be expected to potentially increase the extent of ionization of the prodrug, which could possibly decrease the extent of passive absorption of gabapentin enacarbil. Ingestion of cimetidine is known to increase gastric and duodenal pH, analogous to the effect of ingesting an antacid. However, there was no evidence of a pH-related change in gabapentin enacarbil absorption in this study.
In general, gabapentin enacarbil was well tolerated with or without concomitant dosing of naproxen or cimetidine. Co-administration of these agents did not result in any new TEAEs that were not seen with gabapentin enacarbil, naproxen or cimetidine dosed alone. The most frequent TEAEs, somnolence and dizziness, were reported in subjects receiving gabapentin co-administered with cimetidine as well as in those receiving gabapentin enacarbil alone. The reported TEAEs are similar to previous studies of gabapentin enacarbil administered alone in healthy volunteers, and similar to those reported with gabapentin monotherapy [3, 5].
In summary, no clinically relevant drug–drug interactions were observed with co-administered naproxen or cimetidine. Based on these findings, no clinically relevant pharmacokinetic interactions are expected between gabapentin enacarbil and other substrates of MCT-1 or OCT2.
Acknowledgments
The study was supported by XenoPort, Inc. The authors acknowledge Barbara Wilson, MEd (GlaxoSmithKline, Research Triangle Park, NC, USA) for manuscript co-ordination and editorial assistance and Sarah Brown BSc (Hons) (Caudex Medical Ltd, Oxford, UK) for writing and editorial assistance.
All authors were full-time employees of XenoPort, Inc. at the time this study was conducted. Research funding for design and conduct of this study, collection, management, analysis and interpretation of the data were sponsored by XenoPort, Inc. Preparation, review and approval of the manuscript were sponsored by XenoPort, Inc. and GlaxoSmithKline.
Competing interests
RL, JS, WL, VV, RB and KC are all employees of XenoPort, Inc., and hold stock in the company. JH has been contracted by XenoPort, Inc. to manage various clinical trials.
REFERENCES
- 1.Cundy KC, Branch R, Chernov-Rogan T, Dias T, Estrada T, Hold K, Koller K, Liu X, Mann A, Panuwat M, Raillard SP, Upadhyay S, Wu QQ, Xiang JN, Yan H, Zerangue N, Zhou CX, Barrett RW, Gallop MA. XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J Pharmacol Exp Ther. 2004;311:315–23. doi: 10.1124/jpet.104.067934. [DOI] [PubMed] [Google Scholar]
- 2.Cundy KC, Annamalai T, Bu L, De Vera J, Estrela J, Luo W, Shirsat P, Torneros A, Yao F, Zou J, Barrett RW, Gallop MA. XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: II. Improved oral bioavailability, dose proportionality, and colonic absorption compared with gabapentin in rats and monkeys. J Pharmacol Exp Ther. 2004;311:324–33. doi: 10.1124/jpet.104.067959. [DOI] [PubMed] [Google Scholar]
- 3.Cundy KC, Sastry S, Luo W, Zou J, Moors TL, Canafax DM. Clinical pharmacokinetics of XP13512/GSK1838262, a novel transported prodrug of gabapentin. J Clin Pharmacol. 2008;48:1378–88. doi: 10.1177/0091270008322909. [DOI] [PubMed] [Google Scholar]
- 4.Kushida CA, Becker PM, Ellenbogen AL, Canafax DM, Barrett RW the XP052 Study Group. A randomized, double-blind, placebo-controlled study of XP13512/GSK1838262 in patients with RLS: PIVOT RLS. Neurology. 2009;72:439–46. doi: 10.1212/01.wnl.0000341770.91926.cc. [DOI] [PubMed] [Google Scholar]
- 5.Pfizer. Neurontin® (gabapentin) prescribing information. Available at http://www.pfizer.com/files/products/uspi_neurontin.pdf (last accessed 11 November 2008.
- 6.Choi JS, Jin MJ, Han HK. Role of monocarboxylic acid transporters in the cellular uptake of NSAIDs. J Pharm Pharmacol. 2005;57:1185–9. doi: 10.1211/jpp.57.9.0013. [DOI] [PubMed] [Google Scholar]
- 7.Warren M. Targeting the MCT transporter in drug delivery to increase bioavailability. 2007. AAPS Annual Meeting, November 11–15 San Diego, CA.
- 8.Gill RK, Saksena S, Alrefai WA, Sarwar Z, Goldstein JL, Carroll RE, Ramaswamy K, Dudeja PK. Expression and membrane localization of MCT isoforms along the length of the human intestine. Am J Physiol Cell Physiol. 2005;289:C846–52. doi: 10.1152/ajpcell.00112.2005. [DOI] [PubMed] [Google Scholar]
- 9.Tamai I, Takanaga H, Maeda H, Sai Y, Ogihara T, Higashida H, Tsuji A. Participation of a proton-cotransporter, MCT1, in the intestinal transport of monocarboxylic acids. Biochem Biophys Res Commun. 1995;214:482–9. doi: 10.1006/bbrc.1995.2312. [DOI] [PubMed] [Google Scholar]
- 10.Emoto A, Ushigome F, Koyabu N, Kajiya H, Okabe K, Satoh S, Tsukimori K, Nakano H, Ohtani H, Sawada Y. H+-linked transport of salicylic acid, an NSAID, in the human trophoblast cell line BeWo. Am J Physiol Cell Physiol. 2002;282:C1064–75. doi: 10.1152/ajpcell.00179.2001. [DOI] [PubMed] [Google Scholar]
- 11.Zerangue N. Intestinal absorption. 2003. 3rd International Research Conference: Pharma Conference; Transport and Drugs. August 17–21 Pontresina, Switzerland.
- 12.Grundemann D, Liebich G, Kiefer N, Koster S, Schomig E. Selective substrates for non-neuronal monoamine transporters. Mol Pharmacol. 1999;56:1–10. doi: 10.1124/mol.56.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Feng MR, Ouellet D, Li K, Berna K, Ferreira S, Knowlton P, Fountain S, Randinitis E, Haig G, Wesche D. Pharmacokinetic interaction of gabapentin and naproxen in rats and humans. Clin Pharmacol Ther. 2003;73:P50. abstract. [Google Scholar]
- 14.McLean MJ. Clinical pharmacokinetics of gabapentin. Neurology. 1994;44:S17–22. [PubMed] [Google Scholar]
- 15.Gründemann D, Köster S, Kiefer N, Breidert T, Engelhardt M, Spitzenberger F, Obermüller N, Schömig E. Transport of monoamine transmitters by the organic cation transporter type 2, OCT2. J Biol Chem. 1998;273:30915–20. doi: 10.1074/jbc.273.47.30915. [DOI] [PubMed] [Google Scholar]
- 16.Karbach U, Kricke J, Meyer-Wentrup F, Gorboulev V, Volk C, Loffing-Cueni D, Kaissling B, Bachmann S, Koepsell H. Localization of organic cation transporters OCT1 and OCT2 in rat kidney. Am J Physiol Renal Physiol. 2000;279:F679–87. doi: 10.1152/ajprenal.2000.279.4.F679. [DOI] [PubMed] [Google Scholar]
- 17.Urakami Y, Okuda M, Masuda S, Saito H, Inui KI. Functional characteristics and membrane localization of rat multispecific organic cation transporters, OCT1 and OCT2, mediating tubular secretion of cationic drugs. J Pharmacol Exp Ther. 1998;287:800–5. [PubMed] [Google Scholar]
- 18.Fujita T, Urban TJ, Leabman MK, Fujita K, Giacomini KM. Transport of drugs in the kidney by the human organic cation transporter, OCT2 and its genetic variants. J Pharm Sci. 2006;95:25–36. doi: 10.1002/jps.20536. [DOI] [PubMed] [Google Scholar]
- 19.US Food and Drug Administration. Guidance for industry drug interaction studies – study design, data analysis, and implications for dosing and labelling. Available at http://www.fda.gov/cder/Guidance/6695dft.htm (last accessed 11 November 2008.
- 20.Urban TJ, Brown C, Castro RA, Shah N, Mercer R, Huang Y, Brett CM, Burchard EG, Giacomini KM. Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin Pharmacol Ther. 2008;83:416–21. doi: 10.1038/sj.clpt.6100271. [DOI] [PubMed] [Google Scholar]
- 21.Yabuuchi H, Tamai I, Nezu J-I, Sakamoto K, Oku A, Shimane M, Sai Y, Tsuji A. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J Pharmacol Exp Ther. 1999;289:768–73. [PubMed] [Google Scholar]