

Summary
E3 ligases Cbl-b and Itch have emerged as dominant “tolerogenic” regulators of T cells because their deficiency results in severe autoimmune diseases. Cbl-b and Itch ligase activity regulate T cell anergy and development of Foxp3+ Tregs in the periphery by modulating key components of TCR and TGF-β signaling. Manipulation of Cbl-b and Itch activities may provide unique opportunities to develop future therapies for immune disorders such as autoimmunity and cancer
Keywords: Ubiquitination, tumor Immunity, Tolerance, Tregs, Foxp3
Background
Self-tolerance is a critical component of immune recognition which facilitates elimination of non-self pathogens and minimizes bystander damage to the host. The immune system accomplishes self-tolerance through a series of control elements that operate during both thymic development and peripheral homeostasis. The majority of autoreactive T cells, which express high affinity/avidity antigen receptors for self-antigens, are deleted via negative selection during T cell maturation in the thymus. However, a few pathogenic self-reactive T cells survive thymic selection and populate the periphery. The activation and function of self-reactive T cells are regulated by at least these 4 peripheral tolerance mechanisms; (i) hypo-responsiveness to antigens expressed at low levels, (ii) deletion of activated T cells through activation induced cell death (AICD), (iii) T cell intrinsic functional inactivation (anergy), and (iv) active suppression by immune regulatory cells such as CD4+Foxp3+ regulatory T cells (Tregs) (1).
Multiple molecular pathways have been implicated in the regulation of T cell tolerance. Recently, E3 ligase Itch and Cbl-b have emerged as “gate keepers” of peripheral T cell tolerance. Itch and Cbl-b belong to a class of ubiquitin transferring enzymes involved in the final step of ubiquitin conjugation. Ubiquitin transferring enzymes confer substrate specificity during ubiquitin conjugation (2). A substrate can be tagged with a single ubiquitin (monoubiquitination), or multiple ubiquitins in an elongated ubiquitin chain (polyubiquitination). Ubiquitin molecules in a polyubiquitin chain are generally linked through the lysine residue at position 48 or 63. It was believed that ubiquitin tagged substrates are recognized by (death signal) 26S proteasomes for their degradation. However, recent studies suggest that different polyubiquitin chains have distinct effects on the substrate, including modification of protein function, facilitation of cell-surface-receptor turnover, and control of gene transcription. For example, monoubiquitination and K63 linked polyubiquitination alter protein trafficking and cell-cell interactions whereas K48-linked polyubiquitination target substrates for proteasomal degradation (3). Defects in the ubiquitin conjugation process are linked to several pathological conditions such as autoimmunity, allergy and cancer. Here, the involvement of Cbl-b and Itch in T cell tolerance and their role in immune self reactivity and cancer immunotherapy is discussed.
Cbl-b in T cell anergy
Cbl-b is a member of highly conserved family of Cbl (Casitas B-lineage Lymphoma) proteins and functions as an E3 ligase via its RING domain. The name is derived from retroviral oncoprotein v-Cbl, which promotes B-cell lymphoma in mice. Studies on Cbl-b-/- mice reveal functional alterations in the immune system and a critical negative regulatory role of Cbl-b. Although Cbl-b-/- mice did not show overt alterations in thymic selection or peripheral T cell numbers, they were highly susceptible to spontaneous and antigen-induced experimental autoimmune diseases (4, 5). Cbl-b-/- T cells were hyper-proliferative and, interestingly, bypass the requirement for CD28 costimulation (4). Subsequently, it was hypothesized that Cbl-b may regulate peripheral T cell tolerance. Rao and colleagues demonstrated that T cell anergy inducing stimuli, such as TCR stimulation in the absence of CD28 costimulation, results in upregulation of Cbl-b (6). Weak TCR stimulation induced a Ca+ flux and NFAT activation without the concomitant AP1 activation, resulting in the activation of the Zinc finger transcription factors, early growth response gene (Egr)-2 and Egr-3, which increased Cbl-b transcription (7). We demonstrated that Cbl-b-/- T cells are resistant to anergy induction in vitro and antigen specific anergy in vivo (8, 9). Cbl-b-/- mice developed severe collagen induced arthritis even in the absence of mycobacterial adjuvant (9). T cell anergy was induced in mice carrying p14 TCRVa2Vb8.2 transgene [that recognize lymphocytic choriomeningitis virus (LCMV) p33 peptide presented by MHC class I] by repeated injection of cognate peptide p33. In contrast Cbl-b-/- p14 transgenic mice challenged with p33 resulted in massive activation of CD8+ T cells, a severe cytokine storm, and significant mortality(9). Analogously, LCMV infection of p14/Rip-gp transgenic mice induced diabetes, whereas low agonistic peptide LCMV-LF6 led to diabetes development in <50% of the infected mice. However, Cbl-b-/- P14 TCR transgenic mice infected with LCMV-LF6 triggered rapid diabetes progression and enhanced CTL function in all mice (10). Mechanistically, Cbl-b reduced phosphorylation of phospholipase C-γ1 (PLC-γ1) and, thereby PLC-γ1 activity in anergic T cells (9). These results have highlighted the essential role of Cbl-b in T cell tolerance.
Itch in T cell anergy
Similar to Cbl-b, E3 ligase Itch is upregulated in anergic T cells (8, 11). Itch is a monomeric protein and belongs to the homologous to E6-AP carboxy terminus (HECT) type family of ubiquitin ligases. Itch has an N-terminal protein Kinase C (PKC) related C2 domain, four protein interacting WW domains and a C-terminal HECT domain. The WW domains of Itch recognize the Pro-rich PPXY consensus sequences in target substrates. The conserved cysteine in the HECT domain forms thioester bonds with ubiquitin during ubiquitin conjugation. Itch deficiency in C57BL/6J mice results in progressively lethal systemic autoimmune like lymphoproliferative diseases associated with constant itching of the skin and chronic pulmonary interstitial inflammation. Itch-/- T cells exhibit an activated phenotype, increased production of Th2 cytokines (IL-4, IL-5), and a biased differentiation towards Th2 phenotype. Itch-/- mice also display augmented serum IgE levels (12). We have demonstrated that Itch regulates T cell anergy by modulating PLC-γ1 and PKC-θ, two key signaling molecules induced by Ca++/ calcineurin signaling (8).
Thus, we hypothesized that anergizing stimuli redistributes Itch from the cytosol to the endosomal compartment, where Itch associates with PLC-γ1 and PKC-θ in the adjacent immunologic synapse. Following Itch mediated ubiquitination, PLC-γ1 and PKC-θ undergo endosomal sorting and trafficking into the lysosome for degradation. Reduced levels of PLC-γ1 and PKC-θ are thought to shorten the life span of immunologic synapse resulting in inability to sustain stable APC contact which resulted in T cell unresponsiveness after TCR engagement(8). Itch also regulates anergy and inflammation by ubiquitin dependent degradation of Jun family members, which results in AP-1 inactivation (11). Therefore, the inability of Itch-/- mice to undergo anergy may be one of the mechanisms underlying its autoimmune and inflammatory prone phenotype.
Cbl-b and Itch in Treg development
T cell anergy and immune suppression by Foxp3+ Tregs play critical roles in the maintenance of peripheral tolerance and homeostasis. Tregs are a small population of CD4+ T cells that specifically express transcription factor Foxp3. They develop in the thymus as a functionally distinct mature population of CD4+T cells called natural Tregs (nTregs). In addition, Tregs develop in the periphery by the action of TGF-β on naive CD4+T cells (13). TGF-β initiates signaling fromthe cell surface through transmembrane serine / threonine kinases TGF-β type I receptor (Tβ RI) andTGF-β type II receptor (14). Upon ligand binding, the constitutively active TGF-β type II receptor phosphorylates and activates Tβ RI, leading to phosphorylation of Smad2 or Smad3 on two serines in the carboxyl terminus. Phosphorylated Smad2 and Smad3 associate with Smad4, and translocate to the nucleus, where the Smad complexes, in cooperation with co-activators, participate in transcriptional activation of target genes (14). However, the transcriptional mechanism by which TGF-β induces Foxp3 expression and Treg phenotype in naive CD4 T cells is not clear. A defect in Smad3 phosphorylation and Foxp3 expression occurs in Cbl-b-/- T cells (15). Itch plays a critical role in the regulation of TGF-β signaling and Foxp3 expression in CD4 T cells (16). Itch ablation severely compromises TGF-β induced Foxp3 expression and TGF-β mediated inhibition of T cell proliferation. Furthermore, we demonstrated that Itch targets TGF-β inducible early gene-1 (TIEG1). Itch mediated monoubiquitination of TIEG1 induced its nuclear translocation. TIEG1 binds to GC rich sequences in the Foxp3 promoter (16). The ubiquitination activity of Itch was increased following its phosphorylation by MEKK1-JNK1 (MAPK/ERK kinase kinase1- Jun N-terminal kinase1) downstream of TCR stimulation (17). Whether MEKK1-JNK1 pathway is involved in the regulation of Itch mediated Foxp3 expression awaits further investigation (Figure 1).
Figure 1.
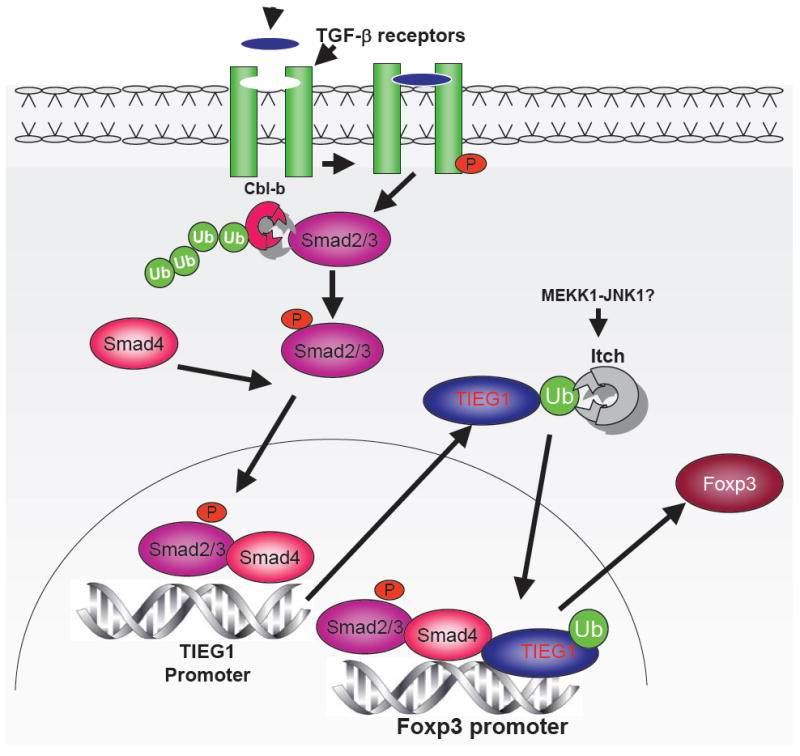
Cbl-b and Itch mediated regulation of TGF-β signaling and Foxp3 expression. Ligation of TCR in the presence of TGF-β results in phosphorylation of Smad2/3. Phosphorylated Smad2/3 in association with Smad4 translocates to the nucleus and induces Foxp3 transcription. Cbl-b regulates Foxp3 expression by modulating Smad2 phosphorylation. Itch targets TIEG1 for monoubiquitination which results in TIEG1 nuclear translocation and Foxp3 expression.
Implications
Development of cancer and autoimmunity is seen as a failure of the immune system to control tumor growth and to regulate autoreactive response. The present therapy for autoimmunity and cancer is mainly based on broad spectrum immunosuppressive or antiproliferative regimens, respectively. The deleterious side effects of prolonged chemotherapy for cancer and immunosuppressive agents for autoimmune diseases are known and highlight the need for more specific and long lasting therapies. Tumor immunotherapy is a promising approach that results in the regression of established primary and metastatic tumors in multiple experimental models. However, tumor vaccines, including dendritic cell vaccines, have been largely ineffective in causing tumor regression in the clinic. The tumors not only survive and disseminate, but, more importantly, they mimic some of the signaling pathways of the immune system, augment conditions that favor tumor immune tolerance, and escape tumor immunity (18). The cytokine milieu of the tumor (TGF-β) inhibits CTL responses, promotes the expansion and de novo generation of tumor antigen-specific Tregs, and contributes to the failure of immunotherapy. Such induced Tregs inhibit the priming and effector function of anti-tumor effector cells, and form a broad and self-amplifying immunosuppressive network through extensive interaction with various antigen presenting cells (APCs) in the tumor microenvironment. Therefore, successful vaccination strategy may need to not only increase the frequency of tumor-specific effector T cells but also prevent Treg induction. Depletion of Tregs by anti-CD25 antibody has been demonstrated to reject several types of transplantable tumors in animal models. However, caution needs to be taken during therapeutic manipulation of Tregs in cancer patients. Since Tregs are required for maintaining self-tolerance, prolonged, global and non-specific depletion of Treg may breach self-tolerance and lead to autoimmune diseases. Therefore, warranted strategies need to selectively curtail the development of locoregional tumor-specific Tregs. A local Treg purging strategy could eliminate the most potent suppressors that inhibit anti-tumor effector cells. Their elimination also can redirect precursor T cells to recognize tumor antigens and differentiate into effector cells that attack tumors. This local Treg purging approach is less likely to affect the thymus derived natural Tregs, and thus minimizes risk of triggering autoimmunity.
The demonstration that Cbl-b and Itch deficiency leads to aggressive T cell response and diminished peripheral development of CD4+CD25+ Tregs suggests that targeting these molecules for tumor immunotherapy may be promising. In a preclinical study, Cbl-b-/- mice rejected implanted TC-1 tumors as well as spontaneous UVB-induced skin tumors (19). Adoptive transfer of Cbl-b-/- CD8 T cells into wild type mice bearing EG7 tumors resulted in tumor regression (20). Moreover, spontaneous lymphoma development, which caused premature death in 50% of ataxia telangiectasia deficient ATM-/- mice, was reduced in ATM-/- Cbl-b-/- double deficient mice. However, care should be taken when extrapolating these results into the clinic as Cbl-b plays an essential critical role in maintenance of immune homeostasis. Nonetheless, the importance of Cbl-b and Itch in immune cell function has been recognized and future efforts to modulate their functions will undoubtedly lead to novel and important therapeutic interventions for a multitude of disease states.
Acknowledgments
I thank Dr. Wei-Zen Wei for support and critical reading of the manuscript.
References
- 1.Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–9. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- 2.Venuprasad K, Yang C, Shao Y, et al. Immune regulation by ubiquitin conjugation. Adv Exp Med Biol. 2006;584:207–17. doi: 10.1007/0-387-34132-3_15. [DOI] [PubMed] [Google Scholar]
- 3.Gao M, Karin M. Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli. Mol Cell. 2005;19:581–93. doi: 10.1016/j.molcel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 4.Chiang YJ, Kole HK, Brown K, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–20. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 5.Bachmaier K, Krawczyk C, Kozieradzki I, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–6. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 6.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 7.Safford M, Collins S, Lutz MA, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–80. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 8.Heissmeyer V, Macian F, Im SH, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–65. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 9.Jeon MS, Atfield A, Venuprasad K, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–77. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Gronski MA, Boulter JM, Moskophidis D, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. 2004;10:1234–9. doi: 10.1038/nm1114. [DOI] [PubMed] [Google Scholar]
- 11.Venuprasad K, Elly C, Gao M, et al. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J Clin Invest. 2006;116:1117–26. doi: 10.1172/JCI26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang D, Elly C, Gao B, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–7. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 15.Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J Immunol. 2006;176:1316–20. doi: 10.4049/jimmunol.176.3.1316. [DOI] [PubMed] [Google Scholar]
- 16.Venuprasad K, Huang H, Harada Y, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–53. doi: 10.1038/niXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao M, Labuda T, Xia Y, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–5. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 18.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 19.Loeser S, Loser K, Bijker MS, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007;204:879–91. doi: 10.1084/jem.20061699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007;117:1029–36. doi: 10.1172/JCI29472. [DOI] [PMC free article] [PubMed] [Google Scholar]