

Abstract
Previously we showed that hypoxia induced mitochondrial respiratory stress in RAW 264.7 macrophages and other cells caused activation of retrograde signaling (also known as mitochondrial respiratory stress signaling), and appearance of TRAP positive cells. In the present study we used N-acetyl cysteine and ascorbate as general antioxidants and Mito-Q, a mitochondria specific antioxidant to investigate the role of intracellular ROS (reactive oxygen species) in osteoclast differentiation. Our results show that hypoxia mediated mitochondrial dysfunction as tested by disruption of mitochondrial transmembrane potential was suppressed by MitoQ as well as the other antioxidants. These agents also suppressed the activation of mitochondrial retrograde signaling. Interestingly, in terms of molar concentrations, MitoQ was more than 1000 fold more effective than general antioxidants in suppressing the RANKL-induced differentiation of RAW264.7 cells into multinucleated and TRAP positive osteoclasts. We propose that mitochondrial function and intramitochondrial ROS play important role in osteoclastogenesis.
Keywords: Mitochondrial ROS production, MitoQ, Respiratory stress signaling, Osteoclast differentiation, RAW 264.7 macrophages, TRAP positive cells
INTRODUCTION
Osteoclastogenesis is a highly regulated process which involves multiple intracellular signaling cascades. Two important signaling molecules essential for inducing osteoclast differentiation are macrophage colony stimulating factor (M-CSF), required for the survival of osteoclast precursor cells and receptor activator of nuclear factor-kB (NF-kB) ligand (RANKL) 1. RANKL binds to the receptor activator of NF-kB (RANK) expressed on macrophages and activates multiple downstream signaling pathways including TRAF6 2,3. The RANK-TRAF6 complex in turn activates the MAPK, Akt and NFkB pathways2,3. RANKL is also responsible for Ca2+ mediated calcineurin activation, which in turn activates NFATc1 by protein dephosphorylation leading to its nuclear translocation. Calcium is also responsible for activation and nuclear translocation of CREB through calmodulin-CaMK pathway4. These transcription factors are believed to play critical role in the expression of various osteoclast specific genes. Previous studies in different cell types have shown that these same factors including calcineurin, NFkB, CREB, Akt and ATF2, are also activated in cells subjected to mitochondrial stress caused by perturbation of mitochondrial transmembrane potential, Δψm 5,6.
Reactive oxygen species (ROS) produced in macrophages play important roles in cellular defense as well as in receptor mediated pathways such as PI3K, Akt and NF-kB7,8. ROS has been shown to mediate RANK signaling in differentiation of osteoclasts 9,10. Although the NADPH oxidase system (Nox1 and Nox2) is a major contributing factor in ROS mediated bone resorption, studies with knockout mouse models failed to show any decrease in ROS production or bone abnormalities 11–13. It is therefore likely that ROS produced at more than one sub cellular site of macrophages is responsible for osteoclast differentiation.
Mitochondria produce ROS both as a normal metabolic byproduct of the electron transport chain as well as during oxidative stress. Several recent studies have shown that ROS are essential intracellular secondary messengers in carrying out many of the normal functions like cell growth, cell adhesion, lymphocyte activation etc14–16. Mitochondrial ROS production is steeply increased under hypoxia, ischemia/reperfusion, chemical stress/drug treatment and under many pathophysiological conditions. Previous studies from our and other laboratories showed that mitochondrial ROS can potentially induce retrograde signaling by way of altered mitochondrial transmembrane potential and activation of calcineurin and Ca2+ dependent kinases 5,6,17,18.
The present study was carried out to determine whether reactive oxygen species formed in the mitochondria of RAW 264.7 murine macrophages has a role in osteoclast formation. Our results show that mitochondrial ROS generation and activation of retrograde signaling are induced under oxidative stress conditions which appear to be important factors in RANKL induced differentiation RAW 264.7 macrophages into TRAP positive multinucleated osteoclasts. Mitochondria targeted antioxidant MitoQ selectively abrogated osteoclastogenesis in RAW 264.7 macrophages.
MATERIALS and METHODS
Cell Culture
RAW 264.7 mouse monocyte macrophages were cultured in alpha-MEM supplemented with batch tested 10% (v/v) heat-inactivated fetal calf serum as described before. Cells at 70–80% confluence were grown under hypoxic conditions (5% O2) or normoxic conditions (21% O2) for 5 days. Osteoclastogenesis was induced by treating cells with RANKL (0, 0.25 or 2.5 ng/ml). To study the effect of antioxidants on osteoclast formation different concentrations of either MitoQ, N-acetyl cysteine or ascorbate were added along with RANKL. Similarly, BAPTA was used as inhibitor of calcium-calcineurin pathway.
Measurement of ROS Production
ROS production was measured by electron paramagnetic resonance (EPR) spin trapping. MitoDEPMPO, a spin trap that specifically targets to mitochondria, was used to detect reactive oxygen radicals in intact mitochondria (Figure 1A). The triphenylphosphonium ion (TPP) was covalently linked to DEPMPO according to the method described in Hardy et al 19. TPP allows Mito-DEPMPO to accumulate in mitochondria in a membrane potential dependent manner to form spin adducts of the reactive oxygen radicals. The spin adducts are subsequently detected by EPR spectroscopy. Reaction mixtures for EPR contained 1mM DTPA and MitoDEPMPO (100mM) in a final volume of 50µl of phosphate buffered saline (pH 7.3). Succinate (10 µM) was added to initiate the electron transport chain and the EPR spectrum was recorded at 37°C using a Bruker EMX spectrometer at 9.5 GHz (X-band) employing a 100 kHz field modulation. The EPR parameters were as follows: Microwave power; 20 mW; modulation amplitude, 1 G; time constant, 1.28 ms; gain 106; sweep time, 335.54; conversion time, 0.163 s, 4 scans. ROS production was measured in mitochondria from cells maintained under normoxic and hypoxic condition with or without treatment with MitoQ (1 µM).
Figure 1.
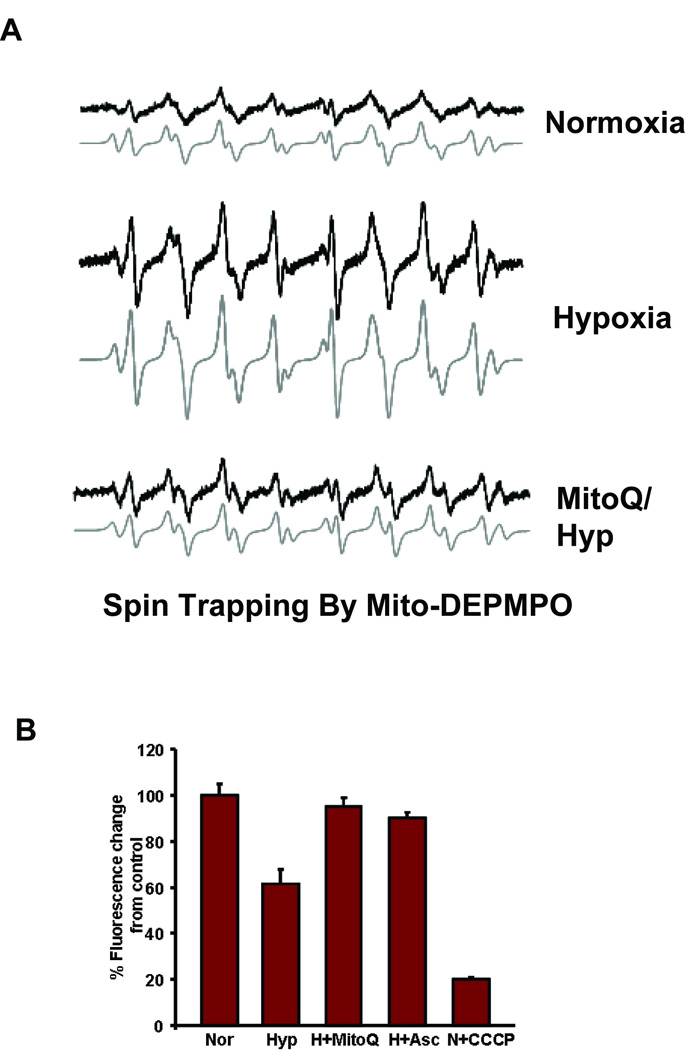
MitoQ effects on ROS production and loss of membrane potential in hypoxic mitochondria. A) EPR spectrum from isolated mitochondria following spin trapping with mito-DEPMPO in normoxic and hypoxic cells. Mitochondria (200 µg) were incubated with Mito-DEPMPO (50 mM) in phosphate buffer at pH 7.3 for 20 min at 37 °C. C. MitoQ was used at 1 µM concentration. B) Mitochondrial membrane potential in normoxic and hypoxic cells. Uptake of Mitotracker orange (50 nM) was measured as described in "Materials and Methods." CCCP-treated cells were used as positive control for membrane potential disruption. Excitation at 525 nm and emission at 575 nm were followed.
Cell fractionation and immunoblot analysis
Cells were homogenized in 70 mM Sucrose, 220 mM Mannitol, 2 mM EDTA, 10 mM HEPES (pH 7.3), supplemented with protease and phosphatase inhibitors (Roche Molecular Biochemicals, Indianapolis, IN, USA) in a Dounce homogenizer and fractionated into nuclear, mitochondrial and cytosolic fractions as described before20. Proteins resolved on polyacrylamide gels were subjected to immunoblot analysis using antibodies. The immunoblots were developed using the Pierce Super signal West Femto maximum sensitivity substrate kit, imaged and quantified in a Bio-Rad Fluor-S imaging system.
TRAP staining
TRAP staining was performed using the SIGMA kit according to the manufacturer’s instructions. RAW 264.7 cells at 30–40% confluence were maintained either at normoxic or at hypoxic (5% O2) condition for 5 days. Fresh medium equilibrated at 5% O2 was replaced every two days. At the end of hypoxia, the adherent cells were fixed with 50% (v/v) ethanol/PBS for 10 min, fixed again with ethanol–acetone (50:50, v:v) for 1 min, and incubated for 10 min at room temperature with the TRAP-staining solution. Cells that stained dark red were counted as TRAP-positive cells.
RESULTS
Mitochondrial ROS disrupts membrane potential and mitochondrial function
As shown in Figure 1A, mitochondrial ROS (both superoxide and hydrogen peroxide), determined by using MitoDEPMPO spin trap probe was markedly increased in mitochondria from hypoxic cells. This was attenuated by treatment with the mitochondria specific antioxidant, MitoQ. Mitochondrial transmembrane potential (Δψm) was measured using mitotracker orange. As shown in figure (1B) normoxic cells exhibited a steady increase in the fluorescence over a period of 20 min of incubation with mitotracker orange. Hypoxic cells on the other hand showed significantly lower mitotracker fluorescence. As a positive control, cells were incubated with CCCP, a mitochondrial ionophore. As seen in the figure 1B, CCCP treatment completely abolished membrane potential. The role of mitochondrial ROS in the disruption of membrane potential was demonstrated by quenching ROS using MitoQ. Membrane potential recovered to nearly normal levels in hypoxic cells treated with ascorbate or MitoQ.
Mitochondrial dysfunction during hypoxia induces calcineurin activity
Mitochondrial respiratory stress signaling (also called mitochondrial retrograde signaling) in mtDNA depleted C2C12 and A549 cells is characterized by the increase in calcineurin activity in response to elevated cytosolic free calcium [Ca2+]c level 5,18. As shown in Fig 2A, in hypoxia grown RAW 264.7 cells, calcineurin Aβ protein level progressively increased with the duration of hypoxia to nearly 3 folds. Inhibition of mitochondrial ROS by MitoQ treatment decreased calcineurin to near control levels. Ascorbate had a similar effect on hypoxic cells (Figure 2A). Calcineurin phosphatase activity was measured by determining the phosphate released from a threonine phosphorylated substrate. Consistent with higher level of calcineurin Aβ subunit, Figure 2B shows that hypoxic cells exhibited four fold higher calcineurin activity compared to normoxic cells. Ascorbate and MitoQ were highly effective in reducing the activity to normal levels. As expected, treatment with FK506, calcineurin inhibitor and the calcium chelator BAPTA inhibited calcineurin activity in both normoxic and hypoxic cells (Figure 2B).
Figure 2.
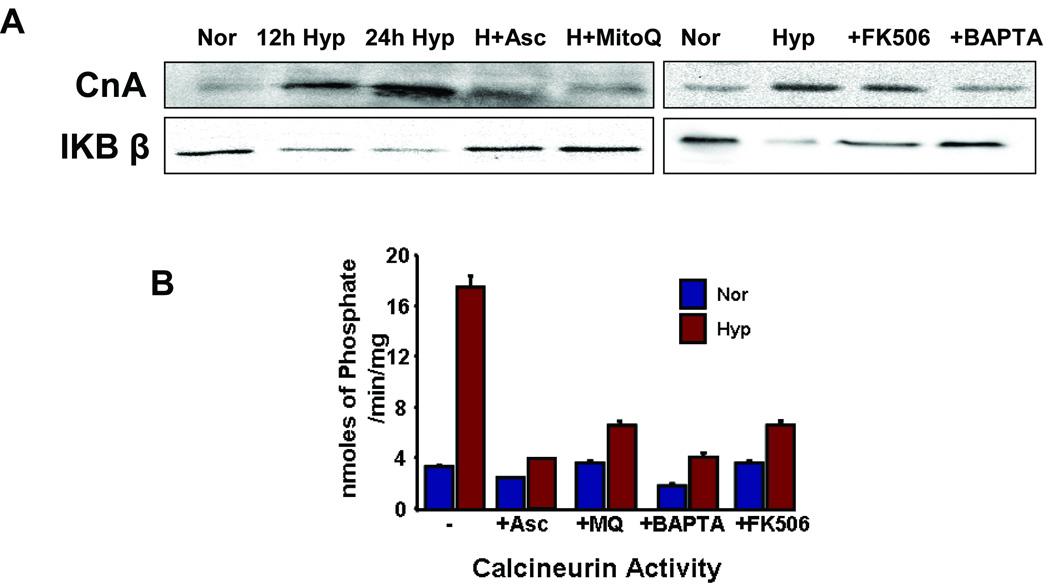
Calcineurin levels in antioxidant treated macrophages. A) Immunoblots of Calcineurin Aβ and IKB-β in normoxic and hypoxic macrophages. Treatments were done in hypoxic cells and are indicated as H+MitoQ (MitoQ: 1 µM), H+Asc (Ascorbate:100 µM), +FK506 (FK506:10 nM) or +BAPTA (BAPTA:2 µM). B) Calcineurin activity was measured in the cytosolic fraction (2 µg) of normoxic and hypoxic cells with treatements as mentioned in A. Calcineurin-specific phosphatase activity was calculated as described in the "Materials and Methods" section.
Activation of NFκB and other transcription factors under hypoxia and reversal by MitoQ
We investigated the activation of the mitochondrial respiratory stress-specific signaling under hypoxia with a view to assess the role of mitochondrial ROS in this activation. As seen in figure (2A), IκBβ level decreased by 50% in the cytoplasmic fraction while there was an increase in the level of calcineurin. In addition, treatment of cells with FK506 or BAPTA during hypoxia, inhibited calcineurin activity and correspondingly increased the levels of IκBβ to that of normoxic cells (Figure 2A). Both ascorbate and MitoQ treatment prevented the hypoxia mediated decrease in IκBβ levels (Figure 2A). This is consistent with the prevailing model of mitochondrial respiratory stress-mediated calcineurin and NFκB activation. Results with MitoQ suggest that the activation of retrograde signaling under hypoxia is mediated by mitochondrial ROS.
Immunoblot analysis of nuclear extract shows three fold accumulation of factors p65 and p50 in hypoxic cells compared to normoxic RAW 264.7 cells (Figure 3). The level of cRel, however did not change under hypoxia. The nuclear level of NFAT2 increased three fold in hypoxic cells as compared to normoxic cells (Figure 3). This is consistent with the increase in calcineurin levels which dephosphorylates and triggers the nuclear translocation of NFAT. Two other calcium dependent nuclear factors, CREB and C/EBPδ were analyzed. About two fold higher level of CREB was also detected in the nuclear extract from hypoxic cells. The effect on C/EBPδ was modest, and corresponded to only 1.5 fold higher in the nuclear extract of hypoxic cells (Figure 3). The role of ROS in the nuclear translocations of these transcription factors was suggested by the effect of ascorbate, a general antioxidant, in reducing the nuclear levels of these factors. Interestingly, MitoQ, a mitochondria-specific antioxidant was fully effective in reducing the nuclear levels of these transcription factors to normoxic levels. These results confirm the role of mitochondria generated ROS in the activation of retrograde stress signaling.
Figure 3.
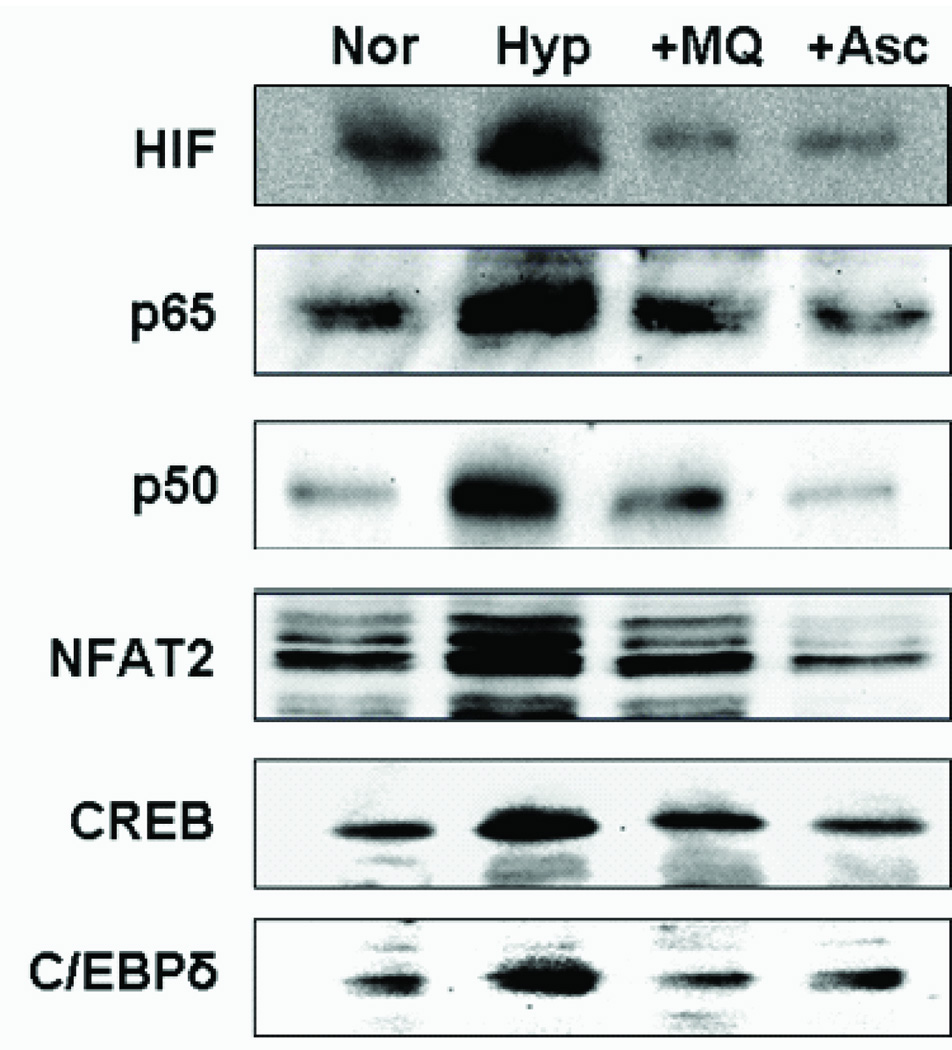
Effect of antioxidants on transcription factors in hypoxic cells. Nuclear extracts (50 µg) from normoxic (Nor), Hypoxic (Hyp), hypoxic cells treated with ascorbate (+Asc) or MitoQ (+MitoQ) were separated on SDS PAGE. Proteins were transferred to PVDF membrane and immunoblotted for the indicated transcription factors.
Role of Mitochondrial ROS in the formation of TRAP positive cells in RAW 264.7 cells
Cells grown under normoxia were treated with different concentrations of RANKL for inducing osteoclastogenesis in the presence or absence of antioxidants. The general antioxidants N-acetyl cysteine, ascorbate and mitochondria-specific antioxidant, MitoQ inhibited RANKL induced osteoclastogenesis in a dose dependent manner. As seen in Figure 4C and D, increasing concentration of MitoQ progressively reduced the number of osteoclasts with complete inhibition at 0.35 µM concentration. This is in accordance with the decrease in ROS formation in MitoQ treated cells (Figure 1A). As expected, treatment with general antioxidant, N-acetyl cysteine inhibited osteoclast formation at >2.5 mM concentration (Figure 4E). Ascorbate, another antioxidant and BAPTA, a chelator of [Ca2+]c also inhibited osteoclast formation, though at higher concentrations than MitoQ (Figure 4F, A and B). In view of our previous results showing that activation of mitochondrial retrograde signaling (also known as mitochondrial respiratory stress signaling), present results suggest that mitochondria generated ROS may be involved in the RANKL mediated osteoclast differentiation. Previously we showed that mild hypoxia alone induced the formation of a small population of TRAP positive cells in RAW 264.7 macrophages 21. Although not shown, morphologically there was no difference in the shapes of RANKL-induced TRAP positive cells formed in normoxic and hypoxic cells. In both cases, however, MitoQ (0.35 µM) effectively prevented the formation of TRAP positive multinucleated cells.
Figure 4.

Measurement of Osteoclasts in antioxidant treated RAW 264.7 cells. Macrophages were plated at 2000 cells per well in a 96 well plate. RANKL (0.25 or 2.5 ng/ml) was added along with the indicated treatments and incubated for 5 days. Number and photographs of TRAP positive osteoclasts in RAW 264.7 cells treated with A & B) BAPTA, C & D) MitoQ, E) N-acetyl cysteine and F) Ascorbate. Cells containing three or more nuclei were counted as osteoclasts.
DISCUSSION
In the physiological milieu, osteoblasts produce RANKL which binds to RANK expressed on the surface of osteoclast precursors and initiates differentiation22. It is widely believed that cells in the macrophage lineage may be the osteoclast precursors. In keeping with this notion, RAW 264.7 macrophages have been shown to differentiate into osteoclasts when stimulated with the physiological stimulus, RANKL Signaling pathways involved in osteoclast differentiation have been extensively studied. Receptor mediated generation of ROS has been implicated in the activation of many downstream signals in target cells. Recent reports show that hypoxia and H2O2 are major stimuli of osteoclast activity23–25. Hypoxia is also shown to be a stimulator of activation of cells derived from bone marrow precursors26. However, the sub cellular site(s) of ROS formation for induction of osteoclastogenesis has not been clearly identified. Although, plasma membrane associated Nox1 and Nox2 are thought to be an important source of RANKL mediated ROS formation, knock out studies in mice have been inconclusive 10–13,27,28.
Some studies suggest that RANKL mediated JNK, p38 and NFκB activation occurs through ROS generated by Nox110. However, Nox1 knockout mice do not show bone abnormalities 12,28. In mature osteoclasts, mRNA levels for Nox4 and Nox2 are elevated, although Nox2 knockout mice were shown to produce superoxide anions and no bone abnormalities have been reported in these mouse models 11,27. Our results using N-acetyl cysteine and ascorbate, general antioxidants indeed suggest that cellular ROS plays a role in the differentiation of macrophages into osteoclasts. Interestingly, mitochondria targeted antioxidant; MitoQ is equally efficient in blocking osteoclastogenesis. This cationic compound is known to accumulate in the mitochondrial compartment with about 1000 fold higher gradient inside mitochondria as compared to extra mitochondrial compartment. Notably, the concentration of MitoQ required to completely inhibit osteoclast formation was nearly 1000 fold less than that of N-acetyl cysteine or ascorbate required for inhibition. Our results therefore suggest that mitochondrial ROS plays an important role in the differentiation of RAW 264.7 macrophages into osteoclasts.
A number of factors including mutagens, drugs, mtDNA depletion and hypoxia are known to induce mitochondrial dysfunction and induce ROS production29. Several laboratories including ours have shown that dysfunctional mitochondria with disrupted mitochondrial transmembrane potential induce a respiratory stress signaling which is also called mitochondrial retrograde signaling. Activation and propagation of mitochondrial stress signaling is known to involve activation of calcineurin and Ca2+ kinase mediated activation of ATF2, NFkB, CREB and C/EBPδ6,29,30 and subsequent transcriptional activation of several target genes 31. This signaling cascade has been shown to modulate the up regulation of about 120 nuclear genes involved in Ca2+ homeostasis, mitochondrial function, cell surface receptors, apoptosis, and oxidative stress 32. Interestingly, the important factors activated under mitochondrial stress signaling such as calcineurin-NFAT pathway, NFKB, and CREB are also key signaling molecules during osteoclastogenesis. Our previous and present results show that hypoxic conditions indeed induce mitochondrial ROS production, disruption of transmembrane potential and activation of retrograde signaling. We therefore propose that intra mitochondrial ROS production due to respiratory stress may be an important contributor to osteoclast differentiation33–35.
In summary, our results show that mitochondria-targeted antioxidant MitoQ suppresses osteoclastogenesis. There is compelling evidence showing that mitochondria generated ROS causes inactivation of mitochondrial electron chain complexes in addition to inducing lipid peroxidation and mitochondrial membrane dysfunction. These results for the first time suggest a role for mitochondrial function and ROS production in osteoclast differentiation. Our results also open up the possibility that RANKL mediated effects may include mitochondrial oxidative and metabolic functions.
Acknowledgements
This work was supported by NIH grant CA-22762. AK was supported by the Merck/NIH summer research program (RR 07065) at the University of Pennsylvania, School of Veterinary Medicine.
References
- 1.Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 2.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 3.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 2006;12:1410–1416. doi: 10.1038/nm1515. [DOI] [PubMed] [Google Scholar]
- 5.Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–7849. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- 6.Biswas G, Guha M, Avadhani NG. Mitochondria-to-nucleus stress signaling in mammalian cells: nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene. 2005;354:132–139. doi: 10.1016/j.gene.2005.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am. J. Physiol Lung Cell Mol. Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 8.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 9.Ha H, Kwak HB, Lee SW, Jin HM, Kim HM, Kim HH, Lee ZH. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp. Cell Res. 2004;301:119–127. doi: 10.1016/j.yexcr.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 10.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 11.Yang S, Zhang Y, Ries W, Key L. Expression of Nox4 in osteoclasts. J. Cell Biochem. 2004;92:238–248. doi: 10.1002/jcb.20048. [DOI] [PubMed] [Google Scholar]
- 12.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Rokutan K. NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J. Med. Invest. 2009;56:33–41. doi: 10.2152/jmi.56.33. [DOI] [PubMed] [Google Scholar]
- 14.Chiarugi P, Fiaschi T. Redox signalling in anchorage-dependent cell growth. Cellular Signalling. 2007;19:672–682. doi: 10.1016/j.cellsig.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 16.Inoue M, Sato EF, Nishikawa M, Park AM, Kira Y, Imada I, Utsumi K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr. Med. Chem. 2003;10:2495–2505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 17.Kalivendi SV, Konorev EA, Cunningham S, Vanamala SK, Kaji EH, Joseph J, Kalyanaraman B. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem. J. 2005;389:527–539. doi: 10.1042/BJ20050285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy M, Rockenbauer A, Vasquez-Vivar J, Felix C, Lopez M, Srinivasan S, Avadhani N, Tordo P, Kalyanaraman B. Detection, characterization, and decay kinetics of ROS and thiyl adducts of mito-DEPMPO spin trap. Chem. Res. Toxicol. 2007;20:1053–1060. doi: 10.1021/tx700101d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Addya S, Anandatheerthavarada HK, Biswas G, Bhagwat SV, Mullick J, Avadhani NG. Targeting of NH2-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J Cell Biol. 1997;139:589–599. doi: 10.1083/jcb.139.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srinivasan S, Avadhani NG. Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann. N. Y. Acad. Sci. 2007;1117:51–61. doi: 10.1196/annals.1402.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 23.Muzylak M, Price JS, Horton MA. Hypoxia induces giant osteoclast formation and extensive bone resorption in the cat. Calcif. Tissue Int. 2006;79:301–309. doi: 10.1007/s00223-006-0082-7. [DOI] [PubMed] [Google Scholar]
- 24.Fukuoka H, Aoyama M, Miyazawa K, Asai K, Goto S. Hypoxic stress enhances osteoclast differentiation via increasing IGF2 production by non-osteoclastic cells. Biochem. Biophys. Res Commun. 2005;328:885–894. doi: 10.1016/j.bbrc.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 25.Arnett TR, Gibbons DC, Utting JC, Orriss IR, Hoebertz A, Rosendaal M, Meghji S. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J Cell Physiol. 2003;196:2–8. doi: 10.1002/jcp.10321. [DOI] [PubMed] [Google Scholar]
- 26.Koller MR, Bender JG, Miller WM, Papoutsakis ET. Reduced oxygen tension increases hematopoiesis in long-term culture of human stem and progenitor cells from cord blood and bone marrow. Exp. Hematol. 1992;20:264–270. [PubMed] [Google Scholar]
- 27.Yang S, Madyastha P, Bingel S, Ries W, Key L. A new superoxide-generating oxidase in murine osteoclasts. J. Biol. Chem. 2001;276:5452–5458. doi: 10.1074/jbc.M001004200. [DOI] [PubMed] [Google Scholar]
- 28.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 29.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol. Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 30.Liao X, Butow RA. RTG1 and RTG2: Two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- 31.Guha M, Pan H, Fang JK, Avadhani NG. Heterogeneous nuclear ribonucleoprotein A2 is a common transcriptional coactivator in the nuclear transcription response to mitochondrial respiratory stress. Mol. Biol. Cell. 2009;20:4107–4119. doi: 10.1091/mbc.E09-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biswas G, Tang W, Sondheimer N, Guha M, Bansal S, Avadhani NG. A distinctive physiological role for IkappaBbeta in the propagation of mitochondrial respiratory stress signaling. J. Biol. Chem. 2008;283:12586–12594. doi: 10.1074/jbc.M710481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol Pathol. 2007;83:84–92. doi: 10.1016/j.yexmp.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Wallace DC. A mitochondrial paradigm for degenerative diseases and ageing. Novartis. Found. Symp. 2001;235:247–263. doi: 10.1002/0470868694.ch20. [DOI] [PubMed] [Google Scholar]
- 35.Puddu P, Puddu GM, Galletti L, Cravero E, Muscari A. Mitochondrial dysfunction as an initiating event in atherogenesis: a plausible hypothesis. Cardiology. 2005;103:137–141. doi: 10.1159/000083440. [DOI] [PubMed] [Google Scholar]