

Abstract
The last ten years have witnessed a surge of interest for the mechanisms underlying the acquisition and extinction of classically conditioned fear responses. In part, this results from the realization that abnormalities in fear learning mechanisms likely participate to the development and/or maintenance of human anxiety disorders. The simplicity and robustness of this learning paradigm, coupled to the fact that the underlying circuitry is evolutionarily well conserved makes it an ideal model to study the basic biology of memory and identify genetic factors and neuronal systems that regulate the normal and pathological expressions of learned fear. Critical advances have been made in determining how modified neuronal functions upon fear acquisition become stabilized during fear memory consolidation and how these processes are controlled in the course of fear memory extinction. With these advances, came the realization that activity in remote neuronal networks must be coordinated for these events to take place. In this paper, we review these mechanisms of coordinated network activity and the molecular cascades leading to enduring fear memory, and allowing for their extinction. We will focus on Pavlovian fear conditioning as a model and the amygdala as a key component for the acquisition and extinction of fear responses.
Keywords: Pavlovian fear conditioning, extinction, prefrontal cortex, hippocampus, synaptic plasticity, oscillations
I. Introduction: The Case for an Animal Model of Fear and Anxiety
Fear and anxiety are adaptive responses generated in anticipation or in the presence of stimuli that threaten to perturb homeostasis. While fear is generally elicited by particular cues or contexts, anxiety can occur in the absence of these triggers (88). Fear and anxiety are exhibited by all mammals, including humans, and appear to be part of a universal survival strategy. Not surprisingly, these states are controlled by a hierarchy of neural systems, which determine the efficacy of the responses and permit dynamic adaptations, thereby ensuring appropriate emotional responses and return to baseline activity once the threat has passed. Extreme variations or perturbations of these mechanisms can lead to prolonged (even irreversible) and disproportional states with respect to the triggering stimulus, persistence of anxiety following withdrawal of the stimulus, or omnipresent generalized anxiety. In their extreme or pathological forms, these states include panic disorders, phobias, and posttraumatic stress disorders (510, 523). These are common diseases with an estimated lifetime prevalence of up to 18% (8, 200), imposing a major challenge to health providers and burden on the economy. A large body of evidence indicates that these states are under genetic and environmental control, during early development as well as later in life, determining inter-individual variations (for review see (143)). Genome-wide linkage analysis and association studies have indeed led to identification of a number of genetic factors that determine the heritability of anxiety disorders, although the wide spectrum of symptoms and restricted sample sizes have limited success so far (143, 163).
The motivation for developing animal models of fear and anxiety is thus twofold. First, animal models allow the study of single-gene modifications in a well-defined genetic background and under controlled environmental conditions, thereby partly overcoming problems inherent to human genomic studies. Second, because fear is well conserved throughout evolution, it is a near-ideal model system to study interactions between genetic factors, operating brain circuits, and behavior, allowing one to unveil the principles regulating the impact of environmental influences, learning and memory.
Of the various models used to investigate emotional behaviors (review in (124), classical “Pavlovian” fear conditioning has proven particularly useful and successful (reviewed in (239)). In this task, subjects learn to associate a previously neutral sensory stimulus [conditioned stimulus (CS), such as a tone, light or odor] or context with a coinciding aversive stimulus [unconditioned stimulus (US), such as a brief electric stimulus]. A memory is formed, so that subsequent exposure to the CS or conditioned context will elicit conditioned fear responses (CRs). These responses involve autonomic components (like hypertension, tachycardia, and hypoalgesia), an overall endocrine arousal, as well as species-specific defensive behaviors, such as freezing and flight (239). Of particular advantage is the possibility to use this task in various species, including humans (369). Furthermore, learning on this task is rapid, robust, readily quantified, and allows for a precise control of major fear memory-modulating parameters, as, for instance, stimulus specificity and predictability, or stress level (for review see (410, 417)). These features make Pavlovian fear conditioning a near-ideal experimental model for identifying critical genetic factors and neuronal systems that drive fear responses and studying how they are regulated by environmental influences. In fact, the last two decades have witnessed an explosion of interest for the mechanisms underlying this relatively simple form of learning. The number of yearly citations returned by PubMed searches using the keywords “Fear Conditioning” rose from ≈50 in the late 80s, to ≈200 at the turn of the century, to ≈1400 in 2006 and 2007. One contributing factor behind this upsurge in interest is the realization that fear learning mechanisms may participate in the etiology of human anxiety disorders. Indeed, the findings of lesion and physiological studies in animals have been confirmed in human work (24, 53, 224, 525). Moreover, human subjects with anxiety disorders exhibit abnormalities in the acquisition and extinction of conditioned fear responses (144, 306, 343). While it remains controversial whether anxiety disorders represent pathological manifestations of normal fear learning mechanisms (302, 312, 313, 383), there is consensus that the structures normally involved in such learning display abnormal activity patterns in anxious subjects (49, 467). Another factor fuelling this sustained level of interest for fear conditioning is the realization that this task is perfectly suited for studying learning and memory formation. Indeed, this model has allowed the identification of key neuronal circuits, neurochemical components, and synaptic events underlying fear memory formation. As a result, the amygdala has been identified as a key region for the processing of aversive signals and fear learning in various species including humans (for reviews see (37, 274, 299, 369, 464, 473)). This knowledge, in turn, provides a strong basis to test the role of particular gene products in a functional context (for review see (486)). For instance, there is consensus now that memory consolidation, the process whereby a memory shifts from a transient state (referred to as short-term memory) to a stable form (referred to as long-term memory), requires gene expression and de novo protein synthesis (193). However, long-term memories are not consolidated in a formal sense, but remain in a labile state, or become labile again after consolidation, susceptible to change and disruption, as for instance after memory retrieval, and therefore require “re-consolidation” (as recently reviewed in (331)). Research on fear conditioning has also paved the way for a better understanding of extinction, a simple form of fear behavior regulation, in which conditioned fear responses decrease when the CS is presented repeatedly in the absence of the US (as reviewed in (274, 327, 389)). The mechanisms of fear extinction have attracted significant interest because of their potential clinical significance (327, 457)).
In recent years, critical advances have been made in determining how the transient synaptic modifications induced during fear conditioning become stabilized during fear memory consolidation (412) and how these processes can be controlled in the course of fear memory extinction (327, 457). With these advances, came the realization that activity in remote neuronal networks must be coordinated for these events to take place. In the present paper, we will review these mechanisms of coordinated network activity and the molecular cascades leading to enduring fear memory on the one hand, and allowing for extinction of these memories on the other. We will focus on Pavlovian fear conditioning as a model and the amygdala as a key component of conditioned fear responses. The reader is referred to a number of excellent reviews for theoretical and behavioral accounts of fear conditioning and extinction (239, 274, 299, 327, 369, 389, 457), the impact of contextual influences (180, 431), the re-consolidation of fear memories (331), the role of the GABAergic system (105) or neuromodulatory systems such as monoamines and stress hormones (299, 410, 417), the neurobiology of anxiety states and disorders (152, 310, 484), experimental models (124, 472) and genetic approaches to these disorders (143, 163, 486).
II. Structure and connectivity of the amygdala
Located in the anterior portion of the temporal lobe, the amygdala is comprised of a dozen or so nuclei and cortex-like structures. Most of these components have been divided in two or more subnuclei that exhibit significant differences in connectivity. Since many comprehensive reviews on the structure and connectivity of the amygdala have been published before (9, 371), we will limit the following account to components of the amygdala that are thought to be involved in the acquisition and extinction of conditioned fear responses. These include the basolateral complex (BLA), the central nucleus (CE), and the intercalated (ITC) cell masses (Fig. 1). Below, we first provide an overview of the structure and cellular composition of these three components and then summarize their connectivity.
Fig. 1.
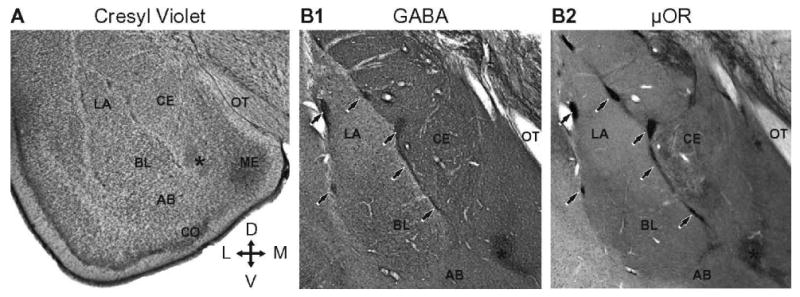
Macroscopic organization of the rat amygdala. Coronal sections. (A) Cresyl violet stain. (B) Two adjacent sections processed to reveal immunoreactivity for GABA (B1) or μ opioid receptors (μOR; B2). Note spatial correspondence between zones expressing high levels of GABA and μOR immunoreactivity. Arrows point to ITC cell clusters. Asterisks indicate main ITC cluster. Cross indicates orientation of the sections where D, V, L, and M respectively stand for dorsal, ventral, lateral, and medial. Abbreviations: AB, accessory basal nucleus; BL, basolateral nucleus; CE, central nucleus; CO, cortical nucleus; LA, lateral nucleus; ME, medial nucleus; OT, optic tract.
A. Structure and cell types
1. Basolateral complex (BLA)
The BLA is comprised of three nuclei: the lateral (LA), basolateral (BL), and basomedial (BM) nuclei. The latter is also known as the accessory basal (AB) nucleus. Moreover, BL and BM are sometimes referred to as the basal nuclei. Morphologically, the neuronal composition of the BLA is similar to that of the cerebral cortex except for the fact that neurons are randomly oriented in the BLA. As in cortex, BLA contains two classes of neurons (reviewed in (285)). The dominant group (≈80%) are glutamatergic projection cells with multipolar dendritic trees covered with spines and axons contributing multiple collaterals to neighboring BLA cells, amygdala nuclei, or other structures of the brain (146, 147, 191, 289). As in cortex, most BLA projection cells express a regular spiking phenotype, with marked cell-to-cell variations in the amount of spike frequency adaptation they exhibit (70, 107, 110, 111, 118, 229, 231, 279, 354, 358, 394, 517).
The second class of BLA neurons consists of local-circuit GABAergic cells with short axons and aspiny to sparsely spiny dendrites (≈20% of the cells). Again as in cortex, local-circuit neurons are heterogeneous morphologically (146, 147, 191, 289), electrophysiologically, and neurochemically, with different subgroups of local-circuit cells expressing neuropeptide Y (NPY), somatostatin (SOM), vasoactive intestinal peptide, or cholecystokinin (CCK; (177, 198, 291-295, 372-374)). In addition, as in cortex, GABA co-localizes with calcium-binding proteins, such as parvalbumin (PV) and calbindin in a high proportion of interneurons (≈ 50-60%; (198, 291-295, 372-374, 478, 479)). Moreover, there is evidence that as in cortex, different sub-types of BLA interneurons target different compartments of projection cells. For instance, PV-immunoreactive interneurons of the BLA tend to contact the soma, initial axonal segment, and/or proximal dendrites of projections cells (298, 325, 477) whereas SOM immunoreactive interneurons preferentially contact their distal dendrites (324). On the input side, there is also evidence that BLA afferents target different subsets of interneurons. For instance, PV interneurons receive few if any cortical inputs but are massively innervated by BLA projection cells (475), suggesting a prevalent involvement in feedback inhibition. At odds with this however, a physiological study (496) reported that most fast-spiking interneurons receive convergent monosynaptic inputs from the cortex and thalamus. Finally, there is physiological and ultrastructural evidence that interneurons belonging to the same neurochemical class are coupled by gap junctions (323, 528), in PV neurons at least.
Compared to cortex, far less data is available on the physiological properties of BLA interneurons but the results obtained so far are generally consistent with the cortical literature. Indeed, the repetitive firing properties of local-circuit cells are extremely diverse, even among neurochemically-homogeneous subgroups (177, 396, 479, 528). For instance, in one study (528), four different subtypes of PV interneurons were observed (fast-spiking, stuttering, delayed firing, and accommodating). Similarly, three subtypes of CCK interneurons were described (177).
2. Central nucleus (CE)
Early accounts identified two divisions in the CE nucleus: lateral (CEl) and medial (CEm; (26, 51, 127, 183, 216). However, the rat CEl was later subdivided further with significant variations between investigators (65, 185, 287, 367, 493). From lateral to medial, these subdivisions include an amygdalostriatal transition area, a lateral sector proper, and interposed between CEl and CEm, an intermediate subnucleus. Because there is scant data indicating that these different subdivisions of CEl form distinct connections, this review will adhere to the initial division of CE in lateral and medial sectors. Finally, a capsular region of CE was identified; it encapsulates CE ventrolaterally (287) and appears to overlap with ITC cell clusters. We will therefore use the latter term for the capsular region.
CEl and CEm each contain one main cell type (64, 65, 146, 191, 287, 500). Although these cells are thought to be GABAergic (288, 290, 359), some might use a different neurotransmitter as many do not stain positively for the GABA synthesis enzyme (glutamic acid decarboxylase-GAD; (373, 491)). In CEl, the main cell type is indistinguishable from medium spiny striatal neurons. Indeed, these cells have multiple primary dendrites that branch profusely and bear a high density of spines. By comparison, the main neuronal type in CEm has a larger soma, dendrites that branch more sparingly, and bear a lower density of dendritic spines. In addition, CEm and CEl contain a low number of aspiny GABAergic local-circuit neurons. Here, it should be mentioned that whereas the GABAergic innervation of the BLA mostly has an intrinsic origin, that of CE includes a significant extrinsic component. This statement is based on a neurochemical study where interruption of the main pathways linking the amygdala with the rest of the brain decreases GAD levels in CE, but not BLA (236).
In terms of electroresponsive properties, the prevalent types of CEl and CEm neurons express a regular spiking firing pattern with variable degrees of spike frequency adaptation and a hyperpolarization-activated cation current (102, 260). Moreover, a proportion of CE neurons are endowed with a t-type calcium current, giving rise to low-threshold spike bursts (102). Because retrograde tracing studies indicate that the vast majority of CEm cells are projection neurons (162), it is likely that these cells are output neurons. Finally, CE also contains a small subgroup of cells with comparatively depolarized resting potentials, higher input resistances, and fast-spiking or burst-firing patterns. These neurons likely correspond to local-circuit cells (102, 260).
3. Intercalated (ITC) cell masses
As a group, ITC cells form a reticulated sheet of neurons that spans the entire rostrocaudal extent of the amygdala (311). ITC neurons occur as small densely packed cell clusters distributed in the main fiber bundles found in and around the amygdala. They are marked by arrows in Fig. 1B. These include the external capsule that borders the BLA laterally as well as the intermediate capsule, the fiber bundle separating the BLA from CE. ITC clusters located in the external and intermediate capsules will hereafter be termed ITC-L and ITC-M, respectively. In addition, in most species, there is a larger ITC cell mass: in cats, it caps the amygdala rostrally (359) whereas in rats is it located dorsomedial to the basal nuclei (339). This larger ITC cluster is labeled with an asterisk in Fig. 1B.
There are two types of ITC neurons. The prevalent type is characterized by a small soma (8-19 μm in diameter), a flattened dendritic tree that mostly remains within the confines of the fiber bundle where its soma is located, and a high density of dendritic spines. These cells are GABAergic (Fig. 1B1; (288, 290, 339, 359)) and express an extremely high density of μ opioid and dopamine type-1 receptors (Fig. 1B2; (156, 176, 381)). In addition, a minute proportion of ITC cells have extremely large somata (>40 μm in diameter) and exceptionally long aspiny dendrites. Little is known about these cells except that they are not GABAergic but perhaps cholinergic (340). They will not be discussed further in this review.
Compared to principal neurons of the BLA and CE, principal ITC cells have a very high input resistance (500-900 MΩ), and can sustain higher firing rates with only modest spike frequency accommodation (137, 277, 426). In guinea pigs, ITC cells exhibit a bistable behavior because they express an unusual voltage-dependent K+ current termed ISD (SD for slowly deinactivating; (426)). ISD activates at subthreshold membrane potentials, inactivates with depolarizations beyond spike threshold, and de-inactivates very slowly upon return to rest. Thus, following periods of firing, ITC cells assume a state of augmented excitability characterized by a sustained membrane depolarization and reduced conductance, thereby increasing the probability that synaptic inputs will trigger spiking.
B. Intrinsic Connectivity
1. Synaptic interactions within the BLA
The BLA is endowed with an extremely divergent system of intrinsic connections. Indeed, principal cells contribute multiple axon collaterals that bear varicosities (146, 147, 191, 289) forming en passant excitatory synapses, usually with other principal neurons (476). Based on the length of the intervaricose segments, it was estimated (476) that each principal cell forms 100-200 excitatory synapses per millimeter of axon, most with other principal cells. Given the presence of a profusely divergent system of excitatory connections between principal BLA neurons, one would expect them to exhibit high firing rates. Yet, single unit recordings in unanesthetized animals have consistently emphasized the opposite (43, 136, 357). As we shall see below, the solution to this paradox resides in the spatial heterogeneity of connections formed by principal cells with interneurons.
Previous work has revealed that several factors reduce the excitability of principal cells. First, they express a calcium-dependent K+ conductance (gKCa) that can be activated when glutamatergic synapses cause Ca2+ entry via NMDA receptors, thereby shunting EPSPs (70, 87, 109, 230). Second, the spontaneous activity of projection cells in vivo is dominated by large amplitude IPSPs mediated by GABA-A and B receptors following GABA release by local-circuit cells (87, 279, 395, 518). We discuss these two mechanisms here in some detail because they interfere with induction of long-term potentiation (LTP) and, as we shall see in section IV, this suppressing effect is relieved by neuromodulators that are released in emotionally arousing conditions (35, 109, 504).
The first clue to the participation of intrinsic gKCa conductances to synaptically evoked inhibition came from intracellular recording studies where it was observed that cortical stimulation triggered EPSPs that were curtailed by large amplitude IPSPs with a reversal potential negative to that expected for chloride mediated GABA-A IPSPs (87, 229). This observation implied that overlapping chloride and K conductances participated to the IPSP. However, this effect was seen at too short a latency for a mediation by GABA-B IPSPs. Furthermore, dialysis of principal cells with a calcium chelator produced a gradual positive shift in IPSP reversal potential toward that expected for pure GABA-A IPSPs, implying a mediation by gKCa (70, 87, 230). Moreover, Ca2+ chelation altered evoked responses within 5 ms of their onset, suggesting that the Ca2+ source (NMDA receptors; (87)) and gKCa channels were in close proximity, possibly in the same dendritic spines or branches (70, 87, 230). Subsequent studies yielded inconsistent results regarding the identity of the gKCa channels involved ((70) IK; (109) SK channels).
The second mechanism reducing the excitability of principal cells, namely GABA-A and B receptor-mediated inhibition, is similar to that seen in neurons throughout the prosencephalon. However, it is expressed differently in principal cells and interneurons of the BLA. Indeed, fast-spiking interneurons are subjected to less inhibition than BLA projection cells. First, they receive a markedly lower proportion of inhibitory synapses (477). Second, IPSPs in BLA interneurons lack a GABA-B component (279). They are comprised of apparently pure GABA-A IPSPs that reverse at more depolarized potentials than in projection cells (by > 15 mV; just under spike threshold). Pharmacological analyses in vitro suggest that this is secondary to a contrasting regulation of intracellular chloride in the two cell types. In projection cells, the main regulators of intracellular chloride are cation-chloride cotransporters that extrude chloride whereas in local-circuit neurons, transporters that accumulate chloride predominate (279).
The various factors listed above should contribute to make interneurons more excitable than projection cells. However, given the extremely divergent excitatory connections that exist between projection cells, their low spontaneous firing rates remains surprising. The key to this paradox resides in the spatial heterogeneity of connections formed by projection cells with each other and interneurons (438, 440). By antidromically activating the axons of LA projection cells ending in the BM nucleus, one study inferred the intra-LA targets of projection cells (438). BM stimuli evoked markedly different synaptic responses depending on the slice orientation with inhibition dominating in coronal slices and excitation in horizontal slices. These results implied that the axon collaterals of projection cells contact different cell types depending on the rostrocaudal position of their targets: inhibitory interneurons at proximity and other projection cells at a distance. A subsequent study (440), using local pressure application of glutamate, revealed that the spatial heterogeneity of connections was not limited to feedback interneurons. Indeed, glutamate application at a distance from the recorded projection cells evoked only inhibitory responses in coronal slices. By contrast, in horizontal slices, the character of the responses depended on the lateromedial position of the glutamate ejection site with respect to the recorded cell. Ejection sites located laterally to the recorded cells evoked mostly excitation whereas inhibition was typically elicited from medial sites. Overall, the ubiquity of inhibition in coronal slices combined with the predominance of excitatory responses in horizontal slices imply that the LA network is designed to allow associative interactions within the rostrocaudal plane while preventing runaway excitation locally.
2. Synaptic interactions within CE
Far less data is available on intrinsic synaptic interactions in CE. As mentioned above, Golgi studies (reviewed in (285)) suggest that CE contains a much lower proportion of local-circuit cells than the BLA. However, projection cells are GABAergic (288, 290) and may inhibit each other via their local axon collaterals. Consistent with this, one study reported that local pressure application of glutamate in CEl evoked IPSPs in CEl neurons (260). In contrast, the same stimuli applied in CEm elicited no responses in CEl cells, in keeping with the lack of connections form CEm to CEl (185). Interestingly, BLA stimulation was reported to elicit an EPSP-IPSP sequence in CE neurons (260, 427, 428). However, local pressure application of glutamate receptor antagonists close to the recorded CE cells (to prevent the excitation of CE interneurons) had little effect on this inhibition (427). This suggests that a significant portion of inhibitory inputs to CE neurons have an extrinsic origin, most likely, ITC and BNST neurons. Finally, it should be mentioned that CE neurons express two types of ionotropic GABA receptors: GABA-A receptors that are blocked by low concentrations of bicuculline and GABA-C receptors that are less sensitive to bicuculline (90, 91). These receptors appear to be expressed differentially at somatic vs. dendritic GABAergic inputs (91).
3. Synaptic interactions within and between ITC cell clusters
Experiments in mice and guinea pigs have revealed that ITC cells are interconnected. Within ITC cell clusters, one study reported that 14% of ITC cell pairs were connected unidirectionally and a much lower proportion bidirectionally (137). These GABAergic synapses exhibited heterogeneous short-term plasticity when presynaptic ITC cells were repeatedly activated with current injection at 0.1-10 Hz. In a roughly equal proportion of cell pairs, release probability increased, decreased, or remained constant. This variability was determined by the properties of the presynaptic neurons since sequential paired recordings revealed that the same presynaptic neuron formed the same type of synaptic connections with different postsynaptic neurons and conversely, that the same postsynaptic neuron was contacted by different types of synapses from different presynaptic neurons (137).
There are also connections between different ITCm clusters (Fig. 2A). So far, this question has only been investigated in guinea pigs (428). In this species, CE is dorsomedial to BLA such that the lateromedial axis in the guinea pig amygdala corresponds to the dorsoventral axis in the rat amygdala. Using local pressure applications of glutamate, it was found that laterally located ITC cell clusters inhibit more medial ones. The same study revealed that this directionality originated from the morphological properties of ITC neurons with their dendrites extending over longer distances in the lateral than the medial direction whereas their axons showed the opposite asymmetry. We will return to the significance of these observations when discussing the interactions between the BLA and CE.
Fig. 2.

Intrinsic connectivity and CS-US input pathways of the amygdala. (A) Scheme showing the directionally polarized connections that exist between different ITCm cell clusters in guinea pigs. These connections prevalently run from lateral to medial. Cross indicates orientation of the sections where D, V, L, and M respectively stand for dorsal, ventral, lateral, and medial. (B) Summary of main internuclear connections between the BLA, CE, and ITC cells. Note that BL and AB also contribute projections to CeL but these were omitted from the scheme for clarity. (C) Scheme illustrating the various routes that exist for the transfer of CS or US information to the amygdala. Note the contrasting termination patterns of PO vs. MGm-PIN in the amygdala. (D) Scheme illustrating the various indirect routes that exist between LA and CeM along with their expected impact on CeM neurons (right). Abbreviations: AB, accessory basal; BL, basolateral; CeL, central lateral; CeM, central medial; ITC, intercalated; LA, lateral; MGm, medial sector of the medial geniculate nucleus; OT, optic tract; PIN, posterior intralaminar nucleus; PO, posterior thalamic nucleus.
C. Internuclear connections
A prominent feature of amygdala organization is the existence of strong and directionally polarized inter-nuclear connections (Fig. 2B; (362, 377)). Within the BLA, there are strong glutamatergic projections from LA to the basal nuclei, particularly massive to BM (216, 360, 375, 378). Some projections from the basal nuclei to LA exist but they are weaker and confined to the most ventral sector of LA (363, 448). Thus, in the rat BLA, the prevalent directionality of inter-nuclear connections is from dorsal to ventral.
Principal BLA cells also project to CE, a projection that is not reciprocated (216, 363, 378, 446, 447, 476). Here, it should be noted that whereas the basal amygdala nuclei project to CEl and CEm, LA only projects to CEl. Given the contrasting projections of CEl and CEm to the brainstem (162, 368), this point will become critical when considering the intra-amygdala pathways participating in fear conditioning.
As BLA axons course toward CE, they form excitatory synapses with ITCm cells (Fig. 2A-B; (189, 427)). In turn, ITCm cells project to CE (361, 427) where they generate feed-forward inhibition (361, 427, 428). Physiological studies in guinea pigs indicate that there is a lateromedial correspondence between the position of ITCm cells, where they derive inputs from BLA, and where they project in CE (427). Assuming that these findings hold in rats, given the differing relative position of BLA and CE in the two species, this would mean a dorsoventral correspondence between the position of rat ITC cells, their BLA inputs and CE outputs. Because this topographical arrangement overlaps with dorsoventral connections between ITC cell clusters, the impact of ITCm activity of different parts of CE will depend on the distribution of activity in the BLA. For instance, even though LA does not project to CEm, it could indirectly affect CEm neurons by exciting laterally located ITCm clusters, which in turn would inhibit more medial ones, leading to a disinhibition of CEm cells.
D. Extrinsic Connectivity
1. Basic organizing principles of amygdala connectivity
The amygdala forms connections with an extremely diverse array of structures including cortex, striatum, some thalamic and hypothalamic nuclei, as well as various basal forebrain structures and brainstem nuclei (reviewed in (9, 371)). As a result, the amygdala is in a position to influence a wide variety of processes from autonomic and motor control to memory formation and neuromodulation. Here, we first highlight basic organizing principles of amygdala connectivity and then consider in more detail the extrinsic and intrinsic pathways thought to participate in the acquisition and expression of conditioned fear responses.
Different amygdala nuclei project to different classes of CNS structures
There is a clear segregation of target structures depending on the amygdala nuclei originating the projections. Indeed, cortical and striatal projections of the amygdala originate from the BLA, not from CE (215, 217, 218). Conversely, BLA has little if any brainstem outputs whereas CE sends strong projections to various brainstem structures (162) involved in generating the behavioral and autonomic correlates of fear (88). However, BLA and CE do send overlapping projections to the lateral hypothalamus, basal forebrain regions containing cholinergic corticopetal neurons as well as to the bed nucleus of the stria terminalis (BNST). The latter target is of particular interest because BNST and CE are reciprocally connected and their brainstem projections overlap extensively (94-99, 162). As to ITC cells, they do not project outside the amygdala, except for a projection from the main ITC group to the substantia innominata and diagonal band of Broca (horizontal limb) (360).
The amygdala receives information about all sensory modalities
Depending on the modality, sensory information can reach the amygdala via the thalamus, cortex, or more direct subcortical routes. Generally, sensory inputs from cortex do not originate from primary sensory areas but reach the amygdala after a cascade of corticocortical projections involving one or more associative cortical areas (286). Consistent with this, sensory information from the thalamus does not originate from specific thalamic nuclei, such as the lateral geniculate or ventrobasal nucleus, but from components of the posterior thalamic complex that tend to receive divergent and typically multisensory sensory inputs (186, 240, 241, 257, 505). We will consider the origin and termination of sensory inputs to the amygdala in more detail below, in the context of fear conditioning.
Many more cortical areas project to the amygdala than targeted by the amygdala
Indeed, a diverse array of associative and polymodal cortical areas project to the amygdala (286, 436). In contrast, the BLA has no cortical projections other than the medial prefrontal cortex (mPFC), insula, rhinal cortices, and a few hippocampal fields (217, 218). Importantly, the latter statement is only valid for lower species (mouse, rat, cat, rabbit). In primates, there is a tremendous expansion of cortical projections including primary sensory and motor areas as well as a number of associative cortical areas (2, 6, 130, 380).
Cortical inputs to the amygdala originate from different layers depending on the target nucleus
Paralleling the cortex-like nature of BLA and striatal-like properties of CE, cortical inputs to BLA and CE mainly originate from layer III and layer V pyramidal cells, respectively (52, 436). Yet, even though cortical cells projecting to BLA and CE tend to be located in different layers, most cortical areas that send axons to BLA also project to CEl (286). In contrast, CEm receives very few cortical inputs, suggesting that inhibition and disinhibition are major determinants of CEm outputs.
The amygdala sends robust projections to neuromodulatory cell groups of the brainstem and basal forebrain
While most prosencephalic structures, including the amygdala (112), receive inputs from neuromodulatory systems (485), relatively few contribute dense projections to these cell groups. The amygdala is a notable exception to this general rule. Via these projections, the amygdala can influence the general excitability of much of the brain, even of structures it is not directly connected to. In turn, because the neuromodulatory inputs often exert facilitating influences on synaptic plasticity (1, 150), these pathways likely enhance the formation of Pavlovian associations and may partly explain how the amygdala facilitates memory formation for emotionally arousing experiences (299). With the exception of substantial BLA projections to the substantia innominata and diagonal band of Broca (184, 215), most amygdala projections to neuromodulatory cell groups originate in CE. These include projections to cholinergic and noradrenergic cell groups located at the junction of the pons and mesencephalon, as well as dopaminergic cells groups of the ventral tegmental area and substantia nigra pars compacta (162, 384).
E. Input and output pathways involved in fear conditioning
Because most data on the cellular and molecular substrates of fear conditioning was obtained using auditory CSs paired with footshocks as USs, the following will focus on sensory pathways relaying auditory and nociceptive inputs to the amygdala. As we shall see, CS and US information can reach the amygdala through multiple routes (Fig. 2C).
CS and US input pathways
First, LA receives auditory inputs from the posterior intralaminar nucleus (PIN) and the medial sector of the medial geniculate nucleus (MGm) (244, 257, 469, 505, 529). Auditory inputs to PIN and MGm originate in the inferior colliculus (IC; (241, 256)). Auditory inputs also reach LA via thalamic projections to temporal auditory cortical fields that innervate LA (280, 415, 416, 466). Importantly, the same posterior thalamic regions that relay auditory information to LA also receive inputs from the spinothalamic tract (243) and may therefore send convergent CS and US inputs to LA.
The above routes of CS and US communication to the amygdala have been studied extensively and figure prominently in most models of auditory fear conditioning. As reviewed in section IV, convergence of CS and US in LA was shown to produce long-term changes in the efficacy of synapses conveying CS information (274). Reversible inactivation of LA during conditioning was found to prevent the acquisition of conditioned fear responses (275, 322, 527), and animals with excitotoxic lesions of the BLA could learn normal contextual fear, but showed substantial forgetting 30 days after training compared to intact controls (382). As a result, LA is thought to be the primary storage site of conditioned CS-US associations (239) and the BLA is thought to be critical for remote fear memories (382). However, other paths exist for the transfer of CS information to the amygdala, but they have received little attention so far. For instance, medial to PIN is the posterior thalamic nucleus (PO). PO could relay CS information to the amygdala. Indeed, PO receives auditory inputs (3) from the external and pericentral nuclei of the IC (221, 256), the dorsal nucleus of the lateral lemniscus (220) and the nucleus of the brachium of the IC (222). However, in contrast with the pathways reviewed above, PO does not project to LA but to CEm and BM (244, 257, 505).
Similarly, there are other routes for US information to reach the amygdala and they too bypass LA. For instance, nociceptive inputs from the spinal cord and trigeminal complex can reach CE (particularly CEl) via the parabrachial nuclear complex of the pons (27-30, 338). In keeping with this, physiological studies have revealed that CE cells respond to both mechanical and thermal noxious stimuli, but rarely to innocuous stimuli (337). Finally, there is evidence that PO relays nociceptive signals from the spinal cord to CEm and BM (186).
Amygdala outputs generating conditioned fear
There is general consensus that the main output station of the amygdala for conditioned fear responses to cues is CE (reviewed in (88); however see (213)). First, CE lesions block or reduce the expression of conditioned fear responses (60, 141). Second, distinct conditioned fear responses can be selectively attenuated by lesioning different targets of CE. For instance, lateral hypothalamic lesions interfere with conditioned changes in arterial pressure, but not conditioned freezing. In contrast, lesions of the periaqueductal gray (PAG) suppress conditioned freezing but not conditioned changes in blood pressure (242). However, not all conditioned fear responses are completely dependent on CE. In contextual fear conditioning for instance, BNST lesions also interfere with conditioned freezing (490). Also, some conditioned avoidance responses do not depend on CE but on BLA outputs (202).
Links between the input and output stations of the amygdala
Behavioral freezing is the most commonly monitored measure of conditioned fear. Importantly, amygdala projections to the brainstem site mediating freezing (PAG) originate exclusively in CEm. This is significant because LA, the presumed storage site of CS-US associations, has no direct projections to CEm (216, 378, 476). However, there are three possible routes for LA activity to influence CEm (Fig. 2D). Indeed, CEl, the basal nuclei, and ITC cells all receive inputs from LA and in turn project to CEm.
Evaluating these various possibilities is complicated by the fact that there is uncertainty regarding the nature of CEm control over conditioned fear. Indeed, CE output cells are thought to use GABA as a transmitter (288, 290) raising the following question: are conditioned fear responses generated by an increase or a decrease in the CS-evoked responses of CEm neurons? Insights in this question can be obtained by considering the effects of CE stimulation and conditioning-induced changes in CE activity. These two lines of evidence are considered in turn below.
Studies that examined the effects of CE stimulation or inactivation yielded somewhat inconsistent results (reviewed in (88)). Yet, the overall pattern of results suggests that an increase in CE activity causes an enhancement in fear expression, as expected given the effects of CE lesions (60, 141). As to conditioning induced changes in CS responsiveness, only three studies have addressed this question. The first, in rabbits (364), reported that the CS-responsiveness of brainstem projecting CE neurons (presumably CEm cells) decreased as a result of fear conditioning. In contrast, the other two studies, in rats (67) and mice (77), reported the opposite, consistent with the effects of lesion and stimulation studies. Therefore, the following will assume that an increase CEm output underlies expression of conditioned fear.
Since CEl activation is expected to inhibit CEm (260), and LA sends glutamatergic projection to CEl (216, 378, 476), it seems unlikely that CEl is the relay station between LA and CEm. Indeed, by enhancing the CS responsiveness of LA neurons, and therefore CEl cells, fear conditioning would be expected to cause a reduction in CEm output.
On the other hand, the two other candidate routes for transmitting LA outputs to CEm appear viable. Indeed, LA sends glutamatergic projections to laterally located ITCm cells (427), which inhibit medially-located ITCm cells (428), therefore causing a disinhibition of CEm output neurons (427). Similarly, LA sends glutamatergic projections to the basal nuclei (363, 476), which form excitatory synapses with CEm output neurons (363). Therefore, when the CS responsiveness of LA neurons increases, both routes are expected to cause an increase in CEm activity, albeit through different mechanisms (disinhibition vs. excitation, respectively).
Consistent with the involvement of the basal nuclei in relaying LA activity to CEm, it was observed that post-training lesions of the basal nuclei abolish conditioned fear responses (12). However, pre-training lesions did not prevent the acquisition of conditioned fear responses (11, 141, 332). This suggests that in an intact brain, the basal nuclei constitute an essential relay of potentiated LA activity to CEm.
However, the fact that animals can learn conditioned fear responses despite pre-training lesions of the basal nuclei indicates that another path exist for the transfer of LA outputs to CEm (ITC cells) or that CEm is not a simple relay station for potentiated LA outputs to the brainstem. Indeed, there is evidence that CE is also a critical site of plasticity for fear conditioning. In particular, local infusions of drugs that affect CE only during fear conditioning are sufficient to prevent the formation of long-term fear memory (526).
Overall, the evidence reviewed above suggest that fear conditioning depends on distributed plasticity in the amygdala. The fact that inactivation of LA or CE during training prevents the acquisition of conditioned fear indicates that both nuclei are essential sites of plasticity but that neither is sufficient. Also, the fact that post-training lesions of basal nuclei block the expression of conditioned fear indicates that, in an intact brain, the basal nuclei are at least required for relaying CS information from LA to CE.
III. Oscillatory Activity During Fear Learning and Emotional Arousal
As mentioned above, LA is thought to be the storage site of CS-US associations. According to this view, fear memory storage would involve an activity-dependent potentiation of synapses conveying CS information to LA neurons (see section IV). This potentiation would result from converging depolarizing inputs about the CS and US during fear conditioning. While in vitro studies have emphasized that tightly correlated pre- and postsynaptic activity is most effective for LTP induction, the paradigm typically used during fear conditioning is not optimal to meet this requirement. Indeed, most fear conditioning experiments involve long tone (CS) presentations (20-30 s) co-terminating with brief (≤ 1 s) footshocks. This is perplexing because the tone responses of LA neurons are strongest at tone onset and quickly diminish with time, nearing pre-tone firing rates toward the end of the CS (for instance see (390)). As a result, it would seem that LA neurons experience comparatively little tone-evoked depolarization when the US occurs, a conclusion that is in apparent contradiction with the findings of in vitro studies on LTP induction mechanisms.
A possible solution to this paradox resides in the ability of BLA neurons to express oscillatory activity (Figs. 3-4). By generating short recurring periods of depolarization during which the activity of pools of BLA neurons is synchronized with that of afferent neurons, oscillations might allow for the facilitated induction of synaptic plasticity with little increases in firing rates. As we shall see below, accumulating evidence indicate that BLA neurons do engage in such oscillatory activity and that these oscillations tend to synchronize BLA neurons with each other and with afferent neurons, with no change in discharge rates.
Fig. 3.
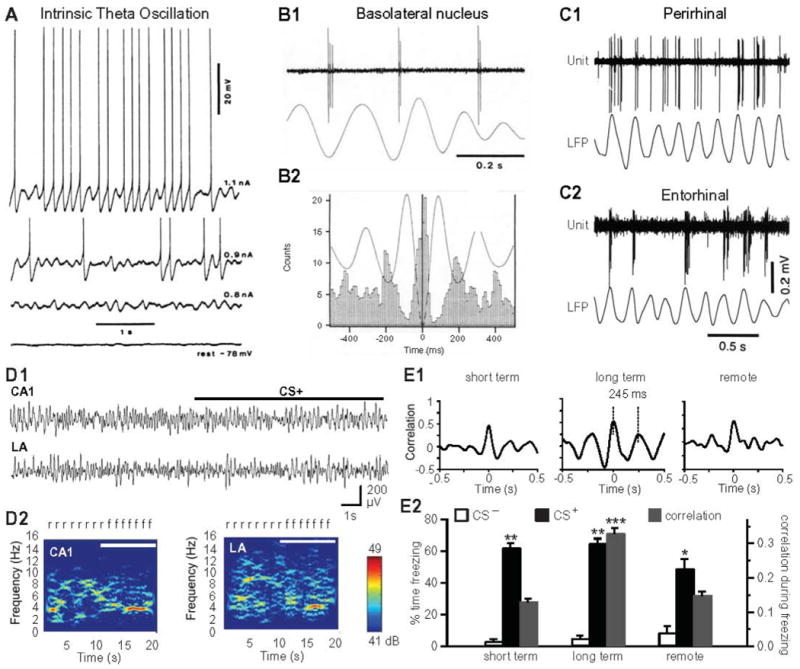
Theta oscillations in the BLA. (A) LA neuron recorded intracellularly in vivo. Near threshold membrane depolarization by intracellular current injection (numbers on right) elicits intrinsic membrane potential oscillations in the theta frequency range. (B) Principal BLA neuron exhibit rhythmic firing at the theta frequency during paradoxical sleep. (B1) Unit activity (top) and LFP (bottom) recorded by the same microelectrode and obtained by high vs. low-pass digital filtering, respectively. (C) Perirhinal (C1) and entorhinal (C2) neurons fire rhythmically at the theta frequency. Traces obtained as in B. (D) Synchronized theta activity in LA and CA1 during retrieval of conditioned fear. LFP recordings (D1) and their color-coded power spectra (D2) demonstrate theta activity in both LA and CA1 during CS+-evoked freezing. White bar in D1 denotes CS+ presentation; f, freezing; r, risk-assessment behavior. (E) LA-CA1 activity during retrieval of conditioned fear at short-term, long-term and remote stages, recorded at 2 hours, 24 hours and 30 days after fear training, respectively. (E1) Crosscorrelograms indicate synchronized theta during long-term (middle; obtained from recordings in D), but not short-term or remote stages. (E2) Significant increase in CS+-evoked freezing (black bars; compared to CS-, white bars) at short, long-term and remote stages is accompanied by synchronized theta in LA-CA1 (grey bars) only at long-term memory stages. * P<0.01, ** P<0.001, ***P<0.0001. Data in D, E modified from (335).
Fig. 4.
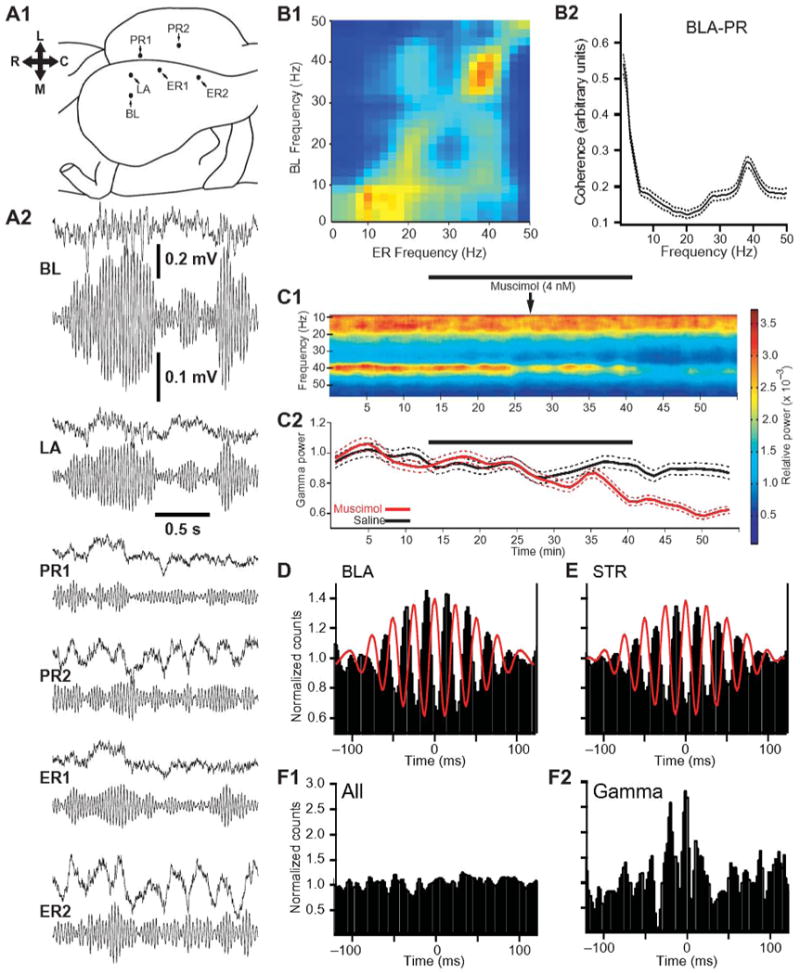
Coherent gamma oscillations in the BLA and its targets. (A) Simultaneous LFP recordings of gamma activity in the BLA and rhinal cortices. (A1) Scheme showing position of recording sites for activity depicted in A2. (A2) Top and bottom traces respectively show raw vs. digitally filtered (35-45 Hz) LFPs. (B) Correlated amygdalo-rhinal gamma activity. (B1) Power fluctuations: long periods of spontaneous field potential activity recorded during the waking state were segmented in one-second windows. Fast-Fourier Transforms were computed for each window and the power in each frequency was correlated with all others for BL and entorhinal (ER) recording sites. (B2) Gamma coherence. Coherence (y-axis) as a function of frequency (x-axis) for recording sites in the BLA and perirhinal cortex. (C) Inhibition of BLA activity by local muscimol infusions produces a selective reduction in striatal gamma activity. (C1) Striatal LFP power (color-coded) in different frequencies (y-axis) plotted as a function of time (x-axis) in experiments where muscimol was slowly infused in the BLA, over a period of 25 min. (C2) Gamma power (y-axis) ± s.e.m. (dashed lines) as a function of time (x-axis) when either saline (black) or muscimol (red) was infused in the BLA. The thick black lines indicate infusion periods. (D-E) Gamma-related unit activity in the BLA (D) and striatum (E). Peri-event histograms of unit activity computed around the positive peaks of high-amplitude gamma cycles recorded by the same electrode as used to record unit activity. (F) Gamma oscillations increase coupling between the activity of BLA and striatal neurons. (F1) Crosscorrelogram that included all spikes generated by a simultaneously recorded couple of BLA and striatal neurons. (F2) Crosscorrelogram of unit activity for the same cell couple after excluding striatal spikes occurring during periods of low amplitude gamma.
A. Theta oscillations
Previous in vitro (351, 354) and in vivo (358) intracellular recordings studies have revealed that BLA neurons have an intrinsic propensity to generate voltage-dependent membrane potential oscillations in the theta frequency range. Two types of intrinsic theta oscillations were identified. The first (Fig. 3A), seen at membrane potentials near firing threshold (354, 358), results from the interplay between a tetrodotoxin-sensitive persistent Na+ conductance and the M-type K+ current (351). The second, seen at supra-threshold membrane potentials, results from the rhythmically alternating influence of high-voltage activated Ca2+ conductances and Ca2+-dependent K+ currents (351). Theta oscillations are regulated by the intracellular AC-cAMP system, in that an increase in intracellular cAMP concentration facilitates generation of oscillatory activity via modulation of SK-type K+ channels (353).
In keeping with this, local field potential (LFP) oscillations and rhythmic unit activity at the theta frequency were seen in the BLA during paradoxical sleep (Fig. 3B; (357)) and periods of intense arousal caused by the anticipation of noxious stimuli (356). Although the intrinsic propensity of principal BLA neurons to oscillate or reverberate at the theta frequency likely played a role in these phenomena, another important contributing factor is the generation of theta oscillations by cortical fields that are reciprocally connected with the BLA such as the hippocampal formation (55), and the rhinal cortices (Fig. 3C; (7, 79, 315)).
In fact, the synchrony of hippocampal CA1 and LA theta increases during consolidation (458) and reconsolidation (336) of fear memories, while theta synchrony decreases at remote memory stages (Fig. 3D,E (335)), and during fear memory extinction (352). Theta activity recorded as LFPs in LA is not likely to be volume conducted from neighboring regions due to the following reasons. First, theta synchrony occurs between LA and CA1 during specific stages of fear memory, but not with CA1 theta during exploratory behavior (335, 458). Second, theta phase relations between regions vary characteristically during different states of fear memory (352). Third, the firing probability of LA neurons fluctuates rhythmically with theta oscillations in LFPs (352). These findings are in line with studies indicating that hippocampal circuits are engaged in the early stages of learning and show only limited activation as memory progresses at remote stages, while the reverse gradient has been documented for prefrontal cortical circuits (42, 128, 129, 283). Theta-entrained activity has indeed been recorded across widespread prefrontal cortical-hippocampal circuits (471, 474). In keeping with this, infralimbic prefrontal cortical activity was phase-locked to CA1-LA theta during retrieval and extinction of fear memory (352). In conclusion, theta synchronization appears to be an important organizing principle for creating time windows of fear memory consolidation within extended hippocampal-amygdala-prefrontal cortical networks.
B. Gamma oscillations
Another type of oscillatory activity that synchronizes principal BLA neurons with each other and with target cells is gamma (35-45 Hz; Fig. 4A). There is reason to believe that theta and gamma oscillations are related in the BLA. Indeed, the theta oscillations seen in the hippocampal formation (47) as well as in the entorhinal (75, 76) and perirhinal (79) cortices are associated with cyclical amplitude modulations of gamma activity at the theta frequency. Given the existence of strong reciprocal connections between these cortical areas and the BLA, it is likely that the two oscillations are similarly related in the BLA. However, this remains to be tested.
Several observations suggest that gamma activity plays a critical role in synchronizing BLA neurons with each other and with target cells. For instance, fluctuations in the power of LFPs recorded simultaneously in the BLA and rhinal cortices are more strongly correlated in the gamma range than other frequencies (Fig. 4B1; (21)). Similarly, the coherence of BLA and rhinal (Fig. 4B2) or striatal LFPs is highest in the gamma range compared to all other frequencies (21, 379). In contrast, other major sources of inputs to the striatum such as intralaminar thalamic nuclei and cortex do not show a preferential coupling at the gamma frequency (379). Thus, there results suggest that coherent gamma activity represents a physiological signature of BLA interactions with target structures.
Two types of evidence indicate that the gamma activity seen in the BLA is not volume conducted from neighboring regions and is perhaps generated within the BLA. First, the firing probability of BLA neurons fluctuates rhythmically with gamma oscillations (Fig. 4D; (21, 379)). Second, local intra-BLA infusions of the GABAA receptor agonist muscimol produce a pronounced and frequency-selective reduction of gamma power in the LFP of target structures (Fig. 4C; (379)). In the BLA, gamma activity typically occurs is short bursts of 2-6 consecutive high amplitude cycles during which there is no overall increase in firing rate, only a change in spike timing (21, 379). Importantly, functional coupling among BLA neurons as well as between BLA and target neurons was shown to increase when gamma power augments (Fig. 4F; (21, 379)).
Although the implication of BLA gamma oscillations in fear conditioning has not been examined so far, these oscillations were shown to coordinate the activity of BLA neurons with target structures during various forms of appetitive learning paradigms. For instance, in an appetitive trace conditioning task, thought to be dependent on the hippocampus, the power of CS-evoked gamma oscillations increased in the BLA and rhinal cortices, in parallel with improvements in behavioral performance (21). Similarly, in a discriminative stimulus-response task, thought to be dependent on the striatum, BLA-striatal gamma coupling increased selectively in relation to the rewarded CS (379), paralleling learning improvements.
Overall, these results suggest that the generation of coherent oscillatory activity in the BLA and related structures might be involved in fear conditioning and extinction. By generating short, recurring time windows during which pools of BLA cells and target neurons, fire synchronously, these oscillations may facilitate the induction of synaptic plasticity, with little or no change in firing rates. Moreover, the fact that coding in the BLA does not necessarily involve global increases in activity but changes in neuronal synchrony, highlight the importance of simultaneously recording multiple neurons to gain insights in the mechanisms that support fear memory and extinction.
IV. Synaptic plasticity in the amygdala related to conditioned fear
Central to the mechanisms of learning and memory are changes in synaptic efficacy, which take place during learning and are stabilized during memory consolidation. The Hebbian postulate (151) and the subsequent discovery of long-term potentiation (LTP) in the hippocampus (38, 39) paved the way for a widely accepted concept of synaptic plasticity, in which temporally correlated pre- and postsynaptic activity results in presynaptic release of glutamate and postsynaptic depolarization. Provided presynaptic activity coincides with a sufficient level of postsynaptic depolarization, postsynaptic NMDA receptors with bound glutamate are relieved from their Mg2+-dependent block and allow a Ca2+ influx into postsynaptic compartments, such as dendritic spines, thereby inducing a lasting increase in synaptic efficacy referred to as LTP (267). NMDA receptors thereby act as coincidence detectors that transform correlated neuronal activity into changes in synaptic strength. A large number of subsequent studies have yielded information on intracellular transduction and signalling pathways related to LTP in unforeseen detail (266). These transient molecular changes must be stabilized in order for the memory to persist (300). There is consensus now that the shift from the transient state of memory (referred to a short-term memory) to the stable form of memory (referred to as long-term memory) requires gene expression and de novo protein synthesis (193), which correlates with structural changes in synaptic morphologies (referred to as structural plasticity; (227)). In analogy to short- and long-term memory in behavioral studies, different phases of LTP have been distinguished based upon the transition from labile to more stable changes in synaptic efficacy. In fact, the maintenance of LTP, like memory storage, depends on intact protein synthesis and thus consists of at least two temporal phases, referred to as transient early-LTP (E-LTP) and protein synthesis-dependent late-LTP (L-LTP; for a review see (131)). Although the two phases of plasticity do not fully match in temporal characteristics at the synaptic and behavioral levels, they seem to share a common set of molecular mechanisms.
In an attempt to link changes in synaptic efficacy to specific learned behaviors, Pavlovian fear conditioning has proven particularly attractive for a number of reasons: a) the training paradigm is relatively simple and results in associative learning, which is rapidly acquired and long-lasting; b) this model allows one to control the induction, expression, and extinction of the memory; and c) the behavioral and autonomic fear-like responses can be reliably measured. While initial studies focused on the thalamus and cortex as possible sites of fear memory storage, subsequent lesion and electrophysiological studies indicated that the amygdala is a site of associative plasticity for Pavlovian fear memories (14, 194, 364). Converging evidence over the last three decades has supported the hypothesis that LTP of synaptic inputs that transmit CS information to the amygdala underlies the increase in fear responsiveness to the CS. Core support for this view comes from three major lines of evidence: a) fear conditioning causes a facilitation of responses to afferents relaying CS information to the amygdala, b) LTP occurs at these afferent inputs, and c) fear conditioning and LTP share a common set of mechanisms affected similarly by a range of experimental manipulations. The extensive literature on these themes is covered by a number of review articles (37, 93, 114, 142, 207, 239, 270, 274, 409, 473). Recently, critical advances have been made in determining how the transient synaptic modifications induced by NMDA receptor activation become stabilized during fear memory consolidation, and how different neuronal input systems must be coordinated for theses events to take place. Here, we will briefly summarize the findings that have laid the groundwork for understanding conditioned fear on a synaptic level, followed by a more extensive review of the molecular cascades of memory stabilization. An overview about the molecular mechanisms is provided in figure 5. The various forms of long-term synaptic plasticity described in amygdala neurons are schematically illustrated in figure 6.
Fig. 5.
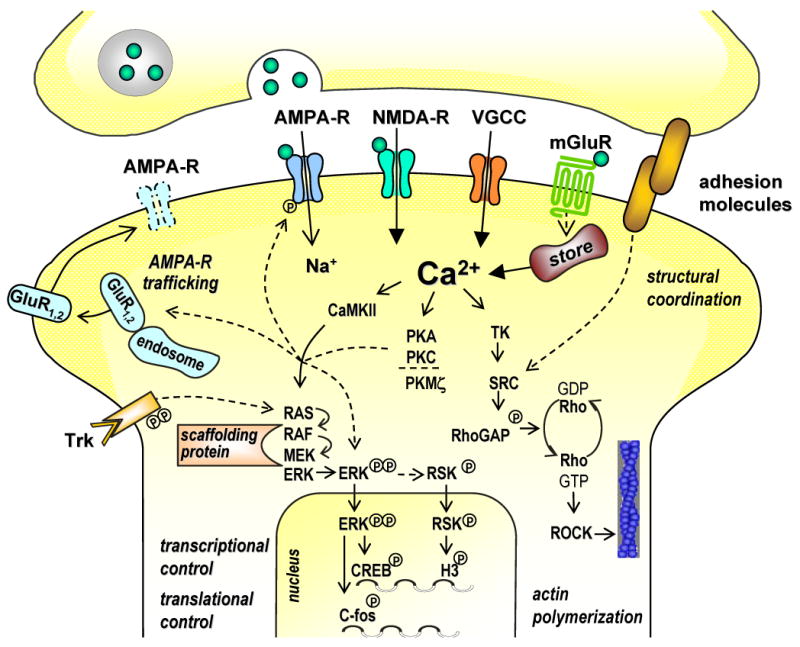
Molecular cascades of fear memory stabilization in the amygdala. A postsynaptic increase in intracellular Ca2+ concentration, mediated through Ca2+ influx via NMDA receptors and voltage-gated Ca2+ channels (VGCCs) and through release from intracellular stores upon activation of metabotropic glutamate receptors (mGluRs), triggers a plethora of signalling steps. Three major, mutually interconnected signalling routes involve Ca2+/calmodulin-dependent protein kinases II (CaMKII), the protein kinase (PK) family of enzymes, and tyrosine kinase (TK) pathways. Signalling cascades can reach the nucleus to induce macromolecular synthesis, and they can control translational processes. Consequently, they can act on cytoskeletal and adhesion molecules to re-organize and stabilize synaptic structures, or regulate AMPA receptor trafficking to the synapse. At intermediate steps, protein kinase signals converge on the mitogen-activated protein kinase (MAPK) signal transduction pathways, including the extracellular regulated kinases (ERK). RAS, RAF, and MEK kinases transduce intra- and extracellular signals, mediated for instance through tyrosin receptor kinases (Trk), to the MAPK/ERK pathway. Scaffolding proteins dictate specificity of activation as well as entry in the nucleus. MAPKs translocated into the nucleus phosphorylate transcription factors, such as cAMP response element binding protein (CREB). Actin rearrangement is under the control of RhoGTPases, whose activation from a GDP- to a GTP-bound form is controlled via Ca2+ or kinase pathways, including tyrosine kinases (TK) and SRC kinases. RhoGTPases control activity of Rho-associated kinases (ROCK), a key molecule for regulation of the cytosekeleton.
Fig. 6.

Long-term synaptic plasticity related to conditioned fear in the basolateral amygdaloid complex. A. Long-term potentiation (LTP) in projection neurons (PN). At thalamic inputs, LTP is homosynaptic upon stimulation of postsynaptic NMDA receptors and/or voltage-gated Ca2+ channels. At cortical inputs, LTP is heterosynaptic upon stimulation of presynaptic NMDA receptors through concurrent activation of thalamic inputs. B. Long-term depression (LTD) in PN can be mediated via stimulation of postsynaptic metabotropic glutamate receptors (mGluRs) at thalamic inputs, or via presynaptic mGluRs at LA-BLA synaptic connections. C. LTP in local GABAergic interneurons (IN) at thalamic and cortical inputs can be homosynaptic upon stimulation of Ca2+ permeable AMPA receptors, or heterosynaptic upon stimulation of NMDA receptors.
A. A classical view
Most of the knowledge about the circuits involved in conditioned fear was derived from experiments on auditory fear conditioning in rodents. The major sensory input station to the amygdala is LA. Therefore, the majority of studies have focused on auditory pathways to LA, and particularly on thalamic inputs to the dorsal part of the LA (LAd). The central idea underlying the cellular hypothesis of fear conditioning is that the convergence of CS and US inputs onto principal LA neurons during Pavlovian fear conditioning results in a lasting increase in synaptic strength at CS inputs, recorded as LTP. This increased activity is relayed to the central amygdala (CE), the main output station for fear responses. This hypothesis is based upon three major assumptions, all of which underwent ample experimental examination, as described below (see also ref (473)).
First, fear conditioning induces changes in efficacy at afferent synaptic inputs to the amygdala
This has been shown, mostly in LAd, by extra- and intracellular recordings of CS-evoked firing in vivo (80, 139, 390, 404, 414, 419) and by recordings of synaptic responses to afferent stimulation in brain slices in vitro obtained from fear-conditioned animals (301, 502, 538). These studies demonstrated the associative nature of the plasticity, established LA as the site of plasticity, and provided support for a causal relationship between LA plasticity and fear memory (reviewed by (274)). For instance, LA responses to the CS+ following conditioning were greater than those after explicitly unpaired presentations of the CS+ and US (390) and were opposite to those evoked by a non-conditioned stimulus (CS-) in a discriminative auditory fear training paradigm (80). These results indicated that LA plasticity is of an associative nature rather than being dominated by non-associative processes, such as sensitization. Importantly, the plastic changes recorded in LA upon fear conditioning preceded increases in responsiveness observed in the auditory thalamus (303) or auditory cortex (386). Moreover, local manipulations of the LA known to interfere with fear conditioning had either no effect on neuronal activity in the auditory thalamus (451) or impaired the development of plasticity in auditory cortex or thalamus (15, 275). These data ruled out the possibility that changes in LA responsiveness simply mirror plasticity occurring upstream of LA, as for instance in the thalamus or cortex (56), and further supported the notion that the LA is a site of associative plasticity.
As fear conditioning gives rise to behavioral changes that could affect CS processing in LA, and fear responses often outlast the stimuli that induce them, it is important to determine whether plastic changes in LA activity are a cause or consequence of conditioned fear behavior. In an elegant study apt to dissociate LA plasticity and fear expression, Maren and colleagues (139) performed discriminative fear conditioning in rats using distinct auditory CSs (CS+ versus CS-), which were then presented in a neutral context and, in a different group of animals, in a context that had been conditioned with an aversive US. Fear conditioning increased both CS+-evoked LA responsiveness and fear behavior, whereas presentation of the CS- did not result in changes in LA responsiveness, even though it evoked high fear behavior in the conditioned context. Further and importantly, inhibiting the behavioral expression of conditioned fear through pharmacological inactivation of CE had no effect on CS+-evoked increases in LA responsiveness. Together these data indicated that LA neurons signal the CS+-US association irrespective of the behavioral expression of fear (as reviewed in (274)).
Second, LTP occurs at synaptic inputs to the amygdala
This has been demonstrated in vivo in anesthetized (78, 413, 531) and freely behaving animals (101), as well as in vitro in slice preparations (69, 164, 166, 170, 497, 503, 519). The focus has been on postsynaptic LTP at thalamic inputs to LA, which was induced by a high frequency train of stimuli allowing summation of depolarizing postsynaptic potentials to unblock NMDA receptors (Fig. 6A). To better model the temporal pattern of CS-US pairing, single presynaptic stimuli have been paired with postsynaptic depolarization in vitro (166, 170, 503, 524). These studies revealed that LTP occurred only at those inputs that underwent paired stimulation, thereby demonstrating input specificity of LA plasticity. More recently, Kwon and Choi (223) in a very clever approach probed a conditioning paradigm in which tetanic microstimulation of the auditory thalamus (MGm) rather than a sensory CS+ was used. Pairing of tetanic stimulation with a US resulted in conditioned fear behavior and LTP-like increases in evoked field potentials in LA, whereas explicitly unpaired protocols or microstimulation of a neighboring thalamic region (MGv) had no effect on behavior or LA responsiveness. These results indicate that LTP induction and associated changes in synaptic efficacy at thalamo-LA inputs are involved in fear learning. Associative LTP has also been shown upon stimulation of both thalamic and cortical inputs to LA in awake rats, with characteristic asymmetries occurring in LTP magnitude and duration between the two inputs (101). Furthermore, paired afferent input or pre- and postsynaptic stimulation revealed the existence of a presynaptic form of LTP at cortical inputs to LA (Fig. 6A) (166, 172, 502). There is agreement that LTP induction at thalamic and cortical inputs to LA involves NMDA receptors, which are predominantly located at postsynaptic and presynaptic sites, respectively (but see (13)). Thalamic and cortical inputs fibres converge onto both projection neurons and local interneurons (496), where they may even converge onto the same dendrites. The question thus arose as to how the two input systems are functionally segregated in a non-layered structure like LA. One answer was provided by Humeau and colleagues (170) who showed that the two inputs contact functionally and morphologically distinct types of dendritic spines, and that this heterogeneity determines Ca2+ influx and thereby the afferent-specific Hebbian plasticity.
Third, fear conditioning and LTP share a common set of mechanisms
The demonstration that intra-amygdala infusion of NMDA receptor antagonists blocks the induction (but not expression) of conditioned fear in vivo and of LTP in vitro provided the basis for the hypothesis that NMDA receptor-mediated LTP represents a cellular substrate of fear conditioning (22, 61, 166, 206, 314). Early studies yielded evidence for the additional contribution of L-type voltage-gated Ca2+ channels (166, 468, 524) or questioned the involvement of NMDA receptors in amygdala LTP (69). Currently, the consensus is that postsynaptic LTP induced by weak stimulation protocols is dependent on NMDA receptors, while stronger induction protocols, such as sustained pre- and postsynaptic pairing, may also require the activation of voltage-gated Ca2+ channels (22, 170, 455) (Fig. 6A). Native NMDA receptors are formed by the heteromeric expression of the NR1 subunit, which is required for the ion channel pore, and one type or a combination of NR2 subunits, which determine the kinetics of the NMDA-mediated currents (reviewed in (350)). In particular, NMDA receptors with NR2B subunits have slow decay kinetics, promoting Ca2+ entry and induction of synaptic plasticity (499). That these receptor subunits are important for conditioned fear is supported by the finding that intra-amygdala infusion of ifenprodil, a NR2B receptor antagonist, disrupts the acquisition – but not the expression - of conditioned fear (411). In keeping with this, NMDA receptors present on principal amygdala neurons (259) and GABAergic interneurons (498) contain NR2B subunits, particularly at thalamo-amygdala synapses (391), and application of ifenprodil blocks LTP at thalamic input pathways to principal LA neurons in vitro (22). These findings do not rule out, however, a contribution of NR2A receptors to synaptic plasticity in LA neurons (320). Inspired by these findings, many pharmacological and genetic studies have targeted molecular processes involved in cellular and behavioral plasticity in the amygdala (reviewed by (412)) and shed light on the mechanisms underlying long-term plasticity in the amygdala. These mechanisms will be reviewed below.
B. Molecular cascades of memory stabilization
As outlined above, induction of synaptic plasticity in the LA involves activation of NMDA receptors, with a critical role played by NR2B receptor subtypes, and voltage-gated Ca2+ channels, both of which mediate an influx of Ca2+ ions into LA neurons. An additional source of Ca2+ is the release from intracellular stores triggered by second messenger systems secondary to stimulation of membrane bound G protein-coupled receptors. Of particular interest here are metabotropic glutamate receptors (mGluRs), of which the group I receptor subtype mGluR5 plays a key role in the modulation of synaptic plasticity. Activation of group I mGluRs may alter the potential for plasticity, a phenomenon referred to as metaplasticity (1). Receptors of the mGluR5 subtype are localized to dendritic shafts and spines in LA neurons, are postsynaptic to thalamic inputs (408), and are blocked through specific antagonists, such as 2-methy-6-(phenyle-ethynyl)-pyridine (MPEP). MPEP impairs the induction of L-LTP at thalamo-LA synapses and the acquisition, but not expression or consolidation, of conditioned fear (119, 246, 408). In keeping with the concept of metaplasticity, infusion into the BLA of a group I mGluR agonist, (R.S.)-3,5-dihydroxyphenylglycine (DHPG), was found to enhance the acquisition of conditioned freezing normally supported by a weak footshock (432). Furthermore, activation of group II mGluRs evokes long-term depression (LTD) of synaptic transmission in the amygdala (153, 255). mGluRs are coupled to Ca2+-cAMP pathways, located postsynaptically at thalamic inputs to principal LA neurons (153), or presynaptically at LA-BLA connections (255) (Fig. 6B). Their significance for conditioned fear remains unclear to date.
The overall rise in intracellular Ca2+ concentration triggers a plethora of signalling steps. There are three major, mutually interconnected signalling routes, which involve Ca2+/calmodulin-dependent protein kinases II and IV (CaMKII, IV), the protein kinase (PK) family of enzymes, and tyrosine kinase (TK) pathways. These signalling cascades eventually can reach the nucleus to induce macromolecular synthesis or control translational processes. Consequently, they can act on cytoskeletal and adhesion molecules to re-organize and stabilize synaptic structures, or target membrane transport systems. These mechanisms may act separately or in concert to consolidate transient changes in synaptic efficacy. They provide the intracellular framework, upon which neuromodulatory systems, such as monoamines and stress hormones, act to regulate memory formation (reviewed in (299, 410)). An overview of these molecular mechanisms is provided in figure 5.
1. Initial protein kinase pathways
One important target of Ca2+ is CaMKII. The α isoform of CaMKII is considered a key mediator of synaptic plasticity and associative learning in a variety of brain regions and species (520). Critical to this function is CaMKII's ability to shift to a constitutively active form, even after Ca2+ has declined to baseline levels, following autophosphorylation of a specific threonine residue (Thr286). Interaction with NMDA receptors, particularly the NR2B subunit, can lock the molecule in this active form (23). Mouse mutants with inducible CaMKII deficiency restricted to the forebrain are impaired at acquiring cued and contextual fear (516). In LA, α CaMKII is postsynaptic to auditory thalamic inputs, and co-localizes with NR2B subunits (409). Fifteen minutes after fear conditioning, CaMKII shifts to the autophosphorylated (active) form, and a CaMKII inhibitor, KN-62, impairs both thalamic-LA LTP in vitro and the acquisition, but not the expression, of auditory cued and contextual fear conditioning (409).
Another route of Ca2+-dependent signalling for stabilization of synaptic plasticity involves the protein kinase family of enzymes. An early study found that infusion into the BLA of H7, a potent albeit rather unspecific blocker of cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC) activity, interfered with long-term but not short-term conditioned fear memory responses (140). These findings were supported by the use of a more specific PKA inhibitor (Rp-cAMPS), which attenuated long-term conditioned fear if administered shortly after fear training into LA (452). Furthermore, a mouse mutant with a deficiency for the β isoform of PKC displayed normal brain anatomy and hippocampal-based electrophysiological responses, but a deficit in cued and contextual fear conditioning (521).
2. Towards protein trafficking
Of eminent importance for synaptic plasticity is the brain-specific, atypical isoform of PKC, termed protein kinase Mzeta (PKMζ; for recent review see (437)). So far, PKMζ is the only molecule identified that is both necessary and sufficient for maintaining LTP. PKMζ consists of the independent catalytic subunit of PKC and is autonomously active to sustain LTP maintenance. LTP induction triggers the synthesis of new PKMζ and the transport of new PKMζ to dendrites, where it increases the number of the AMPA subtype (AMPA-Rs) of glutamate receptors through GluR2 subunit-mediated trafficking to the synapses (for recent review see (106, 199)).
Two lines of evidence support the notion that PKMζ and AMPA-R trafficking are also critical for synaptic plasticity in the amygdala and conditioned fear. First, Serrano and colleagues (462) examined the effects of zeta inhibitory peptide (ZIP), a specific blocker of PKMζ activity. PKMζ inhibition in the BLA, but not in the hippocampus, impaired retention of conditioned associations for both contextual and auditory fear, as well as instrumentally conditioned inhibitory avoidance. Postshock freezing was not affected, indicating that fear expression mediated by the BLA remained intact. Second, Rumpel and colleagues (435) showed that AMPA-R trafficking in LA is essential for cued conditioned fear. They constructed three amplicon vectors to either monitor or perturb AMPA-R trafficking. The first encoded GluR1 fused with green fluorescent protein (GFP) to drive expression of homomeric AMPA-Rs that display electrophysiological properties different from those of endogenous AMPA-Rs, and could be used to tag modified synapses with incorporated GluR1 (“plasticity tag vector”). The second vector encoded the carboxyl cytoplasmic tail of GluR1 fused with GFP that functions as a dominant-negative construct to prevent synaptic incorporation of endogenous GluR1, and which was thus used to block synaptic plasticity (“plasticity block vector”). The third vector drove expression of only GFP and was used as a control (“infection control vector”). After transfection through localized injection into the amygdala, animals were fear-conditioned, and plasticity was examined at the behavioral and synaptic levels in vivo and in vitro, respectively. It was observed that auditory fear conditioning drives GluR1 receptors into synapses onto LA neurons, that this trafficking is specific to thalamic inputs, and that blockade of AMPA receptor incorporation blocks both LTP at thalamo-LA inputs in vitro and retention of conditioned fear in vivo (tested 3 or 24 hours after training). Only about a third of the LA neurons were found to undergo this type of plasticity, thereby supporting the notion that fear memory formation requires coordinated changes in synaptic strength in distributed networks, and perturbing a few plastic units may corrupt integrated function. Of further interest is that the conditioning-induced increase in surface expression of GluR1 depended on the activation of NMDA receptors and protein kinases, and required the synthesis of new proteins (534). Indeed, mice with a genetic deficiency in GluR1 displayed an impairment of both conditioned fear and LTP at thalamo-LA synapses, whereas GluR3-/- mice showed no alteration in conditioned fear, thereby contributing to the view that GluR1-dependent synaptic plasticity predominates in conditioned fear (171). The regulated transport of AMPA-Rs towards exocytosis and endocytosis at synaptic sites seems to be important for balanced plasticity in the amygdala. Blockade of vesicle-mediated exocytosis and endocytosis of AMPA-Rs indeed prevents LTP and LTD at thalamic inputs (535). Conversely, AMPA-R endocytosis is critical for fear extinction (208)(see section V.D).
Recent evidence suggests that regulated trafficking in the amygdala is not restricted to AMPA-Rs. NR2B subunits can be tyrosine-phosphorylated, and mice with a knock-in mutation of the major phosphorylation site (Tyr-1472) show impaired fear learning and reduced amygdala LTP, accompanied by improper localization of the NR2B subunits at amygdala synapses (334). NR2B subunits are downregulated after fear conditioning (539), suggesting that the plastic synaptic events supporting fear learning involve the regulation of NMDA receptor proteins through phosphorylation and/or transport (for review on NMDA-R trafficking see (232)). Moreover, the trafficking of functional molecules at synaptic sites may not be limited to ligand-gated ion channels. One example are small-conductance Ca2+-activated potassium channels (SK channels), which limit postsynaptic responses and plasticity of principal LA neurons (108). Stimulation of β–drenoceptors, known to facilitate fear memory formation (299), results in a PKA-mediated reduction in SK channel activity and their removal from the postsynaptic membrane, thereby enhancing synaptic transmission and facilitating induction of synaptic plasticity (108, 109).
In conclusion, the available evidence suggests that the acquisition of Pavlovian fear involves enduring changes in glutamatergic transmission at thalamic synapses onto LA neurons. These changes are likely maintained by the insertion of AMPA-Rs and other types of ion channels into thalamo-LA synapses. Consistent with this idea, A-kinase anchoring proteins (AKAPs), a family of scaffolding proteins that bind the regulatory subunits of PKA and target PKA to GluR1, are essential for the consolidation of Pavlovian auditory fear memories (316).
3. Towards transcriptional control
The protein kinase signals, including CaMKII and IV, PKA and PKC, are known to converge on the mitogen-activated protein kinase (MAPK) signal transduction pathway, one of the most widespread mechanisms of cell regulation (reviewed in (219)). Six distinct groups of MAPKs have been characterized in mammals, of which the extracellular regulated kinases (ERK) are the best understood. Typical of MAPK is a central three-tiered signalling molecule, consisting of a set of three sequentially acting kinases: an MAPK, an MAPK kinase (MAPKK or MEK) and an MAPKK kinase (MAP3K or MEKK). The ERK/MAPK pathway can be activated by a large number of upstream extracellular and intracellular stimuli, including growth factors, cytokine, and ligands of G-protein-coupled receptors. Their signals are usually transduced to small GTPases, such as RAS, which transmit the signal by recruiting the MAP3K tier-like RAF kinases. Activated RAF binds to and phosphorylates down-stream kinases MEK, which in turn phosphorylate ERK. Of particular importance is that scaffolding proteins of MAPK pathways can dictate the specificity of activation as well as entry in the nucleus. MAPKs translocated into the nucleus phosphorylate transcription factors, such as cAMP response element binding protein (CREB), thereby regulating gene expression and new macromolecular synthesis (mRNA and protein). Examples include the immediate early genes c-jun and c-fos. In fact, tagging of c-fos active neurons allowed the identification of a neuronal subpopulation in BLA that are activated during fear conditioning and are reactivated after during memory retrieval (403).
What evidence indicates that these pathways are involved in long-term synaptic plasticity in the amygdala and conditioned fear? Early studies indicated that pharmacological interference with PKA, MAPK activity, and protein synthesis interferes with the late phase of LTP (L-LTP) at afferent inputs to LA in vitro and with the consolidation of Pavlovian fear in vivo. In contrast, early LTP and short-term fear memory were spared (165, 167, 449, 452). Furthermore, ERK/MAPK is transiently activated/phosphorylated in LA following auditory fear conditioning or high-frequency stimulation of the auditory thalamus (449, 453). Infusions of a MEK (MAPK kinase) inhibitor or of an mRNA synthesis inhibitor into the auditory thalamus before or after fear training yielded impaired long-term memory of conditioned fear and thalamo-LA (13), in line with previous suggestions that thalamic neurons contribute to memory formation by promoting protein-synthesis-dependent plasticity in the LA (272). Of the two ERK isoforms (ERK1, 2), ERK2 seems to contribute critically to conditioned fear, as ERK1 null-mutant mice did not display deficits in the acquisition or retention of either contextual or cued fear (459).
Upstream of ERK/MAPK is the RAS signalling pathway, which has been implicated in fear memory and synaptic plasticity in the amygdala. Mice lacking RAS-GRF, a neuronal-specific factor inducing RAS signalling in response to Ca2+ influx, show impaired consolidation of conditioned fear and BLA LTP, whereas spatial memory tasks and hippocampal LTP were unaffected (48). Mice with a null mutation of RIN1, a RAS effector that competitively inhibits the RAF-MEK-ERK pathway and is preferentially expressed in dendrites, show an enhancement of amygdala LTP and amygdala-dependent aversive memories like fear conditioning (92). Of particular interest here is STEP (for striatal-enriched protein-tyrosine-phosphatase), a molecule that is colocalized with ERK in LA neurons and can prevent their nuclear translocation (366). Fear conditioning induced activation of ERK1/2 in the amygdala as well as a de novo translation of STEP, whereas infusion of a substrate-trapping STEP protein prevented translocation to the nucleus, disrupted LTP in LA, and impaired fear memory consolidation. By contrast, blockade of phosphatidylinositol 3-kinase (PI3-kinase) activity, preventing MAPK activation, CREB phosphorylation and LA LTP, leads to a decrease in conditioned fear (252). Also SRC kinases, nonreceptor kinases downstream to a rise in intracellular Ca2+, seem to be required for the acquisition of conditioned fear (34), particularly upon stimulation of the NR2B subunit (212).
Together, these data support the hypothesis that ERK1/2 signalling and translocation to the nucleus play an important role in the maintenance of synaptic plasticity and consolidation of conditioned fear in the amygdala. Downstream of ERK/MAPK, CREB has been implicated in fear conditioning based upon findings in mice with null mutation in different CREB isoforms or with overexpression of the dominant negative CREB133A (44, 135, 201, 397, 522). In line with this, an increase in phosphorylated CREB and transcription from CRE motifs occur after fear conditioning (174, 483). Expression of a constitutively active form of CREB (VP16-CREB) lowered the induction threshold for late LTP in hippocampal CA1 neurons and increased the intrinsic excitability of CA1 and BA neurons (511). These effects were accompanied by resistance of both cued and contextual fear conditioning to the protein biosynthesis blocker anisomycin, suggesting that de novo protein synthesis can be bypassed by constitutive CREB function (511). Using virus-mediated gene transfer, the critical CREB activity was located to the BLA region and correlated with the strength of the memory trace (187, 513). In particular, LA neurons with increased levels of CREB were preferentially activated by auditory fear memory during training or testing (148). Specific ablation of CREB-overexpressing LA neurons by diphtheria toxin-mediated apoptosis after fear learning abolished the fear memory. These results indicate that CREB function in a subset of LA neurons is critical for the formation and maintenance of the fear memory trace (149). In keeping with this, CREB activation is also linked to histone acetylation through the CREB-binding protein CBP (214), which itself is required for the acquisition of conditioned fear (342). The importance of this mechanism is indicated by the finding that chromatin modifications through increased histone-tail acetylation induce dendritic sprouting, increase the number of synapses, and reinstate hippocampal-dependent learning and access to long-term memories upon exposure to an enriched environment (123). Other rapidly activated transcription factors, like nuclear factor-κB (532, 533), or the potassium channel interacting protein 3 (KChIP3; also known as calsenilin and as the transcription factor DREAM; (5)) also seem to be involved in fear conditioning. However, CREB is the most intensively studied one, found to be bound to at least 6000 genomic loci and to regulate expression of about 1600 transcripts (for review see (173, 258)).
Genes that are transcriptionally regulated upon fear conditioning include immediate early genes, like c-fos (418, 456). As with CREB, a relation has been proposed between expression level and memory strength (393, 483), including an influence of novelty, context, and stress. Other important targets of CREB transcriptional activity are nerve growth factors (NGF) and brain-derived neurotrophic factors (BDNF). A convergent line of evidence indicates that BDNF plays a role in amygdala-dependent learning and memory (reviewed by (36, 346, 400)). BDNF mRNA is elevated during the consolidation of conditioned fear memory (401), and BDNF blockade in the amygdala through expression of a dominant negative isoform or antagonism of the tyrosine kinase receptor B (TrkB) interferes with long-term fear memory (399). Upon fear conditioning, the level of TrkB receptor immunostaining declines in the amygdala, whereas the level of phosphorylated TrK receptors increases, suggesting TrK activation and internalization by BDNF binding (400). The two phosphorylation docking sites of TrkB receptors are specifically linked to the acquisition of cued fear and CaMKII signalling, and to memory consolidation and Akt signalling, respectively (326). Furthermore, in concert with developmental processing of BDNF, cleavage of pro-BDNF by tissue plasminogen activator (tPA) seems to be essential for hippocampal LTP and the formation of contextual fear memory, as tPA null mutation interferes with both processes (10, 349). Particularly interesting are BDNF/TrkB-dependent mechanisms of neuronal plasticitiy that may bypass NMDA-dependent processes (400). One route is via PI3-kinase, a critical intracellular mediator of synaptic plasticity during fear conditioning (253). Another route involves the RAS-RAF-MEK-ERK pathway (reviewed in (400)), thereby suggesting that these intracellular signalling mechanisms likely act in parallel. A recent study in knock-out mice has provided evidence that the immediate early gene vesl-1S (VASP/Ena-related gene upregulated during seizure and LTP, also termed homer-1a) is required for contextual fear memory consolidation and re-consolidation (175). Vesl-1S is the alternatively spliced, short isoform of the vesl-1 gene, the long isoform of which encodes a scaffolding protein modulating intracellular Ca2+ dynamics via metabotropic glutamate receptors, IP3 receptors, and ryanodine receptors. In any case, both fear memory consolidation and re-consolidation were impaired upon vesl-1S knock-out, thereby supporting the view that symmetrical signalling cascades are involved in these two stages of memory stabilization (see (100, 104); reviewed in (331)).
4. Towards post-transcriptional and translational control
Although the transcriptional control of gene expression has received much attention, post-transcriptional and translational mechanisms also participate in memory formation (for recent review see (85)). One of these mechanisms involves the regulation of mRNA stability. The Hu family of RNA-binding proteins is perhaps the most important group of mRNA-stabilizers described so far (196). Recent studies indicate that they are also involved in synaptic plasticity (40, 385), including acquisition and retention of both cued and contextual fear, although their exact role remains unclear (41). One hypothesis is that consolidation involves proteins that are translated from existing mRNA stores, as for instance at synaptic sites in dendrites. One particular example for conditioned fear involves mTOR (155, 355), the mammalian target of rapamycin kinase, which regulates protein synthesis in neurons at the translational level through intracellular phosphorylation. One of its targets, p70s6 kinase, is upregulated after fear training, and prevention of this upregulation by post-training injection of rapamycin into the amygdala, blocked the fear memory formation (155). Interestingly, when rapamycin was infused in the amygdala after fear memory recall, subsequent retention was disrupted, suggesting that local translational control is required for the formation as well as the stability of long-term fear memories. A study in chicks (309) lends support to the hypothesis that re-consolidation is also dependent on dendritically synthesized proteins.
While prevailing models of memory identify transcriptional regulation or post-transcriptional RNA editing as necessary for enduring information storage, consolidation of long-term memory may also occur in the virtual absence of new macromolecular synthesis. The above-mentioned trafficking of AMPA receptors to and from synapses, the processing of BDNF, and the constitutive activity of enzymes at various steps in the intracellular signalling pathways exemplify such a scenario. In fact, an alternative model of long lasting information storage has been proposed (424, 425). According to this model, pre-existing synaptic proteins are modified at a post-translational level upon learning experience, supporting memory formation. One important feature of this model is endogenous, reverberant activity at the respective synaptic interconnections, providing a positive feedback rehearsal mechanism by which proteins are increasingly modified and thereby functionally up-dated for enduring information storage. In this model, protein synthesis is thus a permissive step for the subtle modification of synaptic proteins to occur. The spatiotemporal segregation of the various forms of memory may then be explained through correlated activity in the involved neuronal assemblies. While it is currently unclear how the various transcriptional and post-translational entities interact, it is interesting to note that both types of models require coordinated activity in synaptic networks – for coincidence detection in the Hebbian sense (151) and for rehearsal processes in maintaining memory longevity. The significance of correlated activity in circuits of the amygdala and beyond is discussed in section III of the present review.
5. Towards structural plasticity
How are synaptic changes structurally stabilized? Most excitatory synapses in the mammalian brain end on dendritic spines, which provide an isolated functional compartment for coupling synaptic activity with postsynaptic intracellular signalling pathways. It was proposed that enduring alterations in synaptic transmission depend on changes in the number and/or morphology of spines (as reviewed in (536)), and that spine architecture is an important parameter for the specificity of Hebbian plasticity at thalamic and cortical inputs to LA neurons (170). Spine architecture and spinogenesis, in turn, depend on cytoskeletal filaments, in particular on the dynamics and polymerization of one of their major constituents, actin (103, 398). It was hypothesized that re-organization of actin contributes to the stabilization of spines, thereby providing a mechanism of structural plasticity for memory stabilization. In keeping with this, LTP induces a lasting increase in polymerized actin in dendritic spines (132), and a reduction in actin-based spine motility underlies spine stabilization (282). In fact, fear conditioning alters the expression of cytoskeletal proteins including actin and a-actinin (406, 488) Furthermore, actin dynamics regulate NMDA receptor function, AMPA receptor trafficking, and spinogenesis after contextual fear conditioning in the hippocampus (122). Anchored to the actin cytoskeleton are cadherins, including neuronal (N)-cadherin, which are associated with docking proteins to intracellular pathways and are regulated by extracellular domains mediating cell-cell adhesion (for review see (188, 402)). Much of our current knowledge on N-cadherin involvement in fear conditioning is derived from studies of contextual fear and related hippocampal mechanisms. An N-cadherin antagonistic peptide containing the His-Ala-Val motif (HAV-N) disrupted N-cadherin dimerization in the hippocampus and impaired the formation of long-term contextual fear memory while sparing short-term memory, retrieval, and extinction (454). At the molecular level, HAV-N impaired learning-induced phosphorylation of the cytoskeletally associated fraction of ERK-1/2 in the hippocampus, prevented NMDA-induced dendritic ERK-1/2 phosphorylation in vitro, and caused a relocation of IQGAP1, a scaffold protein linking cadherin-mediated cell adhesion to the cytoskeleton. The N-cadherins may thus enable the translation of cell adhesion signals into long-term cellular responses required for contextual fear in the hippocampus through signalling pathways involving cytoskeletal IQGAP1/ERK signalling.
Actin rearrangement, in turn, is under the control of RhoGTPases, intracellular molecules that can be activated via G protein-coupled receptors, Ca2+ or kinase pathways, and that switch between an active (GTP-bound) and inactive (GDP-bound) form. Indeed, fear conditioning causes the formation of a molecular complex that contains the tyrosine-phosphorylated RhoGTPase-activating protein (RhoGAP), which is located in the dendrites of LA neurons (225). RhoGTPases regulate activity of the Rho-associated kinases (ROCK), whose inhibition in LA impairs long- but not short-term conditioned fear (225). ROCK, in turn, is a key molecule for regulation of the cytoskeleton (reviewed in (262)). A number of other cytoskeletal-regulatory proteins also contribute to synaptic plasticity and fear learning in the amygdala. They include myosin light chain kinase (228), stathmin, an inhibitor of microtubulin formation (470), LIMK-1, a member of a kinase family (LIMK) that induces actin polymerization through the phosphorylation and cofilin, a protein that facilitates de-polymerization of actin (305). Another example is profilin, an actin polymerization-regulatory protein (226). For instance, fear conditioning drives profilin into LA dendritic spines with enlarged postsynaptic densities (226). In line with this, the number of dendritic spines increases in LA after fear conditioning (392).
At the level of cell-cell interactions, various cell recognition molecules seem to translate such cytoskeletal re-arrangements into altered cell-cell and cell-matrix interactions. Fear conditioning-induced expression changes were found for the mRNA of the extracellular matrix molecule tenascin and the cell adhesion molecule neuroligin (406, 488). Persistent expression of neuroligin-1 is indeed required for maintenance of NMDA-R mediated synaptic transmission, enabling normal development of synaptic plasticity and long-term memory in the amygdala (203). Furthermore, interfering with the integrity of the extracellular matrix through null mutation for specific tissue inhibitor of matrix metalloproteinases (TIMPs) interfered with fear-potentiated startle responses (178). One of the most intensively studied cell recognition molecule is the neural cell adhesion molecule (NCAM), which mediates neuromodulatory and hormonal effects on conditioned and unconditioned fear (442, 489). NCAM function is regulated by polysialic acid (PSA). Injection of PSA-NCAM and PSA, but not NCAM, into the hippocampus impaired the formation and consolidation of hippocampus-dependent contextual fear memory (461). The expression of PSA-NCAM increased 24 h after fear conditioning in the amygdala, but only in animals subjected to the highest shock intensity, and intra-amygdala cleavage of PSA-NCAM affected fear extinction rather than acquisition or consolidation of cued fear memory (276). Studies in null mutant mice suggest that NCAM contributes to the stress modulation of long-term context fear memory (4). In any case, many adhesion molecules can initiate signalling pathways that couple the dynamics of extracellular and intracellular events, particularly those that regulate cytoskeletal processes and spine architecture. Together, these processes may then form an interlinked molecular network that regulate structural re-arrangements and morphology between pre- and postsynaptic sites, concomitant with the stabilization of the fear memory trace.
C. Emerging views on distributed synaptic plasticity
In the previous sections, we described converging lines of evidence indicating that Pavlovian fear conditioning depends on mechanisms of enduring synaptic plasticity in the amygdala. However, most studies focused on LTP of thalamic inputs to LA neurons. Although these studies captured synaptic features critical for fear conditioning, it is clear that the underlying molecular changes occur at multiple sites rather than at a single location, a principle referred to as “distributed plasticity”. For instance, studies mapping changes in protein expression, metabolism or electrophysiological activity at multiple sites indicate that learning initiates coordinated patterns of activity in distributed brain areas, reflecting fear memory stabilization (see section IV.B). Furthermore, the induction of synaptic plasticity requires correlated activity to occur in a relatively narrow time window, for instance between pre- and postsynaptic sites or between two afferent input pathways. Fear conditioning, however, does not necessarily require such precise timing, as the US can be applied at the end of the CS with no temporal overlap. This suggests that longer time windows are created for induction of conditioned fear behaviour. One possible solution resides in the ability of BLA neurons to generate oscillatory patterns of activity. These oscillations provide recurring time windows during which groups of BLA neurons are synchronized with afferent inputs signals, thereby facilitating synaptic plasticity with no major increases in activity per se in spatially distributed networks (see section III). These time windows may then facilitate synaptic plasticity in distributed networks involving local neuronal circuits within the amygdala, or neuromodulatory input systems (reviewed in (299)). Outstanding questions therefore relate to the fine-scale organization of these synaptic networks, and the mechanisms of synaptic plasticity within these circuits. Although the detailed mechanisms remain to be identified, some principles have emerged recently, which we discuss in the following section. An overview of the forms of long-term synaptic plasticity detected at the various inputs to types of neurons in the amygdala is provided in figure 6.
1. Long-term pre- and postsynaptic plasticity in principal amygdala neurons
In addition to thalamic inputs, principal LA neurons receive inputs from the cerebral cortex. Postsynaptic NMDA-Rs are expressed at both, thalamic and cortical inputs (116, 117, 265, 498, 524). However, the properties of synaptic plasticity differ considerably at the two inputs, particularly the temporal precision needed for induction of synaptic plasticity. Recent studies have emphasized that the polarity of synaptic plasticity depends on the precise order of pre- and postsynaptic activity, in the millisecond range, a phenomenon referred to as spike-timing-dependent synaptic plasticity (reviewed in (63)). In LA, the standard protocol used to induce spike-timing dependent LTP (presynaptic firing closely followed by postsynaptic depolarization) induces long-term plasticity at thalamic but not cortical afferents in vitro (170). This is in line with previous in vivo findings of a greater LTP magnitude at thalamic compared to cortical inputs (101, 473). Interestingly, the two inputs contact neighbouring but functionally and morphologically distinct types of dendritic spines (170). Spines receiving thalamic inputs are bigger, display larger Ca2+ transients, and express R-type Ca2+ channels, thereby providing reliable Ca2+ influx for postsynaptic LTP induction and expression (170).
In addition to LTP dependent on postsynaptic NMDA receptors, some forms of LTP depend on presynaptic NMDA receptors in the amygdala. For instance, Humeau and colleagues (172) reported an associative form of LTP at cortical inputs to LA neurons that is induced by simultaneous Poisson-train stimulation of thalamic and cortical afferents (Fig. 6A). This LTP is of an associative nature, in that its induction requires simultaneous activation of converging cortical and thalamic inputs to principal LA neurons, whereas stimulation of either input system alone evokes no plasticity (172). Consistent with earlier immuncytochemical observations (115), presynaptic NMDA-Rs, mediate this form of LTP through a persistent increase in transmitter release probability. The intracellular mechanisms involve the cAMP/PKA signalling pathway and a change in the Ca2+ coupling of vesicle release mediated via the active-zone protein and PKA target RIM1α (126). In addition to LA, a presynaptically induced and expressed form of homosynaptic LTP has been discovered at thalamic afferents to CEm neurons (439). Induction was dependent on presynaptic NMDA-Rs since hyperpolarization, chelation of Ca2+ or blockade of NMDA-Rs in the postsynaptic neurons, had no effect.
Consistent with presynaptic sites of plasticity, expression of the synaptic vesicle protein, synaptophysin, increases in the BLA following auditory fear conditioning (341). The gaseous molecule nitric oxide (NO), thought to serve as a retrograde messenger to presynaptic sites of LTP expression, has been shown to contribute to both LTP and consolidation of auditory fear conditioning (450), in part by activating the ERK/MAPK signalling cascade via the cyclicGMP-protein kinase G pathway (344, 365), although this influence has been localized to thalamic rather than cortical inputs.
Overall, the above indicates that thalamic and cortical inputs to LA neurons express different types of plasticity associated with contrasting forms of coincidence detection. The homosynaptic form of thalamo-LA LTP requires pre-synaptic activity coinciding with strong postsynaptic activation and associated Ca2+ influx, thereby apt to detect coincidence at individual inputs in an input-specific manner. By comparison, cortico-LA LTP does not rely on postsynaptic activity, but might be induced by subthreshold activity generated by thalamic and cortical afferents. Through this presynaptic mechanism of coincidence detection, relatively weak cortical inputs may be primed for subsequent induction of homosynaptic Hebbian plasticity at neighbouring synapses, which require stronger afferent activity and/or the induction of postsynaptic action potentials (172). Two observations support this conclusion. First, postsynaptic hyperpolarization reduces but does not abolish LTP in fear-conditioning (421), suggesting that LTP induction independent of postsynaptic activity also occurs in vivo. Second, depletion of RAP1 (a small GTPase involved in AMPA-R trafficking and LTP) in a mouse line with CaMKII-α-Cre-mediated knock-out of rap1a and rap1b genes, results in impaired synaptic plasticity and increased basal transmission of glutamate via presynaptic changes (348). Behaviorally, these mice display impaired fear learning, which could be rescued by training with a more aversive unconditioned stimulus. The gene deletion eliminates 90% of the RAP1 protein in the cortex, suggesting that the deficit in fear learning reflected an impaired interaction between the cortical and thalamic input pathways involving presynaptic priming upon weak training. The importance of network timing has been extracted more directly from patterns of polysynaptic responses within the LAd, where latencies of recurrent activity triggered by thalamic afferent stimulation were found to overlap with cortical afferent latencies (182). The spatio-temporal architecture of the intra-amygdala network may thus be tuned to facilitate coincidence of the two sensory afferent input systems, as for instance, during synaptic plasticity and fear learning (182).
2. GABAergic plasticity
There is ample evidence that GABAergic interneurons regulate signal flow through the amygdala (87, 229), thereby modulating synaptic plasticity in principal cells and influencing fear learning and extinction (reviewed in (105)). The induction of LTP in principal LA neurons can thus be gated by influences that suppress inhibition from local interneurons. Examples include the dopaminergic (35) and noradrenergic (504) transmitter system. Another mechanism of GABAergic influence is via presynaptic GABAB receptors, stimulation of which dampens subsequent transmitter release and thereby mediates short-term plasticity of glutamatergic and GABAergic transmission in LA (496). While presynaptic GABAB receptors exist on glutamatergic afferents to interneurons and principal neurons in LA, they selectively inhibit glutamatergic transmission and suppress LTP in principal neurons (347). This effect is most likely due to a differential local GABA spill over from GABAergic synapses (347). When the extracellular GABA level is decreased in the BLA after fear conditioning (487), the GABAB mediated inhibition of glutamate release may be relieved. Consistent with such a balancing function of presynaptic GABAB on LTP in the amygdala, a genetic deficiency of GABAB(1a) receptors resulted in a shift from the associative, NMDA receptor-dependent form of LTP towards a non-associative, NMDA receptor-independent form of presynaptic LTP at cortico-amygdala afferents (465). The balancing function of the GABAB receptors is dependent on GABAergic activity, and lack of GABAB receptors is associated with generalization of conditioned fear (465). Fear generalization is also observed upon a deficiency of GAD65, the activity-dependent isoform of the GABA synthetizing enzyme (25), indicating the requirement of balanced GABAergic activity for cue specific fear responsiveness.
In addition, glutamatergic inputs to local GABAergic interneurons exhibit activity-dependent synaptic plasticity, although the evidence is sparse compared with that in principal neurons. An overview is provided in figure 6C. LA interneurons receive convergent cortical and thalamic afferents (496), and input-specific LTP can be induced at either input (264, 497). The Ca2+ influx required for induction of input specific LTP is mediated via Ca2+ permeable subtypes of AMPA receptors (264, 497), while NMDA-Rs seem to be involved in a heterosynaptic form of LTP in LA interneurons (20). Fear conditioning results in a decrease in GABAergic plasticity in the LA, reflected by a decrease in the magnitude of GABAergic LTP in principal neurons (497). Furthermore, the extracellular GABA concentration and GAD65 mRNA level are decreased after fear conditioning (25, 487). Changes in GAD expression and decrease in GABAergic plasticity follow a similar time course (497), thereby suggesting the following scenario. Under baseline conditions, the GABAergic influence is high, resulting in dampening of activity and synaptic plasticity in principal neurons, through presynaptic GABAB receptors at afferent inputs and postsynaptic GABAA as well as GABAB receptors. Upon fear conditioning, GAD65 expression and the extracellular GABA concentration decrease, thereby relieving glutamatergic inputs from presynaptic GABAB blockade and facilitating LTP, both the postsynaptic thalamic and the heterosynaptic cortical types. Behaviorally, conditioned fear responses occur with high specificity for the conditioned stimulus. During impaired or blocked function of GAD65 or GABAB receptors (as, for instance, in the respective knock-out mutants), the decreased GABA level or dysfunction of presynaptic GABAB receptors result in a shift from associative to non-associative (NMDA receptor-independent) forms of LTP at cortical inputs, while postsynaptic LTP is preserved at thalamic inputs. Conditioned fear responses occur with reduced stimulus discrimination, i.e. in a generalized manner. Through these mechanisms, GABAergic regulation of synaptic plasticity may help control both the induction of conditioned fear and the CS-specificity of the conditioned responses.
V. Synaptic Plasticity Related to Fear Extinction
Compared to the acquisition and consolidation of conditioned fear, much less is known about the mechanisms of fear extinction. However, extinction is the focus of increasing attention because of its potential clinical significance. Indeed, an approach commonly used by clinicians to treat anxiety disorders (exposure therapy) is similar to that used to extinguish conditioned fear responses in the laboratory. In both cases, the subject is repeatedly presented with the feared object or situation (conditioned stimulus, CS) in the absence of danger (or unconditioned stimulus, US). Thus, it is widely believed that understanding the networks and mechanisms of extinction might ultimately lead to improvements in the treatment of anxiety disorders. Consistent with this, it was proposed that some human anxiety disorders reflect an extinction deficit (84). In fact, this appears to be the case in post-traumatic stress disorder (306). Therefore, this section will review current knowledge and concepts regarding the behavioral properties of extinction as well as the networks and cellular mechanisms participating in extinction.
A. Behavioral properties of extinction
In the Pavlovian fear conditioning paradigm, extinction is studied by repeatedly presenting the CS in the absence of the US, resulting in the decline of CS-evoked fear responses. When considering extinction, it is important to distinguish between the reduction of conditioned fear that takes place within the extinction training session (within-session extinction) from that observed one day or more after extinction training (between-session extinction, extinction retention/retrieval/recall). Indeed, as we shall see below, much evidence suggests that the decrease in behavioral responding seen within an extinction training session depends on mechanisms that partly differ from those underlying the between-session effect.
Whereas conditioned fear responses can persist for the entire adult lifetime of rats (133, 284), the expression of extinction decays with time, a process termed “spontaneous recovery” (407). Similarly, whereas cued conditioned fear responses are expressed even if the training and testing contexts are different, extinction is expressed in a relatively context-specific manner. Indeed, if testing occurs in a different context than the one where extinction training took place, extinction is not expressed as strongly, a phenomenon known as “renewal” (45, 46). Another defining property of extinction is “reinstatement” where presentation of unsignaled USs after extinction training causes a resurgence of conditioned fear responses, provided the USs were presented in the extinction training context (405). Finally, it should be mentioned that the impact of extinction training is relatively specific to the extinguished CS. Indeed, extinction training does not abolish the conditioned fear responses associated to a different CS (157) or with the subsequent acquisition of conditioned responses to a different CS (for instance see (248)). Moreover, extinguishing a generalization stimulus has little effect on fear responding to the CS (for instance, see (509)).
The behavioral properties of extinction suggest that it does not result from the erasure or reversal of the initial fear memory. This statement is based on the fact that conditioned fear responses can re-appear with the passage of time (spontaneous recovery), if the CS is presented in a different context than where extinction training took place (renewal), or if unsignalled USs are presented in the extinction training context prior to testing extinction recall (reinstatement). Thus, the behavioral properties of extinction indicate that this form of safety learning depends on the development of a new inhibitory memory that competes with the initial fear memory for control of behavior. However, as we shall see below, there is also evidence that weakening of the initial CS-US association is involved.
B. Cerebral networks involved in extinction
Three interconnected brain regions have been implicated in extinction: the amygdala, mPFC, and hippocampal formation. Increasing evidence suggests that the amygdala is the critical site of plasticity where the extinction memory is stored. In contrast, the infralimbic component of the mPFC is critical for the consolidation and recall of extinction (387). Finally, the hippocampal formation mediates the context specificity of extinction (180). To understand how the amygdala, mPFC, and hippocampus interact in extinction, we must first consider the connections existing between these structures.
1. Connections between the amygdala and mPFC
Two components of the mPFC are most densely interconnected with the amygdala: the infralimbic and prelimbic areas (286, 436, 463). In the BLA, infralimbic and prelimbic projections show minimal overlap with infralimbic axons focusing on the ventral part of LA and the BM nucleus whereas prelimbic axons mainly target BL (Fig. 7A; (286, 297, 508)). Projections of the mPFC to the BLA are thought to be glutamatergic with mPFC axon terminals forming only asymmetric synapses, usually with the dendritic spines of principal cells, and much less frequently with the dendrites of presumed GABAergic neurons (50, 475). In addition, the infralimbic cortex sends a very dense projection to the medial ITC cell clusters and significant one to CEl (Fig. 7A1; (66, 297)). Although the prelimibc cortex also projects to ITC cells, this projection is significantly weaker than the one originating in the infralimbic region (Fig. 7A2; (286, 297, 508)).
Fig. 7.

Connections between the amygdala, mPFC, and hippocampus. (A) Reciprocal connections of the infralimbic (A1) and prelimbic (A2) components of the mPFC with the amygdala. Solid lines indicate major projections whereas dashed lines indicate weaker ones. (B) Multiples direct and indirect paths for the transfer of contextual influences to the amygdala.
In the context of extinction, the infralimbic projection to ITCm cells is especially significant because electrical infralimbic stimuli that coincide with CS onset reduce conditioned fear responses and accelerate the acquisition of extinction (307). Moreover, infralimbic stimuli block the excitation of CEm neurons by BL inputs (388), an effect thought to depend on the activation of ITCm cells by infralimbic stimuli. Consistent with this, disinhibition of the infralimbic cortex with local picrotoxin infusions enhances c-fos expression by ITCm cells (31).
The amygdala sends return projections to the mPFC (218). However, these projections arise exclusively in the BLA, particularly BL, posterior part of AB and, to a lesser extent, ventral part of LA. The existence of reciprocal connections between the mPFC and BLA has complicated the interpretation of physiological studies, leading to a disagreement regarding the nature of mPFC influences (excitatory vs. inhibitory) over the BLA (247, 420, 422). We consider this issue in some detail as its resolution will impact on how we conceive mPFC involvement in extinction.
Because the BLA and mPFC are reciprocally connected, electrical mPFC stimulation not only recruits mPFC axons ending in the BLA but also antidromically activates BLA axons ending in the mPFC (247). It is important to disentangle the consequences of these two phenomena because the antidromic effects are an unavoidable by-product of electrical stimulation that does not accompany natural mPFC activation. Because the conduction velocity of BLA axons to the mPFC is higher than that of mPFC axons to the BLA (125, 247), the arrival of antidromic impulses precedes that of orthodromic ones. Importantly, because the local axon collaterals of principal BLA neurons recruit feedback interneurons (438), the inadvertent antidromic activation of BLA projections by electrical mPFC stimuli can lead to widespread feedback inhibition in the BLA. This artefactual feedback inhibition can therefore give the impression that mPFC inputs “inhibit” the BLA, as was previously proposed (420, 422). However, given that mPFC axons typically form asymmetric synapses with the dendritic spines of BLA projection cells, this conclusion is probably erroneous. Consistent with this, indirect activation of the mPFC by electrical stimulation of the mediodorsal thalamic nucleus or of the contralateral mPFC elicits a robust synaptic excitation of physiologically identified BLA projection cells (247). Moreover, behavioral studies indicate that the mPFC exerts excitatory influences over the BLA. Indeed, local inactivation of the prelimbic region reversibly inhibits the expression of previously learned cued or contextual fear responses (83, 235).
Overall, the data reviewed above suggests that the impact of mPFC inputs to the amygdala depends on the cortical field at the origin of the projection and the target nuclei. Infralimbic inputs excite ITCm cells that, in turn, inhibit CE neurons and thus, the expression of conditioned fear responses. In contrast, prelimbic inputs excite BLA neurons that send a glutamatergic projection to CE. As a result, prelimbic lesions interfere with the expression of conditioned fear responses (83).
2. Hippocampal projections to the amygdala and mPFC
The results of lesion and/or reversible inactivation studies indicate that the hippocampus is required for the renewal of cued conditioned fear responses after extinction training (reviewed in (180)). However, it is currently unclear how this contextual information is relayed to the amygdala. A number of possible routes exist including direct CA1, subicular, and entorhinal projections to various components of the amygdala (Fig. 7B). In addition, it is possible that one or more of these sources influences the amygdala indirectly (Fig. 7B), via the mPFC projections described above.
The results obtained to date are compatible with all these possibilities. For instance, permanent and/or reversible interference with CA1 (81, 82, 160) or entorhinal activity as well as fornix lesions (181) all prevent the contextual renewal of conditioned fear responses after extinction training. Therefore, it seems that multiple parallel routes convey contextual information to the amygdala and that normal contextual gating of extinction depends on intact coding in theses multiple parallel pathways. However, an alternative interpretation is that in some of these cases at least, the lack of renewal observed following localized lesions or inactivations reflects a disfacilitation of critical amygdala targets rather than the specific signalling of information about the renewal context.
Nevertheless, since the available data is compatible with both interpretations, we now overview the various possible routes through which contextual information from the hippocampus might reach the amygdala. As in section II, we will focus on projections ending in the BLA, CE, and ITC cell clusters. See Refs (286, 376) for projections to other amygdala nuclei. It should be noted that most of the projections described below are reciprocated by the amygdala.
Direct hippocampal projections to the amygdala
Most direct hippocampal projections to the amygdala originate from the temporal subiculum and, to a lesser extent, the adjacent part of CA1 (62, 286, 345, 376, 506, 507). There are no dentate and CA3 outputs to the amygdala. Subicular projections are dense in AB and medial part of BL but moderate in LA and light in CE. CA1 projections to the amygdala are considerably lighter than those originating in the subiculum. They mainly end in BL, with lighter projections to LA and AB (345, 507).
Entorhinal projections to the amygdala
Entorhinal efferents to the amygdala mainly originate from deep (layer V-VI) neurons. Of the various entorhinal fields, the ventrolateral and dorsolateral areas send the densest projections. These entorhinal inputs target much of the BLA but they are heaviest in BL. In contrast, the ventromedial and lateral entorhinal areas contribute the weakest projections.
MPFC transfer of hippocampal outputs to the amygdala
In addition to the direct subicular, CA1, and entorhinal projections described above, contextual information can reach the amygdala via the mPFC. Indeed, CA1 and subicular pyramidal neurons located in the temporal and mid-septotemporal portions of the hippocampus send a heavy projection to the mPFC (17, 161, 179, 434, 494). By comparison, much fewer entorhinal cells project to this region. Double retrograde tracing studies indicate that most hippocampal neurons projecting to the mPFC also have an axon collateral ending in the entorhinal cortex (494). It remains controversial whether the infralimbic cortex, prelimbic cortex or both regions are the main recipients of CA1 and subicular projections (17, 161, 179, 434, 494).
C. Mechanisms of synaptic plasticity underlying extinction learning and consolidation
Overall, the available data indicate that extinction learning and expression relies on a tripartite synaptic circuit, including the amygdala for storing of both conditioned fear and extinction, the hippocampus for processing of contextual information, and the infralimbic region of the mPFC for the consolidation and retrieval of extinction memory (190, 274, 308, 327, 389). In the extinction context, mPFC activity inhibits CE fear output neurons via the glutamatergic activation of GABAergic ITCm neurons, which results in dampening of fear expression (189, 248). Outside the extinction training context, CE neurons are subjected to less inhibition and fear responses re-vive. In addition, different types of neurons in the basal amygdala signal fear memory or extinction, which may shift the balance between the context-dependent expression of fear and or extinction after conditioning (157). Two main classes of synaptic mechanisms and intracellular pathways have been identified in relation to fear extinction: (1) mechanisms underlying the reinforcement of an active inhibitory process that competes with the initial fear memory for the control of behavior; (2) mechanisms that reverse the changes in synaptic efficacy induced during fear conditioning. A most convincing piece of evidence supporting the dual mode of extinction comes from a recent study focusing on the involvement of αCaMKII in extinction (209). First, this study confirmed earlier observations that extinction training conducted 24 hours, but not 15 min, after contextual fear conditioning showed spontaneous recovery. This suggests that depending on the interval between fear conditioning and extinction training, extinction can result from the formation of a new inhibitory learning or from unlearning of the initial CS-US association. However, it should be mentioned that the effects of extinction timing on recovery effects (renewal, spontaneous recovery, reinstatement) are controversial, with contrasting and even completely opposite results between different studies (68, 271, 328). Next, by conducting these tests in heterozygous knock-in mice with partial reduction of αCaMKII activity, this study showed that αCaMKII is required for the formation of the new inhibitory memory, but not for the loss of conditioned fear responses during early extinction, thereby providing molecular evidence for the duality of mechanisms in fear extinction. These two sets of mechanisms seem to be developmentally regulated. In contrast to adult animals, rats at earl postnatal stages (below 3 weeks) do not exhibit reinstatement or re-newal of conditioned fear memories, and extinction has been suggested to reflect an unlearning process leading to erasure of initial fear memories (205). Fear extinction depends on the amygdala at all postnatal stages investigated (205, 274, 327), whereas the mPFC is involved in fear extinction in adult but not young animals (204). In an elegant series of experiments, Gogolla and coworkers (138) have shown that erasure-resistant fear memories are mediated by the formation of perineuronal nets composed of extracellular matrix chondroitin sulphate proteoglycans in the amygdala during a postnatal critical period.
Accordingly, we will use this duality of processes when describing extinction-related synaptic mechanisms. The remainder of this section will focus on signalling pathways and network mechanisms supporting the formation of new extinction-related inhibitory memory. The following section will consider the mechanisms underlying the reversal of conditioning-evoked alterations.
1. NMDA receptors
There is ample evidence that application of NMDA-R antagonists, either systemically or locally into the BLA, just before extinction training, prevents formation of the extinction memory (18, 113, 245, 253), with NR2B receptor subtypes playing a particularly important role (481, 482). Furthermore, the NMDA-R agonist D-cycloserine, a partial agonist acting at the glycine-recognition site of the NMDA-R, facilitates extinction of fear-potentiated startle or conditioned freezing when administered shortly before or after extinction training (237, 238, 512, 530). Importantly, several lines of evidence indicate that effects obtained with experimental manipulation of NMDA-R activity are not due to state-dependent changes in neuronal activity, but that NMDA receptors are specifically involved in learning and consolidation of extinction. First, systemic application of an NMDA-R antagonist during extinction training interfered with extinction recall when tested 24 hours, but not 1.5 or 48 hours later (445). A second application of the antagonist 24 hours after extinction training also affected long-term extinction recall. These results suggest that consolidation of extinction shifts from an NMDA-independent early stage to an NMDA-dependent form. Second, pre- or post-extinction infusion of the relatively selective NR2B antagonist ifenprodil locally into BLA or mPFC indicate that within-session extinction involved NMDA-R activation in the BLA, whereas consolidation depends on NMDA-Rs in the mPFC (445). Interestingly, re-learning of extinction recruits NMDA-Rs in both BLA and mPFC (233, 234). Thus, the development of extinction seems to involve NMDA-Rs in the BLA, whereas its consolidation involves NMDA-Rs in the mPFC (Fig. 8A). Relearning to inhibit fear responses seem to involve NMDA-Rs in both the BLA and mPFC, and consolidation again involves NMDA-Rs in the mPFC. The NR2B receptor subtype is critical for these phase-dependent roles of NMDA-Rs in extinction (482). Third, it was shown that Ca2+-mediated burst firing in infralimbic neurons predicted subsequent recall of extinction, and that this burst activity was dependent on NMDA-R activation (54). Therefore, NMDA-R mediated bursting in infralimbic neurons seems to initiate Ca2+-dependent intracellular cascades that stabilize fear extinction memory.
Fig. 8.

Synaptic plasticity related to fear extinction. A. Activation of NMDA receptors occurs in the basolateral amygdaloid complex (BLA) and the prefrontal cortex (PFC) during within-session and consolidation of extinction, respectively, most likely inducing long-term potentiation (LTP). B. Postsynaptic release of endocannabinoids (eCB) mediates long-term depression of GABAergic transmission (LTDi) via activation of CB1 receptors on cholecystokinin-positive interneurons (CCK-IN). Release of eCB can be stimulated via metabotropic glutamate receptors (mGluRs). C. Increase in glutamatergic transmission to GABAergic mITC neurons mediated through NPS receptors in presynaptic LA principal neurons. D. Both NMDA receptor-dependent LTP and LTD exist at BLA inputs to mITC, which can be induced homo- and heterosynaptically, and which keep the overall synaptic strength in balance.
2. Voltage gated Ca2+ channels
Other sources of intracellular Ca2+ in relation of fear extinction may include voltage-gated Ca2+ channels. However, the evidence remains sparse as compared to that for fear conditioning. There is some evidence for impaired extinction, involving both within-session extinction and extinction recall, upon systemic application of the Ca2+ channel blockers nifedipine and nimodipine (19, 57, 58, 492). However, it was suggested that nifedipine affects fear extinction indirectly, through induction of a stress response (514).
3. Metabotropic glutamate receptors
There is also evidence that mGluRs regulate extinction. Indeed, mGluR7-/- mice exhibit an extinction deficit (59). Moreover, systemic pre-extinction application of a novel mGluR7 allosteric agonist (AMN082) facilitates, whereas mGluR7 knockdown using siRNA prior to aversive training severely attenuates between-session extinction in fear-potentiated startle (120). In addition, acquisition of conditioned fear and thalamo-LA LTP in principal neurons were impaired by application of a mGluR agonist, whereas mGluR knockdown had not effect on the acquisition of conditioned fear. As mGluR7 is localized to presynaptic terminals of glutamatergic neurons in the amygdala and negatively coupled to the adenylyl cyclase/cAMP system (281), a decrease in presynaptic glutamate release may contribute to extinction learning, although it remains unclear how impaired fear acquisition can coincide with facilitated fear extinction. Pre- or postsynaptically located group II mGluRs positively coupled to the adenylyl cyclase/cAMP system have been found to mediate LTD in LA/BLA, although the significance for fear extinction remains unclear to date (153, 255). Further experiments using pharmacological interference with the adenylyl cyclase/cAMP system yielded a somewhat inconsistent picture. Subchronic blockade of phosphodiesterase activity (assuming to raise cAMP levels) resulted in hippocampal CREB activation and increase in freezing behavior throughout extinction training (318), while transgenic mice overexpressing type 1 adenylyl cyclase within the forebrain displayed hippocampal CREB activation and unaltered tone and context fear acquisition but delayed context extinction (515).
4. Protein kinases
Several kinase pathways are involved in fear extinction in the relevant brain regions, including PKA (319, 495), MAPK (159, 168, 169, 261, 423, 530), PI3-kinase (71, 253, 530), SRC kinases (33), and CAMK (32, 495). Pharmacologically interfering with a given kinase pathway before extinction training typically had no effect on within-session extinction but impaired extinction recall, while the same treatments shortly after extinction training resulted in a deficit in extinction recall at later times. These data indicated an involvement of the respective kinase pathway in the consolidation rather than the acquisition of fear extinction. In line with this are reports of an upregulation of phosphorylated MAPK/ERK within the BLA, which occurs in a time-dependent manner at late extinction periods (159) and depends on extinction success (530). Similarly, infusion of MAPK inhibitors prior to or immediately after extinction training into the BLA (159) or the mPFC (168, 169) impaired subsequent retrieval of extinction in later test sessions. A detailed account on kinase pathways comes from the work of Fischer and colleagues (121) on hippocampal ERK/MEK signalling. Both contextual fear conditioning and its extinction triggered an upregulation of phosphorylated ERK-1/2, with conditioning and extinction effects displaying a difference in time course and localization to the cytoplasmic and nuclear compartment of hippocampal neurons, respectively. Pharmacological inhibition of the ERK-activating kinase, MEK, immediately after extinction trials prevented ERK-1/2 activation and impaired extinction recall. Control procedures ruled out actions on fear memory retrieval or consolidation. Hippocampal MEK/ERK signalling may thus serve as one of the key mediators of contextual fear regulation, with specific temporal and compartmental characteristics differentiating between fear conditioning and extinction. Another critical pathway recruits Trk receptors. Blocking BDNF influence in the BLA through lentiviral-induced expression of a dominant-negative truncated TrkB receptor after fear conditioning had no effect on within session extinction, but impaired retention of extinction. This suggests that TrkB activation is required for the consolidation of stable extinction memories (74).
The engagement of kinase pathways suggests that transcriptional modulation of gene expression is involved in extinction consolidation. Indeed, induction of immediate early genes, like c-fos, has been observed in both the BLA and the mPFC following extinction training and has been related to extinction success (158, 321). Increases in c-fos and ERK expression have been observed upon both conditioning and extinction of contextual fear in the hippocampal CA1 area, and have been associated with separate populations of pyramidal neurons (501). Furthermore, infusion of protein synthesis blockers into BLA (253) or mPFC (444) lead to impaired retrieval of extinction in later sessions, suggesting that protein synthesis is required for consolidation of extinction. However, in contrast to the robust involvement of protein synthesis in the consolidation of conditioned fear, its role in extinction appears to vary depending on the conditioning and extinction paradigm. For instance, in a contextual fear paradigm, inhibition of hippocampal protein synthesis after the first extinction trial reduced freezing responses ((122); see also (89)). This effect reflected enhanced extinction rather than loss of stable fear memory, because conditioned freezing could be re-instated by a reminder shock (122). Protein synthesis counteracting extinction during brief extinction trials might thus prevent rapid extinction of conditioned freezing in situations in which the CS does not reliably predict the absence of the US. This possibility is consistent with the downregulation of immediate–early genes such as c-fos, egr-1, and arc (197, 268, 393) with short non-reinforced CS exposures. In fact, there has been some debate as to whether results obtained with protein synthesis blockers relate to effects on extinction or re-consolidation of conditioned fear, given the similar experimental procedures used to examine these two phenomena (as discussed in (327, 331, 333, 389)). Which process predominates in a given retrieval session, and how do the two processes interact? The emerging consensus is that the duration of the re-exposure to the conditioned stimulus determines which process predominates: re-consolidation with very short re-exposure and extinction with long and/or repeated exposure (389). Protein synthesis is involved in both processes (327, 331, 333). During contextual fear conditioning, there is an increased expression of CREB and CREB-dependent Arc in the amygdala and hippocampus after short re-exposure, and in the amygdala and prefrontal cortex after long re-exposure, suggesting that re-activated contextual fear memories undergo CREB-dependent reconsoldation or extinction in distinct brain areas (269).
5. Synaptic remodelling
As discussed above (section IV.B), de novo protein synthesis leads to the persistent activation of a number of protein kinases, that directly, or via downstream targets, lead to synaptic remodelling. One important effector mechanisms is actin stability. Intrahippocampal injections of the actin rearrangement inhibitors cytochalasin D or latrunculin after contextual fear conditioning impaired conditioned freezing, while injection in between extinction trials prevented extinction (122), Notably, the inhibitors were not effective when applied after extinction of conditioned freezing. Supporting these conclusions is the recent finding (441) that Cdk5, a serine/threonine-kinase and important regulator of synaptic function and actin dynamics, regulates between-session extinction of contextual fear. Extinction was found to require a downregulation of Cdk5 and upregulation of p21 activated kinase-1 (PAK-1) activity, which is achieved by a reduced membrane association of the Cdk5 activator p35 and dissociation of p35 from PAK-1, mediated by the small GTPase RAC-1. Actin rearrangement, involving a molecular pathway with counteracting Cdk5, PAK-1, and RAC-1, thus seems to regulate extinction of contextual fear, predominantly during repeated extinction trials (441). As to NCAM, intra-amygdala cleavage of PSA-NCAM did not affect acquisition, consolidation or expression of remote fear memories, nor within-session extinction, but strengthened extinction memory (276). Since NCAM is thought to be involved in stress-modulated contextual fear, its specific contribution to fear extinction remains to be delineated.
6. GABA signalling
Consistent with the role of GABAergic mechanisms in extinction, mRNA and protein levels of the GABAA receptor clustering protein gephyrin are significantly upregulated in the BLA 2 hours after extinction training, together with an increase in the surface expression of GABAA receptors in the BLA (73). In contrast, gephyrin expression is reduced after fear acquisition (254, 406). In fact, the expression of various GABA-related genes seems to be differentially regulated in the amygdala. Three hours after fear training, mRNA levels of the GABAA receptor subtypes α1, α5 and the GABA-synthetizing enzyme GAD were decreased, while after extinction training the mRNA levels of α2, β2, GAD and gephyrin, as well as the GABA transporter GAT1 were increased (154). At the protein level, gephyrin, β2 and γ2 levels were decreased upon extinction of conditioned fear (254). Also, the two isoforms of GAD, GAD 65 and GAD 67, were transiently down-regulated, respectively 3 and 24 hours after fear conditioning (25, 154). Supporting the idea that extinction involves the regulation of GAD65, the activity-dependent GAD isoform, GAD65-deficient mice show impaired extinction of cued fear, both within sessions and during recall (443). In contrast, extinction of contextual fear was unaltered, suggesting functionally or regionally specific differences in GABA-related contributions to fear extinction. In fact, such differences in the regulation of GABA-related genes were reported for LA, BL and CE (154) as a result of fear conditioning and extinction. However, their functional significance remains to be examined.
Together, these findings indicate that the acquisition of conditioned fear induces a downregulation of markers related to GABAergic function in the amygdala, whereas the acquisition of fear extinction produces an upregulation of GABAergic markers. In keeping with this, a decrease in the frequency and amplitude of miniature IPSCs occurring in LA projection neurons one day after fear training returned to baseline levels during retrieval of extinction (254). Furthermore, cell-permeable TAT-conjugated peptide (TAT)-GABA receptor-associated protein (GABARAP) inhibitory peptide that blocked GABAA receptor insertion in the amygdala interfered with both extinction-induced increase in miniature IPSCs and reduction of fear-potentiated startle responses. These results corroborate the view that fear extinction involves GABAergic mechanisms that functionally oppose those recruited during fear acquisition.
One population of GABAergic neurons of critical importance for fear extinction are the paracapsular ITC GABAergic neurons located between the BLA and CE (ITCm; (189, 248)). These cells receive glutamatergic inputs from the BLA, and, in turn, provide GABAergic inhibition to CE neurons. Therefore, they are situated in an ideal position to control signal flow within the amygdala (427). Lesions (248) or modulation through neuropeptide S (NPS) (189) of these GABAergic ITC neurons specifically influences fear extinction with spared fear memory acquisition and consolidation (Fig. 8C). Both NMDA-dependent LTP and LTD occur at BLA inputs to these neurons, and both can be induced homo- and heterosynaptically (Fig. 8D; (429, 430)). Synaptic plasticity seems to be well balanced in ITC cells, as activity-dependent potentiation or depression of particular inputs leads to opposite changes at other inputs ending at different dendritic levels, thereby keeping total synaptic weight constant, although the relative strength of inputs is modified (430). Moreover, ITC neurons display a wide range of short-term presynaptic plasticity, which, in turn, is functionally balanced through synaptic interconnectivity between subpopulations of neurons, thereby stabilizing the pattern of spike firing (137). Therefore, these results suggest that synaptic plasticity in ITCm cells is not a local event engaging a limited group of synapses or neurons, but a distributed event in which the strength of synaptic connections can be affected by the state of other inputs, while keeping the overall weight of the synaptic network and output activity in a stable range. The functional significance of these balanced interactions for conditioned fear and extinction remains to be delineated. Further evidence that GABAergic synaptic plasticity is critical for fear extinction has been obtained by manipulating endocannabinoid signalling (reviewed in (263)). The cannabinoid receptor subtype (CB1) is found presynaptically on the axon terminals of a specific subpopulation of BLA interneurons expressing the anxiogenic peptide CCK (195, 294). CCK exerts a strong depolarizing effect in principal LA neurons via activation CCK2 receptors coupled to transient receptor potential (TRP) type cationic channels (304). CB1 receptor stimulation reduced GABAergic responses in principal neurons (195), and low-frequency afferent stimulation in LA caused the release of endocannabinoids, inducing an LTD of GABAergic synaptic transmission (LTDi) (Fig. 8B; (16)). The consequence of this particular anatomical localization for conditioned fear behavior was investigated in mice with CB1 receptor deficiency (278). These CB1 receptor mutants displayed impaired short- and long-term extinction in auditory fear-conditioning, with spared fear memory acquisition and consolidation (278). Moreover, pharmacological blockade of CB1 receptors led to a similar deficit in wild-type mice (16), which was ameliorated with administration of a CCK2 receptor antagonist (72). This regulation of fear extinction through CB1 receptors was found to be mediated via habituation-like processes rather than associative learning mechanisms (192). Moreover, prior microinjection of a CB1/CB2 receptor agonist into the BLA had no effect by itself on inhibitory avoidance conditioning or extinction, but reversed both the enhancing effects of a stressor on conditioning and its imparing effects on extinction (134). Together these findings underscore the contribution of habituationlike processes and of adaptive components such as stress to fear extinction, and their control by the endocannabinoid system.
Also co-localized with GABA in local-circuit amygdala neurons is NPY (296, 479). Administration of NPY or NPY Y(1) receptor agonists into the BLA inhibits expression of fear-potentiated startle and enhances within-session extinction (145). This effect most likely depends on a decreased excitability of principal neurons secondary to the activation of Y(1)-coupled inwardly rectifying K+ channels (480).
In conclusion, extinction training is followed by a consolidation phase, which recruits much of the same molecular machinery involved in the acquisition of conditioned fear (Fig. 9), and involves a spatially distributed synaptic network including the amygdala, hippocampus, and the mPFC for storing of extinction, processing of contextual information, and determination of extinction retrieval, respectively. Important targets of mPFC influences are GABAergic ITC neurons that, in turn, are capable of synaptic plasticity themselves.
Fig. 9. Molecular mechanisms of unlearning and new learning related to early and late stages of fear extinction.

Reversal of conditioned fear (unlearning) involves activation of the phosphatase calcineurin and regulated AMPA receptor endocytosis. Extinction learning and consolidation (new learning) involve activation of NMDA receptors (in particular the NR2B subtype), kinase pathways (for instance the mitogen-activated protein kinase (MAPK), extracellular regulated kinase (ERK) pathway), transcriptional regulation (via transcription factors, such as cAMP response element binding protein (CREB)), and structural organization (involving cytoskeletal proteins such as actin).
D. Reversal of conditioning-evoked alterations
The most convincing data indicating that extinction can reverse the synaptic changes induced by fear conditioning come from studies of synaptic depotentiation, a physiological reversal of LTP and cellular correlate of unlearning (reviewed in (537)). Depotentiation can be induced in the amygdala by low frequency stimulation in vitro, reverses fear training-induced LTP ex vivo, and is associated with a loss of acquired fear responses. As discussed below, depotentiation shares a common set of mechanisms with extinction, which, together, seem to functionally oppose or invert those underlying LTP and/or conditioned fear.
In a comprehensive set of experiments, Gean and colleagues have identified a key signal in depotentiation and fear extinction: the phosphatase calcineurin (protein phosphatase 2B), which targets and inactivates through de-phosphorylation a number of kinases critical for long-term potentiation and conditioned fear in the amygdala (249-251, 253). Depotentiation in vitro and fear extinction were found to be associated with an upregulation of calcineurin in the BLA, presumably through a Ca2+-regulated process, and both were sensitive to calcineurin inhibitors. Importantly, the fear training-induced phosphorylation of specific substrates, such as MAPK and Akt, was reduced after extinction, and this de-phosphorylation was blocked by calcineurin inhibitors. The exact mechanisms of depotentiation in the amygdala, particularly the involvement of NMDA-Rs, MAPK and protein synthesis, remain to be clarified (253). In addition, Kim and colleagues (208) have found that depotentiation at thalamo-LA synapses and fear extinction were attenuated upon blockade of regulated AMPA-R endocytosis. Indeed, interfering with regulated AMPA-R endocytosis through a GluR2-derived peptide (Tat-GluR23Y) during extinction training disrupted the expression and retention of fear expression, whereas the same treatment during fear conditioning had no effect on the expression or recall of either cue or contextual conditioned fear (86). Because Tat-GluR23Y interferes with LTD, and AMPA-R endocytosis is associated with LTD at thalamic inputs in the amygdala (535), the authors suggested that LTD may be a mechanism that links AMPA-R endocytosis to fear extinction (86). Whether mGluR-dependent forms of LTD in principal BLA neurons (153, 255) are relevant for fear extinction remains to be tested. Furthermore, extinction may involve structural alterations opposing those induced by fear conditioning, as indicated by an increase in expression of PSA-NCAM 24 h post fear training in the amygdala (276). In keeping with this, intra-amygdala cleavage of PSA-NCAM affected fear extinction rather than acquisition or consolidation of cued fear (276).
Together, these data suggest that fear extinction include early processes which may reset fear conditioning-induced plastic changes in the amygdala, through synaptic depotentiation or depression, distributed AMPA-R endocytosis, and kinase de-phosphorylation (Fig. 9).
VI. Conclusions: Relation Between Fear and Extinction Memories
Although it is commonly accepted that extinction training does not abolish the initial fear memory, but rather leads to the formation of a new inhibitory memory, the evidence reviewed in the previous section indicates that extinction does reverse at least some of the increases in synaptic efficacy that embody the fear memory. It is important to reconcile these two views as this may yield clues as to how extinction controls fear expression.
On the one hand, there is incontrovertible behavioral evidence that the CS can still evoke conditioned fear responses after extinction training. In other words, the fear memory is not erased after extinction. For instance, as reviewed above, presentation of unsignaled USs after extinction training causes the reinstatement of conditioned fear responses. Second, extinction memory decays with time allowing a spontaneous recovery of the fear memory. Third, after extinction training, conditioned fear responses can be elicited by the CS if the testing context is different from that used for extinction training.
On the other hand, accumulating data indicates that extinction training leads to a depotentiation of thalamic inputs about the CS in LA (208, 249, 535). These findings, coupled to the preserved ability of the CS to evoke conditioned fear following extinction training, raise the intriguing possibility that different pathways convey CS information to the amygdala before vs. after extinction training. Indeed, phenomena such as renewal and reinstatement are utterly incompatible with the idea that extinction only depends on a reversal of the synaptic alterations induced by fear conditioning. In order for renewal and reinstatement to exist, some pathway still has to convey enhanced CS information to the amygdala after extinction.
Consistent with this idea, single-unit studies have revealed that extinction training does not abolish the increased CS responsiveness of all BLA neurons but rather causes a shift in their spatial distribution. In LAd, where primary thalamic inputs about the CS end, extinction training causes a rapid reduction in the magnitude of CS-evoked responses (390, 404). In contrast, in the ventral part of LA, a region devoid of direct thalamic inputs from PIN and MGm, CS-evoked responses typically persist after extinction training (404). Moreover, a similar situation is seen in BL where around 25% of neurons maintain an increased CS-responsiveness after extinction training and an additional 15% acquire an increased CS-responsiveness as a result of extinction training (157). Finally, a third group of BL neurons, accounting for 13% of the cells, express CS-evoked activity in a context-dependent manner in renewal tests (157).
While the loss of CS-evoked responses in LAd is consistent with an erasure of the fear memory, their persistence in BL and ventral LA is not. Instead, these phenomena suggest that extinction training causes a re-organization of the fear memory; a change in the networks primarily responsible for supporting CS transfer to the amygdala. Additional support for this idea comes from studies that examined the hippocampal-dependence of conditioned fear to cues before vs. after extinction training. Whereas dorsal hippocampal lesions and inactivations do not block expression of conditioned fear responses (82, 370, 460), the same manipulations performed after extinction training do (82, 273). Indeed, dorsal hippocampal lesions and inactivations after extinction training prevented the context-dependent renewal of conditioned fear (82, 273). Moreover, inactivation of the dorsal hippocampus prevented the context-dependent expression of CS-evoked responses in LAd neurons after extinction (273). As suggested by c-Fos expression patterns, the hippocampus has a role in contextual fear memory extinction and renewal, both for presentation of cues in and outside the extinction context (211).
If, as suggested by the depotentiation studies, thalamic inputs are depressed by extinction training, what pathway(s) might support the transfer of CS information to the amygdala? Auditory cortical areas are likely candidates. Indeed, these areas contribute direct projections to the amygdala as well as indirect ones, via the rhinal cortices (286). Consistent with this notion, unit recordings have revealed that many auditory cortical neurons express extinction-resistant CS-evoked responses (386).
A second area of uncertainty pertains to mechanisms supporting the contrasting hippocampal dependence of conditioned fear responses to cues before vs. after extinction training. In the model proposed here, the primary route of CS transmission shifts from the thalamus before extinction training to the auditory cortex after extinction training. In this framework, the differential connectivity of the hippocampus with the thalamus and auditory cortex would account for the changing hippocampal-dependence of fear expression before vs. after extinction training. Indeed, the hippocampus has no projections to MGm-PIN but significant indirect projections to associative auditory cortical areas via the rhinal cortices (286). Therefore, hippocampal output might allow for a contextual regulation of CS-evoked activity in the neocortex.
The view of dual representations of context is particularly interesting in this respect (for review see (431)). According to this view, context can be represented as a set of distinct features, each of which may enter into association with the aversive event via functional links to the amygdala. Alternatively, the distinct features of the situation may be bound into a new representation encoding their co-occurrence or conjunction and, for association with the aversive events, further links to the amygdala. These dual representations have been mapped onto distinct neuroanatomical substrates, in which neocortical systems represent the independent features, whereas the elaboration of features into a unitary conjunctive representation requires that the cortex interacts with the hippocampus (329, 330, 433). These findings raise the intriguing possibility that the two representations of context make a different contribution before versus after extinction, with neocortical/hippocampal interactions and their influence on the amygdala being critically involved in the contextual components of extinction.
From the above, it should be clear that in our view, extinction training does not result from erasure of the initial fear memory, but on its re-organization. True erasure of the fear memory only occurs in special circumstances, as when the fear memory is first reactivated and then the CS is repeatedly presented during the reconsolidation window, or when reconsolidation is disrupted by administration of a β-adrenergic receptor antagonist prior to memory reactivation (210, 317), or perhaps when fear memories are formed at very early postnatal stages and then challenged with repeated unpaired CS presentations (138, 205). True erasure of the fear memory is manifested by a loss of spontaneous recovery, reinstatement, and renewal, and these conditions are not seen following conventional extinction training in adulthood.
Overall, the data reviewed here suggests that extinction training leads to distributed changes in cerebral networks. Besides the system-level alterations in the pathways supporting CS transfer to the amygdala, there are widespread changes in the expression of GABA receptors, in the rate of GABA synthesis, as well as activity-dependent potentiation of BL inputs to ITC cells, resulting in the inhibition of fear output neurons. A major challenge for future studies will be to determine how these various changes cooperate to control fear expression.
Acknowledgments
This review was made possible by grants from the Deutsche Forschungsgemeinschaft (DFG; SFB-TRR 58), the Research Award of the Max Planck Society and the Alexander von Humboldt Foundation, and the Interdisciplinary Centre for Clinical Research Münster (to Hans-Christian Pape) as well as by NIMH grants RO1 MH073610 and RO1 MH-083710 (to Denis Pare).
References
- 1.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 2.Aggleton JP, Saunders RC. The amygdala - What's happened in the last decade? In: Aggleton JP, editor. The Amygdala. Oxford, UK: Oxford University Press; 2000. pp. 1–30. [Google Scholar]
- 3.Aitkin LM, Irvine DR, Nelson JE, Merzenich MM, Clarey JC. Frequency representation in the auditory midbrain and forebrain of a marsupial, the northern native cat (Dasyurus hallucatus) Brain Behav Evol. 1986;29:17–28. doi: 10.1159/000118669. [DOI] [PubMed] [Google Scholar]
- 4.Albrecht A, Bergado J, Pape HC, Stork O. Role of NCAM in amygdalo-hippocampal interactions and stress modulation of context fear memory in revision. 2009. [Google Scholar]
- 5.Alexander JC, McDermott CM, Tunur T, Rands V, Stelly C, Karhson D, Bowlby MR, An WF, Sweatt JD, Schrader LA. The role of calsenilin/DREAM/KChIP3 in contextual fear conditioning. Learn Mem. 2009;16:167–177. doi: 10.1101/lm.1261709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alipour M, Chen Y, Jurgens U. Anterograde projections of the motorcortical tongue area in the saddle-back tamarin (Saguinus fuscicollis) Brain Behav Evol. 2002;60:101–116. doi: 10.1159/000065205. [DOI] [PubMed] [Google Scholar]
- 7.Alonso A, García-Austt E. Neuronal sources of theta rhythm in the entorhinal cortex of the rat. Exp Brain Res. 1987;67:493–501. doi: 10.1007/BF00247282. [DOI] [PubMed] [Google Scholar]
- 8.Alonso J, Angermeyer MC, Bernert S, Bruffaerts R, Brugha IS, Bryson H, de Girolamo G, de Graaf R, Demyttenaere K, Gasquet I, Haro JM, Katz SJ, Kessler RC, Kovess V, Lepine JR, Ormel J, Polidori G, Russo LJ, Vilagut G, Almansa J, rbabzadeh-Bouchez S, Autonell J, Bernal M, Buist-Bouwman MA, Codony M, Domingo-Salvany A, Ferrer M, Joo SS, Martinez-Alonso M, Matschinger H, Mazzi F, Morgan Z, Morosini R, Palacin C, Romera B, Taub N, Vollebergh WAM. Prevalence of mental disorders in Europe: results from the European Study of the Epidemiology of Mental Disorders (ESEMeD) project. Acta Psychiatr Scand. 2004;109:21–27. doi: 10.1111/j.1600-0047.2004.00327.x. [DOI] [PubMed] [Google Scholar]
- 9.Amaral DG, Price JL, Pitkanen A, Carmichael ST. Anatomical organization of the primate amygdaloid complex. In: Aggleton JP, editor. The amygdala: Neurobiological aspects of emotion, memory, and mental dysfunction. New York: Wiley-Liss; 1992. pp. 1–66. [Google Scholar]
- 10.Ammassari-Teule M, Restivo L, Pietteur V, Passino E. Learning about the context in genetically-defined mice. Behav Brain Res. 2001;125:195–204. doi: 10.1016/s0166-4328(01)00301-1. [DOI] [PubMed] [Google Scholar]
- 11.Amorapanth P, LeDoux JE, Nader K. Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci. 2000;3:74–79. doi: 10.1038/71145. [DOI] [PubMed] [Google Scholar]
- 12.Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25:9680–9685. doi: 10.1523/JNEUROSCI.2600-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apergis-Schoute AM, Debiec J, Doyere V, LeDoux JE, Schafe GE. Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: A role for presynaptic plasticity in the fear system. J Neurosci. 2005;25:5703–5709. doi: 10.1523/JNEUROSCI.0096-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Applegate CD, Frysinger RC, Kapp BS, Gallagher M. Multiple unit-activity recorded from amygdala central nucleus during Pavlovian heart-rate conditioning in rabbit. Brain Res. 1982;238:457–462. doi: 10.1016/0006-8993(82)90123-8. [DOI] [PubMed] [Google Scholar]
- 15.Armony JL, Quirk GJ, LeDoux JE. Differential effects of amygdala lesions on early and late plastic components of auditory cortex spike trains during fear conditioning. J Neurosci. 1998;18:2592–2601. doi: 10.1523/JNEUROSCI.18-07-02592.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azuma M, Chiba T. Afferent projections of the infralimbic cortex (area 25) in rats: a WGA-HRP study. Kaibogaku Zasshi. 1996;71:523–540. [PubMed] [Google Scholar]
- 18.Baker JD, Azorlosa JL. The NMDA antagonist MK-801 blocks the extinction of Pavlovian fear conditioning. Behav Neurosci. 1996;110:618–620. doi: 10.1037//0735-7044.110.3.618. [DOI] [PubMed] [Google Scholar]
- 19.Barad M, Blouin AM, Cain CK. Like extinction, latent inhibition of conditioned fear in mice is blocked by systemic inhibition of L-type voltage-gated calcium channels. Learn Mem. 2004;11:536–539. doi: 10.1101/lm.78304. [DOI] [PubMed] [Google Scholar]
- 20.Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–9512. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer EP, Paz R, Pare D. Gamma oscillations coordinate amygdalo-rhinal interactions during learning. J Neurosci. 2007;27:9369–9379. doi: 10.1523/JNEUROSCI.2153-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-Type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 24.Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–1118. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- 25.Bergado-Acosta JR, Sangha S, Narayanan RT, Obata K, Pape HC, Stork O. Critical role of the 65-kDa isoform of glutamic acid decarboxylase in consolidation and generalization of Pavlovian fear memory. Learn Mem. 2008;15:163–171. doi: 10.1101/lm.705408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berman AL, Jones EG. The thalamus and basal telencephalon of the cat. Madison: The University of Wisconsin Press; 1982. [Google Scholar]
- 27.Bernard JF, Alden M, Besson JM. The organization of the efferent projections from the pontine parabrachial area to the amygdaloid complex: A Phaseolus vulgaris leucoagglutinin (PHA-L) study in the rat. J Comp Neurol. 1993;329:201–229. doi: 10.1002/cne.903290205. [DOI] [PubMed] [Google Scholar]
- 28.Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1990;63:473–490. doi: 10.1152/jn.1990.63.3.473. [DOI] [PubMed] [Google Scholar]
- 29.Bernard JF, Huang GF, Besson JM. Effect of noxious stimulation on the activity of neurons of the nucleus centralis of the amygdala. Brain Res. 1990;523:347–350. doi: 10.1016/0006-8993(90)91512-f. [DOI] [PubMed] [Google Scholar]
- 30.Bernard JF, Huang GF, Besson JM. Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1992;68:551–569. doi: 10.1152/jn.1992.68.2.551. [DOI] [PubMed] [Google Scholar]
- 31.Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Pare D. Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience. 2005;132:943–953. doi: 10.1016/j.neuroscience.2005.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevilaqua LR, Bonini JS, Rossato JI, Izquierdo LA, Cammarota M, Izquierdo I. The entorhinal cortex plays a role in extinction. Neurobiol Learn Mem. 2006;85:192–197. doi: 10.1016/j.nlm.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Bevilaqua LR, da Silva WN, Medina JH, Izquierdo I, Cammarota M. Extinction and reacquisition of a fear-motivated memory require activity of the Src family of tyrosine kinases in the CA1 region of the hippocampus. Pharmacol Biochem Behav. 2005;81:139–145. doi: 10.1016/j.pbb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Bevilaqua LRM, Rossato JI, Medina JH, Izquierdo I, Cammarota M. Src kinase activity is required for avoidance memory formation and recall. Behav Pharmacol. 2003;14:649–652. doi: 10.1097/00008877-200312000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Bissière S, Humeau Y, Lüthi A. Dopamine gates LTP induction in lateral amygdala by suppressing. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- 36.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- 37.Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- 38.Bliss TVP, Collingridge GL. A synaptic model of memory - long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 39.Bliss TVP, Lomo T. Plasticity in a monosynaptic cortical pathway. J Physiol (London) 1970;207:61P. [PubMed] [Google Scholar]
- 40.Bolognani F, Merhege MA, Twiss J, Perrone-Bizzozero NI. Dendritic localization of the RNA-binding protein HuD in hippocampal neurons: association with polysomes and upregulation during contextual learning. Neurosci Lett. 2004;371:152–157. doi: 10.1016/j.neulet.2004.08.074. [DOI] [PubMed] [Google Scholar]
- 41.Bolognani F, Qiu SF, Tanner DC, Paik J, Perrone-Bizzozero NI, Weeber EJ. Associative and spatial learning and memory deficits in transgenic mice overexpressing the RNA-binding protein HuD. Neurobiol Learn Mem. 2007;87:635–643. doi: 10.1016/j.nlm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Bontempi B, Laurent-Demir C, Destrade C, Jaffard R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature. 1999;400:671–675. doi: 10.1038/23270. [DOI] [PubMed] [Google Scholar]
- 43.Bordi F, LeDoux J, Clugnet MC, Pavlides C. Single-unit activity in the lateral nucleus of the amygdala and overlying areas of the striatum in freely behaving rats: Rates, discharge patterns, and responses to acoustic stimuli. Behav Neurosci. 1993;107:757–769. doi: 10.1037/0735-7044.107.5.757. [DOI] [PubMed] [Google Scholar]
- 44.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term-memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 45.Bouton ME, Bolles RC. Contextual control of the extinction of conditioned fear. Learn Motiv. 1979;10:455–466. [Google Scholar]
- 46.Bouton ME, Bolles RC. Role of conditioned contextual stimuli in reinstatement of extinguished fear. J Exp Psychol Anim Behav Process. 1979;5:368–378. doi: 10.1037//0097-7403.5.4.368. [DOI] [PubMed] [Google Scholar]
- 47.Bragin A, Jandó G, Nádasdy Z, Hetke J, Wise K, Buzsáki G. Gamma (40-100 Hz) oscillation in the hippocampus of the behaving rat. J Neurosci. 1995;15:47–60. doi: 10.1523/JNEUROSCI.15-01-00047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, RossiArnaud C, Grant SGN, Chapman PF, Lipp HP, Sturani E, Klein R. A role for the Ras signaling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 49.Bremner JD, Elzinga B, Schmahl C, Vermetten E. Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res. 2008;167:171–186. doi: 10.1016/S0079-6123(07)67012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinley-Reed M, Mascagni F, McDonald AJ. Synaptology of prefrontal cortical projections to the basolateral amygdala: an electron microscopic study in the rat. Neurosci Lett. 1995;202:45–48. doi: 10.1016/0304-3940(95)12212-5. [DOI] [PubMed] [Google Scholar]
- 51.Brodal A. The amygdaloid nucleus in the rat. J Comp Neurol. 1947;87:1–16. doi: 10.1002/cne.900870102. [DOI] [PubMed] [Google Scholar]
- 52.Buchanan SL, Thompson RH, Maxwell BL, Powell DA. Efferent connections of the medial prefrontal cortex in the rabbit. Exp Brain Res. 1994;100:469–483. doi: 10.1007/BF02738406. [DOI] [PubMed] [Google Scholar]
- 53.Buchel C, Morris J, Dolan RJ, Friston KJ. Brain systems mediating aversive conditioning: an event-related fMRI study. Neuron. 1998;20:947–957. doi: 10.1016/s0896-6273(00)80476-6. [DOI] [PubMed] [Google Scholar]
- 54.Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 55.Buzsáki G, Leung L, Vanderwolf CH. Cellular bases of hippocampal EEG in the behaving rat. Brain Res Rev. 1983;6:139–171. doi: 10.1016/0165-0173(83)90037-1. [DOI] [PubMed] [Google Scholar]
- 56.Cahill L, Weinberger BR, McGaugh JL. Is the amygdala a locus of “conditioned fear”? Some questions and caveats. Neuron. 1999;23:227–228. doi: 10.1016/s0896-6273(00)80774-6. [DOI] [PubMed] [Google Scholar]
- 57.Cain CK, Blouin AM, Barad M. L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci. 2002;22:9113–9121. doi: 10.1523/JNEUROSCI.22-20-09113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cain CK, Godsil BP, Jami S, Barad M. The L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem. 2005;12:277–284. doi: 10.1101/lm.88805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Callaerts-Vegh Z, Beckers T, Ball SM, Baeyens F, Callaerts PF, Cryan JF, Molnar E, D'Hooge R. Concomitant deficits in working memory and fear extinction are functionally dissociated from reduced anxiety in metabotropic glutamate receptor 7-deficient mice. J Neurosci. 2006;26:6573–6582. doi: 10.1523/JNEUROSCI.1497-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campeau S, Davis M. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2301–2311. doi: 10.1523/JNEUROSCI.15-03-02301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campeau S, Miserendino MJD, Davis M. Intraamygdala infusion of the N-Methyl-D-Aspartate receptor antagonist AP5 blocks acquisition but not expression of fear-potentiated startle to an auditory conditioned-stimulus. Behav Neurosci. 1992;106:569–574. doi: 10.1037//0735-7044.106.3.569. [DOI] [PubMed] [Google Scholar]
- 62.Canteras NS, Swanson LW. Projections of the ventral subiculum to the amygdala, septum, and hypothalamus. J Comp Neurol. 1992;324:180–194. doi: 10.1002/cne.903240204. [DOI] [PubMed] [Google Scholar]
- 63.Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- 64.Cassell MD, Gray TS. Morphology of peptide-imunoreactive neurons in the rat central nucleus of the amygdala. J Comp Neurol. 1989;281:320–333. doi: 10.1002/cne.902810212. [DOI] [PubMed] [Google Scholar]
- 65.Cassell MD, Gray TS, Kiss JZ. Neuronal architecture in the rat central nucleus of the amygdala: a cytological, hodological, and immunocytochemical study. J Comp Neurol. 1986;246:478–499. doi: 10.1002/cne.902460406. [DOI] [PubMed] [Google Scholar]
- 66.Cassell MD, Wright DJ. Topography of projections from the medial prefrontal cortex to the amygdala in the rat. Brain Res Bull. 1986;17:321–333. doi: 10.1016/0361-9230(86)90237-6. [DOI] [PubMed] [Google Scholar]
- 67.Chang CE, Berke JD, Maren S. Simultaneous single-unit recordings in the medial prefrontal cortex and amygdaloid nuclei during the extinction of Pavlovian fear conditioning in rats. Soc Neurosci Abstr. 2008;478:14. [Google Scholar]
- 68.Chang CH, Maren S. Early extinction after fear conditioning yeilds context-independent and short-term suppression of conditional freezing in rats. Learn Mem. 2009;16:62–68. doi: 10.1101/lm.1085009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chapman PF, Bellavance LL. Induction of long-term potentiation in the basolateral amygdala does not depend on NMDA receptor activation. Synapse. 1992;11:310–318. doi: 10.1002/syn.890110406. [DOI] [PubMed] [Google Scholar]
- 70.Chen JC, Lang EJ. Inhibitory control of rat lateral amygdaloid projection cells. Neuroscience. 2003;121:155–166. doi: 10.1016/s0306-4522(03)00430-5. [DOI] [PubMed] [Google Scholar]
- 71.Chen X, Garelick MG, Wang HB, Li V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- 72.Chhatwal JP, Gutman AR, Maguschak KA, Bowser ME, Yang Y, Davis M, Ressler KJ. Functional interactions between endocannabinoid and CCK neurotransmitter systems may be critical for extinction learning. Neuropsychopharmacology. 2009;34:509–521. doi: 10.1038/npp.2008.97. [DOI] [PubMed] [Google Scholar]
- 73.Chhatwal JP, Myers KM, Ressler KJ, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chrobak JJ, Buzsáki G. Gamma oscillations in the entorhinal cortex of the freely behaving rat. J Neurosci. 1998;18:388–398. doi: 10.1523/JNEUROSCI.18-01-00388.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chrobak JJ, Buzsáki G. High-frequency oscillations in the output networks of the hippocampal-entorhinal axis of the freely behaving rat. J Neurosci. 1996;16:3056–3066. doi: 10.1523/JNEUROSCI.16-09-03056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ciocchi S, Herry C, Müller C, Lüthi A. Fear conditioning- and extinction-induced neuronal plasticity in the central amygdala. FENS Abstr. 2008;4:057.010. [Google Scholar]
- 78.Clugnet MC, LeDoux JE. Synaptic plasticity in fear conditioning circuits - induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate-body. J Neurosci. 1990;10:2818–2824. doi: 10.1523/JNEUROSCI.10-08-02818.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Collins DR, Lang EJ, Paré D. Spontaneous activity of the perirhinal cortex in behaving cats. Neuroscience. 1999;89:1025–1039. doi: 10.1016/s0306-4522(98)00396-0. [DOI] [PubMed] [Google Scholar]
- 80.Collins DR, Pare D. Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS+ and CS- Learn Mem. 2000;7:97–103. doi: 10.1101/lm.7.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Corcoran KA, Maren S. Factors regulating the effects of hippocampal inactivation on renewal of conditional fear after extinction. Learn Mem. 2004;11:598–603. doi: 10.1101/lm.78704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corcoran KA, Quirk GJ. Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci. 2007;27:840–844. doi: 10.1523/JNEUROSCI.5327-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Corcoran KA, Quirk GJ. Recalling safety: cooperative functions of the ventromedial prefrontal cortex and the hippocampus in extinction. CNS Spectr. 2007;12:200–206. doi: 10.1017/s1092852900020915. [DOI] [PubMed] [Google Scholar]
- 85.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dalton GL, Wang YT, Floresco SB, Phillips AG. Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology. 2008;33:2416–2426. doi: 10.1038/sj.npp.1301642. [DOI] [PubMed] [Google Scholar]
- 87.Danober L, Pape HC. Mechanisms and functional significance of a slow inhibitory potential in neurons of the lateral amygdala. Eur J Neurosci. 1998;10:853–867. doi: 10.1046/j.1460-9568.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 88.Davis M. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP, editor. The Amygdala: a functional analysis. Oxford; Oxford University Press; 2000. pp. 213–287. [Google Scholar]
- 89.Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 90.Delaney AJ, Sah P. GABA receptors inhibited by benzodiazepines mediate fast inhibitory transmission in the central amygdala. J Neurosci. 1999;19:9698–9704. doi: 10.1523/JNEUROSCI.19-22-09698.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Delaney AJ, Sah P. Pathway-specific targeting of GABA a receptor subtypes to somatic and dendritic synapses in the central amygdala. J Neurophysiol. 2001;86:717–723. doi: 10.1152/jn.2001.86.2.717. [DOI] [PubMed] [Google Scholar]
- 92.Dhaka A, Costa RM, Hu H, Irvin DK, Patel A, Kornblum HI, Silva AJ, O'D TJ, Colicelli J. The RAS effector RIN1 modulates the formation of aversive memories. J Neurosci. 2003;23:748–757. doi: 10.1523/JNEUROSCI.23-03-00748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dityatev AE, Bolshakov VY. Amygdala, long-term potentiation, and fear conditioning. Neuroscientist. 2005;11:75–88. doi: 10.1177/1073858404270857. [DOI] [PubMed] [Google Scholar]
- 94.Dong H, Petrovich GD, Swanson LW. Organization of projections from the juxtacapsular nucleus of the BST: a PHAL study in the rat. Brain Res. 2000;859:1–14. doi: 10.1016/s0006-8993(99)02246-5. [DOI] [PubMed] [Google Scholar]
- 95.Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- 96.Dong HW, Swanson LW. Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol. 2004;468:277–298. doi: 10.1002/cne.10949. [DOI] [PubMed] [Google Scholar]
- 97.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J Comp Neurol. 2006;494:142–178. doi: 10.1002/cne.20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, dorsomedial nucleus: implications for cerebral hemisphere integration of neuroendocrine, autonomic, and drinking responses. J Comp Neurol. 2006;494:75–107. doi: 10.1002/cne.20790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, magnocellular nucleus: implications for cerebral hemisphere regulation of micturition, defecation, and penile erection. J Comp Neurol. 2006;494:108–141. doi: 10.1002/cne.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doyere V, Debiec J, Monfils MH, Schafe GE, LeDoux JE. Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat Neurosci. 2007;10:414–416. doi: 10.1038/nn1871. [DOI] [PubMed] [Google Scholar]
- 101.Doyere V, Schafe GE, Sigurdsson T, LeDoux JE. Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur J Neurosci. 2003;17:2703–2715. doi: 10.1046/j.1460-9568.2003.02707.x. [DOI] [PubMed] [Google Scholar]
- 102.Dumont ÉC, Martina M, Samson RD, Drolet G, Paré D. Physiological properties of central amygdala neurons: species differences. Eur J Neurosci. 2002;15:545–552. doi: 10.1046/j.0953-816x.2001.01879.x. [DOI] [PubMed] [Google Scholar]
- 103.Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci U S A. 1999;96:13438–13443. doi: 10.1073/pnas.96.23.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duvarci S, Nader K, LeDoux JE. De novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learn Mem. 2008;15:747–755. doi: 10.1101/lm.1027208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 106.Esteban JA. Intracellular machinery for the transport of AMPA receptors. Br J Pharmacol. 2008;153:S35–S43. doi: 10.1038/sj.bjp.0707525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faber ES, Callister RJ, Sah P. Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol. 2001;85:714–723. doi: 10.1152/jn.2001.85.2.714. [DOI] [PubMed] [Google Scholar]
- 108.Faber ES, Delaney AJ, Power JM, Sedlak PL, Crane JW, Sah P. Modulation of SK channel trafficking by beta adrenoceptors enhances excitatory synaptic transmission and plasticity in the amygdala. J Neurosci. 2008;28:10803–10813. doi: 10.1523/JNEUROSCI.1796-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- 110.Faber ES, Sah P. Calcium-activated K+ (BK) channel inactivation contributes to spike broadening during repetitive firing in rat lateral amygdala neurons. J Physiol. 2003;552(Pt 2):482–497. doi: 10.1113/jphysiol.2003.050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fallon JH, Ciofi P. Distribution of monoamines with the amygdala. In: Aggleton JP, editor. The amygdala. New York: Wiley-Liss; 1992. pp. 97–114. [Google Scholar]
- 113.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fanselow MS, LeDoux JE. Why we think plasticity underlying pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 115.Farb CR, Aoki C, LeDoux JE. Differential localization of NMDA and AMPA receptor subunits in the lateral and basal nuclei of the amygdala - a light and electron-microscopic study. J Comp Neurol. 1995;362:86–108. doi: 10.1002/cne.903620106. [DOI] [PubMed] [Google Scholar]
- 116.Farb CR, LeDoux JE. Afferents from rat temporal cortex synapse on lateral amygdala neurons that express NMDA and AMPA receptors. Synapse. 1999;33:218–229. doi: 10.1002/(SICI)1098-2396(19990901)33:3<218::AID-SYN6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 117.Farb CR, LeDoux JE. NMDA and AMPA receptors in the lateral nucleus of the amygdala are postsynaptic to auditory thalamic afferents. Synapse. 1997;27:106–121. doi: 10.1002/(SICI)1098-2396(199710)27:2<106::AID-SYN2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 118.Faulkner B, Brown TH. Morphology and physiology of neurons in the rat perirhinal-lateral amygdala area. J Comp Neurol. 1999;411:613–642. [PubMed] [Google Scholar]
- 119.Fendt M, Schmid S. Metabotropic glutamate receptors are involved in amygdaloid plasticity. Eur J Neurosci. 2002;15:1535–1541. doi: 10.1046/j.1460-9568.2002.01988.x. [DOI] [PubMed] [Google Scholar]
- 120.Fendt M, Schmid S, Thakker DRH, Jacobsen LH, Yamamoto R, Mitsukawa K, Maier R, Natt F, Husken D, Kelly PH, McAllister KH, Hoyer D, van der Putten H, Cryan JF, Flor PJ. mGluR7 facilitates extinction of aversive memories amd controls amygdala plasticity. Mol Psychiatry. 2008;13:970–979. doi: 10.1038/sj.mp.4002073. [DOI] [PubMed] [Google Scholar]
- 121.Fischer A, Radulovic M, Schrick C, Sananbenesi F, Godovac-Zimmermann J, Radulovic J. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem. 2007;87:149–158. doi: 10.1016/j.nlm.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 124.Flint J. Animal models of anxiety and their molecular dissection. Semin Cell Dev Biol. 2003;14:37–42. doi: 10.1016/s1084-9521(02)00170-2. [DOI] [PubMed] [Google Scholar]
- 125.Floresco SB, Tse MT. Dopaminergic regulation of inhibitory and excitatory transmission in the basolateral amygdala-prefrontal cortical pathway. J Neurosci. 2007;27:2045–2057. doi: 10.1523/JNEUROSCI.5474-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Luthi A. cAMP/PKA signaling and RIM1+› mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fox CA. Certain basal telencephalic centers in the cat. J Comp Neurol. 1940;72:1–62. [Google Scholar]
- 128.Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- 129.Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- 130.Freese JL, Amaral DG. Synaptic organization of projections from the amygdala to visual cortical areas TE and V1 in the macaque monkey. J Comp Neurol. 2006;496:655–667. doi: 10.1002/cne.20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frey S, Frey JU. ‘Synaptic tagging’ and ‘cross-tagging’ and related associative reinforcement processes of functional plasticity as the cellular basis for memory formation. Prog Brain Res. 2008;169:117–143. doi: 10.1016/S0079-6123(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 132.Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- 133.Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ganon-Elazar E, Akirav I. Cannbinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 2009;29:11078–11088. doi: 10.1523/JNEUROSCI.1223-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp HP, Schutz G. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem. 1998;5:274–288. [PMC free article] [PubMed] [Google Scholar]
- 136.Gaudreau H, Paré D. Projection cells of the lateral nucleus are virtually silent throughout the sleep-waking cycle. J Neurophysiol. 1996;75:1301–1305. doi: 10.1152/jn.1996.75.3.1301. [DOI] [PubMed] [Google Scholar]
- 137.Geracitano R, Kaufmann WA, Szabo G, Ferraguti F, Capogna M. Synaptic heterogeneity between mouse paracapsular intercalated neurons of the amygdala. J Physiol. 2007;585:117–134. doi: 10.1113/jphysiol.2007.142570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gogolla N, Caroni P, Luthi A, Herry C. Perineuronal nests protect fear memories from erasure. SciencE. 2009;325:1258–1261. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- 139.Goosens KA, Hobin JA, Maren S. Auditory-evoked spike firing in the lateral amygdala and Pavlovian fear conditioning: Mnemonic code or fear bias? Neuron. 2003;40:1013–1022. doi: 10.1016/s0896-6273(03)00728-1. [DOI] [PubMed] [Google Scholar]
- 140.Goosens KA, Holt W, Maren S. A role for amygdaloid PKA and PKC in the acquisition of long-term conditional fear memories in rats. Behav Brain Res. 2000;114:145–152. doi: 10.1016/s0166-4328(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 141.Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem. 2001;8:148–155. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goosens KA, Maren S. Long-term potentiation as a substrate for memory: Evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus. 2002;12:592–599. doi: 10.1002/hipo.10099. [DOI] [PubMed] [Google Scholar]
- 143.Gordon JA, Hen R. Genetic approaches to the study of anxiety. Annu Rev Neurosci. 2004;27:193–222. doi: 10.1146/annurev.neuro.27.070203.144212. [DOI] [PubMed] [Google Scholar]
- 144.Grillon C. Startle reactivity and anxiety disorders: Aversive conditioning. Biol Psychiatry. 2002;52:958–975. doi: 10.1016/s0006-3223(02)01665-7. [DOI] [PubMed] [Google Scholar]
- 145.Gutman AR, Yang YL, Ressler KJ, Davis M. The Role of Neuropeptide Y in the Expression and Extinction of Fear-Potentiated Startle. J Neurosci. 2008;28:12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hall E. The amygdala of the cat: A Golgi Study. Z Zellforsch. 1972;134:439–458. doi: 10.1007/BF00307668. [DOI] [PubMed] [Google Scholar]
- 147.Hall E. Some aspects of the structural organization of the amygdala. In: Eleftheriou BE, editor. The Neurobiology of the amygdala. New York: Plenum Press; 1972. pp. 95–121. [Google Scholar]
- 148.Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- 149.Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A, Bontempi B, Neve RL, Frankland PW, Josselyn SA. Selective erasure of a fear memory. Science. 2009;323:1492–1496. doi: 10.1126/science.1164139. [DOI] [PubMed] [Google Scholar]
- 150.Hasselmo ME, Giocomo LM. Cholinergic modulation of cortical function. J Mol Neurosci. 2006;30:133–135. doi: 10.1385/JMN:30:1:133. [DOI] [PubMed] [Google Scholar]
- 151.Hebb DO. The organization of behavior. New York: Wiley; 1949. [Google Scholar]
- 152.Heim C, Nemeroff CB. Neurobiology of posttraumatic stress disorder. CNS Spectr. 2009;14:13–24. [PubMed] [Google Scholar]
- 153.Heinbockel T, Pape HC. Input-specific long-term depression in the lateral amygdala evoked by theta frequency stimulation. J Neurosci. 2000;20:RC68. doi: 10.1523/JNEUROSCI.20-07-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Heldt SA, Ressler KJ. Training-induced changes in the expression of GABAA-associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci. 2007;26:3631–3644. doi: 10.1111/j.1460-9568.2007.05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Helmstetter FJ, Parsons RG, Gafford GM. Macromolecular synthesis, distributed synaptic plasticity, and fear conditioning. Neurobiol Learn Mem. 2008;89:324–337. doi: 10.1016/j.nlm.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Herkenham M, Pert CB. Light microscopic localization of brain opiate receptors: a general autoradiographic method which preserves tissue quality. J Neurosci. 1982;2:1129–1149. doi: 10.1523/JNEUROSCI.02-08-01129.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 158.Herry C, Mons N. Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur J Neurosci. 2004;20:781–790. doi: 10.1111/j.1460-9568.2004.03542.x. [DOI] [PubMed] [Google Scholar]
- 159.Herry C, Trifilieff P, Micheau J, Luthi A, Mons N. Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur J Neurosci. 2006;24:261–269. doi: 10.1111/j.1460-9568.2006.04893.x. [DOI] [PubMed] [Google Scholar]
- 160.Hobin JA, Ji J, Maren S. Ventral hippocampal muscimol disrupts context-specific fear memory retrieval after extinction in rats. Hippocampus. 2006;16:174–182. doi: 10.1002/hipo.20144. [DOI] [PubMed] [Google Scholar]
- 161.Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- 162.Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. 1978;32:529–547. doi: 10.1007/BF00239551. [DOI] [PubMed] [Google Scholar]
- 163.Hovatta I, Barlow C. Molecular genetics of anxiety in mice and men. Ann Med. 2008;40:92–109. doi: 10.1080/07853890701747096. [DOI] [PubMed] [Google Scholar]
- 164.Huang CC, Gean PW. Paired-pulse depression of the N-Methyl-D-Aspartate receptor-mediated synaptic potentials in the amygdala. Br J Pharmacol. 1994;113:1029–1035. doi: 10.1111/j.1476-5381.1994.tb17096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang YY, Kandel ER. Low-frequency stimulation induces a pathway-specific late phase of LTP in the amygdala that is mediated by PKA and dependent on protein synthesis. Learn Mem. 2007;14:497–503. doi: 10.1101/lm.593407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 167.Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hugues S, Chessel A, Lena I, Marsault R, Garcia R. Prefrontal infusion of PD098059 immediately after fear extinction training blocks extinction-associated prefrontal synaptic plasticity and decreases prefrontal ERK2 phosphorylation. Synapse. 2006;60:280–287. doi: 10.1002/syn.20291. [DOI] [PubMed] [Google Scholar]
- 169.Hugues S, Deschaux O, Garcia R. Postextinction infusion of a mitogen-activated protein kinase inhibitor into the medial prefrontal cortex impairs memory of the extinction of conditioned fear. Learn Mem. 2004;11:540–543. doi: 10.1101/lm.77704. [DOI] [PubMed] [Google Scholar]
- 170.Humeau Y, Herry C, Kemp N, Shaban H, Fourcaudot E, Bissiere S, Luthi A. Dendritic spine heterogeneity determines afferent-specific Hebbian plasticity in the amygdala. Neuron. 2005;45:119–131. doi: 10.1016/j.neuron.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 171.Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, Hvalby O, Sprengel R, Luthi A. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci. 2007;27:10947–10956. doi: 10.1523/JNEUROSCI.2603-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- 173.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulation: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 174.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 175.Inoue N, Nakao H, Migishima R, Hino T, Matsui M, Hayashi F, Nakao K, Manabe T, Aiba A, Inokuchi K. Requirement of the immediate early gene vesl-1S/homer-1a for fear memory formation. Mol Brain. 2009;2:7. doi: 10.1186/1756-6606-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jacobsen KX, Hoistad M, Staines WA, Fuxe K. The distribution of dopamine D1 receptor and mu-opioid receptor 1 receptor immunoreactivities in the amygdala and interstitial nucleus of the posterior limb of the anterior commissure: relationships to tyrosine hydroxylase and opioid peptide terminal systems. Neuroscience. 2006;141:2007–2018. doi: 10.1016/j.neuroscience.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 177.Jasnow AM, Ressler KJ, Hammack SE, Chhatwal JP, Rainnie DG. Distinct subtypes of cholecystokinin (CCK)-containing interneurons of the basolateral amygdala identified using a CCK promoter-specific lentivirus. J Neurophysiol. 2009;101:1494–1506. doi: 10.1152/jn.91149.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jaworski DM, Boone J, Caterina J, Soloway P, Falls WA. Prepulse inhibition and fear-potentiated startle are altered in tissue inhibitor of metalloproteinase-2 (TIMP-2) knockout mice. Brain Res. 2005;1051:81–89. doi: 10.1016/j.brainres.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 179.Jay TM, Glowinski J, Thierry AM. Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain Res. 1989;505:337–340. doi: 10.1016/0006-8993(89)91464-9. [DOI] [PubMed] [Google Scholar]
- 180.Ji J, Maren S. Hippocampal involvement in contextual modulation of fear extinction. Hippocampus. 2007;17:749–758. doi: 10.1002/hipo.20331. [DOI] [PubMed] [Google Scholar]
- 181.Ji J, Maren S. Lesions of the entorhinal cortex or fornix disrupt the context-dependence of fear extinction in rats. Behav Brain Res. 2008;194:201–206. doi: 10.1016/j.bbr.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Johnson LR, Hou M, Ponce-Alvarez A, Gribelyuk LM, Alphs HH, Albert L, Brown BL, LeDoux JE, Doyére V. A recurrent network in the lateral amygdale: a mechanism for coincidence detection. Frontiers Neural Circuits. 2009;2:1–19. doi: 10.3389/neuro.04.003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Johnston JB. Further contributions to the study of the evolution of the forebrain. J Comp Neurol. 1923;35:337–481. [Google Scholar]
- 184.Jolkkonen E, Miettinen R, Pikkarainen M, Pitkänen A. Projections from the amygdaloid complex to the magnocellular cholinergic basal forebrain in rat. Neuroscience. 2002;111:133–149. doi: 10.1016/s0306-4522(01)00578-4. [DOI] [PubMed] [Google Scholar]
- 185.Jolkkonen E, Pitkänen A. Intrinsic connections of the rat amygdaloid complex: Projections originating in the central nucleus. J Comp Neurol. 1998;395:53–72. doi: 10.1002/(sici)1096-9861(19980525)395:1<53::aid-cne5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 186.Jones EG. The thalamus. Cambridge, UK: Cambridge University Press; 2007. [Google Scholar]
- 187.Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol Learn Mem. 2004;82:159–163. doi: 10.1016/j.nlm.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 188.Junghans D, Haas IG, Kemler R. Mammalian cadherins and protocadherins: about cell death, synapses and processing. Curr Opin Cell Biol. 2005;17:446–452. doi: 10.1016/j.ceb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 189.Jungling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, Okamura N, Duangdao DM, Xu YL, Reinscheid RK, Pape HC. Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron. 2008;59:298–310. doi: 10.1016/j.neuron.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ. Context-dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci. 2006;26:9503–9511. doi: 10.1523/JNEUROSCI.2021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kamal AM, Tombol T. Golgi studies on the amygdaloid nuclei of the cat. J Hirnforsch. 1975;16:175–201. [PubMed] [Google Scholar]
- 192.Kamprath K, Mariscano G, Tang J, Monory K, Blisogno T, Di Marzo V, Lutz B, Wotjak CT. Cannabinoid CB1 receptor mediates fear extinction via habituation like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kandel ER. Neuroscience - The molecular biology of memory storage: A dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 194.Kapp BS, Frysinger RC, Gallagher M, Haselton JR. Amygdala central nucleus lesions: effect on heart rate conditioning in the rabbit. Physiol Behav. 1979;23:1109–1117. doi: 10.1016/0031-9384(79)90304-4. [DOI] [PubMed] [Google Scholar]
- 195.Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Keene JD. Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc Natl Acad Sci U S A. 1999;96:5–7. doi: 10.1073/pnas.96.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kelly MP, Deadwyler SA. Acquisition of a novel behavior induces higher levels of Arc mRNA than does overtrained performance. Neuroscience. 2002;110:617–626. doi: 10.1016/s0306-4522(01)00605-4. [DOI] [PubMed] [Google Scholar]
- 198.Kemppainen S, Pitkänen A. Distribution of parvalbumin, calretinin, and calbindin-D28k immunoreactivity in the rat amygdaloid complex and colocalization with gamma-aminobutyric acid. J Comp Neurol. 2000;426:441–467. doi: 10.1002/1096-9861(20001023)426:3<441::aid-cne8>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 199.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- 202.Killcross S, Robbins TW, Everitt BJ. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature. 1997;388:377–380. doi: 10.1038/41097. [DOI] [PubMed] [Google Scholar]
- 203.Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ, Kim HS, Choi YB, Scheiffele P, Bailey CH, Kandel ER, Kim JH. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc Natl Acad Sci U S A. 2008;105:9087–9092. doi: 10.1073/pnas.0803448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kim JH, Hamlin AS, Richardson R. Fear extinction across development: the involvement of the medial prefrontal cortex as assessed by temporary inactivation and immunohistochemistry. J Neurosci. 2009;2:9. doi: 10.1523/JNEUROSCI.0596-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kim JH, Richardson R. The effect of temporary amygdala inactivation on extinction and reextinction of fear in the developing rat: unlearning as a potential mechanism for extinction early in development. J Neurosci. 2008;28:1282–1290. doi: 10.1523/JNEUROSCI.4736-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. N-methyl-D-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- 207.Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: A critical review. Neuroscience and Biobehavioral Reviews. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kim JJ, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS, Choi S. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A. 2007;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kimura R, Silva AJ, Ohno M. Autophosphorylation of +›CaMKII is differentially involved in new learning and unlearning mechanisms of memory extinction. Learn Mem. 2008;15:837–843. doi: 10.1101/lm.1049608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- 211.Knapska E, Maren S. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn Mem. 2009;16:486–493. doi: 10.1101/lm.1463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kojima N, Sakamoto T, Endo S, Niki H. Impairment of conditioned freezing to tone, but not to context, in Fyn-transgenic mice: relationship to NMDA receptor subunit 2B function. Eur J Neurosci. 2005;21:1359–1369. doi: 10.1111/j.1460-9568.2005.03955.x. [DOI] [PubMed] [Google Scholar]
- 213.Koo JW, Han JS, Kim JJ. Selective neurotoxic lesions of basolateral and central nuclei of the amygdala produce differential effects on fear conditioning. J Neurosci. 2004;24:7654–7662. doi: 10.1523/JNEUROSCI.1644-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Krettek JE, Price JL. Amygdaloid projections to subcortical structures within the basal forebrain and brainstem in the rat and cat. J Comp Neurol. 1978;178:225–254. doi: 10.1002/cne.901780204. [DOI] [PubMed] [Google Scholar]
- 216.Krettek JE, Price JL. A description of the amygdaloid complex in the rat and cat with observations on intra-amygdaloid axonal connections. J Comp Neurol. 1978;178:255–280. doi: 10.1002/cne.901780205. [DOI] [PubMed] [Google Scholar]
- 217.Krettek JE, Price JL. Projections from the amygdaloid complex and adjacent olfactory structures to the entorhinal cortex and to the subiculum in the rat and cat. J Comp Neurol. 1977;172:723–752. doi: 10.1002/cne.901720409. [DOI] [PubMed] [Google Scholar]
- 218.Krettek JE, Price JL. Projections from the amygdaloid complex to the cerebral cortex and thalamus in the rat and cat. J Comp Neurol. 1977;172:687–722. doi: 10.1002/cne.901720408. [DOI] [PubMed] [Google Scholar]
- 219.Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65:3525–3544. doi: 10.1007/s00018-008-8170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kudo M, Itoh K, Kawamura S, Mizuno N. Direct projections to the pretectum and the midbrain reticular formation from auditory relay nuclei in the lower brainstem of the cat. Brain Res. 1983;288:13–19. doi: 10.1016/0006-8993(83)90077-x. [DOI] [PubMed] [Google Scholar]
- 221.Kudo M, Niimi K. Ascending projections of the inferior colliculus in the cat: an autoradiographic study. J Comp Neurol. 1980;191:545–556. doi: 10.1002/cne.901910403. [DOI] [PubMed] [Google Scholar]
- 222.Kudo M, Tashiro T, Higo S, Matsuyama T, Kawamura S. Ascending projections from the nucleus of the brachium of the inferior colliculus in the cat. Exp Brain Res. 1984;54:203–211. doi: 10.1007/BF00236219. [DOI] [PubMed] [Google Scholar]
- 223.Kwon JT, Choi JS. Cornering the fear engram: long-term synaptic changes in the lateral nucleus of the amygdala following fear conditioning. J Neurosci. 2009;29:9700–9703. doi: 10.1523/JNEUROSCI.5928-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- 225.Lamprecht R, Farb CR, LeDoux JE. Fear memory formation involves p190 RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron. 2002;36:727–738. doi: 10.1016/s0896-6273(02)01047-4. [DOI] [PubMed] [Google Scholar]
- 226.Lamprecht R, Farb CR, Rodrigues SM, LeDoux JE. Fear conditioning drives profilin into amygdala dendritic spines. Nat Neurosci. 2006;9:481–483. doi: 10.1038/nn1672. [DOI] [PubMed] [Google Scholar]
- 227.Lamprecht R, LeDoux JE. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- 228.Lamprecht R, Margulies DS, Farb CR, Hou M, Johnson LR, LeDoux JE. Myosin light chain kinase regulates synaptic plasticity and fear learning in the lateral amygdala. Neuroscience. 2006;139:821–829. doi: 10.1016/j.neuroscience.2005.12.055. [DOI] [PubMed] [Google Scholar]
- 229.Lang EJ, Paré D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- 230.Lang EJ, Paré D. Synaptic and synaptically activated intrinsic conductances underlie inhibitory potentials in cat lateral amygdaloid projection neurons in vivo. J Neurophysiol. 1997;77:353–363. doi: 10.1152/jn.1997.77.1.353. [DOI] [PubMed] [Google Scholar]
- 231.Lang EJ, Paré D. Synaptic responsiveness of interneurons of the cat lateral amygdaloid nucleus. Neuroscience. 1998;83:877–889. doi: 10.1016/s0306-4522(97)00420-x. [DOI] [PubMed] [Google Scholar]
- 232.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 233.Laurent V, Marchand AR, Westbrook RF. The basolateral amygdala is necessary for learning but not relearning extinction of context conditioned fear. Learn Mem. 2008;15:304–314. doi: 10.1101/lm.928208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Laurent V, Westbrook RF. Distinct contributions of the basolateral amygdala and the medial prefrontal cortex to learning and relearning extinction of context conditioned fear. Learn Mem. 2008;15:657–666. doi: 10.1101/lm.1080108. [DOI] [PubMed] [Google Scholar]
- 235.Laurent V, Westbrook RF. Inactivation of the infralimbic but not of the prelimbic cortex impairs consolidation and retrieval of conditioned fear. Learn Mem. 2009;16:520–529. doi: 10.1101/lm.1474609. [DOI] [PubMed] [Google Scholar]
- 236.Le Gal La Salle G, Paxinos G, Emson P, Ben-Ari Y. Neurochemical mapping of GABAergic systems in the amygdaloid complex and bed nucleus of the stria terminalis. Brain Res. 1978;155:397–403. doi: 10.1016/0006-8993(78)91037-5. [DOI] [PubMed] [Google Scholar]
- 237.Ledgerwood L, Richardson R, Cranney J. D-cycloserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci. 2004;118:505–513. doi: 10.1037/0735-7044.118.3.505. [DOI] [PubMed] [Google Scholar]
- 238.Ledgerwood L, Richardson R, Cranney J. D-cycloserine facilitates extinction of learned fear: effects on reacquisition and generalized extinction. Biol Psychiatry. 2005;57:841–847. doi: 10.1016/j.biopsych.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 239.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 240.LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.LeDoux JE, Farb C, Ruggiero DA. Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J Neurosci. 1990;10:1043–1054. doi: 10.1523/JNEUROSCI.10-04-01043.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.LeDoux JE, Ruggiero DA, Forest R, Stornetta R, Reis DJ. Topographic organization of convergent projections to the thalamus from the inferior colliculus and spinal cord in the rat. J Comp Neurol. 1987;264:123–146. doi: 10.1002/cne.902640110. [DOI] [PubMed] [Google Scholar]
- 244.LeDoux JE, Ruggiero DA, Reis DJ. Projections to the subcortical forebrain from anatomically defined regions of the medial geniculate body in the rat. J Comp Neurol. 1985;242:182–213. doi: 10.1002/cne.902420204. [DOI] [PubMed] [Google Scholar]
- 245.Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lee O, Lee CJ, Choi S. Induction mechanisms for L-LTP at thalamic input synapses to the lateral amygdala: requirement of mGluR5 activation. Neuroreport. 2002;13:685–691. doi: 10.1097/00001756-200204160-00030. [DOI] [PubMed] [Google Scholar]
- 247.Likhtik E, Pelletier JG, Paz R, Pare D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Pare D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454:642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lin CH, Lee CC, Gean PW. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol. 2003;63:44–52. doi: 10.1124/mol.63.1.44. [DOI] [PubMed] [Google Scholar]
- 250.Lin CH, Lee CC, Huang YC, Wang SJ, Gean PW. Activation of group II metabotropic glutamate receptors induces depotentiation in amygdala slices and reduces fear-potentiated startle in rats. Learn Mem. 2005;12:130–137. doi: 10.1101/lm.85304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- 253.Lin CH, Yeh SH, Lu HY, Gean PW. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci. 2003;23:8310–8317. doi: 10.1523/JNEUROSCI.23-23-08310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lin HC, Mao SC, Gean PW. Block of gamma-Aminobutyric Acid-A Receptor Insertion in the Amygdala Impairs Extinction of Conditioned Fear. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 255.Lin HC, Wang SJ, Luo MZ, Gean PW. Activation of group II metabotropic glutamate receptors induces long-term depression of synaptic transmission in the rat amygdala. J Neurosci. 2000;20:9017–9024. doi: 10.1523/JNEUROSCI.20-24-09017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Linke R. Differential projection patterns of superior and inferior collicular neurons onto posterior paralaminar nuclei of the thalamus surrounding the medial geniculate body in the rat. Eur J Neurosci. 1999;11:187–203. doi: 10.1046/j.1460-9568.1999.00422.x. [DOI] [PubMed] [Google Scholar]
- 257.Linke R, Braune G, Schwegler H. Differential projection of the posterior paralaminar thalamic nuclei to the amygdaloid complex in the rat. Exp Brain Res. 2000;134:520–532. doi: 10.1007/s002210000475. [DOI] [PubMed] [Google Scholar]
- 258.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 259.Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lopez De Armentia M, Sah P. Firing properties and connectivity of neurons in the rat lateral central nucleus of the amygdala. J Neurophysiol. 2004;92:1285–1294. doi: 10.1152/jn.00211.2004. [DOI] [PubMed] [Google Scholar]
- 261.Lu KW, Walker DL, Davis M. Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci. 2001;21:RC162. doi: 10.1523/JNEUROSCI.21-16-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Ann Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- 263.Lutz B. The endocannabinoid system and extinction learning. Mol Neurobiol. 2007;36:92–101. doi: 10.1007/s12035-007-8004-x. [DOI] [PubMed] [Google Scholar]
- 264.Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- 265.Mahanty NK, Sah P. Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci. 1999;11:1217–1222. doi: 10.1046/j.1460-9568.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 266.Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- 267.Malenka RC, Nicoll RA. Long-term potentiation - a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 268.Malkani S, Rosen JB. Specific induction of early growth response gene 1 in the lateral nucleus of the amygdala following contextual fear conditioning in rats. Neuroscience. 2000;97:693–702. doi: 10.1016/s0306-4522(00)00058-0. [DOI] [PubMed] [Google Scholar]
- 269.Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–413. doi: 10.1523/JNEUROSCI.4639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 271.Maren S, Chang CH. Recent fear is resistant to extinction. Proc Natl Acad Sci U S A. 2006;103:18020–18025. doi: 10.1073/pnas.0608398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Maren S, Ferrario CR, Corcoran KA, Desmond TJ, Frey KA. Protein synthesis in the amygdala, but not the auditory thalamus, is required for consolidation of Pavlovian fear conditioning in rats. Eur J Neurosci. 2003;18:3080–3088. doi: 10.1111/j.1460-9568.2003.03063.x. [DOI] [PubMed] [Google Scholar]
- 273.Maren S, Hobin JA. Hippocampal regulation of context-dependent neuronal activity in the lateral amygdala. Learn Mem. 2007;14:318–324. doi: 10.1101/lm.477007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Maren S, Quirk GJ. Neuronal signaling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- 275.Maren S, Yap SA, Goosens KA. The amygdala is essential for the development of neuronal plasticity in the medial geniculate nucleus during auditory fear conditioning in rats. J Neurosci. 2001;21:RC135. doi: 10.1523/JNEUROSCI.21-06-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Markram K, Lopez Fernandez MA, Abrous DN, Sandi C. Amygdala upregulation of NCAM polysialylation induced by auditory fear conditioning is not required for memory formation, but plays a role in fear extinction. Neurobiol Learn Mem. 2007;87:573–582. doi: 10.1016/j.nlm.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 277.Marowsky A, Yanagawa Y, Obata K, Vogt KE. A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron. 2005;48:1025–1037. doi: 10.1016/j.neuron.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 278.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Marzo VD, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 279.Martina M, Royer S, Paré D. Cell-type-specific GABA responses and chloride homeostasis in the cortex and amygdala. J Neurophysiol. 2001;86:2887–2895. doi: 10.1152/jn.2001.86.6.2887. [DOI] [PubMed] [Google Scholar]
- 280.Mascagni F, McDonald AJ, Coleman JR. Corticoamygdaloid and corticocortical projections of the rat temporal cortex: A Phaseolus vulgaris leucoagglutinin study. Neuroscience. 1993;57:697–715. doi: 10.1016/0306-4522(93)90016-9. [DOI] [PubMed] [Google Scholar]
- 281.Masugi M, Yokoi M, Shigemoto R, Muguruma K, Watanabe Y, Sansig G, van der Putten H, Nakanishi S. Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci. 1999;19:955–963. doi: 10.1523/JNEUROSCI.19-03-00955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- 283.Maviel T, Durkin TP, Menzaghi F, Bontempi B. Sites of neocortical reorganization critical for remote spatial memory. Science. 2004;305:96–99. doi: 10.1126/science.1098180. [DOI] [PubMed] [Google Scholar]
- 284.McAllister WR, McAllister DE, Scoles MT, Hampton SR. Persistence of fear-reducing behavior: relevance for the conditioning theory of neurosis. J Abnorm Psychol. 1986;95:365–372. doi: 10.1037//0021-843x.95.4.365. [DOI] [PubMed] [Google Scholar]
- 285.McDonald AJ. Cell types and intrinsic connections of the amygdala. In: Aggleton JP, editor. The amygdala: Neurobiological aspects of emotion, memory, and mental dysfunction. New York: Wiley-Liss; 1992. pp. 67–96. [Google Scholar]
- 286.McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 287.McDonald AJ. Cytoarchitecture of the central amygdaloid nucleus of the rat. J Comp Neurol. 1982;208:401–418. doi: 10.1002/cne.902080409. [DOI] [PubMed] [Google Scholar]
- 288.McDonald AJ. Immunohistochemical identification of gamma-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci Lett. 1985;53:203–207. doi: 10.1016/0304-3940(85)90186-7. [DOI] [PubMed] [Google Scholar]
- 289.McDonald AJ. Projection neurons of the basolateral amygdala: A correlative Golgi and retrograde tract tracing study. Brain Res Bull. 1992;28:179–185. doi: 10.1016/0361-9230(92)90177-y. [DOI] [PubMed] [Google Scholar]
- 290.McDonald AJ, Augustine JR. Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience. 1993;52:281–294. doi: 10.1016/0306-4522(93)90156-a. [DOI] [PubMed] [Google Scholar]
- 291.McDonald AJ, Betette RL. Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of Calbindin-D(28k) Neuroscience. 2001;102:413–425. doi: 10.1016/s0306-4522(00)00481-4. [DOI] [PubMed] [Google Scholar]
- 292.McDonald AJ, Mascagni F. Colocalization of calcium-binding proteins and GABA in neurons of the rat basolateral amygdala. Neuroscience. 2001;105:681–693. doi: 10.1016/s0306-4522(01)00214-7. [DOI] [PubMed] [Google Scholar]
- 293.McDonald AJ, Mascagni F. Immunohistochemical characterization of somatostatin containing interneurons in the rat basolateral amygdala. Brain Res. 2002;943:237–244. doi: 10.1016/s0006-8993(02)02650-1. [DOI] [PubMed] [Google Scholar]
- 294.McDonald AJ, Mascagni F. Localization of the CB1 type cannabinoid receptor in the rat basolateral amygdala: High concentrations in a subpopulation of cholecystokinin-containing interneurons. Neuroscience. 2001;107:641–652. doi: 10.1016/s0306-4522(01)00380-3. [DOI] [PubMed] [Google Scholar]
- 295.McDonald AJ, Mascagni F. Parvalbumin-containing interneurons in the basolateral amygdala express high levels of the alpha1 subunit of the GABA A receptor. J Comp Neurol. 2004;473:137–146. doi: 10.1002/cne.20101. [DOI] [PubMed] [Google Scholar]
- 296.McDonald AJ, Mascagni F, Augustine JR. Neuropeptide-Y and somatostatin-like immunoreactivity in neurons of the monkey amygdala. Neuroscience. 1995;66:959–982. doi: 10.1016/0306-4522(94)00629-j. [DOI] [PubMed] [Google Scholar]
- 297.McDonald AJ, Mascagni F, Guo L. Projections of the medial and lateral prefrontal cortices to the amygdala: A Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1996;71:55–75. doi: 10.1016/0306-4522(95)00417-3. [DOI] [PubMed] [Google Scholar]
- 298.McDonald AJ, Mascagni F, Mania I, Rainnie DG. Evidence for a perisomatic innervation of parvalbumin-containing interneurons by individual pyramidal cells in the basolateral amygdala. Brain Res. 2005;1035:32–40. doi: 10.1016/j.brainres.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 299.McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- 300.McGaugh JL. Memory - a century of consolidation. Science. 2000;287:208–210. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 301.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 302.McNally RJ. On nonassociative fear emergence. Behav Res Ther. 2002;40:169–172. doi: 10.1016/s0005-7967(01)00049-3. [DOI] [PubMed] [Google Scholar]
- 303.Medina JF, Repa JC, Mauk MD, LeDoux JE. Parallels between cerebellum- and amygdala-dependent conditioning. Nat Rev Neurosci. 2002;3:122–131. doi: 10.1038/nrn728. [DOI] [PubMed] [Google Scholar]
- 304.Meis S, Munsch T, Sosulina L, Pape HC. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to cholecystokinin are mediated by a transient receptor potential-like current. Mol Cell Neurosci. 2007;35:356–367. doi: 10.1016/j.mcn.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 305.Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev Neurosci. 2003;14:233–240. doi: 10.1515/revneuro.2003.14.3.233. [DOI] [PubMed] [Google Scholar]
- 306.Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, Pitman RK. Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J Psychiatr Res. 2008;42:515–520. doi: 10.1016/j.jpsychires.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 308.Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 309.Mileusnic R, Lancashire CL, Rose SPR. Recalling an aversive experience by day-old chicks is not dependent on somatic protein synthesis. Learn Mem. 2005;12:615–619. doi: 10.1101/lm.38005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol. 2003;70:83–244. doi: 10.1016/s0301-0082(03)00087-x. [DOI] [PubMed] [Google Scholar]
- 311.Millhouse OE. The intercalated cells of the amygdala. J Comp Neurol. 1986;247:246–271. doi: 10.1002/cne.902470209. [DOI] [PubMed] [Google Scholar]
- 312.Mineka S, Oehlberg K. The relevance of recent developments in classical conditioning to understanding the etiology and maintenance of anxiety disorders. Acta Psychol (Amst) 2008;127:567–580. doi: 10.1016/j.actpsy.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 313.Mineka S, Ohman A. Born to fear: non-associative vs associative factors in the etiology of phobias. Behav Res Ther. 2002;40:173–184. doi: 10.1016/s0005-7967(01)00050-x. [DOI] [PubMed] [Google Scholar]
- 314.Miserendino MJD, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 315.Mitchell S, Ranck JB. Generation of theta rhythm in medial entorhinal cortex of freely moving rats. Brain Res. 1980;189:49–66. doi: 10.1016/0006-8993(80)90006-2. [DOI] [PubMed] [Google Scholar]
- 316.Moita MA, Lamprecht R, Nader K, LeDoux JE. A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat Neurosci. 2002;5:837–838. doi: 10.1038/nn901. [DOI] [PubMed] [Google Scholar]
- 317.Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Monti B, Berteotti C, Contestabile A. Subchronic rolipram delivery activates hippocampal CREB and arc, enhances retention and slows down extinction of conditioned fear. Neuropsychopharmacology. 2006;31:278–286. doi: 10.1038/sj.npp.1300813. [DOI] [PubMed] [Google Scholar]
- 319.Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Mueller T, Albrecht D, Gebhardt C. Both NR2A and NR2B subunits of NMDA receptor are critical for long-term potentiation and long-term depression in the lateral amygdala of horizontal slices of adult mice. Learn Mem. 2009;16:395–405. doi: 10.1101/lm.1398709. [DOI] [PubMed] [Google Scholar]
- 321.Muigg P, Hetzenauer A, Hauer G, Hauschild M, Gaburro S, Frank E, Landgraf R, Singewald N. Impaired extinction of learned fear in rats selectively bred for high anxiety--evidence of altered neuronal processing in prefrontal-amygdala pathways. Eur J Neurosci. 2008;28:2299–2309. doi: 10.1111/j.1460-9568.2008.06511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Muller J, Corodimas KP, Fridel Z, LeDoux JE. Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci. 1997;111:683–691. doi: 10.1037//0735-7044.111.4.683. [DOI] [PubMed] [Google Scholar]
- 323.Muller JF, Mascagni F, McDonald AJ. Coupled networks of parvalbumin-immunoreactive interneurons in the rat basolateral amygdala. J Neurosci. 2005;25:7366–7376. doi: 10.1523/JNEUROSCI.0899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Muller JF, Mascagni F, McDonald AJ. Postsynaptic targets of somatostatin-containing interneurons in the rat basolateral amygdala. J Comp Neurol. 2007;500:513–529. doi: 10.1002/cne.21185. [DOI] [PubMed] [Google Scholar]
- 325.Muller JF, Mascagni F, McDonald AJ. Pyramidal cells of the rat basolateral amygdala: synaptology and innervation by parvalbumin-immunoreactive interneurons. J Comp Neurol. 2006;494:635–650. doi: 10.1002/cne.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Musumeci G, Sciarretta C, Rodriguez-Moreno A, Al Banchaabouchi M, Negrete-Diaz V, Costanzi M, Berno V, Egorov AV, von Bohlen Und Halbach O, Cestari V, Delgado-Garcia JM, Minichiello L. TrkB modulates fear learning and amygdalar synaptic plasticity by specific docking sites. J Neurosci. 2009;29:10131–10143. doi: 10.1523/JNEUROSCI.1707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- 328.Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Nadel L, Willner J. Context and conditioning: a place for space. Physiol Behav. 1980;8:218–228. [Google Scholar]
- 330.Nadel L, Willner J, Kurz EM. Cognitive maps and environmental context. In: Balsam P, Tomie A, editors. Context and learning. Hillsdale, NJ: Lawrence Erlbaum and Associates; 1985. pp. 385–406. [Google Scholar]
- 331.Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- 332.Nader K, Majidishad P, Amorapanth P, LeDoux JE. Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem. 2001;8:156–163. doi: 10.1101/lm.38101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 334.Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, rima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Narayanan RT, Seidenbecher T, Kluge C, Bergado J, Stork O, Pape HC. Dissociated theta phase synchronization in amygdalo-hippocampal circuits during various stages of fear memory. Eur J Neurosci. 2007;25:1823–1831. doi: 10.1111/j.1460-9568.2007.05437.x. [DOI] [PubMed] [Google Scholar]
- 336.Narayanan RT, Seidenbecher T, Sangha S, Stork O, Pape HC. Theta resynchronization during reconsolidation of remote contextual fear memory. Neuroreport. 2007;18:1107–1111. doi: 10.1097/WNR.0b013e3282004992. [DOI] [PubMed] [Google Scholar]
- 337.Neugebauer V, Galhardo V, Maione S, Mackey SC. Forebrain pain mechanisms. Brain Res Rev. 2009;60:226–242. doi: 10.1016/j.brainresrev.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Neugebauer V, Li WD, Bird GC, Bhave G, Gereau RW. Synaptic plasticity in the amygdala in a model of arthritic pain. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Nitecka L, Ben-Ari Y. Distribution of GABA-like immunoreactivity in the rat amygdaloid complex. J Comp Neurol. 1987;266:45–55. doi: 10.1002/cne.902660105. [DOI] [PubMed] [Google Scholar]
- 340.Nitecka L, Frotscher M. Organization and synaptic interconnections of GABAergic and cholinergic elements in the rat amygdaloid nuclei: single- and double-immunolabeling studies. J Comp Neurol. 1989;279:470–488. doi: 10.1002/cne.902790311. [DOI] [PubMed] [Google Scholar]
- 341.Nithianantharajah J, Murphy M. Auditory specific fear conditioning results in increased levels of synaptophysin in the basolateral amygdala. Neurobiol Learn Mem. 2008;90:36–43. doi: 10.1016/j.nlm.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 342.Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 343.Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol. 2000;109:290–298. [PubMed] [Google Scholar]
- 344.Ota KT, Pierre VJ, Ploski JE, Queen K, Schafe GE. The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn Mem. 2008;15:792–805. doi: 10.1101/lm.1114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Ottersen OP. Connections of the amygdala of the rat. IV: Corticoamygdaloid and intraamygdaloid connections as studied with axonal transport of horseradish peroxidase. J Comp Neurol. 1982;205:30–48. doi: 10.1002/cne.902050104. [DOI] [PubMed] [Google Scholar]
- 346.Ou LC, Gean PW. Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol. 2007;72:350–358. doi: 10.1124/mol.107.034934. [DOI] [PubMed] [Google Scholar]
- 347.Pan BX, Dong YL, Ito W, Yanagawa Y, Shigemoto R, Morozov A. Selective gating of glutamatergic inputs to excitatory neurons of amygdala by presynaptic GABAb receptor. Neuron. 2009;61:917–929. doi: 10.1016/j.neuron.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Pan BX, Vautier F, Ito W, Bolshakov VY, Morozov A. Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J Neurosci. 2008;28:2089–2098. doi: 10.1523/JNEUROSCI.5156-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen SH, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- 350.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 351.Pape HC, Driesang RB. Ionic mechanisms of intrinsic oscillations in neurons of the basolateral amygdaloid complex. J Neurophysiol. 1998;79:217–226. doi: 10.1152/jn.1998.79.1.217. [DOI] [PubMed] [Google Scholar]
- 352.Pape HC, Narayanan RT, Lesting J, Stork O, Seidenbecher T, Kluge C, Sangha S. Distinctive patterns of theta synchronization in amygdalo-hippocampal-prefrontal cortical circuits during fear memory consolidation and extinction. Soc Neurosci Abstr. 2009 in press. [Google Scholar]
- 353.Pape HC, Narayanan RT, Smid J, Stork O, Seidenbecher T. Theta activity in neurons and networks of the amygdala related to long-term fear memory. Hippocampus. 2005;15:874–880. doi: 10.1002/hipo.20120. [DOI] [PubMed] [Google Scholar]
- 354.Pape HC, Paré D, Driesang RB. Two types of intrinsic oscillations in neurons of the lateral and basolateral nuclei of the amygdala. J Neurophysiol. 1998;79:205–216. doi: 10.1152/jn.1998.79.1.205. [DOI] [PubMed] [Google Scholar]
- 355.Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci. 2006;26:12977–12983. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Paré D, Collins DR. Neuronal correlates of fear in the lateral amygdala: Multiple extracellular recordings in conscious cats. J Neurosci. 2000;20:2701–2710. doi: 10.1523/JNEUROSCI.20-07-02701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Paré D, Gaudreau H. Projection cells and interneurons of the lateral and basolateral amygdala: Distinct firing patterns and differential relation to theta and delta rhythms in conscious cats. J Neurosci. 1996;16:3334–3350. doi: 10.1523/JNEUROSCI.16-10-03334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Paré D, Pape HC, Dong JM. Bursting and oscillating neurons of the cat basolateral amygdaloid complex in vivo: Electrophysiological properties and morphological features. J Neurophysiol. 1995;74:1179–1191. doi: 10.1152/jn.1995.74.3.1179. [DOI] [PubMed] [Google Scholar]
- 359.Paré D, Smith Y. Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience. 1993;57:1061–1076. doi: 10.1016/0306-4522(93)90049-l. [DOI] [PubMed] [Google Scholar]
- 360.Paré D, Smith Y. GABAergic projection from the intercalated cell masses of the amygdala to the basal forebrain in cats. J Comp Neurol. 1994;344:33–49. doi: 10.1002/cne.903440104. [DOI] [PubMed] [Google Scholar]
- 361.Paré D, Smith Y. The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience. 1993;57:1077–1090. doi: 10.1016/0306-4522(93)90050-p. [DOI] [PubMed] [Google Scholar]
- 362.Paré D, Smith Y. Intrinsic circuitry of the amygdaloid complex: common principles of organization in rats and cats. Trends Neurosci. 1998;21:240–241. doi: 10.1016/s0166-2236(98)01240-5. [DOI] [PubMed] [Google Scholar]
- 363.Paré D, Smith Y, Paré JF. Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: Phaseolus vulgaris-leucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience. 1995;69:567–583. doi: 10.1016/0306-4522(95)00272-k. [DOI] [PubMed] [Google Scholar]
- 364.Pascoe JP, Kapp BS. Electrophysiological characteristics of amygdaloid central nucleus neurons during Pavlovian fear conditioning in the rabbit. Behav Brain Res. 1985;16:117–133. doi: 10.1016/0166-4328(85)90087-7. [DOI] [PubMed] [Google Scholar]
- 365.Paul C, Schoberl F, Weinmeister P, Micale V, Wotjak CT, Hofmann F, Kleppisch T. Signaling through cGMP-dependent protein kinase I in the amygdala Is critical for auditory-cued fear memory and long-term potentiation. J Neurosci. 2008;28:14202–14212. doi: 10.1523/JNEUROSCI.2216-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, Mills E, Hakim S, Salter MW, Taylor JR, Lombroso PJ. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry. 2007;61:1049–1061. doi: 10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1986. [Google Scholar]
- 368.Petrovich GD, Swanson LW. Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Res. 1997;763:247–254. doi: 10.1016/s0006-8993(96)01361-3. [DOI] [PubMed] [Google Scholar]
- 369.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;20:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 370.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 371.Pitkänen A. Connectivity of the rat amygdaloid complex. In: Aggleton JP, editor. The Amygdala: a functional analysis. Oxford; Oxford University Press; 2000. pp. 31–115. [Google Scholar]
- 372.Pitkänen A, Amaral DG. Distribution of calbindin-D28k immunoreactivity in the monkey temporal lobe: The amygdaloid complex. J Comp Neurol. 1993;331:199–224. doi: 10.1002/cne.903310205. [DOI] [PubMed] [Google Scholar]
- 373.Pitkänen A, Amaral DG. The distribution of GABAergic cells, fibers, and terminals in the monkey amygdaloid complex: An immunohistochemical and in situ hybridization study. J Neurosci. 1994;14:2200–2224. doi: 10.1523/JNEUROSCI.14-04-02200.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Pitkänen A, Amaral DG. Distribution of parvalbumin-immunoreactive cells and fibers in the monkey temporal lobe: The amygdaloid complex. J Comp Neurol. 1993;331:14–36. doi: 10.1002/cne.903310103. [DOI] [PubMed] [Google Scholar]
- 375.Pitkänen A, Amaral DG. Organization of the intrinsic connections of the monkey amygdaloid complex: Projections originating in the lateral nucleus. J Comp Neurol. 1998;398:431–458. doi: 10.1002/(sici)1096-9861(19980831)398:3<431::aid-cne9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 376.Pitkänen A, Pikkarainen M, Nurminen N, Ylinen A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. Ann NY Acad Sci. 2000;911:369–391. doi: 10.1111/j.1749-6632.2000.tb06738.x. [DOI] [PubMed] [Google Scholar]
- 377.Pitkänen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 378.Pitkänen A, Stefanacci L, Farb CR, Go GG, LeDoux JE, Amaral DG. Intrinsic connections of the rat amygdaloid complex: Projections originating in the lateral nucleus. J Comp Neurol. 1995;356:288–310. doi: 10.1002/cne.903560211. [DOI] [PubMed] [Google Scholar]
- 379.Popescu AT, Popa D, Pare D. Coherent gamma oscillations couple the amygdala and striatum during learning. Nat Neurosci. 2009;12:801–807. doi: 10.1038/nn.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Porrino LJ, Crane AM, Goldman-Rakic PS. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J Comp Neurol. 1981;198:121–136. doi: 10.1002/cne.901980111. [DOI] [PubMed] [Google Scholar]
- 381.Poulin JF, Castonguay-Lebel Z, Laforest S, Drolet G. Enkephalin co-expression with classic neurotransmitters in the amygdaloid complex of the rat. J Comp Neurol. 2008;506:943–959. doi: 10.1002/cne.21587. [DOI] [PubMed] [Google Scholar]
- 382.Poulos AM, Li V, Sterlace SS, Tokushige F, Ponnusamy R, Fanselow MS. Persistence of fear memory across time requires the basolateral amygdala complex. Proc Natl Acad Sci U S A. 2009;106:11737–11741. doi: 10.1073/pnas.0905257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Poulton R, Menzies RG. Non-associative fear acquisition: a review of the evidence from retrospective and longitudinal research. Behav Res Ther. 2002;40:127–149. doi: 10.1016/s0005-7967(01)00045-6. [DOI] [PubMed] [Google Scholar]
- 384.Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1:1242–1259. doi: 10.1523/JNEUROSCI.01-11-01242.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Quattrone A, Pascale A, Nogues X, Zhao WQ, Gusev P, Pacini A, Alkon DL. Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA-stabilizing proteins. Proc Natl Acad Sci U S A. 2001;98:11668–11673. doi: 10.1073/pnas.191388398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 387.Quirk GJ, Garcia R, Gonzalez-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 388.Quirk GJ, Likhtik E, Pelletier JG, Pare D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Quirk GJ, Repa JC, LeDoux JE. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: Parallel recordings in the freely behaving rat. Neuron. 1995;15:1029–1039. doi: 10.1016/0896-6273(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 391.Radley JJ, Farb CR, He Y, Janssen WGM, Rodrigues SM, Johnson LR, Hof PR, LeDoux JE, Morrison JH. Distribution of NMDA and AMPA receptor subunits at thalamo-amygdaloid dendritic spines. Brain Research. 2007;1134:87–94. doi: 10.1016/j.brainres.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Radley JJ, Johnson LR, Janssen WG, Martino J, Lamprecht R, Hof PR, LeDoux JE, Morrison JH. Associative Pavlovian conditioning leads to an increase in spinophilin-immunoreactive dendritic spines in the lateral amygdala. Eur J Neurosci. 2006;24:876–884. doi: 10.1111/j.1460-9568.2006.04962.x. [DOI] [PubMed] [Google Scholar]
- 393.Radulovic J, Kammermeier J, Spiess J. Relationship between Fos production and classical fear conditioning: Effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Rainnie DG, Asprodini EK, SG P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- 395.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Inhibitory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:999–1009. doi: 10.1152/jn.1991.66.3.999. [DOI] [PubMed] [Google Scholar]
- 396.Rainnie DG, Mania I, Mascagni F, McDonald AJ. Physiological and morphological characterization of parvalbumin-containing interneurons of the rat basolateral amygdala. J Comp Neurol. 2006;498:142–161. doi: 10.1002/cne.21049. [DOI] [PubMed] [Google Scholar]
- 397.Rammes G, Steckler T, Kresse A, Schutz G, Zieglgansberger W, Lutz B. Synaptic plasticity in the basolateral amygdala in transgenic mice expressing dominant-negative cAMP response element-binding protein (CREB) in forebrain. Eur J Neurosci. 2000;12:2534–2546. doi: 10.1046/j.1460-9568.2000.00108.x. [DOI] [PubMed] [Google Scholar]
- 398.Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 399.Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Rattiner LM, Davis M, Ressler KJ. Brain-derived neurotrophic factor in amygdala-dependent learning. Neuroscientist. 2005;11:323–333. doi: 10.1177/1073858404272255. [DOI] [PubMed] [Google Scholar]
- 401.Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- 402.Redies C. Cadherins in the central nervous system. Prog Neurobiol. 2000;61:611–648. doi: 10.1016/s0301-0082(99)00070-2. [DOI] [PubMed] [Google Scholar]
- 403.Reijmers LG, Perkins BL, Matsuo N, Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- 404.Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci. 2001;4:724–731. doi: 10.1038/89512. [DOI] [PubMed] [Google Scholar]
- 405.Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- 406.Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Robbins SJ. Mechanisms underlying spontaneous recovery in autoshaping. Journal of Experimental Psychology-Animal Behavior Processes. 1990;16:235–249. [Google Scholar]
- 408.Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci. 2002;22:5219–5229. doi: 10.1523/JNEUROSCI.22-12-05219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci. 2009;32:289–313. doi: 10.1146/annurev.neuro.051508.135620. [DOI] [PubMed] [Google Scholar]
- 411.Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 413.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 414.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 415.Romanski LM, LeDoux JE. Information cascade from primary auditory cortex to the amygdala: cortex in the rat. Cereb Cortex. 1993;3:515–532. doi: 10.1093/cercor/3.6.515. [DOI] [PubMed] [Google Scholar]
- 416.Romanski LM, LeDoux JE. Organization of rodent auditory cortex: Anterograde transport of PHA-L from MGv to temporal neocortex. Cereb Cortex. 1993;3:499–514. doi: 10.1093/cercor/3.6.499. [DOI] [PubMed] [Google Scholar]
- 417.Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- 418.Rosen JB, Fanselow MS, Young SL, Sitcoske M, Maren S. Immediate-early gene expression in the amygdala following footshock stress and contextual fear conditioning. Brain Res. 1998;796:132–142. doi: 10.1016/s0006-8993(98)00294-7. [DOI] [PubMed] [Google Scholar]
- 419.Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci. 2002;22:324–337. doi: 10.1523/JNEUROSCI.22-01-00324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Rosenkranz JA, Grace AA. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci. 2001;21:4090–4103. doi: 10.1523/JNEUROSCI.21-11-04090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Rosenkranz JA, Grace AA. Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature. 2002;417:282–287. doi: 10.1038/417282a. [DOI] [PubMed] [Google Scholar]
- 422.Rosenkranz JA, Grace AA. Modulation of basolateral amygdala neuronal firing and afferent drive by dopamine receptor activation in vivo. J Neurosci. 1999;19:11027–11039. doi: 10.1523/JNEUROSCI.19-24-11027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Rossato JI, Bevilaqua LR, Lima RH, Medina JH, Izquierdo I, Cammarota M. On the participation of hippocampal p38 mitogen-activated protein kinase in extinction and reacquisition of inhibitory avoidance memory. Neuroscience. 2006;143:15–23. doi: 10.1016/j.neuroscience.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 424.Routtenberg A. The substrate for long-lasting memory: If not protein synthesis, then what? Neurobiol Learn Mem. 2008;89:225–233. doi: 10.1016/j.nlm.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Routtenberg A, Rekart JL. Post-translational protein modification as the substrate for long-lasting memory. Trends Neurosci. 2005;28:12–19. doi: 10.1016/j.tins.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 426.Royer S, Martina M, Paré D. Bistable behavior of inhibitory neurons controlling impulse traffic through the amygdala: Role of a slowly deinactivating K+ current. J Neurosci. 2000;20:9034–9039. doi: 10.1523/JNEUROSCI.20-24-09034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Royer S, Martina M, Paré D. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci. 1999;19:10575–10583. doi: 10.1523/JNEUROSCI.19-23-10575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Royer S, Martina M, Paré D. Polarized synaptic interactions between intercalated neurons of the amygdala. J Neurophysiol. 2000;83:3509–3518. doi: 10.1152/jn.2000.83.6.3509. [DOI] [PubMed] [Google Scholar]
- 429.Royer S, Pare D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience. 2002;115:455–462. doi: 10.1016/s0306-4522(02)00455-4. [DOI] [PubMed] [Google Scholar]
- 430.Royer S, Pare D. Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature. 2003;422:518–522. doi: 10.1038/nature01530. [DOI] [PubMed] [Google Scholar]
- 431.Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: insights from a two-process model. Neurosci Biobehav Rev. 2004;28:675–685. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 432.Rudy JW, Matus-Amat P. DHPG activation of group 1 mGluRs in BLA enhances fear conditioning. Learn Mem. 2009;16:421–425. doi: 10.1101/lm.1444909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Rudy JW, O'Reilly RC. Conjunctive representations, the hippocampus, and contextual fear conditioning. Cog Affect Behav Neurosci. 2001;1:66–82. doi: 10.3758/cabn.1.1.66. [DOI] [PubMed] [Google Scholar]
- 434.Ruit KG, Neafsey EJ. Hippocampal input to a “visceral motor” corticobulbar pathway: an anatomical and electrophysiological study in the rat. Exp Brain Res. 1990;82:606–616. doi: 10.1007/BF00228802. [DOI] [PubMed] [Google Scholar]
- 435.Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- 436.Russchen FT. Amygdalopetal projections in the cat. I. Cortical afferent connections. A study with retrograde and anterograde tracing techniques. J Comp Neurol. 1982;206:159–179. doi: 10.1002/cne.902060206. [DOI] [PubMed] [Google Scholar]
- 437.Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- 438.Samson RD, Dumont É C, Paré D. Feedback inhibition defines transverse processing modules in the lateral amygdala. J Neurosci. 2003;23:1966–1973. doi: 10.1523/JNEUROSCI.23-05-01966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Samson RD, Pare D. Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci. 2005;25:1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Samson RD, Pare D. A spatially structured network of inhibitory and excitatory connections directs impulse traffic within the lateral amygdala. Neuroscience. 2006;141:1599–1609. doi: 10.1016/j.neuroscience.2006.04.077. [DOI] [PubMed] [Google Scholar]
- 441.Sananbenesi F, Fischer A, Wang X, Schrick C, Neve RL, Radulovic J, Tsai LH. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat Neurosci. 2007;10:1012–1019. doi: 10.1038/nn1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5:917–930. doi: 10.1038/nrn1555. [DOI] [PubMed] [Google Scholar]
- 443.Sangha S, Narayanan RT, Bergado-Acosta JR, Stork O, Seidenbecher T, Pape HC. Deficiency of the 65-kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear in revision. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Santini E, Ge H, Ren K, Pena DO, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Santini E, Muller RU, Quirk GJ. Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci. 2001;21:9009–9017. doi: 10.1523/JNEUROSCI.21-22-09009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Savander V, Go CG, Ledoux JE, Pitkänen A. Intrinsic connections of the rat amygdaloid complex: Projections originating in the accessory basal nucleus. J Comp Neurol. 1996;374:291–313. doi: 10.1002/(SICI)1096-9861(19961014)374:2<291::AID-CNE10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 447.Savander V, Go CG, LeDoux JE, Pitkänen A. Intrinsic connections of the rat amygdaloid complex: Projections originating in the basal nucleus. J Comp Neurol. 1995;361:345–368. doi: 10.1002/cne.903610211. [DOI] [PubMed] [Google Scholar]
- 448.Savander V, Miettinen R, Ledoux JE, Pitkänen A. Lateral nucleus of the rat amygdala is reciprocally connected with basal and accessory basal nuclei: A light and electron microscopic study. Neuroscience. 1997;77:767–781. doi: 10.1016/s0306-4522(96)00513-1. [DOI] [PubMed] [Google Scholar]
- 449.Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Schafe GE, Bauer EP, Rosis S, Farb CR, Rodrigues SM, LeDoux JE. Memory consolidation of Pavlovian fear conditioning requires nitric oxide signaling in the lateral amygdala. Eur J Neurosci. 2005;22:201–211. doi: 10.1111/j.1460-9568.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- 451.Schafe GE, Doyere V, LeDoux JE. Tracking the fear engram: The lateral amygdala is an essential locus of fear memory storage. J Neurosci. 2005;25:10010–10015. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Schafe GE, Swank MW, Rodrigues SM, Debiec J, Doyere V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn Mem. 2008;15:55–62. doi: 10.1101/lm.746808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron. 2007;55:786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Schroeder BW, Shinnick-Gallagher P. Fear memories induce a switch in stimulus response and signaling mechanisms for long-term potentiation in the lateral amygdala. Eur J Neurosci. 2004;20:549–556. doi: 10.1111/j.1460-9568.2004.03517.x. [DOI] [PubMed] [Google Scholar]
- 456.Scicli AP, Petrovich GD, Swanson LW, Thompson RF. Contextual fear conditioning is associated with lateralized expression of the immediate early gene c-fos in the central and basolateral amygdalar nuclei. Behav Neurosci. 2004;118:5–14. doi: 10.1037/0735-7044.118.1.5. [DOI] [PubMed] [Google Scholar]
- 457.Sehlmeyer C, Schoning S, Zwitserlood P, Pfleiderer B, Kircher T, Arolt V, Konrad C. Human fear conditioning and extinction in neuroimaging: a systematic review. PLoS One. 2009;4:e5865. doi: 10.1371/journal.pone.0005865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Seidenbecher T, Laxmi TR, Stork O, Pape HC. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science. 2003;301:846–850. doi: 10.1126/science.1085818. [DOI] [PubMed] [Google Scholar]
- 459.Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Selden NR, Everitt BJ, Jarrard LE, Robbins TW. Complementary roles for the amygdala and hippocampus in aversive conditioning to explicit and contextual cues. Neuroscience. 1991;42:335–350. doi: 10.1016/0306-4522(91)90379-3. [DOI] [PubMed] [Google Scholar]
- 461.Senkov O, Sun M, Weinhold B, Gerardy-Schahn R, Schachner M, Dityatev A. Polysialylated neural cell adhesion molecule is involved in induction of long-term potentiation and memory acquisition and consolidation in a fear-conditioning paradigm. J Neurosci. 2006;26:10888–10898. doi: 10.1523/JNEUROSCI.0878-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, Yin JCP, Sacktor TC, Fenton AA. PKM zeta maintains spatial, instrumental, and classically conditioned long-term memories. Plos Biology. 2008;6:2698–2706. doi: 10.1371/journal.pbio.0060318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- 464.Seymour B, Dolan R. Emotion, decision making, and the amygdala. Neuron. 2008;58:662–671. doi: 10.1016/j.neuron.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 465.Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B, Luthi A. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci. 2006;9:1028–1035. doi: 10.1038/nn1732. [DOI] [PubMed] [Google Scholar]
- 466.Shi CJ, Cassell MD. Cortical, thalamic, and amygdaloid projections of rat temporal cortex. J Comp Neurol. 1997;382:153–175. [PubMed] [Google Scholar]
- 467.Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- 468.Shinnick-Gallagher P, McKernan MG, Xie JG, Zinebi F. L-type voltage-gated calcium channels are involved in the in vivo and in vitro expression of fear conditioning. Amygdala in Brain Function: Bacic and Clinical Approaches. 2003;985:135–149. doi: 10.1111/j.1749-6632.2003.tb07078.x. [DOI] [PubMed] [Google Scholar]
- 469.Shinonaga Y, Takada M, Mizuno N. Direct projections from the non-laminated divisions of the medial geniculate nucleus to the temporal polar cortex and amygdala in the cat. J Comp Neurol. 1994;340:405–426. doi: 10.1002/cne.903400310. [DOI] [PubMed] [Google Scholar]
- 470.Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, Zakharenko SS, Joseph J, Vronskaya S, Yin DQ, Schubart UK, Kandel ER, Bolshakov VY. stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell. 2005;123:697–709. doi: 10.1016/j.cell.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 471.Siapas AG, Lubenov EV, Wilson MA. Prefrontal phase locking to hippocampal theta oscillations. Neuron. 2005;46:141–151. doi: 10.1016/j.neuron.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 472.Siegmund A, Wotjak CT. Toward an animal model of posttraumatic stress disorder. Ann NY Acad Sci. 2006;1071:324–334. doi: 10.1196/annals.1364.025. [DOI] [PubMed] [Google Scholar]
- 473.Sigurdsson T, Doyere V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: A cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 474.Sirota A, Montgomery S, Fujisawa S, Isomura Y, Zugaro M, Buzsaki G. Entrainment of neocortical neurons and gamma oscillations by the hippocampal theta rhythm. Neuron. 2008;60:683–697. doi: 10.1016/j.neuron.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Smith Y, Pare JF, Pare D. Differential innervation of parvalbumin-immunoreactive interneurons of the basolateral amygdaloid complex by cortical and intrinsic inputs. J Comp Neurol. 2000;416:496–508. [PubMed] [Google Scholar]
- 476.Smith Y, Paré D. Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with post-embedding GABA and glutamate immunocytochemistry. J Comp Neurol. 1994;342:232–248. doi: 10.1002/cne.903420207. [DOI] [PubMed] [Google Scholar]
- 477.Smith Y, Paré JF, Paré D. Cat intraamygdaloid inhibitory network: ultrastructural organization of parvalbumin-immunoreactive elements. J Comp Neurol. 1998;391:164–179. doi: 10.1002/(sici)1096-9861(19980209)391:2<164::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 478.Sorvari H, Soininen H, Paljärvi L, Karkola K, Pitkänen A. Distribution of parvalbumin-immunoreactive cells and fibers in the human amygdaloid complex. J Comp Neurol. 1995;360:185–212. doi: 10.1002/cne.903600202. [DOI] [PubMed] [Google Scholar]
- 479.Sosulina L, Meis S, Seifert G, Steinhauser C, Pape HC. Classification of projection neurons and interneurons in the rat lateral amygdala based upon cluster analysis. Mol Cell Neurosci. 2006;33:57–67. doi: 10.1016/j.mcn.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 480.Sosulina L, Schwesig G, Seifert G, Pape HC. Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci. 2008;39:491–498. doi: 10.1016/j.mcn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 481.Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- 482.Sotres-Bayon F, Diaz-Mataix L, Bush DE, LeDoux JE. Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cereb Cortex. 2009;19:474–482. doi: 10.1093/cercor/bhn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Stanciu M, Radulovic J, Spiess J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Mol Brain Res. 2001;94:15–24. doi: 10.1016/s0169-328x(01)00174-7. [DOI] [PubMed] [Google Scholar]
- 484.Stein MB. Neurobiology of generalized anxiety disorder. J Clin Psychiatry. 2009;70 2:15–19. doi: 10.4088/jcp.s.7002.03. [DOI] [PubMed] [Google Scholar]
- 485.Steriade M, Pare D. Gating in cerebral networks. Cambridge, UK: Cambridge University Press; 2007. [Google Scholar]
- 486.Stoppel C, Albrecht A, Pape HC, Stork O. Genes and neurons: molecular insights to fear and anxiety. Genes Brain Behav. 2006;5 Suppl 2:34–47. doi: 10.1111/j.1601-183X.2006.00229.x. [DOI] [PubMed] [Google Scholar]
- 487.Stork O, Ji FY, Obata K. Reduction of extracellular GABA in the mouse amygdala during and following confrontation with a conditioned fear stimulus. Neurosci Lett. 2002;327:138–142. doi: 10.1016/s0304-3940(02)00387-7. [DOI] [PubMed] [Google Scholar]
- 488.Stork O, Stork S, Pape HC, Obata K. Identification of genes expressed in the amygdala during the formation of fear memory. Learn Mem. 2001;8:209–219. doi: 10.1101/lm.39401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Stork O, Welzl H, Wotjak CT, Hoyer D, Delling M, Cremer H, Schachner M. Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J Neurobiol. 1999;40:343–355. doi: 10.1002/(sici)1097-4695(19990905)40:3<343::aid-neu6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 490.Sullivan GM, Apergis J, Bush DE, Johnson LR, Hou M, Ledoux JE. Lesions in the bed nucleus of the stria terminalis disrupt corticosterone and freezing responses elicited by a contextual but not by a specific cue-conditioned fear stimulus. Neuroscience. 2004;128:7–14. doi: 10.1016/j.neuroscience.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 491.Sun N, Yi H, Cassell MD. Evidence for a GABAergic interface between cortical afferents and brainstem projection neurons in the rat central extended amygdala. J Comp Neurol. 1994;340:43–64. doi: 10.1002/cne.903400105. [DOI] [PubMed] [Google Scholar]
- 492.Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Swanson LW. Brain Maps: structure of the rat brain. Amsrterdam: Elsevier; 1992. [Google Scholar]
- 494.Swanson LW. A direct projection from Ammon's horn to prefrontal cortex in the rat. Brain Res. 1981;217:150–154. doi: 10.1016/0006-8993(81)90192-x. [DOI] [PubMed] [Google Scholar]
- 495.Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- 496.Szinyei C, Heinbockel T, Montagne J, Pape HC. Putative cortical and thalamic inputs elicit convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci. 2000;20:8909–8915. doi: 10.1523/JNEUROSCI.20-23-08909.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Szinyei C, Narayanan RT, Pape HC. Plasticity of inhibitory synaptic network interactions in the lateral amygdala upon fear conditioning in mice. Eur J Neurosci. 2007;25:1205–1211. doi: 10.1111/j.1460-9568.2007.05349.x. [DOI] [PubMed] [Google Scholar]
- 498.Szinyei C, Stork O, Pape HC. Contribution of NR2B subunits to synaptic transmission in amygdaloid interneurons. J Neurosci. 2003;23:2549–2566. doi: 10.1523/JNEUROSCI.23-07-02549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu GS, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 500.Tombol T, Szafranska-Kosmal A. A Golgi study of the amygdaloid complex in the cat. Acta Neurobiol Exp (Wars) 1972;32:835–848. [PubMed] [Google Scholar]
- 501.Tronson NC, Schrick C, Guzman YF, Huh KH, Srivastava DP, Penzes P, Guedea AL, Gao C, Radulovic J. Segregated populations of hippocampal principal CA1 neurons mediating conditioning and extinction of contextual fear. J Neurosci. 2009;29:3387–3394. doi: 10.1523/JNEUROSCI.5619-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300. doi: 10.1016/s0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- 503.Tsvetkov E, Shin RM, Bolshakov VY. Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron. 2004;41:139–151. doi: 10.1016/s0896-6273(03)00800-6. [DOI] [PubMed] [Google Scholar]
- 504.Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Turner BH, Herkenham M. Thalamoamygdaloid projections in the rat: A test of the amygdala's role in sensory processing. J Comp Neurol. 1991;313:295–325. doi: 10.1002/cne.903130208. [DOI] [PubMed] [Google Scholar]
- 506.van Groen T, Wyss JM. The connections of presubiculum and parasubiculum in the rat. Brain Res. 1990;518:227–243. doi: 10.1016/0006-8993(90)90976-i. [DOI] [PubMed] [Google Scholar]
- 507.van Groen T, Wyss JM. Extrinsic projections from area CA1 of the rat hippocampus: olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J Comp Neurol. 1990;302:515–528. doi: 10.1002/cne.903020308. [DOI] [PubMed] [Google Scholar]
- 508.Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- 509.Vervliet B, Vansteenwegen D, Baeyens F, Hermans D, Eelen P. Return of fear in a human differential conditioning paradigm caused by a stimulus change after extinction. Behav Res Ther. 2005;43:357–371. doi: 10.1016/j.brat.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 510.Victor AM, Bernstein GA. Anxiety disorders and posttraumatic stress disorder update. Psychiatr Clin North Am. 2009;32:57–69. doi: 10.1016/j.psc.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 511.Viosca J, Lopez de Armentia M, Jancic D, Barco A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learn Mem. 2009;16:193–197. doi: 10.1101/lm.1254209. [DOI] [PubMed] [Google Scholar]
- 512.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Wallace TL, Stellitano KE, Neve RL, Duman RS. Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol Psychiatry. 2004;56:151–160. doi: 10.1016/j.biopsych.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 514.Waltereit R, Mannhardt S, Nescholta S, Maser-Gluth C, Bartsch D. Selective and protracted effect of nifedipine on fear memory extinction correlates with induced stress response. Learn Mem. 2008;15:348–356. doi: 10.1101/lm.808608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- 516.Wang H, Shimizu E, Tang YP, Cho M, Kyin M, Zuo WQ, Robinson DA, Alaimo PJ, Zhang C, Morimoto H, Zhuo M, Feng RB, Shokat KM, Tsien JZ. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc Natl Acad Sci U S A. 2003;100:4287–4292. doi: 10.1073/pnas.0636870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Washburn MS, Moises HC. Inhibitory responses of rat basolateral amygdaloid neurons recorded in vitro. Neuroscience. 1992;50:811–830. doi: 10.1016/0306-4522(92)90206-h. [DOI] [PubMed] [Google Scholar]
- 519.Watanabe Y, Ikegaya Y, Saito H, Abe K. Roles of GABA(A) NMDA and muscarinic receptors in induction of long-term potentiation in the medial and lateral amygdala in-vitro. Neurosci Res. 1995;21:317–322. doi: 10.1016/0168-0102(94)00867-f. [DOI] [PubMed] [Google Scholar]
- 520.Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR. Calmodulin-kinases: Modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Weber JT, Rzigalinski BA, Willoughby KA, Moore SF, Ellis EF. Alterations in calcium-mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium. 2000;26:289–299. doi: 10.1054/ceca.1999.0082. [DOI] [PubMed] [Google Scholar]
- 522.Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calcium-calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002;5:573–579. doi: 10.1038/nn0602-855. [DOI] [PubMed] [Google Scholar]
- 523.Weisberg RB. Overview of generalized anxiety disorder: epidemiology, presentation, and course. J Clin Psychiatry. 2009;70:4–9. [PubMed] [Google Scholar]
- 524.Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- 525.Whalen PJ, Rauch SL, Etcoff NL, McInerney SC, Lee MB, Jenike MA. Masked presentations of emotional facial expressions modulate amygdala activity without explicit knowledge. J Neurosci. 1998;18:411–418. doi: 10.1523/JNEUROSCI.18-01-00411.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci. 2006;26:12387–12396. doi: 10.1523/JNEUROSCI.4316-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Wilensky AE, Schafe GE, LeDoux JE. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci. 1999;19:RC48. doi: 10.1523/JNEUROSCI.19-24-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Woodruff AR, Sah P. Networks of parvalbumin-positive interneurons in the basolateral amygdala. J Neurosci. 2007;27:553–563. doi: 10.1523/JNEUROSCI.3686-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Woodson W, Farb CR, Ledoux JE. Afferents from the auditory thalamus synapse on inhibitory interneurons in the lateral nucleus of the amygdala. Synapse. 2000;38:124–137. doi: 10.1002/1098-2396(200011)38:2<124::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 530.Yang YL, Lu KT. Facilitation of conditioned fear extinction by D-cycloserine is mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase cascades and requires de novo protein synthesis in basolateral nucleus of amygdala. Neuroscience. 2005;134:247–260. doi: 10.1016/j.neuroscience.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 531.Yaniv D, Schafe GE, LeDoux JE, Richter-Levin G. A gradient of plasticity in the amygdala revealed by cortical and subcortical stimulation, in vivo. Neuroscience. 2001;106:613–620. doi: 10.1016/s0306-4522(01)00312-8. [DOI] [PubMed] [Google Scholar]
- 532.Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kappa B in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]
- 533.Yeh SH, Lin CH, Lee CF, Gean PW. A requirement of nuclear factor-kappa B activation in fear-potentiated startle. J Biol Chem. 2002;277:46720–46729. doi: 10.1074/jbc.M206258200. [DOI] [PubMed] [Google Scholar]
- 534.Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- 535.Yu SY, Wu DC, Liu L, Ge Y, Wang YT. Role of AMPA receptor trafficking in NMDA receptor-dependent synaptic plasticity in the rat lateral amygdala. J Neurochem. 2008;106:889–899. doi: 10.1111/j.1471-4159.2008.05461.x. [DOI] [PubMed] [Google Scholar]
- 536.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Ann Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 537.Zhou Q, Poo MM. Reversal and consolidation of activity-induced synaptic modifications. Trends Neurosci. 2004;27:378–383. doi: 10.1016/j.tins.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 538.Zinebi F, McKernan M, Shinnick-Gallagher P. Expression of fear-conditioning is accompanied by increased paired-pulse depression within the amygdala. Pharmacol Biochem Behav. 2002;71:393–400. doi: 10.1016/s0091-3057(01)00684-0. [DOI] [PubMed] [Google Scholar]
- 539.Zinebi F, Xie JG, Liu J, Russell RT, Gallagher JP, McKernan MG, Shinnick-Gallagher P. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci. 2003;23:10283–10291. doi: 10.1523/JNEUROSCI.23-32-10283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]