

Abstract
Prion diseases are neurodegenerative disorders of the central nervous system that are associated with the misfolding of the prion protein (PrP). PrP is glycosylphosphatidylinositol-anchored, and therefore the hydrophobic membrane environment may influence the process of prion conversion. This study investigates how the morphology and mechanism of growth of prion aggregates on membranes are influenced by lipid composition. Atomic force microscopy is used to image the aggregation of prions on supported lipid bilayers composed of mixtures of the zwitterionic lipid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and the anionic lipid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS). Circular dichroism shows that PrP interactions with POPS membranes result in an increase in β-sheet structure, whereas interactions with POPC do not influence PrP structure. Prion aggregation is observed on both zwitterionic and anionic membranes, and the morphology of the aggregates formed is dependent on the anionic phospholipid content of the membrane. The aggregates that form on POPC membranes have uniform dimensions and do not disrupt the lipid bilayer. The presence of POPS results in larger aggregates with a distinctive sponge-like morphology that are disruptive to membranes. These data provide detailed information on the aggregation mechanism of PrP on membranes, which can be described by classic models of growth.
Introduction
Prion diseases are a group of fatal neurodegenerative disorders that include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, and scrapie in sheep. Such diseases can manifest as infectious, sporadic, or familial, and are all associated with the misfolding of the normal cellular form of the prion protein, PrPC, into the disease-associated isoform, PrPSc (1). This misfolding event involves the conformational rearrangement of PrPC from a soluble, protease-sensitive form that is rich in α-helical structure to an aggregated form, PrPSc, that is rich in β-sheet structure (2) and is proteinase K-resistant (3). Aggregated deposits of the misfolded PrP are found in the brain on autopsy and are accompanied by vacuolation, astrocytosis, and neuronal loss (4). The aggregates that form are morphologically diverse and include ordered amyloid fibers and amorphous material (5,6). Although the mechanisms of toxicity are unknown, it is established that PrPC expression is required in the host for neuronal cell death (7) and disease development (8), which indicates that the aggregation of host-encoded PrP results in cell death. However, since detectable levels of PrPSc are not always associated with disease (9,10), smaller oligomeric species that are more difficult to detect may be the toxic PrP species.
Understanding the biochemical processes of prion misfolding and aggregation will help us to understand the initiation and progression of prion disease. Therefore, various studies have attempted to mimic the structural transition and associated fibrillization of PrP in vitro (11–14). Such studies have highlighted the multiple pathways of protein misfolding and the formation of structurally distinct misfolded PrP isoforms. The majority of studies have focused on fibrillization of PrP in solution; however, since PrP is glycosylphosphatidylinositol (GPI)-anchored, it is closely associated with lipid membranes (15), and therefore it is important to study the influence of the lipid membrane environment on the aggregation of PrP.
Folding studies of PrP in solution have indicated that the α-helical and β-sheet-rich isoforms of PrP are separated by an energy barrier involved in the unfolding and subsequent oligomerization of PrP (16). Our hypothesis is that the hydrophobic membrane environment may lower this energy barrier to promote prion conversion. This hypothesis is supported by the fact that acidic phospholipids have been shown to promote protein misfolding and amyloid formation for a number of amyloidogenic and nonamyloidogenic proteins (17). This property has been attributed to 1), the hydrophobic nature of the lipid membrane core, which promotes protein unfolding; 2), the surface properties of membranes that enable them to effectively concentrate protein, which promotes protein-protein interactions; and 3), the ability of acidic membranes to neutralize positively charged clusters on the surface of proteins, which prevents repulsion between protein molecules and promotes oligomerization.
We previously showed that recombinant, truncated PrP binds to model membranes composed of palmitoyloleoylphosphatidyglycerol (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol) (POPG). This binding event results in a structural rearrangement of PrP to a conformation that is rich in β-sheet and prone to aggregation (18). In the study presented here, we used 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), a zwitterionic phospholipid, and palmitoyloleoylphosphatidyserine (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine) (POPS), an anionic phospholipid. These phospholipids are highly representative of the two major phospholipids in neuronal cell membranes (19). Under native conditions, phosphatidylserines are exclusively located on the cytoplasmic side of the membrane and therefore should not come into contact with PrP. However, under pathological conditions, exposure of POPS to the outer leaflet of the membrane could result in an abnormal interaction with PrP. In this study, we used atomic force microscopy (AFM) to image the assembly of prion aggregates on POPC and mixtures of POPC/POPS supported lipid bilayers (SLBs). SLBs consist of a lipid bilayer deposited on a solid support. The membrane is separated from the support by a nanometer-thick layer of water, which enables diffusion of lipid molecules within the plain of the membrane such that the bilayer retains the lateral fluidity of an unsupported free bilayer (20).
For this study, the truncated prion domain PrP(90-231), refolded to the native α-helical conformation of PrPC, was used. The sequence of this fragment corresponds to the proteinase K-resistant fragment of PrPSc that is associated with the infectious core of PrP. Therefore, the use of this protein domain is highly pertinent and biologically relevant. Because PrP(90-231) lacks the GPI anchor, the observed interactions result from the affinity of the polypeptide chain for the membrane. The use of AFM enabled us to follow in situ the aggregation events of PrP on SLBs. This technique was previously used to image the aggregation of amyloidogenic proteins, such as Aβ filaments (21), prion fragments (22), and islet amyloid polypeptide (23).
The morphology of prion aggregates observed by AFM on SLBs is correlated to protein structure changes in vesicle systems. The results show that PrP aggregation is directed down distinct pathways by the types of lipid present in the membrane, and we propose growth models to describe this aggregation process.
Materials and Methods
Details of the expression and purification of PrP and vesicle preparations are provided in the Supporting Material.
Binding assay
The binding of PrP to POPC lipid vesicles was assayed by sucrose gradient centrifugation. PrP in 5 mM MES, pH 5, was mixed with vesicles to obtain a lipid/protein molar ratio of 400, and the sample was then incubated for >1 h. This sample was adjusted to 40% (w/v) sucrose to obtain a total volume of 800 μL, which was then overlaid with a 30% sucrose layer (3 mL) followed by a 5% sucrose layer (1 mL) to form a discontinuous sucrose gradient. The samples were centrifuged at 140,000 g in a Beckman SW50.1 rotor at 4°C for 16 h. Fractions (0.5 mL) spanning the entire gradient were taken from the top and analyzed by Western dot blotting to detect protein. For this purpose, 150 μL of each fraction were taken, and the protein was precipitated with acetone and then resuspended in 30 μL of resuspension buffer (62.5 mM Tris-HCL, pH 6.8, 2% SDS). Then 3 × 10 μL aliquots of the resuspended protein were spotted onto a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Bucks, UK) and immunostrained using mAb1562 IgG (Chemicon International, Watford, UK) at 1:8000 dilution followed by alkaline phosphatase conjugated anti-mouse IgG at 1:5000 dilution. The immunostained protein was visualized using 2 mL of Western blue stabilized substrate for alkaline phosphatase (Promega, Southampton, UK).
Light scattering
Lipid-containing fractions from the sucrose density gradient were identified by light scattering on a Photon Technology International (Ford, UK) spectrofluorometer, using a 4 mm pathlength quartz cuvette. Synchronous scans (200–300 nm) were recorded with excitation light (1 nm bandwidth) and emission light (0.5 nm bandwidth) set to the same wavelengths. Spectra were collected with a step size of 1 nm and an integration time of 0.5 s, and values were taken at 280 nm as a measure of light scattering.
Circular dichroism
For each measurement, a concentrated stock of vesicles was titrated into a 250 μL aliquot of 8 μM PrP. Far-UV (190–260 nm) circular dichroism (CD) spectra were measured on a JASCO J-815 spectropolarimeter using a 1 mm path length quartz cuvette. Typically, a scanning rate of 100 nm /min, a time constant of 1 s, and a bandwidth of 1.0 nm were used. Spectra were measured at 20°C with a resolution of 0.5 nm, and typically eight or 16 scans were averaged per spectrum. The corresponding appropriate backgrounds (buffer for protein solutions, or lipid alone at corresponding concentrations for lipid-protein samples) were subtracted from the final spectra.
Preparation of SLBs and AFM
AFM measurements were performed with a Nanoscope V atomic force microscope (Veeco, Santa Barbara, CA) equipped with a J scanner (120 μm) and a tapping-mode fluid cell. The measurements were obtained using triangular silicon nitride cantilevers (spring constant ∼0.12 N/m) with oxide-sharpened tips (Veeco). Mica, mounted on a BYTAC-coated metal support disk, was freshly cleaved and installed on the scanner. The fluid cell containing the cantilever and an S-shaped silicon O-ring was assembled on top. The fluid cell, O-ring, and tubing were cleaned with 2% SDS solution and rinsed with copious amounts of nanopure water before each experiment.
SLBs were prepared directly in the fluid cell of the atomic force microscope by exposing the freshly cleaved mica to the vesicle solution at a lipid concentration of 0.5 mg/mL in 10 mM HEPES, pH 7.4, containing 2 mM CaCl2 and 150 mM NaCl. After an incubation period of 30–40 min, excess vesicles and salt were removed by exchanging the buffer with 10 mM HEPES, pH 7.4. The bilayer was then equilibrated with 5 mM MES, pH 5, and imaged to ensure that the bilayer was intact. Freshly thawed PrP(90-231) in 5 mM MES, pH 5, was injected over the bilayer at concentrations between 0.3 and 2 μM. Exposure to air was carefully avoided at all times.
Results
PrP binds to POPC vesicles and retains its native structure
We previously reported fluorescence and CD data indicating that PrP does not bind to POPC membranes (18). However, a recent study detected an interaction by separating a mixture of PrP and POPC vesicles on a sucrose density gradient (24), and data obtained by dual-polarization interferometry show that the full-length PrP binds to POPC SLBs (25). It is therefore apparent that fluorescence and CD measurements may not be able to detect low levels of binding, due to the large proportion of free protein that dominates the spectrum. To confirm that PrP binds to POPC membranes, we performed a sucrose gradient binding assay (Fig. 1). PrP alone localizes to the bottom of the gradient (fractions 9 and 10), but on incubation with POPC vesicles, PrP becomes distributed across the higher fractions (1–5), with free protein remaining in fractions 8–10. The location of PrP in fractions 1–5 correlates with the presence of the lipid vesicles as detected by light scattering (Fig. 1 B). Estimates from an ultracentrifugation binding assay suggest that ∼30% of PrP binds to the lipid vesicles (data not shown).
Figure 1.
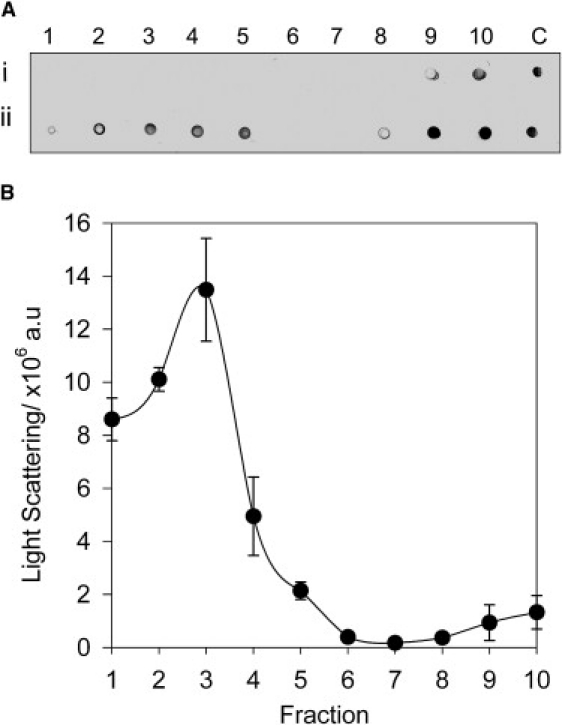
Sucrose density separation of PrP bound to POPC lipid vesicles. (A) Western dot blot analysis of fractions taken from a sucrose density gradient containing (i) PrP alone and (ii) PrP incubated with POPC lipid vesicles. Numbers 1–10 indicate the fractions from the top to the bottom of the gradient (C is the PrP-positive control). (B) Light-scattering measurements of corresponding fractions indicating the location of lipid vesicles. Error bars represent the standard error of three independent light-scattering measurements.
The far-UV CD spectrum of PrP contains two defined minima at 208 and 222 nm that are characteristic of α-helical structure. On incubation with POPC vesicles, the spectra retains its shape (Fig. 2 A), which shows that there is no detectable change in the secondary structure of PrP on binding to POPC vesicles.
Figure 2.
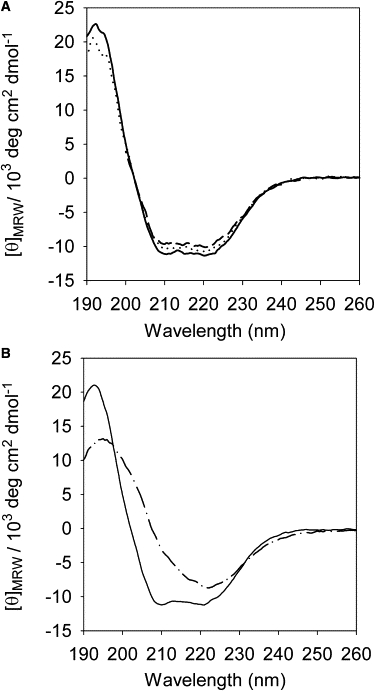
Secondary structure of PrP in solution and on incubation with POPC and POPS. Far-UV CD spectra of PrP in solution (solid line) and on incubation with (A) POPC at lipid/protein ratios of 100 (dotted line) and 500 (dashed line), and (B) in the presence of POPS (dash-dot line) at a lipid/protein ratio of 3.75.
PrP binds to POPS lipid membranes and results in an increase in β-sheet structure
In contrast to POPC, the presence of POPS results in a dramatic change in the shape of the CD spectrum (Fig. 2 B). The spectrum becomes more rounded, showing a single minimum around 220 nm, disappearance of signal at 208 nm, and attenuation of the band around 190–195 nm. The shape of this spectrum represents a protein that is rich in β-sheet structure. The band at ∼216 nm, which is typical of β-sheet structure in solution, red-shifts to ∼220 nm because the β-sheet structure is present in a lipid membrane environment. Examples of this effect can be found for other proteins in lipid membranes (26), including PrP (18).
PrP forms flat clusters on POPC membranes
The growth of prion aggregates on POPC SLBs was characterized by AFM. After the first injection of PrP onto the POPC SLB, small protein clusters could be visualized on the surface (Fig. 3 A). After further injections of protein, these nuclei grew across the surface of the membrane to form larger round, flat structures (Fig. 3, B–D). The growth of clusters is clearly illustrated in Fig. 3, B′ and C′, where the same clusters at different stages of growth can be visualized between panels. The aggregates grow until they fuse with each other (Fig. 3 D) and the protein layer begins to saturate the lipid membrane.
Figure 3.
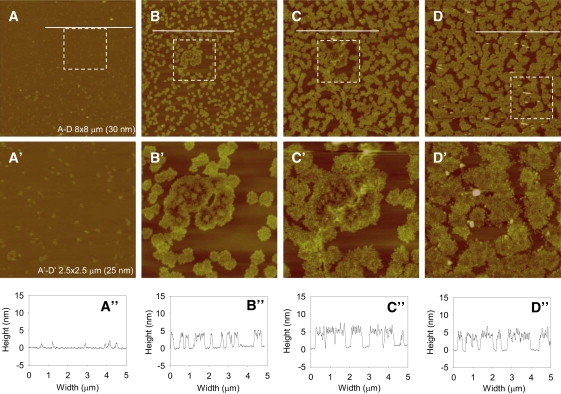
Growth of prion aggregates on POPC SLBs. Each panel shows a POPC SLB with successive 0.5 mL additions of 0.5 μM PrP (A–C) and (D) 2 μM PrP. The image size (Z-scale) of panels A–D is 8 × 8 μm (30 nm). A′–D′ are zoomed-in versions of panels A–D as indicated by the dashed boxes. The image size (Z-scale) of panels A′–D′ is 2.5 μm × 2.5 μm (25 nm). Cross-section analysis (A″–D″) reveals the height profiles across the surface along the corresponding white lines in panels A–D.
A section analysis reveals that the aggregates grow to a stable height of 5 nm (Fig. 3, B″–D″), which is equivalent to ∼1 molecule of PrP, judging by the NMR structure (27). Once they have reached this height, they do not grow vertically in height despite further injections of PrP. The section analysis also shows that the aggregates form and spread across the surface without penetrating and disrupting the bilayer. This is highlighted in the section analysis by the maintenance of a horizontal bilayer that surrounds the clusters (Fig. 3, A″–D″).
Within each condition, the size of the aggregates on the POPC SLB appear uniform in size (Fig. 3), which indicates an ordered mechanism of growth. Therefore, a size distribution of the particles was calculated for each data set (Fig. 4). After the first injection of PrP, nuclei form with a mean diameter of 94 nm, and after the next two injections the mean diameter increases to 200 and 307 nm, respectively. The diameters of the clusters correspond to ∼18, 39, and 61 molecules of PrP. The clusters grow in a protein concentration-dependent manner (Fig. 4, inset) until they fuse with neighboring aggregates.
Figure 4.
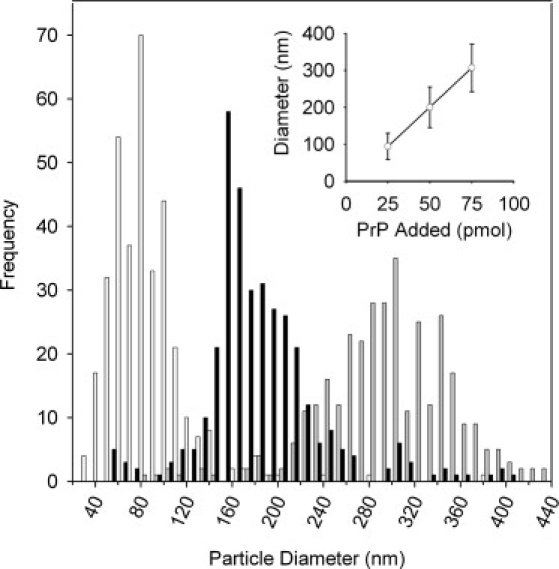
Size distribution of prion clusters formed on POPC SLBs. Each data set shows the size distribution of 350 particles selected from the corresponding images that make up Fig. 3, A–C. The size distribution of the particles is shown after successive additions of 0.5 mL of 0.5 μM PrP (white), a further 0.5 μM PrP (black), and a further 0.5 μM PrP (gray). Inset shows the mean size of the aggregates plotted against the amount of protein added, with error bars representing the standard deviation.
PrP forms holey, sponge-like aggregates on POPC/POPS membranes
SLBs prepared on mica from vesicles containing anionic lipids show an asymmetric distribution of anionic lipids between bilayer leaflets (28), with the amount of POPS exposed to solution lower than the lipid composition of the vesicles. A previous study showed that for vesicles containing 50%, 40%, and 20% POPS, only 20%, 15%, and 7% POPS, respectively, is exposed in the outer leaflet of SLB (28).
SLBs were formed from vesicles containing 20% POPS, resulting in an upper lipid leaflet containing ∼7% POPS. On injection of PrP, aggregates form that are similar in morphology to those grown on POPC alone (Fig. 5). A comparison of the clusters shown in Fig. 5, A and B, with those in Fig. 3, C and D, reveals that they are very similar in appearance. Section analysis reveals that these aggregates have the same height (5 nm) as those grown on POPC alone (Fig. 5, A′ and B′). Again these clusters grow across the membrane as more protein is added, and do not disrupt the bilayer. Fig. 5 C shows the presence of larger and thicker protein aggregates that form between existing clusters. These domains are ∼10 nm in height and could therefore represent a second layer of protein. As these aggregates are not observed in the POPC lipid system, they are likely to result from the presence of POPS in the membrane. Section analysis of the aggregates over the membrane reveals minor membrane disruptions (Fig. 5 C′).
Figure 5.
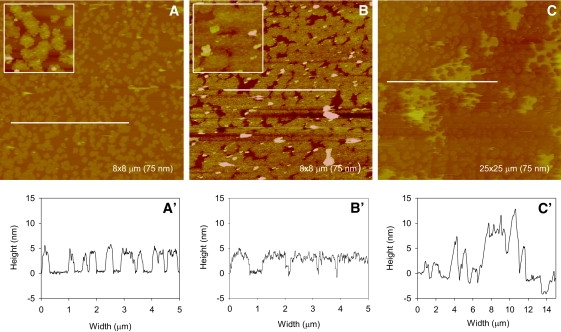
Growth of prion aggregates on POPS/POPC bilayers (∼7% POPS). Each panel shows a POPS/POPC SLB (∼7% POPS in the outer leaflet) with successive additions of 0.5 mL of 0.5 μM PrP (A and B), and (C) incubation after the previous addition of protein. The image size (Z-scale) of panels A and B is 8 × 8 μm (75 nm), and the insets are 1.5 × 1.5 μm (25 nm). The image size (Z-scale) of panel C is 25 × 25 μm (75 nm). Cross-section analysis (A′–C′) reveals the height profiles taken at areas indicated by white lines in the corresponding panels.
We further increased the concentration of POPS in the mixed membrane system to see how it would affect prion aggregation. SLBs were formed from vesicles of 40% POPS (Fig. 6), resulting in an upper lipid leaflet containing ∼15% POPS. The aggregates formed in this bilayer system were strikingly different from those grown on POPC alone. Fig. 6 A shows the formation of protein aggregates that contain holes, which makes them sponge-like in appearance. The same area of panel A is imaged in panels B and C, and therefore the growth of the aggregates can be observed as more protein is injected and the sample is incubated. Distinct nucleation sites are visible in panel A, which can be identified in panels B and C. On addition of protein, these grow outward, away from the nucleation sites and across the surface of the membrane. On further incubation (Fig. 6 C), the aggregates grow further and saturate the membrane.
Figure 6.
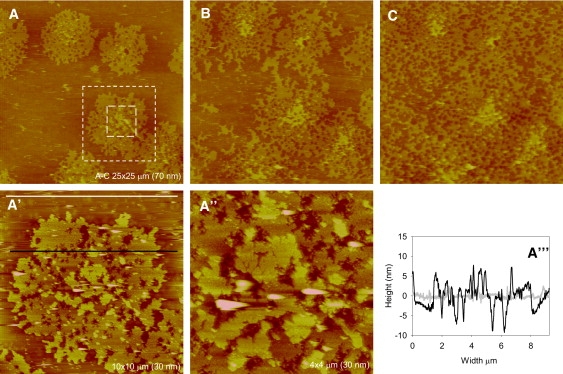
Growth of PrP aggregates on POPC/POPS bilayers (∼15% POPS). Each panel shows an SLB with the addition of (A) 0.5 mL of 1 μM PrP, (B) a further 0.5 mL of 1 μM PrP, and (C) incubation for 1 h after the second injection of protein. The image size (Z-scale) of panels A–C is 25 × 25 μm (70 nm). Panels A′ and A″ are zoomed-in versions of panel A as indicated by dashed lines. The image size (Z-scale) of A′ is 10 × 10 μm (30 nm) and the image size (Z-scale) of A″ is 4 × 4 μm (30 nm). Cross-section analysis reveals the height profiles taken at areas indicated by black and white lines.
Section analysis reveals the formation of pores that penetrate into the membrane (Fig. 6 A″′). Therefore, in the presence of POPS, PrP aggregation clearly disrupts the lipid membrane, in contrast to the aggregation of PrP on POPC bilayers.
Discussion
PrP is a GPI-anchored protein and therefore is localized to the outer leaflet of the plasma membrane (29). This close association with lipid membranes indicates that prion conversion is likely to occur in a membrane environment. We previously showed that PrP(90-231) interacts with membranes containing the anionic phospholipid POPG, promoting an increase in β-sheet structure (18) and amorphous aggregation (30). In this study, we investigated how phospholipid content affects the growth and morphology of prion aggregates on membranes composed of the zwitterionic phospholipid POPC and the anionic phospholipid POPS.
PrP binds peripherally to POPC membranes
Previous data obtained by fluorescence, CD, and attenuated total reflection Fourier transform infrared (ATR-FTIR) indicated that PrP does not interact with POPC lipid vesicles (18,30,31). In contrast, Re et al. (24) recently detected binding by separating a mixture of PrP and POPC vesicles on a sucrose density gradient. To confirm whether PrP(90-231) binds to POPC lipid membranes, we employed a sucrose gradient centrifugation binding assay (Fig. 1), which clearly showed evidence of binding. The interaction between PrP and POPC is therefore detectable by separation techniques, but not by spectroscopic techniques that rely on significant protein structure changes or insertion into the membrane. As a result, the interaction with POPC does not induce a detectable change in PrP structure, and thus there is no change in the CD spectrum (Fig. 2). Overall, the fact that PrP binds to POPC lipid membranes with no significant penetration into the membrane or change in protein structure indicates that the interaction is peripheral.
Further evidence for the peripheral interaction between PrP and POPC comes from the formation of PrP clusters on POPC SLBs, as observed by AFM. These clusters grow laterally across the membrane in a consistently ordered fashion that is dependent on protein concentration. The height of the clusters (∼5 nm) is consistent with the thickness of layers of full-length PrP on POPC SLBs (4.6 ± 0.7 nm), as measured by dual-polarization interferometry (25). In addition, previous studies have shown that the small hydrophobic peptide PrP(106-126) forms similar structures on the surface of POPC SLBs (22). It is difficult to compare those structures with the ones presented here, because the former were obtained with a much shorter PrP fragment. However, they appear to form aggregates that are dense and clustered in appearance. A noticeable difference is that PrP(106-126) forms pores in the POPC membrane, whereas the PrP(90-231) aggregates are clearly not disruptive to the membrane. The ability of this peptide to disrupt membranes in contrast to the complete globular domain of PrP may be related to the neurotoxic properties of the PrP(106-126) fragment (32).
In vivo POPC is present on the exoplasmic face of the plasma membrane, which is the same leaflet where all GPI-anchored proteins, including PrP, are located. Since GPI anchors do not extend across the whole bilayer, GPI-anchored proteins are not bound as strongly to membranes as transmembrane proteins (33). It is therefore possible that the protein-lipid interactions between PrP and POPC contribute to the association of PrP with lipid membranes and help retain the association of PrP with the bilayer. PrP forms ordered aggregates on POPC SLBs but does not disrupt the membrane. This is consistent with previous fluorescence dye release assays (18) that showed that PrP does not disrupt POPC lipid vesicles. This evidence suggests that an interaction between POPC and PrP is unlikely to initiate cell death via membrane destabilization in vivo, and therefore these clusters are unlikely to be toxic.
PrP aggregation on membranes containing POPS
In the presence of 7% POPS, aggregates formed on the SLB have a morphology similar to that of aggregates grown on POPC alone. Therefore, at this level of POPS, the greater content of POPC drives the aggregation process, even though PrP binds with higher affinity to POPS (31). Only on incubation do we see the formation of larger aggregates that are distinctly different in morphology from those formed on POPC. This suggests that the majority of protein on the membrane aggregates via the same pathway observed on the POPC bilayer, and only the smaller amounts of protein bound to POPS initiate an alternative aggregation pathway that results in the formation of the larger aggregates over time.
When the POPS concentration is increased to ∼15%, localized protein aggregates form with a sponge-like appearance as a result of membrane-disrupting pores that are visible as holes within the aggregates (Fig. 6). These aggregates are distinctly different in morphology from those formed on POPC bilayers (Fig. 3), and therefore the increased concentration of POPS, which gives the membrane surface a negative charge, significantly alters the PrP aggregation pathway. Binding to POPS results in increased β-sheet structure, and such β-sheet aggregates induce membrane disruption, as previously shown (30). Therefore, the alternative pathway is likely to result from the altered protein structure and disruption to the membrane. The fact that the aggregates formed on the 15% POPS membrane are distinctly different from those grown on 7% POPS indicates that a threshold level of POPS has to be reached for the charge density to be high enough to influence prion aggregation. This observation correlates with studies claiming that clustered negative charges are required on the membrane surface for β-sheet formation (34) and PrP-induced membrane disruption (35).
In addition to lipids, a variety of other negatively charged molecules have been shown to influence prion conversion, including glycosaminoglycans (36–38), DNA (39), and RNA (40,41). A large amount of evidence therefore suggests that anionic molecules may be crucial cofactors for prion conversion. Furthermore, in a previous study (42), infectious prions were formed in the absence of exogenous PrPSc through protein misfolding cyclic amplification (PMCA) by the addition of polyanionic molecules to native PrPC. This reaction contained lipid molecules that copurified with the protein, which led to the conclusion that the minimal components required to form infectious prions are PrPC, lipids, and anionic molecules. This finding, combined with the fact that anionic phospholipids clearly induce the misfolding of PrP and influence its aggregation pathway, suggests that anionic phospholipids may be cofactors for the prion conversion process.
PrP aggregation on POPS membranes induces membrane disruption
A variety of amyloidogenic proteins have been shown to form pore-like structures in lipid bilayers (43), and this is a possible mechanism of cell death in prion disease. We previously showed that membrane disruption occurs on interaction between PrP and anionic lipid membranes (18), and that membrane permeability is an early event in PrP-induced neurotoxicity (44). On SLBs with low POPS content (i.e., 7% POPS (Fig. 5)), there is no evidence for membrane disruption, which correlates with the fact that the morphology of the aggregates is similar to that observed on POPC alone. When POPS content is increased to 15%, the distinctly different PrP aggregates induce membrane disruption.
Disruption is likely to be induced by PrP through interactions with the hydrophobic core of the membrane. This is supported by studies conducted in transgenic mice and cultured cells, which showed that cytosolic PrP (cyPrP) interacts with the hydrophobic core of the lipid membrane in cells from the cerebellum, correlating with the location of neuronal loss (45). Therefore, there is a strong link between PrP interactions with the hydrophobic core of the membrane and neurotoxicity.
Because interactions between PrP and POPS clearly result in protein misfolding and membrane disruption, this interaction in vivo is likely to lead to cell death. POPS flip-flops through diffusion in both directions across the membrane, but the action of amino phospholipid flipase enzymes, which make internal transport more efficient, result in an asymmetrical POPS distribution (46,47). As a result, POPS is located exclusively on the cytoplasmic side of the membrane, and thus under normal circumstances PrP and POPS should be located on different sides of the membrane. However, a number of physiologically relevant events have been documented that can result in membrane scrambling and the presence of POPS on the exoplasmic side of the membrane. These include cellular activation, the presence of amphiphilic drugs, and the effects of anesthetics (46,47). A loss of POPS symmetry also occurs during apoptosis (48), which is then a signal for macrophages to engulf the cell. Therefore, in normal, healthy cells, PrP and POPS should be efficiently separated, preventing a pathological PrP-POPS interaction. However, the loss of POPS asymmetry in vivo is likely to result in a PrP-POPS interaction, which would result in protein misfolding and membrane disruption, leading to a spontaneous prion disease.
Aggregation mechanisms of PrP on SLBs
It is apparent that the shape of any growth form depends on the mechanism by which it originates. Therefore, it is possible to deduce a mechanism of aggregation from the structure of the aggregates formed. When we compare the morphology of the aggregates that form on POPC (Fig. 3) and POPC/POPS (Fig. 6) systems, it is clear that they grow through different mechanisms. In an attempt to describe these mechanisms of aggregation, we researched models that have been used to describe the growth of natural phenomena. The Eden model was originally used to describe the growth of bacterial cells on a substrate (49), and the growth of PrP aggregates on POPC membranes seems to fit this model. If so, then a site on the membrane is randomly occupied by the PrP molecule (Fig. 7), and the nearest neighbors to this point (i.e., the surrounding membrane) become potential sites for the next molecule of PrP to bind. At the next time point, one of the potential sites is randomly occupied by another PrP molecule, and again the nearest neighbors of the newly occupied site become potential growth sites. This process is repeated until a large cluster of occupied sites is formed and the PrP aggregate can be visualized on the surface of the membrane. This process continues until the PrP aggregates fuse with their nearest neighbors.
Figure 7.
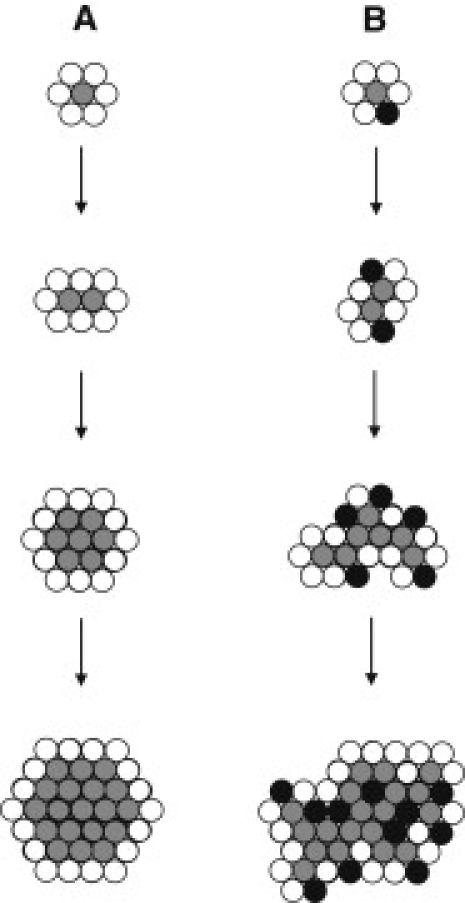
Proposed mechanisms of growth of PrP aggregates on lipid membranes. (A) Diagram illustrating the Eden model of growth, with gray circles representing bound PrP and white circles representing potential growth sites. (B) Diagram following the epidemic model of growth, with gray circles representing bound PrP, white circles representing potential growth sites, and black circles representing sites of membrane disruption, where growth cannot occur.
In 7% POPS membranes, no disruption of the bilayer is observed and the aggregates are similar to those formed on POPC membranes (Fig. 5, A and B). These aggregates appear to grow according to the Eden model of growth. This suggests that the dominating factor driving the aggregation process in this lipid system is the interaction between PrP and POPC. On incubation, larger aggregates are detectable, which shows that POPS is influencing the growth of a subpopulation of aggregates, resulting in a mixture of aggregates. When the content of POPS in the SLBs is increased (Fig. 6), the growth of the aggregates fits the epidemic model of growth (Fig. 7). In this model, sites on the membrane are randomly occupied by PrP; then, the nearest neighbors either become potential sites of occupancy or are killed so that they can never be occupied. In membranes where 15% POPS is present, PrP is converted to β-sheet structure, disrupting the lipid membrane and resulting in a site that can no longer become occupied. However, the other sites adjacent to the PrP aggregate where the membrane is still intact are still potential sites of growth. The aggregates therefore grow around holes in the membrane, resulting in the formation of large networks of sponge-like aggregates across the membrane.
Overall, the data presented in this study provide new insights into how lipid membranes can induce prion aggregation, and how the constituents of the membrane can direct prions down alternative aggregation pathways. Zwitterionic lipid preserves the α-helical structure of PrP and promotes a continuous mode of aggregate growth. Negatively charged lipid induces β-sheet structure, which disrupts the membrane and results in holey aggregates. To our knowledge, this is the first time that such aggregates have been imaged at the level of detail shown here, revealing two distinct mechanisms of growth that are dependent on the lipid content of the membrane.
Supporting Material
Additional methods are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00005-6.
Supporting Material
Acknowledgments
We acknowledge the CIC biomaGUNE (San Sebastián, Spain) for providing AFM facilities and expertise. We also thank Robin Ball, University of Warwick (Coventry, UK), for discussions on growth models of aggregates.
This project was funded by the Biotechnology and Biological Sciences Research Council (grant BB/D524516/1) and a studentship (88/DTA19176) to P.J.R.
References
- 1.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caughey B.W., Dong A., Caughey W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 3.Oesch B., Westaway D., Weissmann C. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 4.Bell J.E., Ironside J.W. Neuropathology of spongiform encephalopathies in humans. Br. Med. Bull. 1993;49:738–777. doi: 10.1093/oxfordjournals.bmb.a072645. [DOI] [PubMed] [Google Scholar]
- 5.Giaccone G., Verga L., Tagliavini F. Prion protein preamyloid and amyloid deposits in Gerstmann-Sträussler-Scheinker disease, Indiana kindred. Proc. Natl. Acad. Sci. USA. 1992;89:9349–9353. doi: 10.1073/pnas.89.19.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeffrey M., Goodbrand I.A., Goodsir C.M. Pathology of the transmissible spongiform encephalopathies with special emphasis on ultrastructure. Micron. 1995;26:277–298. doi: 10.1016/0968-4328(95)00004-n. [DOI] [PubMed] [Google Scholar]
- 7.Brandner S., Isenmann S., Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 8.Büeler H., Aguzzi A., Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 9.Collinge J., Palmer M.S., Lantos P. Transmission of fatal familial insomnia to laboratory animals. Lancet. 1995;346:569–570. doi: 10.1016/s0140-6736(95)91405-6. [DOI] [PubMed] [Google Scholar]
- 10.Lasmézas C.I., Deslys J.P., Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997;275:402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- 11.Swietnicki W., Morillas M., Surewicz W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry. 2000;39:424–431. doi: 10.1021/bi991967m. [DOI] [PubMed] [Google Scholar]
- 12.Baskakov I.V., Legname G., Cohen F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 13.Bocharova O.V., Breydo L., Baskakov I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc) J. Mol. Biol. 2005;346:645–659. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 14.Baskakov I.V., Bocharova O.V. In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry. 2005;44:2339–2348. doi: 10.1021/bi048322t. [DOI] [PubMed] [Google Scholar]
- 15.Stahl N., Borchelt D.R., Prusiner S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 16.Baskakov I.V., Legname G., Cohen F.E. Folding of prion protein to its native α-helical conformation is under kinetic control. J. Biol. Chem. 2001;276:19687–19690. doi: 10.1074/jbc.C100180200. [DOI] [PubMed] [Google Scholar]
- 17.Zhao H., Tuominen E.K., Kinnunen P.K. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 18.Sanghera N., Pinheiro T.J. Binding of prion protein to lipid membranes and implications for prion conversion. J. Mol. Biol. 2002;315:1241–1256. doi: 10.1006/jmbi.2001.5322. [DOI] [PubMed] [Google Scholar]
- 19.van Meer G. Lipid traffic in animal cells. Annu. Rev. Cell Biol. 1989;5:247–275. doi: 10.1146/annurev.cb.05.110189.001335. [DOI] [PubMed] [Google Scholar]
- 20.Sackmann E. Supported membranes: scientific and practical applications. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L., Zhong J., Sha Y. Parallel-oriented fibrogenesis of a β-sheet forming peptide on supported lipid bilayers. J. Phys. Chem. B. 2008;112:8950–8954. doi: 10.1021/jp802424h. [DOI] [PubMed] [Google Scholar]
- 22.Zhong J., Zheng W., Sha Y. PrP106-126 amide causes the semi-penetrated poration in the supported lipid bilayers. Biochim. Biophys. Acta. 2007;1768:1420–1429. doi: 10.1016/j.bbamem.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Domanov Y.A., Kinnunen P.K. Islet amyloid polypeptide forms rigid lipid-protein amyloid fibrils on supported phospholipid bilayers. J. Mol. Biol. 2008;376:42–54. doi: 10.1016/j.jmb.2007.11.077. [DOI] [PubMed] [Google Scholar]
- 24.Re F., Sesana S., Masserini M. Prion protein structure is affected by pH-dependent interaction with membranes: a study in a model system. FEBS Lett. 2008;582:215–220. doi: 10.1016/j.febslet.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Sanghera N., Swann M.J., Pinheiro T.J. Insight into early events in the aggregation of the prion protein on lipid membranes. Biochim. Biophys. Acta. 2009;1788:2245–2251. doi: 10.1016/j.bbamem.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Knight J.D., Hebda J.A., Miranker A.D. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 27.James T.L., Liu H., Cohen F.E. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. USA. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richter R.P., Bérat R., Brisson A.R. Formation of solid-supported lipid bilayers: an integrated view. Langmuir. 2006;22:3497–3505. doi: 10.1021/la052687c. [DOI] [PubMed] [Google Scholar]
- 29.Caughey B., Raymond G.J. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- 30.Kazlauskaite J., Sanghera N., Pinheiro T.J. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry. 2003;42:3295–3304. doi: 10.1021/bi026872q. [DOI] [PubMed] [Google Scholar]
- 31.Morillas M., Swietnicki W., Surewicz W.K. Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 1999;274:36859–36865. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- 32.Forloni G., Angeretti N., Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 33.Paulick M.G., Bertozzi C.R. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry. 2008;47:6991–7000. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miura T., Yoda M., Takeuchi H. Clustered negative charges on the lipid membrane surface induce β-sheet formation of prion protein fragment 106-126. Biochemistry. 2007;46:11589–11597. doi: 10.1021/bi700939j. [DOI] [PubMed] [Google Scholar]
- 35.Henriques S.T., Pattenden L.K., Castanho M.A. PrP(106-126) does not interact with membranes under physiological conditions. Biophys. J. 2008;95:1877–1889. doi: 10.1529/biophysj.108.131458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaked G.M., Meiner Z., Gabizon R. Reconstitution of prion infectivity from solubilized protease-resistant PrP and nonprotein components of prion rods. J. Biol. Chem. 2001;276:14324–14328. doi: 10.1074/jbc.M007815200. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Zaken O., Tzaban S., Taraboulos A. Cellular heparan sulfate participates in the metabolism of prions. J. Biol. Chem. 2003;278:40041–40049. doi: 10.1074/jbc.M301152200. [DOI] [PubMed] [Google Scholar]
- 38.Caughey B., Race R.E. Scrapie-associated PrP accumulation and its inhibition: revisiting the amyloid-glycosaminoglycan connection. Ann. N. Y. Acad. Sci. 1994;724:290–295. doi: 10.1111/j.1749-6632.1994.tb38918.x. [DOI] [PubMed] [Google Scholar]
- 39.Nandi P.K., Leclerc E., Takahashi M. DNA-induced partial unfolding of prion protein leads to its polymerisation to amyloid. J. Mol. Biol. 2002;322:153–161. doi: 10.1016/s0022-2836(02)00750-7. [DOI] [PubMed] [Google Scholar]
- 40.Deleault N.R., Lucassen R.W., Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 41.Adler V., Zeiler B., Grossman A. Small, highly structured RNAs participate in the conversion of human recombinant PrP(Sen) to PrP(Res) in vitro. J. Mol. Biol. 2003;332:47–57. doi: 10.1016/s0022-2836(03)00919-7. [DOI] [PubMed] [Google Scholar]
- 42.Deleault N.R., Harris B.T., Supattapone S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanghera N., Wall M., Pinheiro T.J. Globular and pre-fibrillar prion aggregates are toxic to neuronal cells and perturb their electrophysiology. Biochim. Biophys. Acta. 2008;1784:873–881. doi: 10.1016/j.bbapap.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 45.Wang X., Wang F., Ma J. The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J. Biol. Chem. 2006;281:13559–13565. doi: 10.1074/jbc.M512306200. [DOI] [PubMed] [Google Scholar]
- 46.Boon J.M., Smith B.D. Chemical control of phospholipid distribution across bilayer membranes. Med. Res. Rev. 2002;22:251–281. doi: 10.1002/med.10009. [DOI] [PubMed] [Google Scholar]
- 47.Zachowski A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem. J. 1993;294:1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin S.J., Reutelingsperger C.P., Green D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eden, M. 1960. A two-dimensional growth process. Proc. Berkeley Symp. Math. Stat. Prob., 4th, Berkeley. 223–239.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.