

Abstract
Kinetic investigation on the wild-type apomyoglobin and its 12 mutants with substitutions of hydrophobic residues by Ala was performed using stopped-flow fluorescence. Characteristics of the kinetic intermediate I and the folding nucleus were derived solely from kinetic data, namely, the slow-phase folding rate constants and the burst-phase amplitudes of Trp fluorescence intensity. This allowed us to pioneer the ϕ-analysis for apomyoglobin. As shown, these mutations drastically destabilized the native state N and produced minor (for conserved residues of G, H helices) or even negligible (for nonconserved residues of B, C, D, E helices) destabilizing effect on the state I. On the other hand, conserved residues of A, G, H helices made a smaller contribution to stability of the folding nucleus at the rate-limiting I→N transition than nonconserved residues of B, D, E helices. Thus, conserved side chains of the A-, G-, H-residues become involved in the folding nucleus before crossing the main barrier, whereas nonconserved side chains of the B-, D-, E-residues join the nucleus in the course of the I→N transition.
Introduction
The protein folding pathways include transition states and, in most cases, accumulation of intermediate states. The characterization of the folding mechanism requires our examining the structures and energetics of the stable intermediate (I) states and of unstable transition states (TS).
For the last decade and a half, structural studies of the transition states were mainly focused on small proteins with a two-state folding/unfolding mechanism, i.e., without accumulated intermediate states (1–13).
The transition states of proteins with a multistep folding/unfolding pathway are not very popular objects of study, because they require determination of the rates of all folding/unfolding steps over a wide range of denaturant concentrations. Besides, early intermediates are formed too rapidly to be measured directly; rather, their presence affects the visible rate constant of the native (N) state formation (14).
Currently, there is only one tool for experimental structural studies of the transition state (Φ-analysis by Fersht; see (1)), which uses Tanford's chevron plots (15) and point mutations for identification of residues whose replacement proves to be crucial for the protein folding rate and leaves its unfolding rate almost unaffected. Namely, these residues form the protein-folding nucleus, i.e., the structured part of the transition state (1,2). It should be emphasized that the Φ analysis has nothing to do with the transition state topology but only informs us about the strength of interactions between side chains of the residues in the transition state. In addition, it should be noted that the energy barrier may be counted from the I state (for the rate-limiting I→N transition) or from the unfolded (U) state (in the U→I→N transition). Depending on the transition type (I→N or U→I→N), Φ-values are different for a given amino acid in one and the same transition state, because for the I→N transition, the interactions, already fixed in the I state, do not contribute much to the residue's Φ-value. However, these interactions do contribute to Φ-values estimated for the U→I→N transition—an important peculiarity that arises in interpreting the folding nuclei of multistate proteins.
To overcome this difficulty, some modifications of the initial approach from Fersht (1,2) have been used. For example, some authors (16–21) analyzed the transition state structure, taking
whereas the initial form from Fersht (1,2) is
Here, ΔΔG(TS-N) is a change in TS free energy after mutation relative to N yielded by unfolding kinetics experiments (whereas ΔΔG(TS-U) is obtained from folding kinetics), and
is a change in stability of U relative to N after the introduced mutations, as derived from equilibrium unfolding experiments. However, this approach is applicable only when almost no accumulation of the intermediate state is observed in the mutant during equilibrium unfolding. If otherwise, the value of ΔΔG(U-N), and hence the Φ value, is erroneous.
Importantly, the determination of ΔG from equilibrium experiments can contain rather large errors. These result from the presence of an equilibrium intermediate distorting the observed transition curve, which is responsible for inaccurate determination of the equilibrium ΔG.
Another approach was reported in our previous article (22). It allows estimating energetic parameters of the intermediate state and the main transition state for a protein with one rapidly forming intermediate from kinetics data, i.e., from the rate constants and amplitudes of the burst kinetic phase. When applied to mutant forms, it derives the Φ parameter solely from folding/unfolding kinetics data. This approach, unlike the traditional analysis, allows an independent determination of fI, and consequently of kIN values (see details in Materials and Methods).The only prerequisite to its applicability is a general condition of validity of the Φ-analysis (1,2): the absence of mutation-induced crucial changes in the folding pathway or folding nucleus or in the native state structure.
The stopped-flow analysis of apomyoglobin (apoMb) folding/unfolding kinetics is focused on the apoMb transition state that forms on the barrier between the native state and the intermediate state. Apomyoglobin is a single-domain protein with a molecular weight of 17 kDa. Its helical structure is formed by A–E, G, and H helices, whereas its F helix remains unfolded until the heme becomes bound. Properties of apoMb are well studied (23–27). ApoMb has a tertiary structure typical of holomyoglobin; it is capable of undergoing a heat-induced cooperative transition that is accompanied by significant changes in its enthalpy and heat capacity. The folding process of urea-unfolded apomyoglobin involves one kinetic intermediate state that is structurally close to an equilibrium intermediate I accumulating at pH 4.2, which includes A, G, H helices, as shown by nuclear magnetic resonance (NMR) (28). In addition, the I state may also include fragments of B, C, D, and E helices under certain conditions (29,30). The existence of such an obligatory on-pathway intermediate was reported previously (31,32).
This article describes folding/unfolding kinetics of apoMb and its 12 mutant forms over a wide range of urea concentrations, which allowed identifying residues responsible for protein stability and for the rate of its native structure formation. It shows a strong destabilization of the native (folded) state of the mutant proteins as compared with the wild-type form, and a relatively minor influence of the mutations on stability of the intermediate state relative to the unfolded one. It is shown that residues belonging to the A, G, H helical complex contribute to stability of the intermediate state and to that of the folding nucleus in the U→I→N transition, whereas in the I→N transition, the apoMb folding nucleus is formed after formation of the A, G, H helical complex mostly by residues pertaining to B, C, E helices.
In this article, we are the first to report an estimate of Φ values obtained for the same rate-limiting barrier that exists in both I→N and U→I→N transitions of apomyoglobin.
Materials and Methods
Apomyoglobin and its investigated point mutants
We used wild-type (WT) apoMb and its 12 mutants. Six of the substituted (by Ala) residues are conserved in the globin family, but not involved in heme binding: V10, W14 (A helix), I111, L115 (G helix), M131, and L135 (H helix). These have been pointed out by Shaknovitch et al. (33) and Ptitsyn and Ting (34) as being important for initiation of apoMb folding. The other six are nonconserved residues, but they play a key role in free energy landscape-based computer simulations of apoMb folding (35): I28, F33 (B helix), L40 (C helix), M55 (D helix), and L61, L76 (E helix). They have a large number of contacts in the native structure comparable with that of conserved residues (see Samatova et al. (36) and Fig. 4).
Figure 4.
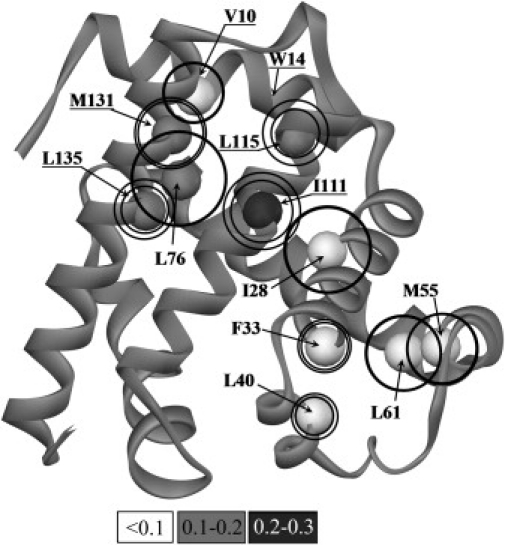
The sperm whale apomyoglobin fold (adapted from (48)). The studied residues are shown as spheres; their color intensity (see the scale below the figure) is proportional to the ΦI value, i.e., to the extent of their side-chain involvement in the intermediate state I (according to both kinetic and equilibrium data). The outer circles are proportional to the ΦTS(UN) value, the extent of involvement of side chains of these residues in the folding nucleus in the U→N transition; the inner circles are proportional to the ΦTS(IN) value, the extent of involvement of side chains in the folding nucleus in the I→N transition. Labels of conserved nonfunctional residues of A, G, H helices are underlined.
Expression and isolation
Plasmids with mutant apoMb genes were constructed by polymerase chain reaction with the plasmid pET17 (a kind gift of P.E. Wright) containing the sperm whale apoMb gene, appropriate primers, and using a QuikChange (Stratagene, La Jolla, CA) kit (37). WT apoMb and its mutants were isolated and purified as described previously (22,37) after expression of appropriate plasmids in Escherichia coli BL21 (DE3) cells.
The protein purity was checked by SDS/PAGE electrophoresis. Protein concentration was determined spectrophotometrically from absorbance at 280 nm using the extinction coefficient = 0.80 for all samples (36,38), except for one Trp-lacking mutant with substitution W14A; for this one, the used extinction coefficient was determined from nitrogen-based measurements according to Jaenicke (39) and appeared = 0.56 (36).
Equilibrium studies of urea-induced denaturing transitions
All fluorescence and far ultraviolet (UV) circular dichroism (CD) measurements were carried out at 11°C; 0.01 M sodium acetate, pH 6.2, was used as a buffer system (40).
Equilibrium protein fluorescence measurements were taken on a model No. RF-5301 PC spectrofluorimeter (Shimadzu, Kyoto, Japan) using standard 1-cm path-length quartz cuvettes. The excitation wavelength was 293 nm. The emission spectra were recorded between 300 and 500 nm (with a special attention to the emission intensity at 335 nm (I335)) at a protein concentration of 0.03 mg/mL.
A model No. J-600 spectropolarimeter (JASCO, Oklahoma City, OK) was used to perform CD experiments. Far UV CD spectra were measured between 200 and 250 nm using 0.1-mm path-length cells. The protein concentration was 1.0 mg/mL. Molar ellipticity [θ] was calculated as
where θ is measured ellipticity (degree, 10−3); MRW is the average molecular weight of a residue calculated from the sequence; l is the path length (mm); and c is protein concentration (mg/mL).
Kinetic studies
Kinetic measurements were taken using a home-made spectrofluorimeter equipped with a stopped-flow attachment as described earlier (22). The excitation wavelength was 293 nm, and emission spectra were recorded using 320-nm cut-off glass optical filter. The dead time of the equipment was <<20 ms. All measurements were taken at 11°C, pH 6.2. The final protein concentration was 0.03 mg/mL. The initial urea concentration was 5.5 M for folding experiments, and 0.0 M for unfolding ones. The initial protein solution was mixed (1:6) with buffer of various urea content using the stopped-flow attachment. All obtained kinetic curves were well described (after the dead-time burst phase) by a single-exponential approximation. Kinetic data were repeated twice for two independently prepared solutions, and there were 3–4 sequential mixings for each point.
Determination of the population of the kinetic intermediate I
The kinetic intermediate population among the nonnative states, i.e., fI(M) = [I]/([I] + [U]), where [I] and [U] are concentrations of the I and U states, was determined at various urea concentrations M from the burst phase fluorescence amplitude,
| (1) |
where AU(M) and AI(M) are the integral fluorescence intensities for U and I states, and A(M) is the integral fluorescence intensity achieved as a result of the first, burst (U→I) phase in the (U→I) transition in protein folding at the given final urea concentration M.
Calculation of folding rates, free energies, and Φ-values for the transition and intermediate states
The rate constant kIN of the second (slow as compared to the U→I) folding transition I→N across the major (rate-limiting) barrier between the native and intermediate states was calculated according to the literature (22,41,42), as follows:
| (2) |
Here fI(M) is the kinetic intermediate population calculated from Eq. 1; kobs is the observed rate constant for approaching equilibrium; and kNI is the rate constant for the unfolding transition N→I.
To calculate free energies of the intermediate (GI) and native (GN) states relative to that of the unfolded (GU) state from the folding/unfolding kinetics, the following equations were used (22):
| (3) |
| (4) |
Here kNI is the rate constant for the unfolding transition N→I dictated solely by the highest free energy barrier between N and I states (i.e., kNI = kNU) (22), kIN is the rate constant for the folding transition I→N given by Eq. 2, and
| (5) |
Involvement of a side chain of a given residue in the folding intermediate or folding nucleus was calculated using Φ-analysis (43). The value of
| (6) |
(where Δ before parentheses means a difference between values in parentheses for WT apoMb and its mutant form) reflects involvement of the substituted residue in the folding intermediate I. The value of
| (7) |
describes the increment of involvement of the substituted residue side chain in the folding nucleus corresponding to the I→N transition, and the value of
| (8) |
shows total involvement of the side chain of this residue in the same folding nucleus corresponding to the whole the U→N transition.
We also used the well-known three-state expressions (see, e.g., the article by Parker et al. (44)) on chevron plot fitting.
Results and Discussion
Equilibrium urea-induced denaturing transitions of apomyoglobin and its mutants
Urea-induced equilibrium unfolding of the designed mutants was studied by far UV CD and by Trp fluorescence to learn the effect of these mutations on conformational stability of apomyoglobin and to find conditions favorable for formation of the native, unfolded, and intermediate states.
Fig. 1 shows changes in molar ellipticity at 222 nm (Fig. 1 a) and in Trp fluorescence at 335 nm (Fig. 1 b) for WT apoMb and for its representative mutants in dependence on urea concentration. The complete set of experimental data for all the mutants is presented in Fig. S1 in Supporting Material. As seen, the N→U transition of the WT form appears more cooperative than that of the mutants and occurs at a higher urea concentration.
Figure 1.
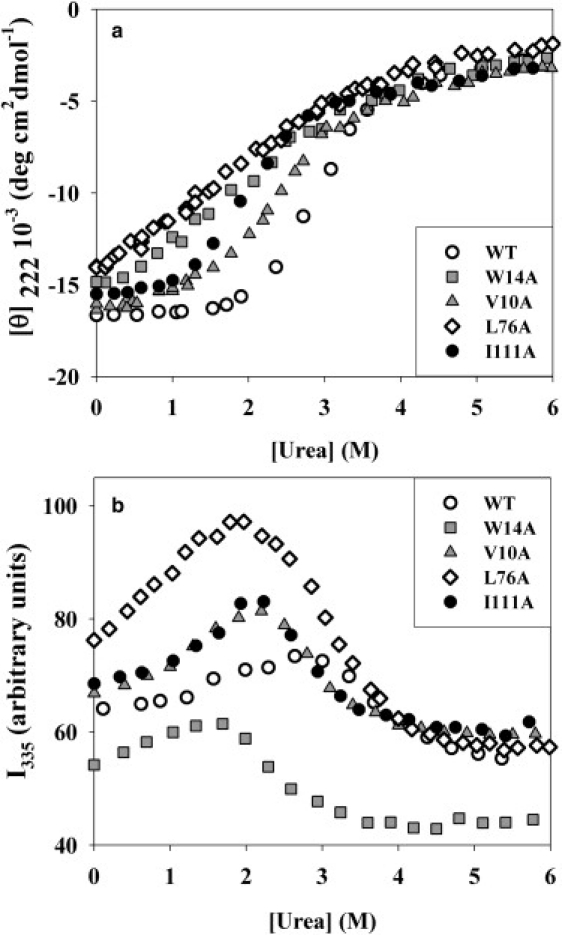
Equilibrium urea-induced unfolding of WT apomyoglobin and some of its representative mutant forms at pH 6.2: (a), as detected by molar ellipticity at 222 nm ([θ]222); (b), as detected by changing intensity of tryptophan fluorescence at 335 nm (I335). The complete set of data is given in Supporting Material.
A shift of the molar ellipticity midtransition toward lower urea concentrations demonstrates destabilization of the native (folded) structure of all the mutants, as compared to WT apoMb. The observed urea dependence of molar ellipticity [θ] at 222 nm does not provide a reliable quantitative estimate for the intermediate state in this process (22), thereby preventing us from using the equilibrium urea-induced denaturing transitions to measure a relative stability of the native and intermediate states of apoMb and its mutants. However, the CD-monitored decrease in cooperativity of transitions may suggest some accumulation of the intermediate state (45), which is more clearly demonstrated by the increase in Trp fluorescence at the intermediate urea concentrations.
Fig. 1 b shows the urea-induced equilibrium unfolding of the same mutants, as demonstrated by varying intensity of their Trp fluorescence at 335 nm. As we reported previously, at this wavelength, the presence of the intermediate state I and of the N→I and I→U transitions are most distinct (22). This is important for the analysis of equilibrium denaturing transitions. As follows from Fig. 1 b, for all the proteins studied, urea concentration dependence of fluorescence intensity shows a maximum, which demonstrates a certain accumulation of an intermediate state by displaying a more intensive fluorescence than both the native and unfolded states (22,46). On the curves for mutant proteins this maximum is more distinct and shifted toward lower urea concentrations, as compared to that of WT apoMb; the latter also indicates a drop in stability of structures of the mutant proteins. The increase in Trp fluorescence intensity at the intermediate urea concentrations can be explained by the increased intermediate state population (36).
Similar but even more pronounced results were obtained from equilibrium experiments on pH-induced denaturing transitions of apomyoglobin mutants (36).
Folding/unfolding kinetics of apomyoglobin and its mutants
As it follows from equilibrium measurements, Trp fluorescence intensity increased upon transition from the N to I state and decreased during transition from the I to U state (see Fig. 1 b). In kinetic studies, the previous finding was the existence of two consecutive fluorescence steps occurring in apoMb folding (renaturation): its increase during the stopped-flow dead time (<<20 ms) reflecting formation of an early kinetic intermediate (28) bearing a close structural similarity to the well-studied thermodynamically stable intermediate state (36,45) and subsequent decrease during transition from the intermediate to native state. The final step, a fluorescence intensity decrease, takes place in the measurable time interval (seconds) and denotes formation of the native state of apomyoglobin (28). A similar pattern is typical for folding kinetics of the apomyoglobin mutants.
Fig. 2 shows folding (a) and unfolding (b) kinetics of the mutant with L115A substitution, as a representative example. As seen, in the folding process, a low final concentration of urea induces increased burst-phase fluorescence depicting a burst appearance of the early intermediate state. Hence, two important parameters can be derived from the folding kinetics: the burst-phase amplitude A of fluorescent intensity, which is proportional to a fraction fI of the intermediate state formed after the initial folding phase, and the observed rate constant kobs for the last slow phase of folding. The folding/unfolding rate constants kobs observed at various final urea concentrations M allow constructing the so-called chevron plots (1,2,15), i.e., plots of logarithm of the observed rate constant for the process of approaching equilibrium (lnkobs) versus denaturant concentration M in solution. Knowing the rate constants kobs(M), intermediate state fractions fI(M), and rate constants kNI(M) obtained from the unfolding branch (due to very fast U↔I transition, kNI = kNU), one can estimate the rate of the I→N folding kIN(M) at a given final urea concentration (22) (see Materials and Methods). Thus, the three-state transition U↔I↔N can be divided into two two-state transitions, U↔I and I↔N. For all the mutant forms of apomyoglobin, the folding/unfolding process was studied over a wide range of final urea concentrations. In the folding process, the relationship between increasing population of the intermediate state (fI) and decreasing final urea concentration can be derived from the urea dependence of the initial burst phase amplitude (of U→I transition) for all the mutant forms. Urea concentration dependences of the I→N and N→I transition constants (kIN and kNI, respectively) have been obtained as well. Fig. 3 gives as an example fitting of kIN (M) and kNI (M) for WT apomyoglobin (see also (22)) and for two of its representative mutant forms (L115A and I28A) showing different behavior of the chevron plots.
Figure 2.
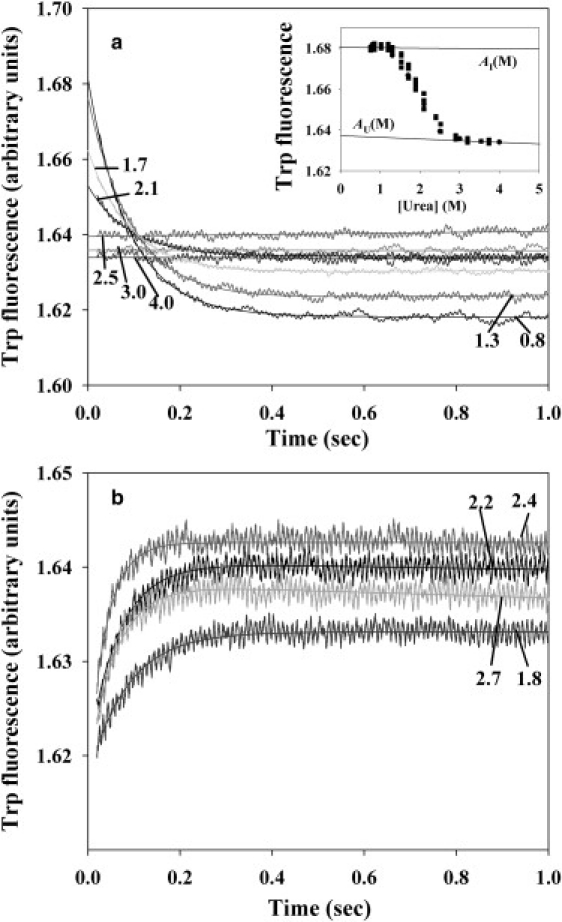
Kinetic curves for apomyoglobin mutant with Leu115 replaced by Ala in the process of its folding (a) and unfolding (b) at various final urea concentrations (indicated by numbers near the curves). The process was monitored at pH 6.2 by integral Trp fluorescence intensity. (Solid lines) Single-exponential approximations of the slow-phase folding and of unfolding kinetics. (Inset) Burst-phase amplitude A versus urea concentration M in the folding process and baselines AU(M), AI(M) for amplitudes of unfolded and intermediate states.
Figure 3.
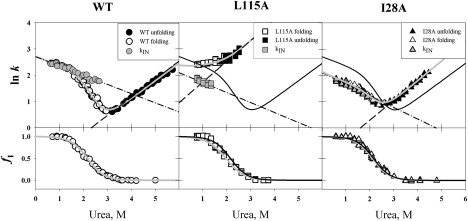
(Upper panels) Observed folding/unfolding rate constant (kobs) versus the final urea concentration M for apomyoglobin mutants. All rate constants are measured in s−1 and given in a logarithmic scale. The left part of each chevron corresponds to folding experiments (open symbols), the right part to unfolding experiments (solid symbols). (Shaded symbols) Folding rate constants kIN(M) for the I→N transition; these are calculated from Eq. 2 using the corresponding kobs(M), extrapolations of unfolding rates kNI(M) = kNU(M), and fI(M) plots shown for each protein (lower panel). The kNI(M) values were calculated from the folding data in the region where the fI(M) > 0.1. (Dashed lines) Linear approximations of ln(kNI(M)), and (dash-dot lines) linear approximations of ln(kIN(M)). (Dark-shaded lines) Chevron plots calculated using an equation kobs = kNI + fI × kIN. (Solid lines, upper panels) Chevron plots for WT protein (see WT panel). (Shaded lines, lower panels) Two-state fitting of fI versus urea concentration. (Solid lines, lower panels) The fI plots for WT protein (see WT panel).
To fit the experimental chevron plots, the approach developed in our laboratory (22) was used. We also tried to fit the whole chevron plot by the well-known three-state expression (see, e.g., (44)), which does not use amplitudes A(M) and is based on kobs(M) only, but it gives a reasonable description only for those chevron plots that show visible rollover at low urea concentrations. In other cases, this approach gives described kIN(M) values with very large errors. The examples of such calculations are shown in Fig. S3. Thus, to obtain reliable kinetic parameters, we used, in addition to chevron plots of ln(kobs(M)), the plots of fI(M), independently obtained from the burst phase amplitudes, and applied Eq. 2 to compute kNI(M). It should be emphasized that Eq. 2 is applicable in practice only within the denaturant content range where kobs(M) is larger than kNI(M) and fI(M) reliably differs from zero. The folding rate constants kIN calculated from Eq. 2 are marked by shaded symbols in Fig. 3. Based on the linear approximations of logarithms of the folding and unfolding rate constants and Eqs. 3–8, kinetic and thermodynamic parameters of folding/unfolding reactions were determined and presented in Table 1. The complete set of experimental data for kinetics of de- and renaturing transitions of apomyoglobin mutants is presented in Supporting Material. For each mutant, linear approximations of logarithms of the unfolding (kNI) and folding (kIN) branches of the chevron plots are also given.
Table 1.
Thermodynamic parameters on folding/unfolding kinetics in apomyoglobin and its mutant forms
| Parameter | Substitutions in conserved nonfunctional positions |
Substitutions in nonconserved positions |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | V10A | W14A | I111A | L115A | M131A | L135A | I28A | F33A | L40A | M55A | L61A | L76A | |
| C1/2 | 2.2 ± 0.1 | 2.2 ± 0.2 | 2.0 ± 0.1 | 2.0 ± 0.1 | 2.0 ± 0.2 | 2.0 ± 0.1 | 2.0 ± 0.2 | 2.1 ± 0.1 | 2.1 ± 0.2 | 2.1 ± 0.1 | 2.1 ± 0.2 | 2.2 ± 0.1 | 2.0 ± 0.1 |
| mIU | 1.47 ± 0.05 | 1.43 ± 0.06 | 1.50 ± 0.06 | 1.5 ± 0.1 | 1.45 ± 0.05 | 1.46 ± 0.05 | 1.40 ± 0.07 | 1.49 ± 0.05 | 1.48 ± 0.06 | 1.49 ± 0.08 | 1.45 ± 0.09 | 1.5 ± 0.1 | 1.49 ± 0.05 |
| ln kIN∗ | 2.70 ± 0.09 | 2.4 ± 0.1 | ? | 2.3 ± 0.1 | 2.1 ± 0.2 | 1.4 ± 0.3 | 2.4 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.2 | 2.5 ± 0.2 | 1.6 ± 0.2 | 1.4 ± 0.2 | 0.0 ± 0.2 |
| mIN | −0.35 ± 0.02 | −0.38 ± 0.04 | ? | −0.41 ± 0.03 | −0.37 ± 0.07 | −0.5 ± 0.2 | −0.41 ± 0.06 | −0.44 ± 0.03 | −0.35 ± 0.02 | −0.40 ± 0.04 | −0.4 ± 0.1 | −0.40 ± 0.08 | −0.41 ± 0.06 |
| ln kNI∗ | −1.6 ± 0.4 | −1.1 ± 0.3 | 0.2 ± 0.3 | −0.7 ± 0.4 | 1.4 ± 0.6 | 0.8 ± 0.3 | 1.3 ± 0.4 | −1.4 ± 0.4 | 1.1 ± 0.8 | 0.0 ± 0.2 | 0.1 ± 0.6 | 0.8 ± 0.8 | −0.2 ± 0.3 |
| mNI | 0.68 ± 0.04 | 0.81 ± 0.04 | 1.67 ± 0.08 | 0.87 ± 0.04 | 0.6 ± 0.2 | 0.81 ± 0.06 | 0.71 ± 0.08 | 0.79 ± 0.04 | 0.7 ± 0.2 | 0.75 ± 0.03 | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.75 ± 0.03 |
C1/2 is the denaturant concentration in the middle of the fI urea dependence in the U-I transition, in [M]; mIU is the slope of the urea dependence of the intermediate state stability in relation to the unfolded state, in kcal mol−1 [M]−1.
Extrapolated to the absence of denaturant; ln kIN and ln kNI are logarithms of the folding and unfolding rate constants linearly extrapolated to water; mIN and mNI are dependences of logarithms of the folding and unfolding rate constants with urea, respectively, in [M]−1.
As it is seen from the Table 1, values of mIN and mNI (dependences of logarithms of folding and unfolding rate constants on urea concentration) are close to the value for WT protein within the experimental error. This means that used substitutions do not crucially change the folding pathway except for W14A, for which mNI value is rather high, but in this case the largest aromatic residue is substituted by the smallest aliphatic one, and such a great difference makes the Φ-analysis for this mutant questionable.
All the mutant forms have been analyzed (see Fig. 3 and Materials and Methods) by the method developed for WT apomyoglobin using the three-state kinetics analysis (22). This method was applied to determine energetic parameters of folding and unfolding processes which are presented in Table 2, and was used to assess involvement of various residues in formation of the intermediate (see also (36)) and transition states.
Table 2.
Parameters derived from kinetic data on folding/unfolding of apomyoglobin and its mutant forms
| Parameter∗ | Substitutions in conserved nonfunctional positions |
Substitutions in nonconserved positions |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | V10A | W14A | I111A | L115A | M131A | L135A | I28A | F33A | L40A | M55A | L61A | L76A | |
| ΔGIU | −3.19 ± 0.03 | −3.17 ± 0.07 | −2.93 ± 0.06 | −2.9 ± 0.2 | −2.86 ± 0.09 | −2.92 ± 0.06 | −2.86 ± 0.09 | −3.16 ± 0.05 | −3.1 ± 0.1 | −3.1 ± 0.1 | −3.1 ± 0.1 | −3.2 ± 0.2 | −2.91 ± 0.05 |
| ΔGNI | −2.50 ± 0.01 | −1.80 ± 0.02 | ? | −1.54 ± 0.01 | −0.64 ± 0.04 | −0.22 ± 0.04 | −0.74 ± 0.01 | −1.86 ± 0.01 | −0.61 ± 0.03 | −1.47 ± 0.03 | −0.69 ± 0.02 | −0.46 ± 0.01 | −0.20 ± 0.03 |
| ΔGNU | −5.69 ± 0.03 | −4.97 ± 0.07 | ? | −4.5 ± 0.2 | −3.6 ± 0.1 | −3.14 ± 0.07 | −3.60 ± 0.09 | −5.02 ± 0.05 | −3.7 ± 0.1 | −4.5 ± 0.1 | −3.8 ± 0.2 | −3.8 ± 0.2 | −3.11 ± 0.05 |
| ΦTS(IN) | — | 0.3 ± 0.2 | ? | 0.3 ± 0.2 | 0.16 ± 0.07 | 0.35 ± 0.07 | 0.10 ± 0.04 | 0.8 ± 0.5 | 0.17 ± 0.07 | 0.12 ± 0.09 | 0.3 ± 0.1 | 0.34 ± 0.09 | 0.67 ± 0.08 |
| ΦI | — | 0.0 ± 0.1 | ? | 0.2 ± 0.1 | 0.16 ± 0.05 | 0.11 ± 0.02 | 0.15 ± 0.05 | 0.04 ± 0.09 | 0.05 ± 0.06 | 0.0 ± 0.1 | 0.04 ± 0.07 | 0.0 ± 0.1 | 0.10 ± 0.02 |
| ΦTS(UN) | — | 0.3 ± 0.2 | ? | 0.5 ± 0.2 | 0.32 ± 0.08 | 0.42 ± 0.07 | 0.24 ± 0.06 | 0.8 ± 0.5 | 0.2 ± 0.1 | 0.1 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.71 ± 0.08 |
Identification of residues forming the I state and the I→N transition state
With the values of lnkIN(M) and lnkNI(M) known (and extrapolated to the absence of denaturant) for the mutants and for WT apomyoglobin, the Φ-analysis proposed by Fersht in Matouschek et al. (1,2) can be applied to assess involvement of the substituted residues in the structure of the transition state on the barrier between the I and N states. With ΦTS(IN) close to zero, such a substitution disturbs stability of the transition state as much as that of the intermediate state, and hence, the substituted residue is involved in the folding nucleus only to the same extent as in the intermediate state. In contrast, with ΦTS(IN) close to unity, the substitution equally disturbs stabilities of the transition state and the native state, and hence, the substituted residue participates in the folding nucleus of the I→N transition.
Table 2 shows that all these mutant forms with substituted conserved residues have a ΦTS(IN) value of 0.2–0.3, which suggests that, in the transition state of the I→N transition, they participate in ≤30% of additional (relative to the I state) native interactions. However, one should note that most of these residues have ΦI = 0.1–0.2, which means that they have already formed 10–20% of the native contacts in the state I. For the mutants with substitutions in the nonconserved residues of B, D, E helices (among these L76 has ∼10% of its native contacts in the state I, whereas the others have much less), ΦTS(IN) varies from 0.1 to 0.8. It is as high as 0.8 for Ile28 (located in the middle of B helix) and 0.7 for Leu76 (in the C-terminus of E helix), which indicates their key role in the I→N transition. For Met55 and Leu61, ΦTS(IN) is ∼0.3, whereas Phe33 and Leu40 have low ΦTS(IN) values. Thus, the majority but not all of the studied nonconserved residues play a more important role in the I→N transition than the conserved ones.
The difference between the two classes of mutants is striking as to their contribution to the intermediate state (ΦI-values) and not so large when it comes to the transition state ΦTS(IN), except for two positions, I28 and L76, whose contribution is large. Different behavior of L111 and L115, as well as M131 and L135, may be explained by nonnative contacts of L115 and L135 in the intermediate state, as shown by Nishimura et al. (30). It is quite plausible that these nonnative interactions become native only after the transition state is formed, with residues M55 and L61, and C, CD, D, and partly E helices involved in the hydrophobic cluster. Apparently, the residue F33 of B helix and L40 of C helix dock later because they have lower Φ-values.
The mutational analysis gives information on interactions of side chains rather than on the backbone behavior studied in detail by Nishimura et al. (30) using NMR. From this point of view, interactions of side chains in the molten globulelike intermediate are not so strong, but nevertheless, the conserved residues contribute markedly more than nonconserved ones, except for, possibly, L76. We can speculate that this residue may form some nonnative interactions in the intermediate state (because such interactions were reported in (30)). The same may be a reason for low ΦTS(IN) values of L115A and L135A.
Another picture is seen if we consider ΦTS(UN) values calculated for the three-state U→I→N transition. In this transition ΦTS(UN) values for the conserved residues and for three mutants from nonconserved ones are in the same range, but side chains of the latter start to contribute to overall interactions only during the I→N transition, whereas side chains of the conserved residues participate in both I state and I→N transitions. F33, at the C-end of the B helix, and L40 of the C helix (showing fairly low Φ-values), probably dock after they have crossed the main rate-limiting barrier. I28A and L76A create the main contribution to side-chain interactions in the I→N transition state, as their docking to the already formed A, G, H helical complex makes the N-end of the B helix and the C-end of the E helix join this complex, which stabilizes it (i.e., these nonconserved residues and thereby the corresponding helices consolidate using the A, G, H complex as a nucleation point). However, the complete set of side-chain interactions can be expected only in the native state. It should be stressed that all of these side-chain interactions are observed when the backbone interactions have been, for practical purposes, fully realized (our far UV CD spectra clearly showed the presence of substantial secondary structure even at moderate denaturant concentrations, in the state I).
We see that the role of conserved residues is more significant than that of nonconserved residues specifically in the U→I transition, which supports Ptitsyn's supposition (47) that conserved residues are of special importance in forming the intermediate structure at the initial stage of apoMb folding.
Combining both sets of results gives a more accomplished picture of the intermediate and transition states. Residues from different helices are involved in the secondary structure at an early step of folding. However, as it follows from Φ-analysis, the fraction of interactions of side chains increases step by step, from 20% or less in the intermediate state to 30–70% in the I-N transition state, to reach its maximum in the apoMb native state.
A detailed investigation of apomyoglobin mutants by NMR was performed by Nishimura et al. (29,30) and Tsui et al. (32), who studied the folding pattern and mobility of the apomyoglobin backbone using the technique of amide H-D exchange in the I and N states. Here, we considered interactions of the apomyoglobin side chains and strength of their interactions in the I and TS states. Therefore, experimental results of the two teams are complementary and allow a more-detailed understanding of interactions occurring in apomyoglobin folding.
Conclusions
Fig. 4 shows the sperm whale apomyoglobin fold, where the studied residues are marked out and their role in the I state and in the I↔N and U↔I↔N transitions is outlined. As seen, the residues whose side chain form the kinetic intermediate state I are positioned mainly in G, H helices (in accordance with data on the equilibrium intermediate state (28,36)) whereas those limiting the apomyoglobin folding rate in the I↔N transition are in another part of the protein, namely, in B, D, E helices. The resulting folding nucleus for the three-state U↔I↔N transition is a sum of contributions of all these residues. Thus, the developed approach taking into account only kinetic experiments allowed us to estimate contribution of various residues at different steps of folding of a multistate protein. Additional studies using other ultrafast techniques are undoubtedly required to shed light upon the U↔I transition in apoMb.
Supporting Material
One table and three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00098-6.
Supporting Material
Acknowledgments
The study was supported by the Program on “Molecular and Cell Biology” from the Russian Academy of Sciences; by an International Research Scholar Award from the Howard Hughes Medical Institute (Grant No. 55005607) to A.V.F.; by INTAS (Grant No. 05-1000004-7747) and, in part, by award No. RB2-2022 from the US Civilian Research & Development Foundation for the Independent States of the Former Soviet Union (to E.N.S., V.E.B., and D.A.D.) and by a grant from the Russian Federal Agency for Science and Innovation (02.740.11.0295).
The authors thank P. E. Wright for providing a plasmid with sperm whale apomyoglobin gene, I. A. Kashparov and N. B. Ilyina for assistance in protein isolation and determination of protein extinction coefficients, O. V. Galzitskaya, D. N. Ivankov, and S. A. Garbuzynskiy for their help in modeling of myoglobin unfolding, and E. V. Serebrova for assistance in manuscript preparation.
References
- 1.Matouschek A., Kellis J.T., Fersht A.R. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 2.Matouschek A., Kellis J.T., Fersht A.R. Transient folding intermediates characterized by protein engineering. Nature. 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 3.Itzhaki L.S., Otzen D.E., Fersht A.R. The structure of the transition state for folding of chymotrypsin inhibitor 2 analyzed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 4.Fulton K.F., Main E.R., Jackson S.E. Mapping the interactions present in the transition state for unfolding/folding of FKBP12. J. Mol. Biol. 1999;291:445–461. doi: 10.1006/jmbi.1999.2942. [DOI] [PubMed] [Google Scholar]
- 5.Chiti F., Taddei N., Dobson C.M. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 1999;6:1005–1009. doi: 10.1038/14890. [DOI] [PubMed] [Google Scholar]
- 6.Villegas V., Martinez J.C., Serrano L. Structure of the transition state in the folding process of human procarboxypeptidase A2 activation domain. J. Mol. Biol. 1998;83:1027–1036. doi: 10.1006/jmbi.1998.2158. [DOI] [PubMed] [Google Scholar]
- 7.Grantcharova V.P., Riddle D.S., Baker D. Important role of hydrogen bonds in the structurally polarized transition state for folding of the Src SH3 domain. Nat. Struct. Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 8.Kragelund B.B., Osmark P., Poulsen F.M. The formation of a native-like structure containing eight conserved hydrophobic residues is rate limiting in two-state protein folding of ACBP. Nat. Struct. Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 9.Jackson S.E. How do small single-domain proteins fold? Fold. Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 10.Martínez J.C., Serrano L. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 1999;6:1010–1016. doi: 10.1038/14896. [DOI] [PubMed] [Google Scholar]
- 11.Riddle D.S., Grantcharova V.P., Baker D. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 12.Perl D., Welker C., Schmid F.X. Conservation of rapid two-state folding in mesophilic, thermophilic and hyperthermophilic cold shock proteins. Nat. Struct. Biol. 1998;5:229–235. doi: 10.1038/nsb0398-229. [DOI] [PubMed] [Google Scholar]
- 13.Steensma E., van Mierlo C.P.M. Structural characterization of apoflavodoxin shows that the location of the stable nucleus differs among proteins with a flavodoxin-like topology. J. Mol. Biol. 1998;282:653–666. doi: 10.1006/jmbi.1998.2045. [DOI] [PubMed] [Google Scholar]
- 14.Clarke A.R., Waltho J.P. Protein folding and intermediates. Curr. Opin. Biotechnol. 1997;8:400–410. doi: 10.1016/s0958-1669(97)80060-2. [DOI] [PubMed] [Google Scholar]
- 15.Tanford C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 16.Serrano L., Matouschek A., Fersht A.R. The folding of an enzyme. III. Structure of the transition state for unfolding of barnase analyzed by a protein engineering procedure. J. Mol. Biol. 1992;224:805–818. doi: 10.1016/0022-2836(92)90563-y. [DOI] [PubMed] [Google Scholar]
- 17.López-Hernández E., Serrano L. Structure of the transition state for folding of the 129 aa protein CheY resembles that of a smaller protein, CI-2. Fold. Des. 1996;1:43–55. [PubMed] [Google Scholar]
- 18.Cota E., Clarke J. Folding of β-sandwich proteins: three-state transition of a fibronectin type III module. Protein Sci. 2000;9:112–120. doi: 10.1110/ps.9.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schymkowitz J., Rousseau F., Itzhaki L. The folding pathway of the cell-cycle regulatory protein p13suc1: clues for the mechanism of domain swapping. Struct. Fold. Des. 2000;8:89–100. doi: 10.1016/s0969-2126(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 20.Nölting B., Golbik R., Fersht A.R. The folding pathway of a protein at high resolution from microseconds to seconds. Proc. Natl. Acad. Sci. USA. 1997;94:826–830. doi: 10.1073/pnas.94.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowler S.B., Clarke J. Mapping the folding pathway of an immunoglobulin domain: structural detail from ϕ-value analysis and movement of the transition state. Structure. 2001;9:355–366. doi: 10.1016/s0969-2126(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 22.Baryshnikova E.N., Melnik B.S., Bychkova V.E. Three-state protein folding: experimental determination of free-energy profile. Protein Sci. 2005;14:2658–2667. doi: 10.1110/ps.051402705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliezer D., Wright P.E. Is apomyoglobin a molten globule? Structural characterization by NMR. J. Mol. Biol. 1996;263:531–538. doi: 10.1006/jmbi.1996.0596. [DOI] [PubMed] [Google Scholar]
- 24.Gast K., Damaschun H., Damaschun G. Compactness of protein molten globules: temperature-induced structural changes of the apomyoglobin folding intermediate. Eur. Biophys. J. 1994;23:297–305. doi: 10.1007/BF00213579. [DOI] [PubMed] [Google Scholar]
- 25.Griko Y.V., Privalov P.L. Thermodynamic puzzle of apomyoglobin unfolding. J. Mol. Biol. 1994;235:1318–1325. doi: 10.1006/jmbi.1994.1085. [DOI] [PubMed] [Google Scholar]
- 26.Lecomte J.T., Kao Y.H., Cocco M.J. The native state of apomyoglobin described by proton NMR spectroscopy: the A-B-G-H interface of wild-type sperm whale apomyoglobin. Proteins. 1996;25:267–285. doi: 10.1002/(SICI)1097-0134(199607)25:3<267::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 27.Pfeil W. Stability and co-operative properties of partially folded proteins. Biochemistry (Mosc.) 1998;63:294–302. [PubMed] [Google Scholar]
- 28.Jennings P.A., Wright P.E. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura C., Dyson H.J., Wright P.E. The apomyoglobin folding pathway revisited: structural heterogeneity in the kinetic burst phase intermediate. J. Mol. Biol. 2002;322:483–489. doi: 10.1016/s0022-2836(02)00810-0. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura Ch., Dyson H.J., Wright P.E. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol. 2006;355:139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 31.Jamin M., Baldwin R.L. Two forms of the pH 4 folding intermediate of apomyoglobin. J. Mol. Biol. 1998;276:491–504. doi: 10.1006/jmbi.1997.1543. [DOI] [PubMed] [Google Scholar]
- 32.Tsui V., Garcia C., Wright P.E. Quench-flow experiments combined with mass spectrometry show apomyoglobin folds through an obligatory intermediate. Protein Sci. 1999;8:45–49. doi: 10.1110/ps.8.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shakhnovich E., Abkevich V., Ptitsyn O. Conserved residues and the mechanism of protein folding. Nature. 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 34.Ptitsyn O.B., Ting K.-L.H. Non-functional conserved residues in globins and their possible role as a folding nucleus. J. Mol. Biol. 1999;291:671–682. doi: 10.1006/jmbi.1999.2920. [DOI] [PubMed] [Google Scholar]
- 35.Garbuzinskii S.A., Finkelstein A.V., Galzitskaia O.V. [On prediction of folding nuclei in globular proteins] Mol. Biol. (Mosk.) 2005;39:1032–1041. [PubMed] [Google Scholar]
- 36.Samatova E.N., Katina N.S., Finkelstein A.V. How strong are side chain interactions in the folding intermediate? Protein Sci. 2009;18:2152–2159. doi: 10.1002/pro.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jennings P.A., Stone M.J., Wright P.E. Overexpression of myoglobin and assignment of its amide, Cα and Cβ resonances. J. Biomol. NMR. 1995;6:271–276. doi: 10.1007/BF00197808. [DOI] [PubMed] [Google Scholar]
- 38.Harrison S.C., Blout E.R. Reversible conformational changes of myoglobin and apomyoglobin. J. Biol. Chem. 1965;240:299–303. [PubMed] [Google Scholar]
- 39.Jaenicke L. A rapid micromethod for the determination of nitrogen and phosphate in biological material. Anal. Biochem. 1974;61:623–627. doi: 10.1016/0003-2697(74)90429-1. [DOI] [PubMed] [Google Scholar]
- 40.Baryshnikova E.N., Sharapov M.G., Bychkova V.E. Investigation of apomyoglobin stability depending on urea and temperature at two different pH values. Mol. Biol. (Mosc.) 2005;39:292–297. [PubMed] [Google Scholar]
- 41.Parker M.J., Spencer J., Clarke A.R. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J. Mol. Biol. 1995;253:771–786. doi: 10.1006/jmbi.1995.0590. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin R.L. On-pathway versus off-pathway folding intermediates. Fold. Des. 1996;1:R1–R8. doi: 10.1016/S1359-0278(96)00003-X. [DOI] [PubMed] [Google Scholar]
- 43.Fersht A.R. 3rd Ed. W. H. Freeman; New York: 2000. Structure and Mechanism in Protein Science. [Google Scholar]
- 44.Parker M.J., Lorch M., Clarke A.R. Thermodynamic properties of transient intermediates and transition states in the folding of two contrasting protein structures. Biochemistry. 1998;37:2538–2545. doi: 10.1021/bi972459k. [DOI] [PubMed] [Google Scholar]
- 45.Hughson F.M., Baldwin R.L. Use of site-directed mutagenesis to destabilize native apomyoglobin relative to folding intermediates. Biochemistry. 1989;28:4415–4422. doi: 10.1021/bi00436a044. [DOI] [PubMed] [Google Scholar]
- 46.Hargrove M.S., Krzywda S., Olson J.S. Stability of myoglobin: a model for the folding of heme proteins. Biochemistry. 1994;33:11767–11775. doi: 10.1021/bi00205a012. [DOI] [PubMed] [Google Scholar]
- 47.Ptitsyn O.B. Structures of folding intermediates. Curr. Opin. Struct. Biol. 1995;5:74–78. doi: 10.1016/0959-440x(95)80011-o. [DOI] [PubMed] [Google Scholar]
- 48.Vojtechovský J., Chu K., Schlichting I. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys. J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.