

Abstract
Gating-modifier toxins inhibit voltage-gated ion channels by binding the voltage sensors (VS) and altering the energetics of voltage-dependent gating. These toxins are thought to gain access to the VS via the membrane (i.e., by partitioning from water into the membrane before binding the VS). We used serial multiscale molecular-dynamics (MD) simulations, via a combination of coarse-grained (CG) and atomistic (AT) simulations, to study how the toxin VSTx1, which inhibits the archeabacterial voltage-gated potassium channel KvAP, interacts with an isolated membrane-embedded VS domain. In the CG simulations, VSTx1, which was initially located in water, partitioned into the headgroup/water interface of the lipid bilayer before binding the VS. The CG configurations were used to generate AT representations of the system, which were subjected to AT-MD to further evaluate the stability of the complex and refine the predicted VS/toxin interface. VSTx1 interacted with a binding site on the VS formed by the C-terminus of S1, the S1-S2 linker, and the N-terminus of S4. The predicted VS/toxin interactions are suggestive of toxin-mediated perturbations of the interaction between the VS and the pore domain of Kv channels, and of the membrane. Our simulations support a membrane-access mechanism of inhibition of Kv channels by VS toxins. Overall, the results show that serial multiscale MD simulations may be used to model a two-stage process of protein-bilayer and protein-protein interactions within a membrane.
Introduction
Gating-modifier toxins inhibit voltage-gated ion channels by binding the voltage-sensing domain of their target channels and altering the energetics of voltage-dependent gating (1). One such toxin is voltage sensor toxin 1 (VSTx1), a 34-residue peptide toxin from tarantula venom that inhibits the archeabacterial voltage-gated potassium (Kv) channel KvAP by binding the voltage sensors (VS) (2, 3). Gating-modifier toxins such as VSTx1 are small globular proteins with an amphipathic molecular surface. One-half of the surface is predominantly hydrophobic, and the other half consists of polar and charged residues (1, 4). It is thought that VS toxins gain access to the VS via the membrane, i.e., by first partitioning into the headgroup/water interface of the membrane (4, 5, 6, 7, 8). This is consistent with the amphipathic nature of these toxins. Once at the interface, VS toxins diffuse laterally in the plane of the membrane before binding the VS.
Which regions of the VS do VS toxins target? The VS domain of Kv channels is comprised of four putative transmembrane (TM) α-helices, termed S1–S4. The S4 helix, with its array of positively charged residues (primarily arginines), is thought to be the principal voltage-sensing element. Together with the C-terminus of S3 (S3b), the N-terminus of S4 (S4a) forms a helix-turn-helix structural motif termed the VS paddle (9, 10). The paddle motif has been proposed in structural models of Kv channel gating (9, 10, 11, 12) to move ∼15 Å as a semirigid body from the intracellular (IC) to the extracellular (EC) side of the membrane during membrane depolarization to open the pore of the channel (12). Scanning mutagenesis data suggest that VS toxins target the paddle motif (2). When the paddle region (i.e., S3b/S4a) of the KvAP VS was scanned, a combination of polar and hydrophobic residues located primarily on S3b was found to be important for binding VSTx1 (2). Systematic mutagenesis of SGTx1, a closely related gating-modifier toxin that inhibits the Kv2.1 channel, identified a number of polar and hydrophobic residues that are crucial for binding the VS (13). The discoveries of a voltage-sensitive enzyme with a VS domain (14) and a voltage-gated proton channel with a fold similar to that of an isolated VS (15, 16) suggest the VS domain is an evolutionary-conserved, voltage-sensing module (2, 12). The VS domain of KvAP has been shown to form a stable TM protein when in isolation in a lipid bilayer (17, 18). VS toxins, which are known to interact promiscuously with different families of voltage-gated ion channels (1), appear to have evolved to target the VS. It is therefore of considerable interest to better understand how VS toxins interact with the VS.
We recently used molecular-dynamics (MD) simulations to study how VSTx1 interacts with lipid bilayers (19, 20). On the basis of atomistic (AT) simulations over nanosecond timescales, we showed that VSTx1 preferred to be positioned at the headgroup/tail interface of the membrane. Nishizawa and Nishizawa (22) used AT-MD simulations to study how Hanatoxin (HaTx), the first VS toxin to be identified (21), interacts with lipid bilayers. Recent coarse-grained (CG) simulation methodologies (23, 24, 25, 26, 27, 28, 29) permit simulations of larger systems over extended (approximately microsecond/millisecond) timescales. Despite the inherent simplification, CG simulations appear to be robust and can be used to make predictions of protein-bilayer (23, 30, 31, 32, 33) and protein-protein (34, 35) interactions. CG simulations suggest that when VSTx1 interacts in isolation with lipid bilayers, it is thermodynamically favorable (with a ΔG of −26 to −34 kcal/mol depending on the lipid species) for VSTx1 to partition from water into the headgroup/water interface, where the polar and charged residues of the toxin are positioned to interact with the lipid headgroups, with the hydrophobic residues exposed to the lipid tails (20). The amphipathic nature of VS toxins is consistent with the headgroup/tail interface of lipid bilayers, and it has been suggested that a significant fraction of the apparent free energy of VS/toxin binding could be accounted for by the free energy of toxin partitioning into the membrane (5), especially if the VS/toxin interface consists of only several residue pairs (2).
In this study, we performed serial multiscale MD simulations (36) with explicit membrane and solvent CG and AT simulations to investigate how VSTx1, initially located in water and outside the membrane, interacts with an isolated membrane-embedded VS molecule of KvAP. We investigated the mechanism by which VSTx1 binds the VS, and whether a combination of CG and AT simulations can be used to predict the VS/toxin interface. Thus, CG simulations were used to model the longer-timescale dynamics of membrane partitioning and eventual VS binding, and AT simulations were used to refine the predicted VS/toxin interface. Our results suggest that VSTx1 binds to one major site on the VS of KvAP, at least when the toxin interacts with the VS in isolation (i.e., without the pore domain). The VS/toxin interface as derived from CG and AT simulations is consistent and is suggestive of toxin-mediated perturbations of the interaction between the VS and the pore domain of Kv channels, and the membrane.
Materials and Methods
CG-MD simulations
We modeled a KvAP VS molecule in a palmitoyl-oleoyl phosphatidylcholine (POPC) membrane using a self-assembly protocol (30). A VS molecule was placed in the center of a simulation box with 549 randomly placed POPC lipids. A lipid bilayer with dimensions ∼135 × 135 Å2 formed with the VS adopting a TM orientation, consistent with AT (17) and CG (37) simulations of the isolated VS, and a model of KvAP in lipid bilayers (38). One molecule of VSTx1 was placed in the EC solvent, ∼25 Å from the membrane surface (Fig. 1). Five repeat, unrestrained CG-MD simulations (each of 3 μs duration, with different initial velocities) were performed. For the CG simulations, we used a local modification of the MARTINI force field, which was originally developed for semiquantitative lipid simulations (24) and subsequently extended to protein simulations (23, 39), in a similar spirit to other recent MARTINI developments (25, 26, 27, 28). It is based on a 4-to-1 mapping of heavy atoms, with each amino acid modeled with one CG backbone particle, and one to two CG side-chain particle(s) depending on the residue size. The CG protein model has been parameterized on experimental partitioning free energies of side-chain analogs (23), and successfully used to predict the orientation of membrane proteins in lipid bilayers (30). We used AT coordinates from the Protein Data Bank (KvAP VS, PDB code: 1ORS (40); VSTx1, PDB code: 1S6X (4)) to build the CG VS and toxin models. The seventh member of the NMR-derived ensemble of VSTx1 structures was used because it was annotated as being the most representative conformer in the ensemble (4). Protein secondary and tertiary structures were maintained in the CG simulations by means of an elastic network model utilizing a distance cutoff of 7 Å (force constant of 10 kJ mol−1 Å−2). Further details of the CG parameters are described in Bond et al. (23) and Marrink et al. (24).
Figure 1.
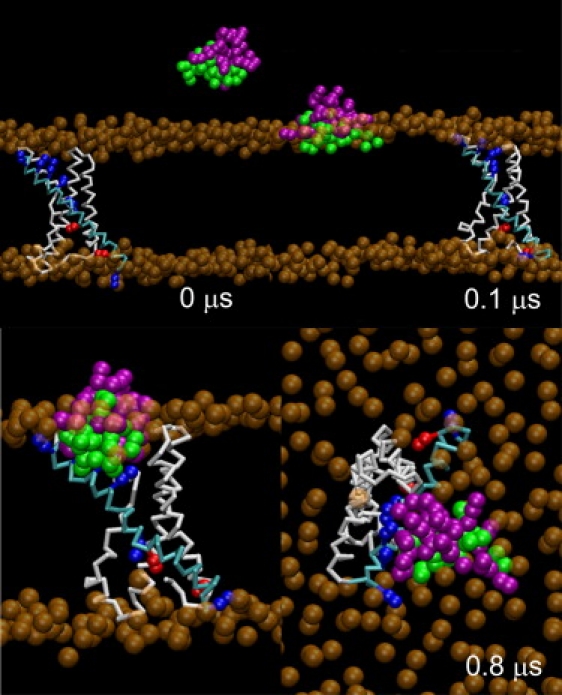
Snapshots from CG simulation 5. VSTx1 partitioned from water into the headgroup/water interface of the POPC bilayer before binding the isolated VS. The backbone of helices S1–S3 and S4 of the VS are colored white and cyan, respectively, with Arg and Lys side chains of S4 shown in blue and red, respectively. The polar and charged, and hydrophobic residues of VSTx1 are colored purple and green, respectively. POPC lipid phosphates are colored orange. All other particles are omitted for clarity. The snapshots at 0.8 μs depict side (left) and EC (right) views.
AT-MD simulations
We converted the CG systems to an AT representation using a fragment-based approach (P. J. Stansfeld and M. S. P. Sansom, unpublished results). We clustered each CG trajectory over 1–3 μs (the equilibrated period, i.e., after VSTx1 had partitioned into the membrane and bound the VS; see Results) using the full linkage algorithm (41). Only the CG backbone particles of the VS and toxin were clustered. The median of the cluster, which corresponds to system with the most frequently observed VS/toxin complex, was converted to AT. To convert the CG lipid to an AT model, energy-minimized AT lipid fragments were aligned to the CG particles, and the missing atoms between each particle were extracted and appended to the CG lipids. The AT lipids were then energy-minimized to remove any clashes that resulted from the conversion. We used Pulchra (42) to build initial AT models of each peptide using the CG protein backbone and side-chain coordinates as the template. Modeler v9 (43) was used to optimize secondary structure in the AT models, on the basis of the experimental structures (4, 40). Each converted system was subjected to 1 ns equilibration AT-MD, with positional restraints (force constant of 10 kJ mol−1 Å−2) applied on the VS and toxin to allow the lipids and solvent to relax around the complex. This was followed by an unrestrained production simulation of 20 ns duration. AT simulations utilized the GROMOS96 force field (44) and Berger parameters for POPC lipids (45, 46).
Simulation protocols
MD simulations were performed using GROMACS 3.3.3 (http://www.gromacs.org). The AT systems were solvated with SPC waters (47). Cl− counterions were added to keep all systems electrically neutral. In the AT simulations, electrostatic interactions were calculated using the particle mesh Ewald method (48), employing a grid spacing of ∼1 Å−1 and an interpolation order of 4. A cutoff of 12 Å was used for the real space portion of the Ewald sum and the Lennard-Jones interactions. The LINCS algorithm (49) was used to constrain all covalent bonds, and the SETTLE algorithm (50) was used to maintain the geometry of the water molecules. In the CG simulations, electrostatic interactions utilized a relative dielectric constant of 20, which was smoothly shifted to zero between 0 and 12 Å, and van der Waals interactions were smoothly shifted to zero between 9 and 12 Å (24). Each system was temperature-coupled with a Berendsen thermostat (51) to a reference temperature of 310 K, with a coupling constant of 0.1 and 1.0 ps in AT and CG, respectively. Semiisotropic pressure coupling in x and y (membrane normal along z) at 1 bar with a coupling constant of 1.0 ps and a compressibility value of 4.6 × 10−5 and 5.0 × 10−6 bar−1 in AT and CG, respectively, was used. Ionizable side chains were kept in the default protonation (i.e., charged) state for pH 7.
Results
CG simulations of the encounter between the toxin and VS
The interaction between the toxin and the lipid bilayer was monitored by measuring the distance between the centers of mass (COMs) of VSTx1 and the POPC bilayer, as projected onto the bilayer normal (the bilayer center is at ∼0 Å; Fig. 2 A). As anticipated from previous simulation studies (20), VSTx1 partitioned into the headgroup/water interface of the membrane, at a distance of 19–21 Å from the bilayer center within the first 0.2 μs, where it remained until the end of each simulation. While VSTx1 was located at the water/bilayer interface, its polar and charged residues were exposed to lipid headgroups and waters, with the hydrophobic residues directed toward the lipid tails.
Figure 2.
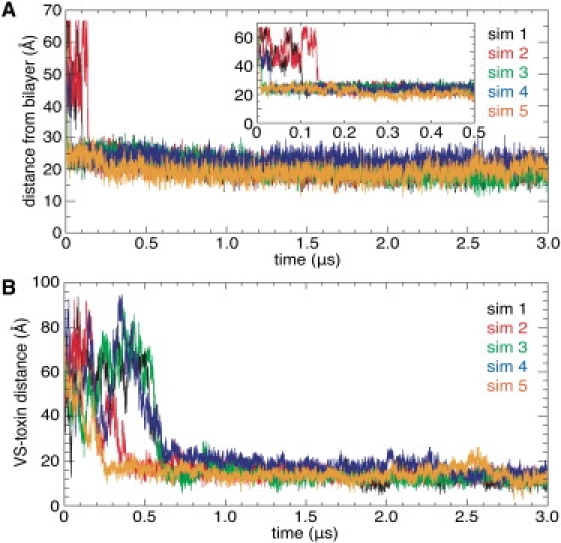
(A) Distance between the COMs of VSTx1 and the POPC bilayer projected along the bilayer normal in the CG simulations. The inset shows the distances over the first 0.5 μs. (B) Distance between the COMs of VSTx1 and VS projected along the plane of the membrane surface in the CG simulations.
To follow the encounter between the toxin and the VS, we measured the distance between the COMs of VSTx1 and the VS projected onto the plane of the membrane (Fig. 2 B). VSTx1 did not encounter the VS immediately upon binding to the membrane in any of the simulations. Instead, the toxin diffused laterally within the plane of the membrane for up to ∼0.6 μs after binding to the membrane/water interface, before binding to the VS. Each VS/toxin complex stayed intact for the remainder of the simulation.
To characterize the VS/toxin interface, we calculated the frequency of contacts between VS and toxin residues over 1–3 μs of each simulation (i.e., after VSTx1 binds the VS). We used a distance cutoff of 7 Å to define a contact, as in recent CG simulations of protein/protein docking (34). In simulations 1, 3, and 5, the pattern of contacts between the VS and toxin were similar in that VSTx1 interacted predominantly with VS residues on the C-terminus of S1, the S1-S2 linker, and S4a (Fig. 3). The VS/toxin interface was comprised of a combination of charged and hydrophobic residues. The COMs of the VS and toxin in the complex, projected along the plane of the membrane, were 13–14 Å apart (Fig. 2 B; simulations 1, 3, and 5).
Figure 3.
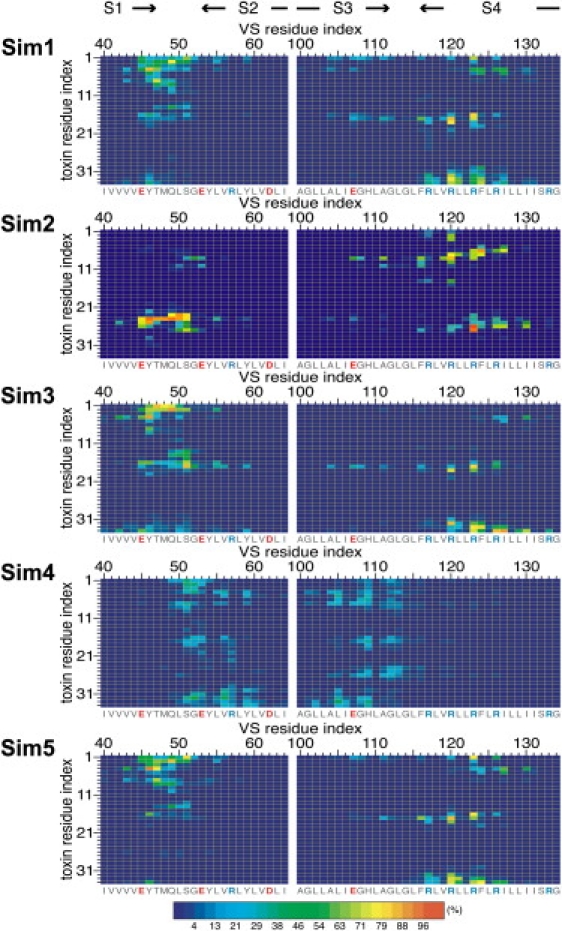
Analyses of the VS/toxin interface in the CG simulations, showing the frequency of contacts (expressed as a percentage) between VS and toxin residues over 1–3 μs (i.e., after VSTx1 binds the VS). A distance cutoff of 7 Å was used to define a contact. Basic and acidic residues are labeled in blue and red, respectively.
In simulation 2, the VS residues that contacted VSTx1 were similar to those in simulations 1, 3, and 5, but the residues on the toxin were different. In contrast, in simulation 4, VSTx1 interacted with a different region of the VS corresponding to the S1-S2 linker, the N-terminus of S2, and S3b. The frequencies of interactions in simulation 4 were substantially lower than in the other simulations, and the distance between the COMs of the VS and toxin (∼17 Å; Fig. 2 B) was higher than in the other simulations (∼14 Å), indicative of weaker interactions. Of interest, in simulation 4 the toxin did not bind as deeply into the membrane (∼23 Å vs. ∼19 Å in simulations 1–3 and 5; Fig. 2 A). Overall, this suggests that in simulation 4, the toxin and VS domain did not form a stable complex.
AT simulations of the toxin/sensor complex
To refine the toxin/sensor complexes and compare the complexes from the various CG simulations, we converted the complexes and the lipid bilayer to AT resolution and used the resulting configurations as the starting point for relatively short (20 ns) AT-MD simulations. We monitored the structural stability of the AT models of the VS and toxin by calculating the root mean-squared deviation (RMSD) of the Cα atoms of the complex and its components (i.e., the VS and the toxin) with respect to the initial structure (i.e., at 0 ns; see Fig. S1 in the Supporting Material). The RMSDs of the complexes did not exceed ∼3.5 Å, and those of the VS and toxin individually did not exceed ∼3.5 Å and ∼3 Å, respectively. Thus, there were no large changes in conformation over the course of the 20 ns simulations, indicating that the CG-to-AT conversion had resulted in locally stable structures.
Visualization of the complexes (Fig. 4) and analysis of the toxin-sensor COM distances (Fig. S2) suggest two types of behavior. For simulations 1–3 and 5, the distance is fairly constant at ∼16 Å for the duration of the simulation. In contrast, for simulation 4, the toxin/sensor distance is up to ∼28 Å over the course of the simulation. Visualization of simulation 4 does not show complete dissociation of the complex. However, examination of residue-residue contacts for simulation 4 reveals almost none closer than 3.5 Å (Fig. S3). Thus, we may conclude that the complex formed in CG simulation 4 is relatively unstable, and we may exclude it from further consideration.
Figure 4.
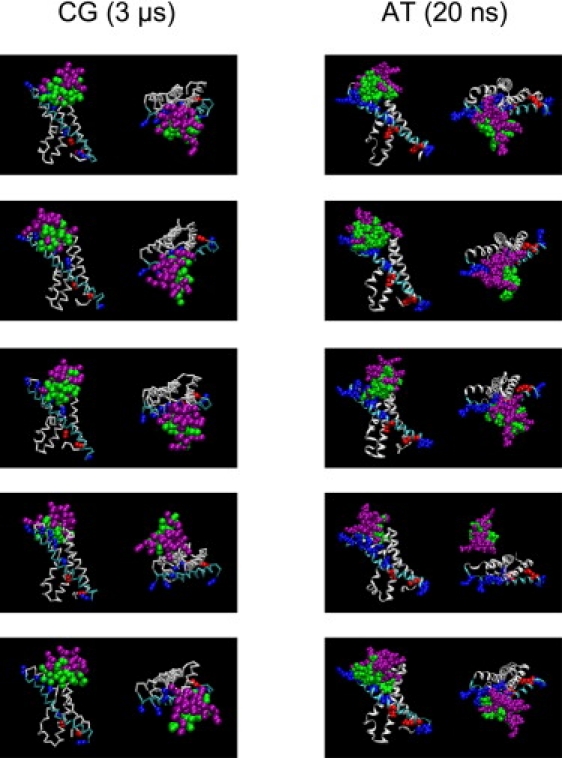
Snapshots of the VS/VSTx1 complex in the CG and AT simulations. (top to bottom: simulations 1 to 5) Views from the side of the membrane and from the EC solvent are provided. The color scheme identical is to that in Fig. 1. The lipid bilayer is not shown. In simulations 1– 3 and 5, the toxin bound to the C-terminus of S1, the S1-S2 linker, and S4a of the VS.
Turning to the remaining four AT-MD simulations (simulations 1–3 and 5), secondary structure analyses of the VS over the time course of each simulation showed that helices S1, S2, S3b, and S4 remained α-helical, whereas S3a showed some degree of flexibility (Fig. S4). In each case, the VS remained embedded in the membrane, with evidence of local membrane deformation around the S4 helix and the formation of water-filled crevices in the VS, consistent with previous simulations of the KvAP VS in lipid bilayers (17, 37). In each of the four simulations, the toxin remained at the bilayer headgroup/water interface (∼20 Å from the bilayer center) and the VS/toxin complex remained intact, with inter-COM distances of 12–16 Å (Fig. S2).
We analyzed the VS/toxin interface by calculating the frequency of contacts (using a distance cutoff of 3.5 Å) between VS and toxin residues over the latter half (i.e., 10–20 ns) of each of the four simulations (Fig. 5). VSTx1 shows a consistent pattern of contacts with VS residues on the C-terminus of S1, the S1-S2 linker, and S4a. On S3b, the toxin interacted with E107 in simulations 1 and 2. Overall, the pattern of contacts observed in the AT simulations was consistent with those in the preceding CG simulations.
Figure 5.
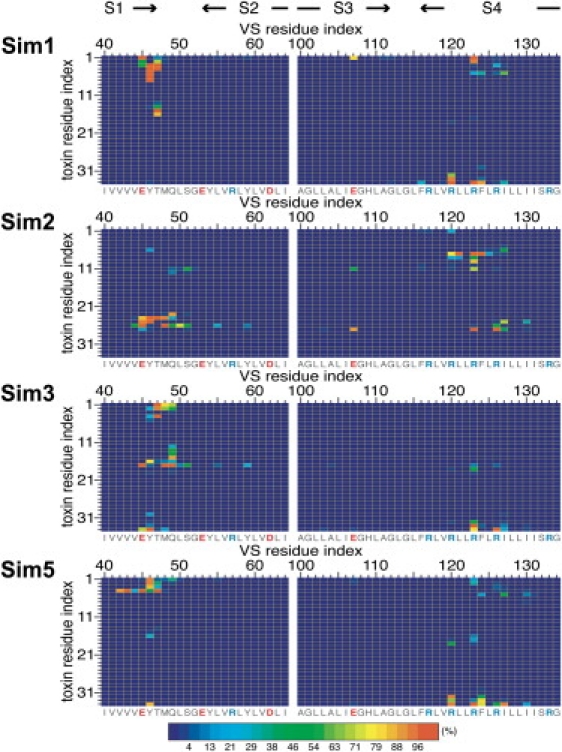
Analyses of the VS/toxin interface in AT simulations 1–3 and 5, showing the frequency of contacts (expressed as a percentage) between VS and toxin residues over 10–20 ns. A distance cutoff of 3.5 Å was used to define a contact. Basic and acidic residues are labeled in blue and red, respectively.
An example of a refined VS/VsTX1 complex structure (the 20 ns snapshot of simulation 5) is shown in Fig. 6. It can be seen that the VS and toxin locations within the bilayer (Fig. 6 A) are the same as those observed in simulations of the isolated components (17, 19, 20, 37, 52), and that the toxin contacts S1/S2 and S4. The toxin binds between the C-terminus of S1 and the S3b-S4a paddle (Fig. 6 B). This is somewhat in contrast to the proposal of Alabi and Swartz (53) based on alanine-scanning experiments, but appears to be broadly consistent with more recent data (54), as discussed in more detail below.
Figure 6.
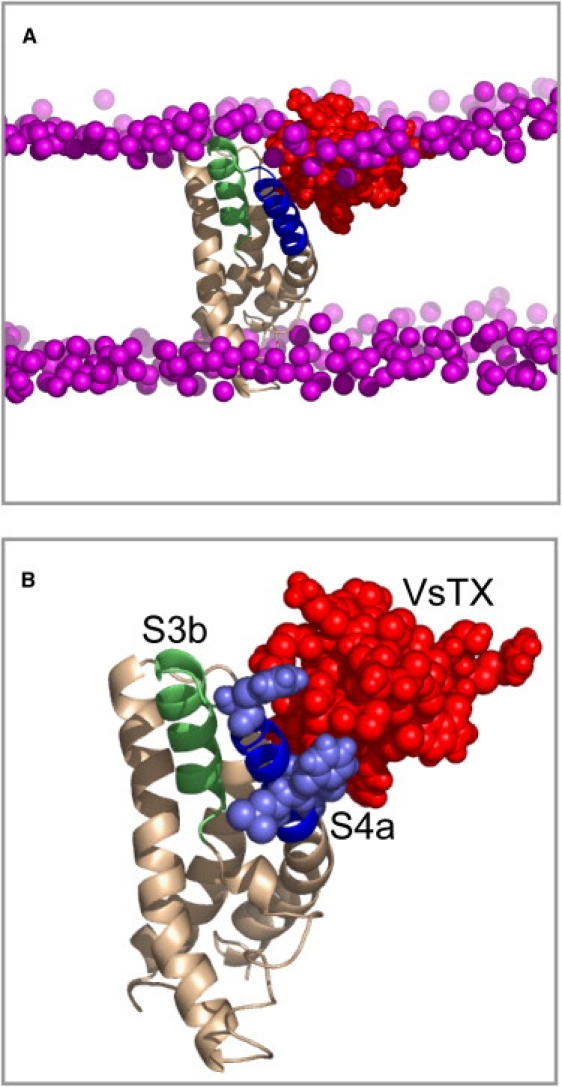
Example of a refined VS/VsTX1 complex structure, showing the 20 ns snapshot of simulation 5. (A) Complex in a bilayer showing the VS in beige (with the S3b and S4a helices of the paddle in green and blue, respectively), VsTx1 in red, and the phosphorus atoms of the lipid headgroups in purple. (B) View of the complex with the consensus interaction side chains of S4a (i.e., consensus between simulation and experiment; see text for details) in pale blue in space-filling format.
Discussion and Conclusions
In this work, we combined CG and AT simulations serially to study how a VS toxin, initially located in water, binds to a membrane-embedded VS domain of KvAP. The CG simulations showed VSTx1 partitioning from water into the headgroup/water interface of the membrane (20) before binding to the VS. An interfacial location for VSTx1 is consistent with a variety of experimental data (4, 5, 6, 7) and with free-energy calculations of toxin-membrane interactions (20). Using a CG simulation as a starting point, we performed multinanosecond AT simulations to further evaluate the stability of the toxin when bound to the membrane-embedded VS, and to refine the interactions at the VS/toxin interface.
In four of the five simulations (simulations 1–3 and 5), we see a consistent pattern of interactions between the toxin and the VS involving the C-terminus of S1, the S1-S2 linker and S3b, and especially S4a of the voltage-sensing paddle. In particular, the first four arginines of S4a (R117, R120, R123, and R126) interacted with the toxin. This differs somewhat from the earlier results of alanine-scanning mutagenesis of the S3b-S4a paddle (2), but is in broad consensus with more recent data (54).
It is important to consider the origin of the differences between the contacts between toxin and the isolated VS identified in the simulations, and the residues that result in functional perturbations in alanine-scanning mutagenesis (2). There are several possibilities. It could be that the CG method does not reveal the physiologically relevant interaction. However, it has been shown that CG simulations position proteins correctly relative to a lipid bilayer (30), and that they can reveal aspects of protein-protein interactions. For example, CG-MD revealed correctly the two interaction modes of the cohesin/dockerin complex (34). Furthermore, when CG simulations generated an unstable mode of interaction of the toxin and VS (i.e., simulation 4), this was revealed as such in the subsequent AT-MD simulations. Alternatively, one might consider possible complications in the interpretation of the alanine-scanning data for the S3b-S4a paddle (53). For example, the effects of the mutations could be indirect (i.e., allosteric) in the intact channel protein, rather than revealing directly the accessibility of residues. However, a more likely possibility is that the environment (broadly construed) of the VS may be linked to the mode of interaction of VsTX1. In this regard, we would include considerations of the isolated VS domain (in this study, and also recently characterized experimentally (18)) versus the intact channel (which can adopt different conformational/gating states), although it is known that the toxin binds to isolated VS experimentally (3), as well as the nature (both chemical and physical) of the lipid bilayer, which is known to modulate Kv channel gating (8, 55, 56). We noted that two or three lipid molecules were able to bridge the toxin/VS complex in channel snapshots. It is therefore of considerable interest that recent experiments (54) suggest that lipids play a key role in VS paddle-toxin interactions.
A consensus view would perhaps favor the third (environmental) explanation, indicating the need for further simulation studies of toxin-VS interaction as a function of the channel state and bilayer composition. In particular, by comparing those residues in the KvAP S4a that show significant coupling between the effects of channel mutations and lipid modifications (54) with those that show significant toxin/sensor contacts during the simulations (Fig. 5), one can arrive at four consensus residues in S4a (F116, F124, L125, and I127) that are implicated by both experiment and simulations (Fig. 6 B). Significantly, these consensus residues of S4a interact not only with the toxin, but also with lipid molecules during the AT-MD simulations.
It might be argued that the proposed docking surface of the VS is not likely to be available to a toxin in the membrane when the VS forms part of the intact ion channel, as a comparison with the x-ray structure of Kv1.2/2.1 (PDB code: 2R9R (57)) suggests that this surface would be largely hidden from the surrounding membrane by the pore domain. However, given the ability of the isolated VS domain to exist as an independent, stable TM protein (18), it is important to model toxin interactions with the isolated VS. Furthermore, it is likely that the packing of the VS domain and the pore domain of Kv channels in the membrane differs from that in the crystal structure, depending on the functional state of the channel.
Thus, we may conclude that the pore domain and the tetrameric arrangement of monomers in KvAP may place additional constraints on toxin access to the VS. Indeed, if we superimpose the VS of the VS/VSTx1 complex seen in simulations 1–3 and 5 on the VS domain of the Kv1.2/2.1 chimera (PDB code: 2R9R (57)), the toxin sits between the VS and the pore domain, partly overlapping in space with the latter. This indicates that toxin binding could perturb the VS-pore interaction, thus altering the gating properties of the channel. Recent experimental studies have shown that the membrane environment is important for Kv channel gating (8, 55, 56). It has been proposed that VSTx1 inhibits Kv channels by perturbing the interaction forces that exists between the channel and the membrane (8). This is consistent with our simulations, which support a membrane-access mechanism of inhibition. Overall, our results suggest that VSTx1 targets the S4a helix of the VS paddle motif of KvAP, and the VS/toxin interface is made up of both charged and hydrophobic residues.
More generally, serial multiscale MD simulations (36), with initial CG simulations (which are well sampled but use an approximate force field) subsequently refined by AT simulations (where sampling is more of an issue, but for which the force field is more accurate), may be used to model the binding of a small peptide toxin to the VS of a Kv channel, and to make predictions about the subsequent complex. It may be of interest to use such a multiscale approach to simulate the whole channel (i.e., with the pore domain) with the toxin bound.
Acknowledgments
We thank our colleagues in the Structural Bioinformatics and Computational Biochemistry Unit (Oxford University, Oxford, UK), especially Phillip Stansfeld.
C.L.W. is funded by the Oxford Centre for Integrative Systems Biology. Research in the M. S. P. Sansom laboratory is funded by the Biotechnology and Biological Sciences Research Council and the Wellcome Trust.
Editor: Peter C. Jordan.
Footnotes
Four figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00089-5.
Supporting Material
References
- 1.Swartz K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49:213–230. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alabi A.A., Bahamonde M.I., Swartz K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 2007;450:370–375. doi: 10.1038/nature06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruta V., MacKinnon R. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry. 2004;43:10071–10079. doi: 10.1021/bi049463y. [DOI] [PubMed] [Google Scholar]
- 4.Jung H.J., Lee J.Y., Kim J.I. Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry. 2005;44:6015–6023. doi: 10.1021/bi0477034. [DOI] [PubMed] [Google Scholar]
- 5.Lee S.Y., MacKinnon R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature. 2004;430:232–235. doi: 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- 6.Phillips L.R., Milescu M., Swartz K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 2005;436:857–860. doi: 10.1038/nature03873. [DOI] [PubMed] [Google Scholar]
- 7.Milescu M., Vobecky J., Swartz K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007;130:497–511. doi: 10.1085/jgp.200709869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt D., MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. USA. 2008;105:19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swartz K.J. Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 2004;5:905–916. doi: 10.1038/nrn1559. [DOI] [PubMed] [Google Scholar]
- 10.Jiang Y., Ruta V., MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 11.Ruta V., Chen J., MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 12.Swartz K.J. Sensing voltage across lipid membranes. Nature. 2008;456:891–897. doi: 10.1038/nature07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J.M., Roh S.H., Swartz K.J. Molecular surface of tarantula toxins interacting with voltage sensors in K(v) channels. J. Gen. Physiol. 2004;123:455–467. doi: 10.1085/jgp.200309005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murata Y., Iwasaki H., Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 15.Ramsey I.S., Moran M.M., Clapham D.E. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki M., Takagi M., Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 17.Sands Z.A., Sansom M.S.P. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure. 2007;15:235–244. doi: 10.1016/j.str.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krepkiy D., Mihailescu M., Swartz K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bemporad D., Wee C.L., Sansom M.S.P. VSTx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry. 2006;45:11844–11855. doi: 10.1021/bi061111z. [DOI] [PubMed] [Google Scholar]
- 20.Wee C.L., Gavaghan D., Sansom M.S.P. Lipid bilayer deformation and the free energy of interaction of a Kv channel gating-modifier toxin. Biophys. J. 2008;95:3816–3826. doi: 10.1529/biophysj.108.130971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swartz K.J., MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 22.Nishizawa M., Nishizawa K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006;35:373–381. doi: 10.1007/s00249-006-0044-z. [DOI] [PubMed] [Google Scholar]
- 23.Bond P.J., Wee C.L., Sansom M.S.P. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry. 2008;47:11321–11331. doi: 10.1021/bi800642m. [DOI] [PubMed] [Google Scholar]
- 24.Marrink S.J., de Vries A.H., Mark A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B. 2004;108:750–760. [Google Scholar]
- 25.Monticelli L., Kandasamy S.K., Marrink S.J. The MARTINI coarse grained force field: extension to proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 26.Shih A.Y., Arkhipov A., Schulten K. Coarse grained protein-lipid model with application to lipoprotein particles. J. Phys. Chem. B. 2006;110:3674–3684. doi: 10.1021/jp0550816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shelley J.C., Shelley M.Y., Klein M.L. A coarse grain model for phospholipid simulations. J. Phys. Chem. B. 2001;105:4464–4470. [Google Scholar]
- 28.Izvekov S., Voth G.A. A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B. 2005;109:2469–2473. doi: 10.1021/jp044629q. [DOI] [PubMed] [Google Scholar]
- 29.Voth G.A. CRC Press; Boca Raton, FL: 2008. Coarse-Graining of Condensed Phase and Biomolecular Systems. 456 p. [Google Scholar]
- 30.Scott K.A., Bond P.J., Sansom M.S.P. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure. 2008;16:621–630. doi: 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Bond P.J., Holyoake J., Sansom M.S. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 2007;157:593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Periole X., Huber T., Sakmar T.P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- 33.Reynwar B.J., Illya G., Deserno M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 2007;447:461–464. doi: 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- 34.Hall B.A., Sansom M.S.P. Coarse-grained MD simulations and protein-protein interactions: the cohesin-dockerin system. J. Chem. Theory Comput. 2009;5:2465–2471. doi: 10.1021/ct900140w. [DOI] [PubMed] [Google Scholar]
- 35.Periole X., Cavalli M., Ceruso M.A. Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 2009;5:2531–2543. doi: 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- 36.Ayton G.S., Noid W.G., Voth G.A. Multiscale modeling of biomolecular systems: in serial and in parallel. Curr. Opin. Struct. Biol. 2007;17:192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Bond P.J., Sansom M.S.P. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc. Natl. Acad. Sci. USA. 2007;104:2631–2636. doi: 10.1073/pnas.0606822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S.Y., Lee A., MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bond P.J., Sansom M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang Y., Lee A., MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 41.Krznaric D., Levcopoulos C. Fast algorithms for complete linkage clustering. Discrete Comput. Geom. 1998;19:131–145. [Google Scholar]
- 42.Rotkiewicz P., Skolnick J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008;29:1460–1465. doi: 10.1002/jcc.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 44.van Gunsteren W.F., Kruger P., Tironi I.G. Biomos & Hochschulverlag AG an der ETH Zurich; Groningen/Zurich: 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. [Google Scholar]
- 45.Berger O., Edholm O., Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marrink S.J., Berger O., Jähnig F. Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations. Biophys. J. 1998;74:931–943. doi: 10.1016/S0006-3495(98)74016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hermans J., Berendsen H.J.C., Postma J.P.M. A consistent empirical potential for water-protein interactions. Biopolymers. 1984;23:1513–1518. [Google Scholar]
- 48.Darden T., York D., Pedersen L. Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 49.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 50.Miyamoto S., Kollman P.A. Settle—an analytical version of the Shake and Rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 51.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 52.Wee C.L., Bemporad D., Sands Z.A., Gavaghan D., Sansom M.S.P. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007;92:L07–L09. doi: 10.1529/biophysj.106.098681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alabi A.A., Swartz K.J. VSTx1 stabilizes a closed state in K(v)2.1-K(v)AP paddle chimeras. Biophys. J. 2007;S:471A. [Google Scholar]
- 54.Milescu M., Bosmans F., Swartz K.J. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt D., Jiang Q.X., MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt D., Cross S.R., MacKinnon R. A gating model for the archeal voltage-dependent K(+) channel KvAP in DPhPC and POPE:POPG decane lipid bilayers. J. Mol. Biol. 2009;390:902–912. doi: 10.1016/j.jmb.2009.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.