

Abstract
Human telomere contains guanine-rich (G-rich) tandem repeats of single-stranded DNA sequences at its 3′ tail. The G-rich sequences can be folded into various secondary structures, termed G-quadruplexes (G4s), by Hoogsteen basepairing in the presence of monovalent cations (such as Na+ and K+). We developed a single-molecule tethered particle motion (TPM) method to investigate the unfolding process of G4s in the human telomeric sequence AGGG(TTAGGG)3 in real time. The TPM method monitors the DNA tether length change caused by formation of the G4, thus allowing the unfolding process and structural conversion to be monitored at the single-molecule level. In the presence of its antisense sequence, the folded G4 structure can be disrupted and converted to the unfolded conformation, with apparent unfolding time constants of 82 s and 3152 s. We also observed that the stability of the G4 is greatly affected by different monovalent cations. The folding equilibrium constant of G4 is strongly dependent on the salt concentration, ranging from 1.75 at 5 mM Na+ to 3.40 at 15 mM Na+. Earlier spectral studies of Na+- and K+-folded states suggested that the spectral conversion between these two different folded structures may go through a structurally unfolded intermediate state. However, our single-molecule TPM experiments did not detect any totally unfolded intermediate within our experimental resolution when sodium-folded G4 DNA molecules were titrated with high-concentration, excess potassium ions. This observation suggests that a totally unfolding pathway is likely not the major pathway for spectral conversion on the timescale of minutes, and that interconversion among folded states can be achieved by the loop rearrangement. This study also demonstrates that TPM experiments can be used to study conformational changes in single-stranded DNA molecules.
Introduction
Telomeres, the ends of chromosomes, generally consist of many tandem repeats of guanine-rich (G-rich) motifs, such as the hexameric repeats of TTAGGG/CCCTAA in vertebrate telomeres (1). Of special interest is the finding that a short 3′-end single-stranded overhang of telomeres containing a G-rich sequence can adopt G-quadruplex (G4) structures using Hoogsteen hydrogen bonding of G nucleotides under physiological conditions both in vitro and in vivo (2,3). More importantly, folding of telomeric DNA into G4 structures has been proposed to protect the chromosome ends (4) and to inhibit telomerase activity in cancer cells (5,6). Therefore, the telomere and telomerase have become targets for anticancer therapy, and knowledge of the telomeric structure is essential for the development of anticancer drugs. However, the structural diversity of G4s has posed a problem for structural analysis. For example, the G4 structure of human telomeric sequence d[AG3(T2AG3)3] (h22) in K+ solution remains undetermined. Although several groups have attempted to modify the original sequence to obtain a single G4 structure (7–9), different sequences may lead to very different structures. Careful studies on the rich diversity of the G4 conformations and the possible conversion of the G4 structures are required.
Recent studies based on circular dichroism (CD) measurements showed a spectral conversion of CD band signatures of a Na+-containing folded G4 structure to that of a K+-containing structure upon addition of K+ ions (7,9,10). Considering the basket-type form of h22 in Na+ solution determined by NMR (11), and the possibly mixed type-I folded form inferred from experiments in K+ solution (7–9), several studies (7,9,12,13) suggested a mechanism by which the spectral conversion results from an actual structural conversion through the switch of the orientation on the 5′- or 3′-end GGG strand from the basket-type form to a mixed type-I form and mixed type-II form, respectively. Scheme 1 shows the proposed structural conversion mechanism responsible for the spectral conversion. On the other hand, the CD and ligand binding-induced CD studies carried out by Chang et al. (10) suggested that the spectral conversion of d[(T2AG3)4] (h24) occurs on a fast timescale (on the order of seconds). In contrast to the structural conversion model, where conversion between different types of the folded G4 structures requires the unfolding intermediate, Chang et al. proposed a loop rearrangement mechanism for the spectral conversion. In their model, the spectral change of h24 is attributed to the fast ion exchange resulting in different loop-base interactions and various hydrogen-bonding effects upon K+ titration (10).
Scheme 1.
Is there any direct evidence that an unfolding intermediate, a prediction of the structural conversion model, accompanies the spectral conversion process? The structural morphology of G4s has been proposed to play a critical role in their biological function (14–16). Understanding the folding and unfolding of G4s and their structural flexibility is essential not only for elucidating the biological role of G4s, but also for designing new anticancer drugs.
In contrast to conventional ensemble-averaged measurements, single-molecule methods can be used to measure the time course of individual molecules during a biochemical process and provide novel information about the reaction mechanisms. The tethered particle motion (TPM) method measures the DNA length change by monitoring the Brownian motion (BM) of beads attached to individual DNA molecules tethered at the coverglass surface (17). TPM has been used to study the length change of duplex DNA molecules resulting from DNA-modifying enzymes (18–22) or from DNA looping (23). In this work, we developed a novel (to our knowledge) single-molecule TPM experiment based on single-stranded DNA (ssDNA) molecules. Since the structural conversion between the folded and unfolded states of human telomeric sequence h22 leads to a DNA tether length change, it is feasible to use TPM experiments to directly monitor the structural conversion process in real time. Here, we demonstrate that TPM experiments can distinguish between the folded and unfolded states of single-stranded G4 substrates. Upon addition of antisense DNA, TPM is able to detect the unfolding process of h22-containing DNA in real time. Furthermore, our TPM experiments indicate that a totally unfolding intermediate is not observed in most traces upon Na/K ion exchange at our experimental resolution, which suggests that an unfolding intermediate is likely not the major pathway of interconversion among G4 structures.
Materials and Methods
DNA preparation
A 113 nt long ssDNA (h22L) containing human telomeric sequence was labeled with digoxigenin at its 5′ end. First, a 93 nt 5′ phosphate-labeled ssDNA containing 22 nt human telomeric DNA (AG3(T2AG3)3, h22), 48 nt CA repeat linker ((CA)24) to avoid any secondary structure, and two flanking ends for annealing (23 nt; 5′-phosphate-ATGTGTAACAG(CA)24 AG3(T2AG3)3ATAGTGCCTCTT-3′, purified by high-performance liquid chromatography) was purchased from Bio Basic (Ontario, Canada). This 93 nt oligo was then T4 ligased (New England Biolabs, Ipswich, MA) with a p9 primer (20 nt, 5′-digoxigenin-CGT CAC CCT GGA TGC TGT AG-3′) with a p81 sprinter (35 nt, TGT GCT GTT ACA CAT CTA CAG CAT CCA GGG TGA CG-3′) to generate a 113 nt long template (h22L) with a 5′-digoxigenin label. The h22L template was amplified using a forward primer p9 (digoxigenin labeled) and a reversed primer p80 (18 nt, 5′-phosphate-AAG AGG CAC TAT CCC TAA-3′) in a 30-cycle polymerase chain reaction (PCR) with a mixture of Phusion and Vent DNA polymerases (New England Biolabs). The double-stranded DNA products from PCR were cleaned up (PCR purification kit; Qiagen, Valencia, CA) and then subjected to λ-exonuclease digestion (New England Biolabs) at 37°C for 90 min to remove the 5′-phosphate-labeled strands. The digested ssDNA products were verified by 2% agarose gel electrophoresis and gel-extracted (gel extraction kit; Qiagen) for future use. Before single-molecule TPM experiments were conducted, the purified h22L was annealed with a 5′-biotin-labeled oligo3 (12 nt, 5′-biotin-AAG AGG CAC TAT-3′) to allow the attachment of streptavidin-coated latex beads. The sequence of antisense c12 oligo (5′-CCC TAA CCC TAA-3′) is complementary to h22, whereas that of t16 oligo (5′-TTT TTT TTT TTT TTT T-3′) is not, and h24 has a sequence of 5′-(TTAGGG)4-3′. All primers were purchased from Bio Basic.
Slide preparation
Coverglasses (24 × 60 mm, #1.5; Marienfeld, Lauda-Königshofen, Germany) were sonicated in 2 M KOH for 15 min, followed by sonication in ethanol for 10 min, then in MillQ water, and finally dried by nitrogen gas. The flow chambers were made of a long coverglass and a smaller coverglass (22 × 22 mm) sealed by heated paraffin wax to make three chambers on one slide (enabling three experiments). All buffers used were filtered with a 0.22 μm filter (Millex GS; Millipore, Cork, Ireland). Then 20 μg/mL of antidigoxigenin were incubated for 30 min at room temperature followed by blocking buffer (pH = 7.4, 10 mM Tris-HCl, 2 mg/mL bovine serum albumin, in the presence of 150 mM NaCl, KCl, LiCl, if applicable and specified in each experiment) incubation for 15 min. Annealed 1 nM h22L (113 nt) was flown onto the reaction chamber for 1 h incubation at room temperature. After any free DNA substrates were washed away by reaction buffer (10 mM Tris-HCl, 2 mg/mL bovine serum albumin, 150 mM NaCl or KCl, 0.1% Tween-20, pH = 7.4), the chamber was ready for imaging.
Streptavidin-coated beads
Covalently labeled streptavidin beads were prepared by directly coupling carboxylated modified latex beads (200 nm diameter; Bangs Laboratories, Fishers, IN) with streptavidin (SA20; Prozyme, San Leandro, CA) using 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (Merck, Darmstadt, Germany) and sulfo-N-hydroxysulfosuccinimide (Fluka) The coupling reaction was quenched by excess glycine (Merck). Excess streptavidin was removed by spin and wash processes (6 min and 140 Krpm spin) to pellet and resuspend the beads at least seven times. These purified streptavidin-coated beads were stored in phosphate-buffered saline containing 0.05% Tween-20 (Merck). Before each use, the beads were sonicated for several minutes to remove aggregation. In our experiments, 40 pM streptavidin-coated beads were incubated for 10 min in the chamber before being washed out.
Microscope setup, imaging acquisition, and analysis
W observed the BM of the tethered beads by means of an inverted light microscope (Olympus IX-71, Tokyo, Japan) using the differential interference contrast (DIC) method. The microscope setup was similar to that described previously (24), except that we used a Newvicon camera (NC-70; DAGE-MTI, Michigan City, IN) with a frame interval of 33 ms. The images were directly digitalized through a frame-grabber (PCI-1411; National Instrument, Austin, Texas) and stored in computers. The pixel size in our setup was calibrated to be 75 × 75 nm. The centroid position of the chosen tethered bead was fitted to a two-dimensional Gaussian in each frame with nanometer precision. The stage drift was corrected by subtracting the average movement of the best-correlated pair of beads that preadsorbed to the coverglass surface in the same field of view. The precision of the centroid determination was 3.7 nm as calculated from beads preabsorbed to the coverglass. The amplitude of bead BM was quantified using the mean-square displacement (MSD; 〈Δx2〉 = 〈x2〉 − 〈x〉2) of the consecutive 20 frames (0.66 s) of drift-correlated centroid position of the tether beads in our TPM assay. The use of more frame numbers does not alter the MSD values. For time-course measurements, 20 frame intervals were used. For population measurements, MSD values were calculated from 1000 consecutive frames to obtain the average BM amplitude. To ensure that only one DNA molecule was attached to each bead, only the beads with symmetrical BM were used, and the MSD values of the x/y ratio were limited to 1.1–0.9 for further analysis.
CD measurements
For CD measurements, we used 10 μM h24 samples in a 200 μL volume. The spectra were averages for 10 scans on a Jasco J-715 spectropolarimeter (Tokyo, Japan) with a 2 nm bandwidth at room temperature. The scan speed was 50 nm/min with a 0.2 nm step resolution. The CD spectra were measured under nitrogen gas over a range of 210–350 nm to monitor the G4 structures.
Results and Discussion
Folded/unfolded conformations in TPM experiments
For our TPM experiments, the 5′ digoxigenin-labeled 113 nt long ssDNA h22L containing 22 nt human telomeric DNA sequence (see Materials and Methods) was specifically attached to the antidigoxigenin-decorated coverglass surface (Fig. 1 a). To visualize it using the TPM, we annealed surface-anchored ssDNA with a 12 nt biotin-labeled oligo, so each DNA molecule can be attached to a streptavidin-labeled bead. In the presence of beads, the annealed DNA molecules appear as tethers, and the BM of the beads is limited by the DNA tether length. At a high salt condition of 150 mM Na+ solution, the h22 sequence has been shown to fold into an antiparallel G4 structure as determined by NMR assay (11). This was also verified in the CD spectra with signature peaks at 265 nm and 295 nm (see Fig. 3). When these h22L DNA molecules were incubated under the high Na+ condition at the single-molecule level, the BMs of the DNA-tethered beads were measured individually. The distribution of BM amplitude can be fitted into a single Gaussian curve, with a mean BM amplitude of 12.75 ± 2.62 nm (full width at half maximum; N = 144, where N is the number of effective tethers; Fig. 1 b). We assigned this BM amplitude to be the G4 folded state.
Figure 1.
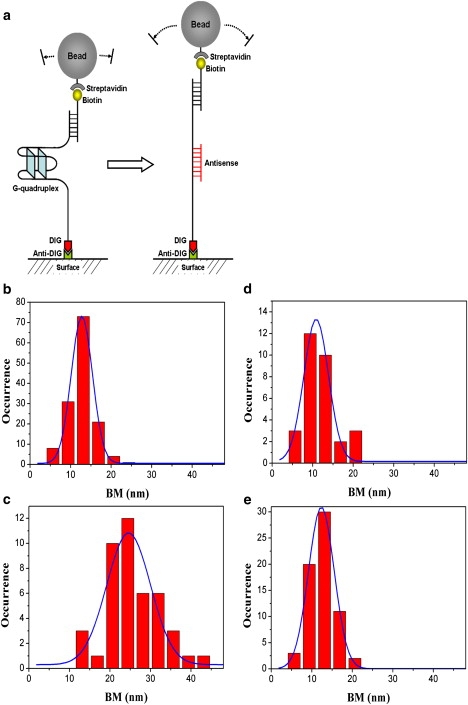
Observing the folded and unfolded states of 113 nt h22L ssDNA using the TPM method. (a) Our experimental geometry contains human telomeric sequence AGGG(TTAGGG)3 and 48 nt AC copolymer (no secondary structure). DNA is immobilized on the surface with a streptavidin-decorated polystyrene bead (200 nm) attached on the other end as a reporter. (b) Histogram of the BM amplitude of h22L under the folded-state condition (150 mM NaCl). Gaussian fitting returns the peak at 12.75 ± 2.62 nm (±σ) (N = 144). (c) BM histogram of h22L interacting with 12 nt, antisense c12 DNA. The peak at 24.52 ± 5.40 nm (N = 44) is assigned to the unfolded state. (d) BM histogram of h22L with noncomplementary sequence t16. The peak stays at the folded state, 10.80 ± 3.00 nm (N = 30). (e) BM histogram of h22L at 100 mM KCl condition with the peak at the folded state, 12.30 ± 3.15 nm (N = 67).
Figure 3.
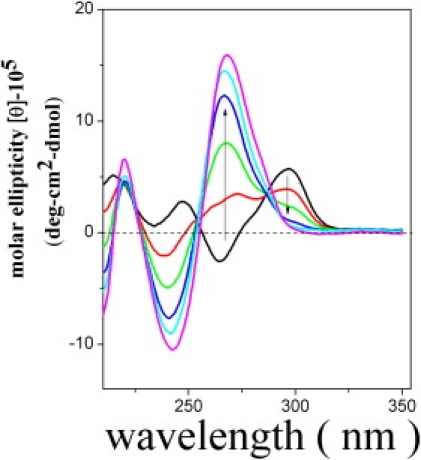
Antisense c12 oligo stabilizes the unfolded states of G4. CD spectra of h24 mixed with various concentrations of c12, indicating a decrease of the 295 nm band and an increase of the 268 nm band with increasing c12 concentrations.
To observe the G4 unfolded state, we challenged the h22L molecules by antisense c12 oligos, which has been found to stabilize the unfolded state of the G4 by its sequence complementarity to the telomeric h22 DNA (25,26), as illustrated in Fig. 1 a. The h22L and 100-fold excess antisense c12 mixture was incubated in 150 mM Na+ solution offline for 4 h before it was anchored on the coverglass surface for observation. The bead BM was found to be a single distribution with the peak amplitude shift to a larger value of 24.52 ± 5.20 nm (N = 44), as shown in Fig. 1 c. We assigned this to the G4 unfolded state. The BM distribution of this unfolded state was rather broad compared to that of the folded state. This was expected since there are multiple binding possibilities between a 12 nt c12 and complementary 22 nt h22 sequence. A controlled experiment using 100-fold excess noncomplementary oligos t16 returned a bead BM of 10.80 ± 3.00 nm (N = 30; Fig. 1 d), the same as in the G4 folded state assigned earlier. This further confirms our assignment of the folded and unfolded states of the h22L G4 DNA.
The crystal structure of the telomeric DNA sequence under the high K+ condition (100 mM) showed a propeller-folded G4 structure (27). TPM experiments carried out under this K+ condition returned a BM amplitude of 12.30 ± 3.15 nm (N = 67; Fig. 1 e), the same as in the experiments done under 150 mM Na+ salt. This again verifies the assignment of the folded state of h22L DNA. Although our TPM experiment is not sensitive enough to distinguish fine structural differences among various folded states, such as parallel or antiparallel conformations, it can clearly differentiate the folded and unfolded states of h22L DNA based on the end-to-end distance of the DNA tether length. We use this difference to investigate the structural conversion of telomeric DNA at the single-molecule level.
Salt-dependent folded equilibrium
It is known that monovalent cations such as sodium and potassium can stabilize G4 structures (28,29). Furthermore, the concentrations of these cations are known to modulate the equilibrium constant of G4s. We used TPM to directly sample the distribution of the folded and unfolded states of h22L DNA substrates in different sodium salt concentrations (15 mM and 5 mM; Fig. 2, a and b). In contrast to the high sodium salt concentration at 150 mM (Fig. 1 b), the bead BM displays a distribution that can be fitted by two Gaussians at lower salt concentrations: one centered around 10–12 nm, and the other around 24 nm. The first distribution coincides with the folded state of h22L studied at 150 mM sodium salt, and the latter coincides with the unfolded state studied with a mixture of h22L and antisense c12 oligos. The area under each Gaussian represents the population of each conformation under a specific salt concentration. Therefore, it is possible to unambiguously determine the folding equilibrium constant at different salt concentrations. For example, under the 5 mM sodium condition, the folding equilibrium constant is 1.75 ± 0.04 (N = 41). The folding equilibrium constant increases to 3.40 ± 0.11 (N = 74) with the 15 mM sodium concentration, and eventually becomes folding dominant at the high salt condition. The differences in Gibbs free energy between the unfolded and folded states are −0.72 kcal/mol and −0.33 kcal/mol for 15 mM and 5 mM Na+ salts, respectively. The difference between the two salt concentrations is on the order of the strength of hydrogen bonding.
Figure 2.
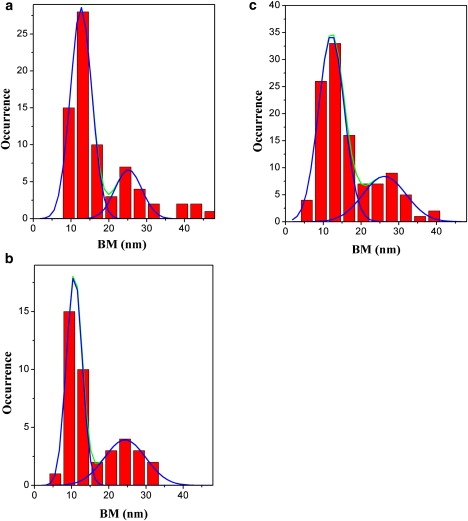
The stability of the G4 structure depends on the salt concentration. (a) BM histogram of h22L at 15 mM NaCl, with peaks at 12.60 ± 2.77 and 25.12 ± 3.52 nm (N = 74); this leads to a folding equilibrium constant of 3.40 ± 0.11. (b) BM histogram at 5 mM NaCl, with peaks at 10.65 ± 2.10 and 24.37 ± 5.55 nm (N = 41). Kfolding = 1.75 ± 0.04. (c) The BM histogram at 150 mM LiCl also shows two populations, peaks at 12.15 ± 3.37 and 26.17 ± 5.70 nm (N = 110). Kfolding = 2.48 ± 0.16.
Potassium and sodium ions are known to be the best cations for maintaining human telomeric G4 stability because of their suitable size (radius: 1.33 Å and 0.95 Å, respectively) and their relatively strong dehydration ability, which enables electrostatic interactions with four O6 within purine on the G-quartet (30). Can smaller-sized cations, such as lithium (radius: 0.60 Å), stabilize the G4 as well? TPM experiment carried out under the 150 mM lithium condition shows two distinct populations centered at 12.15 ± 3.37 nm and 26.17 ± 5.70 nm. This allows us to determine a folding equilibrium constant of 2.48 ± 0.16 (N = 110). Under the same high salt concentrations (150 mM), sodium salt leads mostly to a G4 folded state, whereas the smaller lithium ion leads to only a 70% folded population, as shown in Fig. 2 c. The less-stable folded state under lithium salt can be explained by the smaller size and higher dehydration energy of lithium, which cannot provide a sufficient chelating framework in the presence of Hoogsteen basepairing.
Antisense DNA interaction
CD spectra measure the absorption of two different circularly polarized lights to sensitively probe the secondary structure of biopolymers, and have been routinely applied to characterize G4 structures (31–34). Antiparallel G4 structures are normally characterized by two positive bands around 295 and 240 nm, and a negative band around 265 nm. This specific CD pattern has not been found for other DNA structures, and therefore has been used as a diagnostic tool specifically for the antiparallel G4 structure. The CD spectra of h24 in 150 mM Na+ salt (Fig. 3, black curve) show characteristic peaks indicative of the antiparallel G4 structure. When h24 was challenged by increasing amounts of antisense c12 oligo (c12/h24 ratio of 0.5, 1, 1.5, 2, and 3; Fig. 3, red, green, blue, cyan, pink line, respectively) for 15 min, the CD spectra clearly show a gradual disappearance of the 295 nm band as a function of antisense c12 concentration, as well as an increase in the 268 nm band (arrow indicates increasing c12 concentration). An interpretation based on the increase in the 268 nm band alone could be complicated, but the decrease in the 295 nm band unambiguously reveals the collapse of the antiparallel folded G4 state. The change in this CD pattern suggests the disruption of the antiparallel G4 structure upon c12 addition within 15 min, likely due to unfolding. To further confirm that antisense c12 oligos can indeed induce the unfolding of an antiparallel G4 structure, and shift the population to the unfolded state, we also carried out a native DNA gel-shift experiment. In the presence of a 1:3 molar ratio of h24 and antisense c12 (data not shown), the h24 band was mostly shifted up to a higher-molecular-weight band under the folding condition (150 mM Na+). It is consistent with the model that the presence of antisense DNA stabilized the unfolded population.
Single-molecule TPM experiments can also be used to study the unfolding kinetics of h22L DNA upon the addition of antisense c12 DNA in real time. We carried out TPM experiments at a rate of 30 Hz and further analyzed the data using an average of 20 frames. Therefore, the TPM experiments had a time resolution of ∼0.66 s. The G4-containing h22L substrate was first incubated on the coverslip under the 150 mM Na+ condition, followed by the bead attachment. Under this salt condition, the h22L tethers were found in the folded state, as shown in Fig. 1 b. The BMs of h22L tethers were first recorded for a short duration to verify their DNA tether lengths. The solution containing antisense c12 DNA (400, 4000, and 10,000 folds excess to the surface-bound h22L DNA) in 150 mM Na+ salt was then flown into the reaction chamber. The recording dead time for solution exchange and restabilization of the coverslip for imaging was ∼80 s (gray shaded region in Fig. 4). The images were continuously recorded throughout the solution exchange to follow the bead motion of the same individual tethers for up to another 8 min after the dead time. Fig. 4 a shows the typical time trace for monitoring the unfolding process of the folded h22L DNA in the presence of a 400–10,000 fold excess of c12 DNA. In fact, only one of 26 tethers unfolded within 8 min under 400-fold excess of c12. When the c12 concentration increased to a 4000-fold excess, the percentage of unfolding events increased (16 out of 122) to 13% within the observation time. This percentage increased to 25% (27 out of 107 tethers) when the c12 was in 10,000-fold excess. Different percentages of unfolding events upon c12 concentrations may be due to the effective collision probability. For tethers that unfolded within the 8-min recording time, 60% (26 out of 43 tethers) were found to unfold immediately after the buffer exchange (Fig. 4 b, inset). The distribution of the unfolding time of all 43 observed unfolding tethers can be fitted into a single exponential decay (Fig. 4 b, inset), returning an apparent unfolding time of 5 s. Since there is a dead time for buffer exchange and stage restabilization of ∼80 s, the expected unfolding time on this timescale is ∼85 s.
Figure 4.
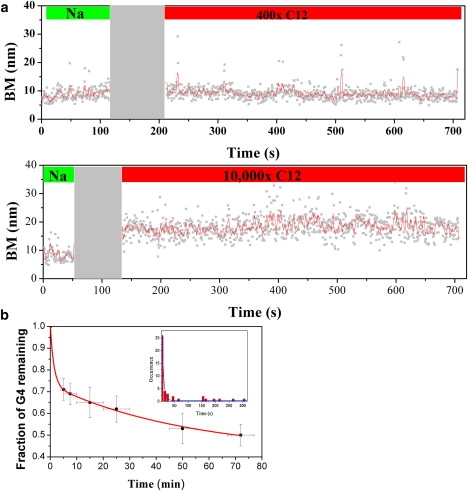
Monitoring G4 structures disrupted by antisense c12 oligo using single-molecule TPM in real time. (a). The h22L substrates were prepared at 150 mM NaCl (folded state, green bar) and subjected to buffer change (gray area) with various concentrations of c12. The recordings were continuous during the buffer change, so it is certain that the same DNA molecules were followed. The dead time of the experiment (∼80 s) was due to the buffer change and restabilization of the stage for imaging. The recordings were then taken for another 8 min (red bar) to monitor the unfolding process. We rarely observe unfolding process in the presence of 400-fold excess c12 within 8 min of recording (1/26, 3.8%). More unfolded events were observed under high c12 concentrations: 27/107, 25.2% for 10,000-fold. (b) The averaged folded fraction of h22L at various time points after adding 10,000-fold antisense c12 shows biexponential decay kinetics. Different fields of DNA tethers were sampled at specified time points to estimate the folded fraction. The unfolding lifetimes were fitted to 82 ± 43 s (46%) and 3152 ± 1138 s (54%), with R2 = 0.99. (b, inset) Histograms of the unfolding times observed in the time-course experiments shown in panel a, where the unfolding time is defined as the dwell time between the end of buffer exchange (gray area) and the beginning of the unfolded state. Note that it does not contain the ∼80 s dead time mentioned above. The histogram is fitted to a single exponential decay of 5.5 s unfolding time. Considering the experimental dead time, this is consistent with the 82 s unfolding time measured from panel b.
The fact that only ∼20% of the h22L DNA unfolded within 8 min of antisense c12 addition suggests that there must exist additional unfolding pathways when h22L DNA is challenged with antisense DNA. Since it is not computationally feasible to observe many individual h22L tethers for a longer time period, we measured the averaged folded fraction (Xfolded) of h22L DNA tethers on the coverslip at various time points after adding 10,000-fold c12, as shown in Fig. 4 b. At each time point, we repeated the experiment at least six times, sampling >30 tethers each time, to calculate the mean fraction of tethers that remained folded and its error. Confirming our earlier prediction, the unfolding kinetics showed biexponential decays, with an unfolding lifetime of 82 ± 43 s (46%; the percentage was determined by the weighting factor of curve-fitting) and 3152 ± 1138 s (54%). Similar biexponential unfolding kinetics were previously observed using a gel-shift assay on a mixture of (T4G4)4 and (C4A4)8, which showed a 30% fast initial burst followed by a much slower unfolding (3 h) (35). The multiple unfolding times reported here suggest that there must exist multiple unfolding pathways, as observed in other studies (36). It was previously proposed that the initial burst could result from the partially unfolded form of folded G-quartets, which are quickly stabilized into the unfolded form by the antisense DNA, whereas the slower unfolding time is the mean time required to convert the folded state into the unfolded state (35). We consider that antisense DNA can stabilize the unfolded state by hybridizing to the G4 DNA sequence and preventing the reformation of the folded structure. G4 DNA may go through a few cycles of folded/unfolded equilibrium before the unfolded form can hybridize with the antisense DNA to be observed in the stabilized unfolded form. However, in the presence of a great excess of antisense DNA (10,000-fold used here), it is more likely that the long unfolding time is dominated by the equilibrium between the folded and unfolded states of G4 DNA. Even though it has been suggested that the dynamics of DNA conformational change could occur on a much faster timescale (37), the data shown here, as well as data from other works (25,35,38), support the idea that the conversion between the folded and unfolded states of G4 states may take place much more slowly (on the order of tens of minutes, or even longer) depending on the strand concentration, pH value, and temperature.
Na/K-induced structural conversion
Our unfolding measurement upon addition of antisense DNA to unfolded G4 h22L DNA demonstrates that our TPM setup allows us to monitor the folded/unfolded structural conversion in real time, and to successfully exchange buffers as desired. As discussed above, G4 folded states can exist in an antiparallel state (as seen in the 150 mM Na+ solution NMR structure) (11) or in mixed type-I, mixed type-II forms (in 100 mM K+) (7–9,12,13). How these structures interconvert remains a key question. Earlier studies by Lee et al. (36) indicated that conversion between the two G4 folded states at 2 mM potassium requires unfolding intermediate states. In addition, several studies using NMR and CD spectroscopy suggested a required unfolded intermediate resulting from the guanine polarity change during the structural conversion between antiparallel and mixed type structures (7–9). On the other hand, Chang et al. (10) proposed that the fast spectral conversion of CD patterns under potassium titration of a sodium folded structure does not display the characteristic pattern of any unfolding intermediate. We aimed to use TPM experiments to directly test whether the spectral conversion process accompanies structural conversion, which requires an unfolding intermediate.
To that end, h22L DNA molecules were prepared under 150 mM Na+ salts and confirmed to tether in the folded state (green bar in Fig. 5). Then 100 mM K+ salts (buffer exchange, gray area) were introduced into the reaction chamber. These two different salt conditions have been suggested to be in different structurally folded states: one antiparallel (11) and the other with possibly a mixed type-I structure (7–9). Our earlier experiment showed that the unfolded state tethers have BM of ∼24 nm; therefore, we can follow the time course of the bead BM of the folded tethers to ascertain whether they go through any totally unfolded intermediate. Since the results of our antisense experiment (Fig. 4) suggest that the buffer exchange occurs quickly, we assume that the structure of h22L will be at the K+ condition after the buffer exchange. Three exemplary time courses of this Na/K-induced structural conversion are shown in Fig. 5. Obviously, there is little change in the bead BM after the buffer exchange (cyan bar). The trace at the bottom shows some BM fluctuations around the folded state, but the maximum BM of the trace is <15 nm. In fact, 61 of the 63 time-course traces we studied showed no change in bead BM after 8 min of buffer change. Only two traces showed a larger fluctuation in BM amplitude (up to 20 nm; see Fig. S1 in the Supporting Material) before they were stable around the tether value of the folded state. To further prove that the buffer exchange does happen on a fast timescale, we carried out a control experiment at 15 mM Na+ salt condition (data not shown). As shown in Fig. 2 a, under the low Na+ salt concentration, there exists a mixture of folded and unfolded states. Upon buffer exchange of 100 mM K+, the initial tethers at the unfolded state convert into the folded state within a few seconds due to the exchange of high-concentration potassium ions. Meanwhile, those tethers that were initially in the folded state in the same field of view stayed in the folded state with no apparent change in bead BM.
Figure 5.
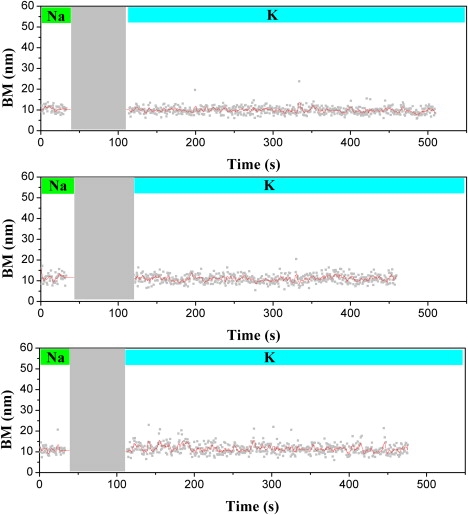
The time courses of Na/K-induced structural conversion does not show any totally unfolded intermediate states. The h22L substrates were first prepared at 150 mM NaCl (green bar). The DNA molecules were then subjected to buffer change with 100 mM KCl (gray area, experimental dead time < 80 s). The experiments were continued for another 8 min (cyan bar). Three exemplary time traces are shown here. With 63 tethers observed, no apparent totally unfolded intermediate (BM > 25 nm) is seen within our experimental time resolution of 1 s and dead time of 80 s. Two of the 63 tethers showed larger BM fluctuations, but not large enough to reach the totally unfolded state assigned earlier (see Fig.S1 in Supporting Material).
Recently, Gray et al. (39) monitored the G4 conformational change process of an Na+-based solution under potassium titration by tracing the CD intensity change of 291 and 261 nm. They proposed that the interconversion upon K+ titration could occur on a timescale of minutes. Thus, our TPM experiments should be capable of detecting an appreciable percentage of unfolded intermediate state in the totally unfolded intermediate model. The fact that we saw no change in bead BM in 97% of the tethers (N = 63) implies that the interconversion process does not involve a totally unfolded intermediate as a major, dominant pathway during the conversion between antiparallel and possibly either mixed type-I or mixed type-II folded states at the time resolution of our experiments.
To test whether the totally unfolded intermediate state occurred within our TPM dead time (∼80 s), we carried out the same Na/K conversion experiment using a stopped-flow CD measurement of 100 ms time resolution and 200 ms dead time. If a totally unfolded intermediate is required for a pathway during this conversion, an apparent decrease in the 291 nm CD band, which signals the quadruplex structure, is expected (40). Our experiment (shown in Fig. S2) did not show any decrease in the 291 nm band on a millisecond timescale, and therefore does not supporting the unfolded intermediate model on the subsecond scale.
On the basis of CD spectra and singular value decomposition analysis, Gray et al. (39) suggested that ion exchange happens within 5 ms, and subsequent K+-induced folding occurs through an intermediate on a timescale of 40–50 s and further to a final structure on a timescale of 600–800 s. Their model suggests that the folding process is much slower than what was proposed previously, and the timescale is on the order of minutes. These numbers offer reference points to estimate the lifetime of the unfolding state, if it exists. Since the folding equilibrium constant at 15 mM NaCl is 3.40 (Fig. 2), it is reasonable to assume that the equilibrium constant of 50 mM K+ condition used by Gray and Chaires (37) is much larger. The 40–50 s folding timescales imply that the unfolding timescale has to be, at least, much longer than 3 min. Therefore, our TPM experiments are capable of detecting an appreciable amount of unfolding intermediates on a timescale > 1 min. In addition, Renciuk et al. (41) used CD spectra to show that K+-induced folded states can adopt several different structures, including antiparallel, hybrid (31), and propeller parallel structures, depending on the salt and DNA concentration used. They found that under the low DNA concentration used in our single-molecule studies, both Na+- and K+-induced folded states have the same antiparallel structures. Since the reactants and the products can have the same structures, it is possible that the G4 structure stays the same throughout the Na/K exchange, with only a minor difference in structural compactness due to the larger-sized potassium ions. This model is consistent with our experimental observation of no appreciable change in the BM upon Na/K exchange.
The NMR structure of G4 of d[(G3T2A)3G3T] recently solved by Lim et al. (42) under K+ salt shows a new K+-based, basket-type folded structure with only two G-tetrad layers, with even higher stability. Therefore, it is feasible that the three G-tetrad-layer, antiparallel Na+ basket structure could be converted into the two G-tetrad-layer, antiparallel K+ basket structure (42). The loop arrangements are basically the same in these three and two G-tetrad-layer structures, and also dispense with the requirement of an unfolding intermediate during the structural conversion. A structure of the wild-type telomeric sequence under the K+-based folded condition will provide critical information.
Apparently, a simple model that attempts to describe G4 structures and their interconversion is not sufficient to explain all existing experimental observations. It is also evident that minor modifications of G4 sequences resulted in very different structures in recent crystallography and NMR studies. The underlying complexity and diversity of these G4 structures suggests that more caution is required in experimental interpretation. In the single-molecule study presented here, TPM experiments were successfully applied to study the conformational change of ssDNA, for the first time to our knowledge. The single-molecule TPM method initiated in this study directly measures the tether length change of individual telomeric DNA molecules, and provides a new approach to monitor structural conversion among various G4 structures and protein-G4 interactions in real time.
Supporting Material
Three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00123-2.
Supporting Material
Acknowledgments
We thank Dr. Shu-Chuan Jao (Biophysics Core Facility in Institute of Biological Chemistry, Academia Sinica) and Dr. Yuan-Chao Lou (Institute of Biomedical Sciences, Academia Sinica) for kindly providing the stopped-flow CD instrument for our measurements.
This work was supported by the Thematic Research Program of Academia Sinica (AS-98-TP-A04) and grants from the National Science Council of Taiwan to T.-C.C. and H.-W.L.
References
- 1.Morin G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M., Lipsett M.N., Davies D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang C.C., Kuo I.C., Chang T.C. Detection of quadruplex DNA structures in human telomeres by a fluorescent carbazole derivative. Anal. Chem. 2004;76:4490–4494. doi: 10.1021/ac049510s. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn E.H., Greider C.W. Cold Spring Harbor Laboratory Press; New York: 1996. Telomeres. [Google Scholar]
- 5.Williamson J.R. G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 6.Bodnar A.G., Ouellette M., Wright W.E. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 7.Ambrus A., Chen D., Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luu K.N., Phan A.T., Patel D.J. Structure of the human telomere in K+ solution: an intramolecular (3 + 1) G-quadruplex scaffold. J. Am. Chem. Soc. 2006;128:9963–9970. doi: 10.1021/ja062791w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y., Noguchi Y., Sugiyama H. The new models of the human telomere d[AGGG(TTAGGG)3] in K+ solution. Bioorg. Med. Chem. 2006;14:5584–5591. doi: 10.1016/j.bmc.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 10.Chang C.C., Chien C.W., Chang T.C. Investigation of spectral conversion of d(TTAGGG)4 and d(TTAGGG)13 upon potassium titration by a G-quadruplex recognizer BMVC molecule. Nucleic Acids Res. 2007;35:2846–2860. doi: 10.1093/nar/gkm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Patel D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 12.Dai J., Carver M., Yang D. Structure of the hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007;35:4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai J., Punchihewa C., Yang D. Structure of the intramolecular human telomeric G-quadruplex in potassium solution: a novel adenine triple formation. Nucleic Acids Res. 2007;35:2440–2450. doi: 10.1093/nar/gkm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bates P., Mergny J.L., Yang D. Quartets in G-major. The First International Meeting on Quadruplex DNA. EMBO Rep. 2007;8:1003–1010. doi: 10.1038/sj.embor.7401073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson W.D., Sugiyama H. First international meeting on quadruplex DNA. ACS Chem. Biol. 2007;2:589–594. doi: 10.1021/cb7001686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane A.N., Chaires J.B., Trent J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008;36:5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin H., Landick R., Gelles J. Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule. Biophys. J. 1994;67:2468–2478. doi: 10.1016/S0006-3495(94)80735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dohoney K.M., Gelles J. χ-Sequence recognition and DNA translocation by single RecBCD helicase/nuclease molecules. Nature. 2001;409:370–374. doi: 10.1038/35053124. [DOI] [PubMed] [Google Scholar]
- 19.Schafer D.A., Gelles J., Landick R. Transcription by single molecules of RNA polymerase observed by light microscopy. Nature. 1991;352:444–448. doi: 10.1038/352444a0. [DOI] [PubMed] [Google Scholar]
- 20.Mumm J.P., Landy A., Gelles J. Viewing single λ site-specific recombination events from start to finish. EMBO J. 2006;25:4586–4595. doi: 10.1038/sj.emboj.7601325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerra R.F., Imperadori L., Finzi L. DNA compaction by the nuclear factor-Y. Biophys. J. 2007;93:176–182. doi: 10.1529/biophysj.106.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pouget N., Turlan C., Chandler M. IS911 transpososome assembly as analysed by tethered particle motion. Nucleic Acids Res. 2006;34:4313–4323. doi: 10.1093/nar/gkl420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finzi L., Gelles J. Measurement of lactose repressor-mediated loop formation and breakdown in single DNA molecules. Science. 1995;267:378–380. doi: 10.1126/science.7824935. [DOI] [PubMed] [Google Scholar]
- 24.Fan H.F., Li H.W. Studying RecBCD helicase translocation along χ-DNA using tethered particle motion with a stretching force. Biophys. J. 2009;96:1875–1883. doi: 10.1016/j.bpj.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan A.T., Mergny J.L. Human telomeric DNA: G-quadruplex, i-motif and Watson-Crick double helix. Nucleic Acids Res. 2002;30:4618–4625. doi: 10.1093/nar/gkf597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W., Miyoshi D., Sugimoto N. Structural competition involving G-quadruplex DNA and its complement. Biochemistry. 2003;42:11736–11744. doi: 10.1021/bi034168j. [DOI] [PubMed] [Google Scholar]
- 27.Parkinson G.N., Lee M.P.H., Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 28.Sen D., Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 29.Williamson J.R., Raghuraman M.K., Cech T.R. Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell. 1989;59:871–880. doi: 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- 30.Hud N.V., Plavec J. The role of cations in determining quadruplex structure and stability. In: Neidle S., Balasubramanian S., editors. Quadruplex Nucleic Acids. Royal Society of Chemistry; Cambridge, UK: 2006. p 100. [Google Scholar]
- 31.Balagurumoorthy P., Brahmachari S.K., Sasisekharan V. Hairpin and parallel quartet structures for telomeric sequences. Nucleic Acids Res. 1992;20:4061–4067. doi: 10.1093/nar/20.15.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giraldo R., Suzuki M., Rhodes D. Promotion of parallel DNA quadruplexes by a yeast telomere binding protein: a circular dichroism study. Proc. Natl. Acad. Sci. USA. 1994;91:7658–7662. doi: 10.1073/pnas.91.16.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hazel P., Huppert J., Neidle S. Loop-length-dependent folding of G-quadruplexes. J. Am. Chem. Soc. 2004;126:16405–16415. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 34.Qi J., Shafer R.H. Covalent ligation studies on the human telomere quadruplex. Nucleic Acids Res. 2005;33:3185–3192. doi: 10.1093/nar/gki632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raghuraman M.K., Cech T.R. Effect of monovalent cation-induced telomeric DNA structure on the binding of Oxytricha telomeric protein. Nucleic Acids Res. 1990;18:4543–4552. doi: 10.1093/nar/18.15.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J.Y., Okumus B., Ha T. Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. USA. 2005;102:18938–18943. doi: 10.1073/pnas.0506144102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray R.D., Chaires J.B. Kinetics and mechanism of K+- and Na+-induced folding of models of human telomeric DNA into G-quadruplex structures. Nucleic Acids Res. 2008;36:4191–4203. doi: 10.1093/nar/gkn379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ying L., Green J.J., Balasubramanian S. Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2003;100:14629–14634. doi: 10.1073/pnas.2433350100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gray R.D., Li J., Chaires J.B. Energetics and kinetics of a conformational switch in G-quadruplex DNA. J. Phys. Chem. B. 2009;113:2676–2683. doi: 10.1021/jp809578f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monchaud D., Yang P., Mergny J.L. A metal-mediated conformational switch controls G-quadruplex binding affinity. Angew. Chem. Int. Ed. Engl. 2008;47:4858–4861. doi: 10.1002/anie.200800468. [DOI] [PubMed] [Google Scholar]
- 41.Renciuk D., Kejnovská I., Vorlícková M. Arrangements of human telomere DNA quadruplex in physiologically relevant K+ solutions. Nucleic Acids Res. 2009;37:6625–6634. doi: 10.1093/nar/gkp701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim K.W., Amrane S., Phan A.T. Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 2009;131:4301–4309. doi: 10.1021/ja807503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.