

Abstract
The 5-HT3 receptor is a member of the Cys-loop family of transmitter receptors. It can function as a homopentamer (5-HT3A-only subunits) or as a heteropentamer. The 5-HT3AB receptor is the best characterized heteropentamer. This receptor differs from a homopentamer in its kinetics, voltage dependence, and single-channel conductance, but its pharmacology is similar. To understand the contribution of the 5-HT3B subunit to the binding site, we created homology models of 5-HT3AB receptors and docked 5-HT and granisetron into AB, BA, and BB interfaces. To test whether ligands bind in any or all of these interfaces, we mutated amino acids that are important for agonist and antagonist binding in the 5-HT3A subunit to their corresponding residues in the 5-HT3B subunit and vice versa. Changes in [3H]granisetron binding affinity (Kd) and 5-HT EC50 were determined using receptors expressed in HEK-293 cells and Xenopus oocytes, respectively. For all A-to-B mutant receptors, except T181N, antagonist binding was altered or eliminated. Functional studies revealed that either the receptors were nonfunctional or the EC50 values were increased. In B-to-A mutant receptors there were no changes in Kd, although EC50 values and Hill slopes, except for N170T mutant receptors, were similar to those for 5-HT3A receptors. Thus, the experimental data do not support a contribution of the 5-HT3B subunit to the binding pocket, and we conclude that both 5-HT and granisetron bind to an AA binding site in the heteromeric 5-HT3AB receptor.
Introduction
The 5-HT3 receptors are members of the Cys-loop receptor superfamily of ligand-gated ion channels, which includes the nicotinic acetylcholine (nACh), GABAA, and glycine receptors (1). These receptors are membrane proteins and function as a pentameric arrangement of subunits. Five 5-HT3 receptor subunits (5-HT3A–5-HT3E; the nomenclature employed in this work adopts the recent recommendations of the NC-IUPHAR (2)) have been identified to date, although only homomeric 5-HT3A and heteromeric 5-HT3AB receptors have been extensively characterized (3,4). The 5-HT3B subunit shares 45% sequence identity with the 5-HT3A subunit, but, in contrast to the 5-HT3A subunit, fails to produce homomeric functional receptors (5). However, 5-HT3B subunits coexpress with 5-HT3A subunits to yield functional heteromeric complexes with a proposed subunit stoichiometry of 2A:3B, and a proposed subunit arrangement of B-B-A-B-A (6).
Studies of 5-HT3AB receptors in heterologous systems have revealed that the 5-HT3B subunit alters several biophysical properties of the 5-HT3 receptor, including the EC50 for 5-HT, receptor desensitization kinetics, current-voltage relationship, Hill slope, and permeability to Ca2+ (7,8). In addition, heteropentameric 5-HT3AB receptors display a large single-channel conductance (16 pS), which contrasts with the almost nondetectable single-channel conductance of homopentameric 5-HT3A receptors (0.4 pS) (3,9). Further studies led to the discovery of a cytoplasmic region (HA-stretch) that determines the single-channel conductance in 5-HT3 receptors. Replacement of three Arg residues unique to the HA-stretch of the 5-HT3A subunit by their 5-HT3B subunit counterparts increased single-channel conductance 28-fold (9–12).
The agonist-binding domain of 5-HT3 receptors can reasonably be extrapolated from crystal structures of acetylcholine-binding proteins (AChBPs) (13), and a range of homology models for the homopentameric 5-HT3A receptor have been constructed (14–17). Amino acid residues from six discontinuous regions of the extracellular domain contribute to the binding site. Three of these regions are on the primary (+) face of the subunit (loops A–C), and three are on the complementary (−) face of the adjacent subunit (loops D–F) (Fig. 1). Investigators have achieved a good understanding of the residues involved in the ligand binding in the homopentameric receptor, but it is still unclear which residues are responsible in the heteromeric 5-HT3AB receptor. This is particularly important because recent studies suggest that the efficacy of antiemetic drugs depends critically on the 5-HT3B subunit (18). The recently presented architecture of the heteropentameric 5-HT3AB receptor (6) offers three possibilities for the heteromeric binding site: A+B−, B+A−, or B+B− (A = 5-HT3A subunit, B = 5-HT3B subunit). In this work, we used a range of techniques (mutagenesis, modeling, radioligand binding, and functional assays) to examine which, if any, of these binding sites is correct. More specifically, we docked 5-HT and granisetron into the possible heteromeric binding sites to determine potential interactions with specific binding-site amino acids, and, to test the resulting predictions, we substituted a range of amino acid residues that are important for ligand binding in the 5-HT3A subunit to the corresponding 5-HT3B subunit residues and vice versa, and expressed and tested the resulting mutant receptors. The results suggest that the 5-HT3B subunit does not contribute to the binding site in heteromeric 5-HT3AB receptors.
Figure 1.
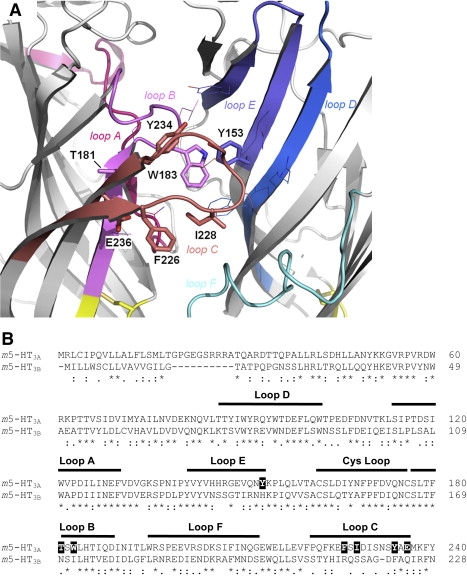
(A) Location of the 5-HT3A subunit residues (stick representation) that were mutated to the corresponding 5-HT3B subunit residues. Two adjacent subunits (principal and complementary) show the positions of the six binding loops, A–F. Other residues of the putative ligand-binding site are shown in line representation. (B) ClustalW sequence alignment of the murine 5-HT3A (accession number: Q6J1J7) and 5-HT3B subunits (Q9JHJ5). 5-HT3A subunit residues that were mutated to 5-HT3B subunit residues in this study are highlighted as white text on black background. The six binding loops and the Cys loop are indicated by black lines above the text. Numbering of residues and structural features are taken from the AChBP protein crystal structure (13).
Materials and Methods
Modeling and docking studies
Modeling and docking studies were performed as described previously (14,19). For the heteromeric 5-HT3AB open-state receptor model, the protein sequences of the mouse 5-HT3A (accession number: Q6J1J7) and 5-HT3B receptor subunits (accession number: Q9JHJ5) were coaligned with the sequence of AChBP from Lymnaea stagnalis (accession number: P58154) using FUGUE (20). A three-dimensional homology model with a 2A:3B subunit stochiometry and a B-B-A-B-A subunit arrangement around the receptor rosette was generated using MODELER 6v2 (21) based on the crystal structure of AChBP at 2.7 Å resolution (PDB ID: 1I9B). The pentamer was generated by superimposing 5-HT3A or 5-HT3B subunits onto each protomer of AChBP, and was then energy-minimized using the force field implemented in MODELER 6v2. The best model was selected after Ramachandran plot analysis of all the generated models. For the heteromeric 5-HT3AB closed-state receptor model, the protein sequences of the 5-HT3A and 5-HT3B subunits were coaligned with the sequence of the nACh receptor α1 subunit from Torpedo californica (accession number: P02710). In a procedure similar to that described above, a three-dimensional homology model was generated based on the cryo-electron microscopy structure of the nACh receptor at 4 Å resolution (PDB ID: 2BG9).
The three-dimensional, protonated structure of 5-HT was extracted from the Cambridge Structural Database (reference code: SERHOX) and the counter anion was removed for the docking. The protonated form of granisetron was constructed in Chem3D Ultra 7.0 (CambridgeSoft, Cambridge, UK) based on the crystal structure of a related indazole carboxamide (reference code: FIZXUH), and energy-minimized using the MM2 force field.
Docking of the protonated ligands into the heteromeric 5-HT3AB receptor homology models was carried out using GOLD 3.0 (Cambridge Crystallographic Data Centre, Cambridge, UK). 5-HT was docked into the A+B−, B+A−, and B+B− interfaces of the open-state homology model (+ and − denote the principal and complementary faces of the heteromeric binding site, respectively), whereas granisetron was docked into the A+B−, B+A−, and B+B− interfaces of the closed-state homology model. The following atoms were used as reference points for ligand docking: Cδ2 atom of W183 and Cζ atom of Y234 for A+ face, Cδ1 atom of W90 and Cγ atom of Y153 for A− face, Cβ atom of A219 and Cζ atom of F222 for B+ face, and Cɛ2 atom of W79 and Cγ atom of H142 for B− face. The amino acid residues were chosen based on the preferred binding-site models of Reeves et al. (14) and Thompson et al. (19). Ten genetic algorithm runs were performed on each docking exercise and the structures were analyzed using the implemented GOLDScore fitness function.
Cell culture
Human embryonic kidney (HEK) 293 cells were maintained on 90 mm tissue culture plates at 37°C and 7% CO2 in a humidified atmosphere. They were cultured in DMEM:F12 (Dulbecco's modified Eagle's medium/nutrient mix F12 ; Gibco BRL, UK) with GlutaMAX I media containing 10% fetal calf serum. For radioligand binding studies, cells in 90 mm dishes were transfected using calcium phosphate precipitation at 80–90% confluency and incubated for 3–4 days before use (22,23).
Harvested stage V–VI Xenopus oocytes were washed in four changes of ND96 (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.5), defolliculated in 1.5 mg mL−1 collagenase Type 1A for ∼2 h, washed again in four changes of ND96, and stored in ND96 containing 2.5 mM sodium pyruvate, 50 mM gentamycin, 0.7 mM theophylline. Mouse 5-HT3A and 5-HT3B subunit cDNA was cloned into pGEMHE for oocyte expression (24). cRNA was in vitro transcribed from linearized (NheI) plasmid cDNA template using the mMessage mMachine T7 Transcription kit (Ambion, Austin, TX). Stage V and VI oocytes were injected with 20 ng cRNA, and currents were recorded 1–4 days postinjection. A ratio of 1:3 (5-HT3A/5-HT3B) was used for the expression of heteromeric 5-HT3 receptors.
Site-directed mutagenesis
Mutagenesis reactions were performed according to the method described by Kunkel (25) using mouse 5-HT3A receptor subunit cDNA (accession number: AY605711) or mouse 5-HT3B receptor subunit cDNA (accession number: AF155045) in pcDNA3.1 (Invitrogen, Paisley, UK) as described previously (26). Oligonucleotide primers were designed according to the recommendations of Sambrook et al. (27) and some suggestions from the Primer Generator (28). A silent restriction site was incorporated into each primer to assist rapid identification.
Radioligand binding
Radioligand binding was performed as previously described, with minor modifications (26). Briefly, transfected HEK 293 cell membranes were incubated in 0.5 mL HEPES buffer containing the 5-HT3 receptor antagonist [3H]granisetron (63.5 Ci/mmol; PerkinElmer, UK). Nonspecific binding was determined using 1 μM quipazine. Incubations were terminated by filtration, which limited our determination of Kd values to ≤∼10 nM because separation of bound from free ligand occurs too slowly to determine Kd values greater than that (29). Data were analyzed by iterative curve-fitting (Prism v3.0; GraphPad Software, San Diego, CA) according to the equation B = (Bmax · [L])/(Kd + [L]), where B is bound radioligand, Bmax is maximum binding at equilibrium, Kd is the equilibrium dissociation constant, and [L] is the free concentration of radioligand. Values are presented as the mean ± SE. Statistical analysis was performed using analysis of variance in conjunction with Dunnett's posttest.
Immunofluorescence
Immunofluorescence was achieved as described previously (30,31). Briefly, transfected cells were fixed (4% paraformaldehyde) and incubated overnight at 4°C with a 5-HT3A or 5-HT3B specific antisera at 1:1000 in Tris-buffered saline (0.1 M Tris, pH 7.4, 0.9% NaCl). Biotinylated anti-rabbit IgG (Vector Laboratories, Burlingame, CA) and fluorescein isothiocyanate avidin D (Vector Laboratories) were used to detect bound antibody as instructed by the manufacturer. Coverslips were mounted in Vectashield HardSet mounting medium (Vector Laboratories). Immunofluorescence was observed using an UltraVIEW LCI confocal imaging system (Perkin Elmer, Boston, MA).
Electrophysiology
With the use of two electrode voltage clamps, Xenopus oocytes were clamped at −60 mV using an OC-725 amplifier (Warner Instruments, Hamden, CT), Digidata 1322A, and the Strathclyde Electrophysiology Software Package (Department of Physiology and Pharmacology, University of Strathclyde, Glasgow, UK; http://www.strath.ac.uk/Departments/PhysPharm/). Currents were filtered at a frequency of 1 kHz and sampled at 350 Hz. Microelectrodes were fabricated from borosilicate glass (GC120TF-10; Harvard Apparatus, Kent, UK) using a two-stage horizontal pull (P-87; Sutter Instrument Co., Novato, CA) and filled with 3 M KCl. Pipette resistances ranged from 0.5 to 1.5 MΩ. Oocytes were perfused with saline at a rate of 15 mL min−1. Drug application was achieved via a simple gravity-fed system calibrated to run at the same rate. Extracellular saline contained (mM) 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.4. Analysis and curve-fitting was performed using Prism v3.0 (GraphPad Software, San Diego, CA; http://www.graphpad.com). Concentration-response data for each oocyte were normalized to the maximum current for that oocyte. The mean and mean ± SE for a series of oocytes were plotted against agonist concentration and iteratively fitted to the following equation: where Imin is the baseline current, Imax is the peak current evoked by agonist, EC50 is the concentration of agonist needed to evoke a half-maximum response, [L] is the concentration of agonist, and nH is the Hill coefficient. Statistical analysis was performed using analysis of variance in conjunction with Dunnett's posttest.
Results
Homology modeling and docking
Docking of 5-HT into the open-state model, and granisetron into the closed-state model at the A+B−, B+A−, and B+B− interfaces revealed ligand orientations that were not only quite distinct from each other, but also different from orientations found at the A+A− interface (14,19) (Fig. 2). The latter was expected, since the 5-HT3B subunit shares only ∼45% sequence identity with the 5-HT3A subunit, resulting in heteromeric subunit interfaces with unique architectures. The data show that in the A+B− binding pocket, the orientation of 5-HT is broadly similar to that proposed for the A+A− interface: the ammonium is located between Trp-183 and Tyr-234, forming a hydrogen bond with the backbone of Ser-182, and within cation-π interaction distance of Trp-183. The hydroxyl group is in a pocket lined by hydrophilic residues. In the A+B− binding pocket, there are hydrogen bonds with Glu-129, Tyr-234, and the backbone of Leu-184. The ammonium is no longer within cation-π interaction distance of Trp-183. At the B+A− interface, 5-HT has a different orientation. It predominantly interacts with residues from the complementary 5-HT3A subunit, forming hydrogen bonds with the backbones of Tyr-153 and Tyr-141, and the side chain of Gln-151, and potentially undergoing a cation-π interaction with Tyr-143. At the B+B− interface, 5-HT forms hydrogen bonds with Ser-217, Ser-218, and the backbone of Ser-171 (all B+).
Figure 2.
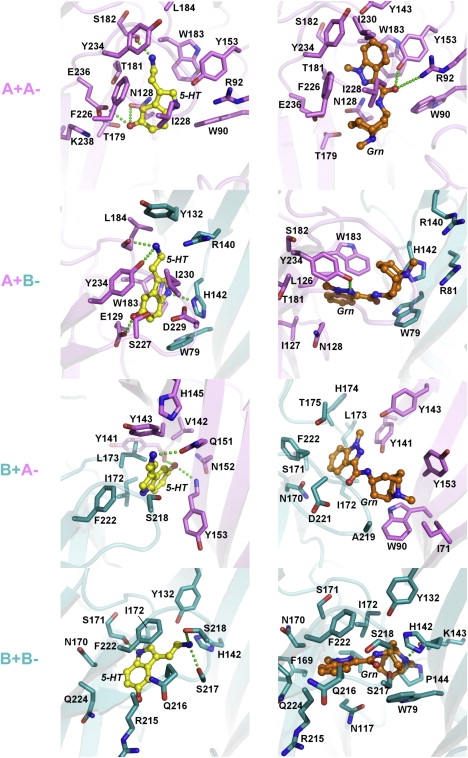
Docking of 5-HT (yellow, left panels) and granisetron (Grn, orange, right panels) into homomeric 5-HT3A (14,19) and heteromeric 5-HT3AB receptor homology models. The 5-HT3A subunit is shown in violet, and the 5-HT3B subunit is shown in teal. Residues within 4 Å of docked ligand are rendered in stick representation, color-coded according to the corresponding subunits, and numbered according to Fig. 1B. The + and − signs denote the principal and complementary face of the binding site, respectively. Proposed hydrogen bonds are shown as green dotted lines. The orientation of the docking models is the same as in Fig. 1A.
Differences were also observed for granisetron, which at the A+A− interface is oriented such that the aromatic ring of granisetron stacks face-to-face with Trp-183 and the azabicyclic ring is close to Trp-90. In the A+B− binding pocket, granisetron undergoes an edge-to-face interaction with the Trp-183 side chain and forms a hydrogen bond with Tyr-234. Granisetron does not form any hydrogen bonds in the B+A− model, and appears relatively shallowly bound between the A and B subunits. At the B+B− interface, granisetron forms a hydrogen bond with the backbone of His-142 (B−).
A closer analysis of the docking poses reveals that the majority of the residues examined in this study are within similar distances of the docked ligands in the heteromeric models as compared to the homomeric model (Table 1). This is to be expected because the structures used to generate the models are the same for the A and B subunits; however, since they are only homology models, they must be viewed with some caution, unless there is experimental evidence to verify specific interactions (e.g., the cation-π interaction with Trp-183 (32)). Nevertheless, the similarities do support our use of the A subunit to probe potential interactions of B subunit residues (see below).
Table 1.
Shortest distances between docked ligands and specific residues in the A or B subunit
| A: Shortest distances (Å) between docked 5-HT and examined residues | |||||
|---|---|---|---|---|---|
| Residue | Loop | A+A− site | A+B− site | B+A− site | B+B− site |
| Y153/H142 | E | 2.4 | 2.9 | 3.0 | 3.5 |
| T181/N170 | B | 4.0 | 5.6 | 8.4 | 4.2 |
| W183/I172 | B | 3.2 | 2.8 | 2.5 | 3.0 |
| F226/R215 | C | 4.1 | 4.2 | 8.4 | 3.0 |
| I228/S217 | C | 3.8 | 5.8 | 6.7 | 2.8 |
| Y234/F222 | C | 4.1 | 3.1 | 3.5 | 3.0 |
| E236/Q224 | C | 3.4 | 7.0 | 10.3 | 3.8 |
| B: Shortest distances (Å) between docked granisetron and examined residues | |||||
|---|---|---|---|---|---|
| Residue | Loop | A+A− site | A+B− site | B+A− site | B+B− site |
| Y153/H142 | E | 2.0 | 3.1 | 3.3 | 2.5 |
| T181/N170 | B | 3.0 | 4.1 | 3.5 | 2.9 |
| W183/I172 | B | 2.9 | 3.1 | 2.8 | 3.5 |
| F226/R215 | C | 3.9 | 6.2 | 6.3 | 3.5 |
| I228/S217 | C | 3.2 | 8.6 | 7.2 | 3.6 |
| Y234/F222 | C | 3.9 | 3.4 | 3.7 | 3.2 |
| E236/Q224 | C | 4.0 | 5.8 | 7.0 | 4.0 |
B-to-A mutant receptors
We created a series of A-like mutations in the 5-HT3B subunit, where we substituted potential binding-site residues in the 5-HT3B subunit into their equivalent locations in the 5-HT3A subunit. These were N170T and I172W (loop B), F222Y (loop C), and H142Y (loop E). Loops A and D have the same residues in 5-HT3A and 5-HT3B receptor subunits, and loop F is currently poorly defined and thus it is not yet clear which residues face into the binding pocket. Expression of these mutant receptors with wild-type (WT) 5-HT3A receptor subunits in HEK cells revealed no significant changes in [3H]granisetron binding affinity compared to WT heteromeric or homomeric receptors (Table 2), although probing with a 5-HT3B selective antisera revealed that the B subunits were expressed at the cell surface (Fig. 3). This was as expected, since previous studies have shown that the affinity of granisetron and many other 5-HT3 selective ligands is similar in A-only and AB receptors.
Table 2.
Effects of B-to-A substitutions on [3H]granisetron-binding affinities
| Mutants | Kd mean ± SE | n |
|---|---|---|
| WT A | 0.41 ± 0.08 | 6 |
| WT AB | 0.55 ± 0.12 | 4 |
| A+BH142Y | 0.44 ± 0.09 | 4 |
| A+BN170T | 0.34 ± 0.08 | 4 |
| A+BI172W | 0.65 ± 0.12 | 4 |
| A+BF222Y | 0.71 ± 0.20 | 4 |
Figure 3.

HEK cells labeled with a 5-HT3B-selective antisera reveal that both WT and mutant 5-HT3B receptor subunits reach the cell surface when coexpressed with WT 5-HT3A receptor subunits. Scale bar: 20 μm.
Functional expression of WT heteromeric receptors revealed an ∼2-fold decrease in the Hill slope compared to homomeric receptors, as previously reported (33), and we also observed an ∼2 fold increase in EC50 (Table 3). However, expression of the mutant B subunits I172W, F222Y, and H142Y with WT A receptor subunits revealed EC50 and/or Hill slopes similar to those of homomeric receptors. Because the B subunits did reach the cell surface (Fig. 3), these data suggest that exchanging these residues allows the B subunits to act as an A subunit and bind ligand. However, we cannot exclude the possibility that homomeric receptors are also expressed, and are responsible for the A-like responses. Thus, to more specifically probe the potential role of these and other amino acid residues in the B subunits, we used the A subunit, in which case we could ensure that all of the data related specifically to changes in these residues.
Table 3.
Functional effects of B-to-A substitutions at the 5-HT3 receptor
| Mutants | pEC50 mean ± SE | EC50 (μM) | nH mean ± SE | n |
|---|---|---|---|---|
| WT A | 6.12 ± 0.02 | 0.75 | 2.22 ± 0.23 | 13 |
| WT AB | 5.85 ± 0.09∗ | 1.42 | 1.12 ± 0.17∗ | 4 |
| A+BH142Y | 6.25 ± 0.08† | 0.57 | 1.89 ± 0.35 | 4 |
| A+BN170T | 5.80 ± 0.15 | 1.60 | 1.01 ± 0.15∗ | 6 |
| A+BI172W | 6.15 ± 0.08† | 0.70 | 2.31 ± 0.23† | 4 |
| A+BF222Y | 6.04 ± 0.05† | 0.92 | 2.30 ± 0.32† | 4 |
Significantly different from WT A.
Significantly different from WT AB.
A-to-B mutant receptors
We created a series of B-like mutations in the 5-HT3A receptor subunit (which has five A+A− binding sites) to mimic the effects of an adjacent 5-HT3B subunit. Loop B mutations (T181N and W183I) and loop C mutations (F226R, I228S, F234Y, and E236Q) are on the + face, whereas the mutation in loop E (Y153H) is on the − face (Fig. 1 A). In addition, we created seven double mutants (T181NW183I, T181NF226R, Y153HW183I, Y153HI228S, W183IF226R, F226RI228S, and F234YE236Q). Functional expression of these mutant receptors revealed that exchanging even one of the 5-HT3A subunit residues for its equivalent 5-HT3B subunit residue has a considerably larger effect on the receptor characteristics than has been observed in comparisons of a homomeric 5-HT3A and a heteromeric 5-HT3AB receptor. The difference in EC50 between a 5-HT3A and a 5-HT3AB receptor is ∼2-fold, but single mutations of 5-HT3A to 5-HT3B subunit binding-site residues resulted in EC50 increases of 4- to 15-fold, with functional double mutants causing >100-fold increases (see Fig. 5). Most double mutants and one single mutant (F226R) were nonfunctional (Table 4).
Figure 5.
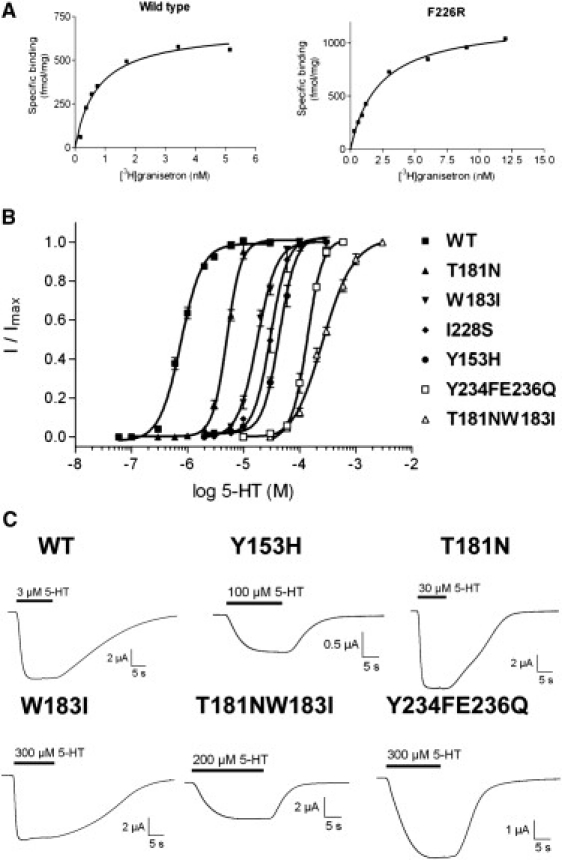
Example data for radioligand binding and functional studies of WT and mutant receptors. (A) Kd values were estimated using the 5-HT3 receptor antagonist [3H]granisetron. The examples show binding curves for single experiments, fitted with a one-site binding equation. Kd values for a series of experiments were averaged for each mutant and are presented in Tables 2 and 5. (B) Concentration response curves in 5-HT3 WT and mutant receptors measured using two-electrode voltage clamp and fitted with a four-parameter logistic equation. The calculated EC50 values are shown in Tables 3 and 4. (C) Typical 5-HT responses from oocytes expressing WT and A-to-B mutant receptors.
Table 4.
Functional effects of A-to-B substitutions at the 5-HT3 receptor
| Mutants | pEC50 mean ± SE | EC50 (μM) | nH mean ± SE | n |
|---|---|---|---|---|
| WT A | 6.12 ± 0.02 | 0.75 | 2.22 ± 0.23 | 13 |
| Y153H | 4.36 ± 0.02∗ | 43.4 | 2.94 ± 0.33 | 7 |
| T181N | 5.30 ± 0.01∗ | 5.07 | 3.38 ± 0.22∗ | 8 |
| W183I | 4.75 ± 0.02∗ | 17.7 | 2.50 ± 0.22 | 14 |
| F226R | NR∗ | - | - | > 30 |
| I228S | 4.52 ± 0.02∗ | 30.4 | 2.87 ± 0.39 | 9 |
| Y234F | 5.02 ± 0.09∗ | 9.6 | 1.82 ± 0.42 | 6 |
| E236Q | 3.95 ± 0.06∗ | 111 | 1.54 ± 0.34 | 7 |
| T181NW183I | 3.60 ± 0.03∗ | 254 | 1.54 ± 0.18 | 8 |
| F226RI228S | NR∗ | - | - | > 30 |
| Y234FE236Q | 3.86 ± 0.01∗ | 138 | 3.20 ± 0.20 | 8 |
Significantly different from WT. NR = no response (up to 300 μM 5-HT).
All of the A-to-B mutant receptors, except T181N, had a lower [3H]granisetron binding affinity than the WT receptors (Table 5). Bmax values varied considerably for different experiments (as expected for transient transfections) and ranged from ∼200 to ∼6000 fmol/mg protein. However, only T181N (365 ± 91 fmol/mg protein) mutant receptors had significantly lower expression levels compared to WT receptors (1516 ± 378 fmol/mg protein), which suggests that expression levels are not related to functional characteristics. Mutations W183I, Y153H, T181NW183I, Y153HW183I, Y153HI228S, T181NF226R, and W183IF226R caused ablation of [3H]granisetron binding. Studies with a 5-HT3A selective antisera revealed that all of the nonfunctional and nonbinding receptors were expressed at the cell surface (examples are shown in Fig. 4).
Table 5.
Effects of A-to-B substitutions on [3H]granisetron-binding affinities
| Single mutants | Kd (nM) | n | Double mutants | Kd (nM) | n |
|---|---|---|---|---|---|
| WT A | 0.41 ± 0.08 | 6 | WT A | 0.41 ± 0.08 | 6 |
| Y153H | NB∗ | 3 | Y153HW183I | NB∗ | 5 |
| T181N | 0.38 ± 0.06 | 3 | Y153HI228S | NB∗ | 5 |
| W183I | NB∗ | 5 | T181NW183I | NB∗ | 5 |
| F226R | 2.44 ± 0.49∗ | 5 | T181NF226R | NB∗ | 5 |
| I228S | 2.53 ± 0.60∗ | 5 | W183IF226R | NB∗ | 4 |
| Y234F | 0.72 ± 0.18 | 3 | F226RI228S | 7.88 ± 1.70∗ | 4 |
| E236Q | 1.51 ± 0.21∗ | 3 | Y234FE236Q | 13.5 ± 1.2∗† | 4 |
Significantly different from WT. NB = no binding.
Kd > 10 nM may be inaccurate (see text for details).
Figure 4.
Examples of HEK cells labeled with 5-HT3A-selective antisera, which reveals that both WT and mutant 5-HT3A receptors subunits are expressed on the cell surface. Scale bar: 20 μm.
Discussion
The data presented here strongly suggest that the 5-HT3B subunit does not contribute to the binding site in the mouse heteromeric 5-HT3AB receptor. Because the binding site is constituted from two adjacent subunits, residues from the 5-HT3B subunit could potentially contribute to an A+B−, B+A−, or B+B− binding site. The homology models of 5-HT3AB receptors indicate that both 5-HT and granisetron could dock into all of these types of binding pockets, where their interactions would be distinct from those that occur at the A+A− interface. The functional and radioligand binding data, however, do not support a contribution from the B subunit.
A major difference in the docked 5-HT at the A+B− compared to the A+A− binding site is the lack of a cation-π interaction at Trp-183. Such an interaction is found in neuronal nACh receptors between Trp-149 and nicotine, but does not occur in the muscle nACh receptor, a loss that results in such a major decrease in affinity for nicotine that it can no longer act as an agonist (32). Thus, we would anticipate that the loss of such an interaction in the 5-HT3AB receptor would lead to a similar loss of ligand-binding affinity, whereas the data show only a twofold change in 5-HT EC50, and no change in granisetron-binding affinity. In addition, we might anticipate that removal of the hydroxyl at Tyr-234, as in the Y234F substitution, would have a major effect on receptor function, as the hydroxyl is predicted to hydrogen bond with 5-HT, whereas such a substitution causes only a relatively small increase in EC50.
In the B+A− interface, 5-HT docks close to the A− face, but the cation-π interaction again is lost, which should lead to a much greater loss of efficacy of 5-HT than is observed. In addition, although substitution of Tyr-153 (with which 5-HT forms a hydrogen bond) with His should allow this bond to be retained, it results in a large change in the EC50 and ablation of granisetron binding as described above. Finally, at the B+B− interface, there is again no cation-π interaction with 5-HT, which we would anticipate would lead to a large decrease in binding affinity. There is, however, a predicted hydrogen bond between granisetron and His-142 (equivalent to Tyr-153), but as we observed no change in granisetron Kd when His-142 was substituted, we propose that this ligand does not bind in a B+B− pocket. It is also striking that residues Thr-181, Phe-226, Ile-228, and Glu-236, which are predicted to be much farther away from the docked ligands in the B subunit-containing binding sites, have such a significant effect on ligand binding and receptor function. Thus, overall, the docking data show that agonists and antagonists could potentially bind in A+B−, B+A−, and B+B− interfaces, but the experimental data suggest that they do not.
This conclusion is supported by the functional and radioligand binding data. We observed that incorporation of even one B residue into the A-binding pocket has a much more dramatic effect on the function of the receptor than is observed in the WT 5-HT3AB receptor. [3H]Granisetron-binding data reveal a large difference in affinities or ablation of binding in heteromeric receptors with a B-like mutation in their A subunits, but no change in affinity for B subunits with A-like mutations.
The 5-HT3B subunit was the second 5-HT3 receptor subunit to be identified, and it immediately raised great interest because although it cannot form functional receptors alone, when coexpressed with the 5-HT3A subunit, it alters a number of biophysical properties. The most striking of these is an increase in single-channel conductance of 1–2 orders of magnitude, which is caused by specific residues in the M3–M4 loop. Other effects include changes in the Hill slope, desensitization kinetics, and the shape of the current-voltage relationship, all of which could be explained by changes to the transmembrane region (3,7–10,33). The pharmacological characteristics of the 5-HT3AB receptors, however, are almost identical to those of the homomeric 5-HT3A receptor. Therefore, since the binding sites lie at subunit interfaces, 5-HT3AB receptors presumably contain at least one A+A− interface. It was therefore surprising that incompatible data were obtained in a recent study. Barrera and co-workers (6) used atomic force microscopy to image the heteromeric receptor, which was expressed after distinct epitope tags were engineered onto the two subunits. Images of receptors that were doubly liganded by anti-epitope antibodies revealed none where the antibodies were separated at an angle that would indicate two 5-HT3A subunits were adjacent, although they did observe this for tagged 5-HT3B subunits. Thus, they concluded that the subunit arrangement around the receptor rosette is B-B-A-B-A. At this time, we cannot explain the discrepancy between their data and ours. It is possible that there are distinct 5-HT3AB subunit arrangements in different species (we used mouse and they used human). This is supported by the fact that the difference in EC50 is much greater between A and AB receptors in humans (10- to 20-fold) than in mice (1- to 2-fold) (33,34). However, opposing this argument is the fact that human 5-HT3AB receptors, like those in the mouse, have a pharmacological profile similar to that of human homomeric 5-HT3A receptors. It may be possible that different cell types express different heteromeric receptors, or that intracellular AB receptors (which would be characterized by the atomic force microscopy study in addition to those expressed extracellularly) are of the wrong stoichiometry. Considerably more work needs to be performed to determine whether any of these possibilities are correct.
Our data also reveal some interesting features of the 5-HT3A receptor-binding site. The most surprising is that substituting Trp-183 for Ile results in a functional receptor, albeit with a ∼20-fold increase in EC50 and no observable granisetron binding. As described above, Trp-183 is involved in a cation-π interaction with 5-HT (32), and thus we speculate that in the W183I mutant receptor there may be such an interaction with another aromatic binding-site residue. Molecular-dynamics studies in the GABAC receptor, for example, have shown that there is a potential for cation-π interactions with two of the binding-site aromatics (35), and in the 5-HT-gated MOD-1 receptor, the cation-π interaction can move from the residue equivalent to Trp-183 (Tyr-180) to the residue equivalent to Tyr-234 (Trp-226), because a Trp has the potential for a stronger cation-π interaction than a Tyr (36).
Exchanging Phe-226 for Arg results in nonfunctional receptors, whereas substitution of nearby Ile-228 for Ser does not, although both mutations have a similar effect on ligand binding (an ∼5-fold increase in Kd). These data support previous studies suggesting that different regions of loop C have differential effects on ligand binding and gating (15,19,37).
Conclusions
In this work, we mutated binding-site residues in the 5-HT3A subunit to the corresponding 5-HT3B subunit residues and demonstrated that almost all of the resulting mutant receptors had a significantly reduced antagonist-binding affinity and increased 5-HT EC50. Conversely, we did not observe any significant changes when we introduced A-like mutations in the 5-HT3B subunit. Our docking models show that it is possible for 5-HT and granisetron to bind at A+B−, B+A−, and B+B− interfaces; however, the experimental data do not support the location of these ligands in these heteromeric binding sites. We therefore conclude that the 5-HT3B subunit does not contribute to the binding site, and that 5-HT and granisetron bind to an A+A− interface in heteromeric 5-HT3AB receptors.
Acknowledgments
This study was supported by a grant from the Wellcome Trust (to S.C.R.L.). M.L. received a postdoctoral fellowship from the Swiss National Science Foundation (PA00A-105073). S.C.R.L. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Studies.
References
- 1.Reeves D.C., Lummis S.C.R. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel (review) Mol. Membr. Biol. 2002;19:11–26. doi: 10.1080/09687680110110048. [DOI] [PubMed] [Google Scholar]
- 2.Collingridge G.L., Olsen R.W., Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies P.A., Pistis M., Kirkness E.F. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- 4.Niesler B., Frank B., Rappold G.A. Cloning, physical mapping and expression analysis of the human 5-HT3 serotonin receptor-like genes HTR3C, HTR3D and HTR3E. Gene. 2003;310:101–111. doi: 10.1016/s0378-1119(03)00503-1. [DOI] [PubMed] [Google Scholar]
- 5.Hanna M.C., Davies P.A., Kirkness E.F. Evidence for expression of heteromeric serotonin 5-HT3 receptors in rodents. J. Neurochem. 2000;75:240–247. doi: 10.1046/j.1471-4159.2000.0750240.x. [DOI] [PubMed] [Google Scholar]
- 6.Barrera N.P., Herbert P., Edwardson J.M. Atomic force microscopy reveals the stoichiometry and subunit arrangement of 5-HT3 receptors. Proc. Natl. Acad. Sci. USA. 2005;102:12595–12600. doi: 10.1073/pnas.0503253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubin A.E., Huvar R., Erlander M.G. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J. Biol. Chem. 1999;274:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- 8.Hapfelmeier G., Tredt C., Rammes G. Co-expression of the 5-HT3B serotonin receptor subunit alters the biophysics of the 5-HT3 receptor. Biophys. J. 2003;84:1720–1733. doi: 10.1016/S0006-3495(03)74980-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley S.P., Dunlop J.I., Peters J.A. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- 10.Peters J.A., Kelley S.P., Lambert J.J. The 5-hydroxytryptamine type 3 (5-HT3) receptor reveals a novel determinant of single-channel conductance. Biochem. Soc. Trans. 2004;32:547–552. doi: 10.1042/BST0320547. [DOI] [PubMed] [Google Scholar]
- 11.Peters J.A., Hales T.G., Lambert J.J. Molecular determinants of single-channel conductance and ion selectivity in the Cys-loop family: insights from the 5-HT3 receptor. Trends Pharmacol. Sci. 2005;26:587–594. doi: 10.1016/j.tips.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Hales T.G., Dunlop J.I., Peters J.A. Common determinants of single channel conductance within the large cytoplasmic loop of 5-hydroxytryptamine type 3 and α4β2 nicotinic acetylcholine receptors. J. Biol. Chem. 2006;281:8062–8071. doi: 10.1074/jbc.M513222200. [DOI] [PubMed] [Google Scholar]
- 13.Brejc K., van Dijk W.J., Sixma T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 14.Reeves D.C., Sayed M.F.R., Lummis S.C. Prediction of 5-HT3 receptor agonist-binding residues using homology modeling. Biophys. J. 2003;84:2338–2344. doi: 10.1016/S0006-3495(03)75039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi P.R., Suryanarayanan A., Bikádi Z. Interactions of granisetron with an agonist-free 5-HT3A receptor model. Biochemistry. 2006;45:1099–1105. doi: 10.1021/bi051676f. [DOI] [PubMed] [Google Scholar]
- 16.Maksay G., Bikádi Z., Simonyi M. Binding interactions of antagonists with 5-hydroxytryptamine3A receptor models. J. Recept. Signal Transduct. Res. 2003;23:255–270. doi: 10.1081/rrs-120025568. [DOI] [PubMed] [Google Scholar]
- 17.Yan D., White M.M. Spatial orientation of the antagonist granisetron in the ligand-binding site of the 5-HT3 receptor. Mol. Pharmacol. 2005;68:365–371. doi: 10.1124/mol.105.011957. [DOI] [PubMed] [Google Scholar]
- 18.Tremblay P.B., Kaiser R., Brockmoller J. Variations in the 5-hydroxytryptamine type 3B receptor gene as predictors of the efficacy of antiemetic treatment in cancer patients. J. Clin. Oncol. 2003;21:2147–2155. doi: 10.1200/JCO.2003.05.164. [DOI] [PubMed] [Google Scholar]
- 19.Thompson A.J., Price K.L., Lummis S.C. Locating an antagonist in the 5-HT3 receptor binding site using modeling and radioligand binding. J. Biol. Chem. 2005;280:20476–20482. doi: 10.1074/jbc.M413610200. [DOI] [PubMed] [Google Scholar]
- 20.Shi J., Blundell T.L., Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 21.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 22.Chen C.A., Okayama H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 23.Jordan M., Schallhorn A., Wurm F.M. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liman E.R., Tytgat J., Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 25.Kunkel T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price K.L., Lummis S.C.R. The role of tyrosine residues in the extracellular domain of the 5-hydroxytryptamine3 receptor. J. Biol. Chem. 2004;279:23294–23301. doi: 10.1074/jbc.M314075200. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. Molecular Cloning. A Laboratory Manual. [Google Scholar]
- 28.Turchin A., Lawler J.F. The primer generator: a program that facilitates the selection of oligonucleotides for site-directed mutagenesis. Biotechniques. 1999;26:672–676. doi: 10.2144/99264st02. [DOI] [PubMed] [Google Scholar]
- 29.Bylund D.B., Toews M.L. Radioligand binding methods: practical guide and tips. Am. J. Physiol. 1993;265:L421–L429. doi: 10.1152/ajplung.1993.265.5.L421. [DOI] [PubMed] [Google Scholar]
- 30.Spier A.D., Wotherspoon G., Lummis S.C.R. Antibodies against the extracellular domain of the 5-HT3 receptor label both native and recombinant receptors. Brain Res. Mol. Brain Res. 1999;71:369. doi: 10.1016/s0169-328x(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 31.Reeves D.C., Lummis S.C.R. Detection of human and rodent 5-HT3B receptor subunits by anti-peptide polyclonal antibodies. BMC Neurosci. 2006;7:27–34. doi: 10.1186/1471-2202-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beene D.L., Brandt G.S., Dougherty D.A. Cation-π interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: the anomalous binding properties of nicotine. Biochemistry. 2002;41:10262–10269. doi: 10.1021/bi020266d. [DOI] [PubMed] [Google Scholar]
- 33.Das P., Dillon G.H. Molecular determinants of picrotoxin inhibition of 5-hydroxytryptamine type 3 receptors. J. Pharmacol. Exp. Ther. 2005;314:320–328. doi: 10.1124/jpet.104.080325. [DOI] [PubMed] [Google Scholar]
- 34.Thompson A.J., Lummis S.C.R. Antimalarial drugs inhibit human 5-HT(3) and GABA(A) but not GABA(C) receptors. Br. J. Pharmacol. 2008;153:1686–1696. doi: 10.1038/bjp.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melis C., Lummis S.C.R., Molteni C. Molecular dynamics simulations of GABA binding to the GABAC receptor: the role of Arg104. Biophys. J. 2008;95:4115–4123. doi: 10.1529/biophysj.107.127589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mu T.W., Lester H.A., Dougherty D.A. Different binding orientations for the same agonist at homologous receptors: a lock and key or a simple wedge? J. Am. Chem. Soc. 2003;125:6850–6851. doi: 10.1021/ja0348086. [DOI] [PubMed] [Google Scholar]
- 37.Suryanarayanan A., Joshi P.R., Schulte M.K. The loop C region of the murine 5-HT3A receptor contributes to the differential actions of 5-hydroxytryptamine and m-chlorophenylbiguanide. Biochemistry. 2005;44:9140–9149. doi: 10.1021/bi050661e. [DOI] [PubMed] [Google Scholar]