

Abstract
Hexameric helicases are molecular motor proteins that utilize energy obtained from ATP hydrolysis to translocate along and/or unwind nucleic acids. In this study, we investigate the dynamic behavior of the Simian Virus 40 hexameric helicase bound to DNA by performing molecular dynamics simulations employing a coarse-grained model. Our results elucidate the two most important molecular features of the helicase motion. First, the attractive interactions between the DNA-binding domain of the helicase and the DNA backbone are essential for the helicase to exhibit a unidirectional motion along the DNA strand. Second, the sequence of ATP binding at multiple binding pockets affects the helicase motion. Specifically, concerted ATP binding does not generate a unidirectional motion of the helicase. It is only when the binding of ATP occurs sequentially from one pocket to the next that the helicase moves unidirectionally along the DNA. Interestingly, in the reverse order of sequential ATP binding, the helicase also moves unidirectionally but in the opposite direction. These observations suggest that in nature ATP molecules must distinguish between different available ATP binding pockets of the hexameric helicase in order to function efficiently. To this end, simulations reveal that the binding of ATP in one pocket induces an opening of the next ATP-binding pocket and such an asymmetric deformation may coordinate the sequential ATP binding in a unidirectional manner. Overall, these findings may provide clues toward understanding the mechanism of substrate translocation in other motor proteins.
Introduction
Helicases are ubiquitous enzymes involved in many aspects of nucleic acid metabolism, such as replication, recombination, and repair (1–4). The main function of helicases is to unwind double-stranded (dsDNA) DNA or translocate single-stranded DNA (ssDNA), using energy derived from ATP hydrolysis. Despite several structural and biochemical studies, the details of how ATP binding and hydrolysis are coupled to conformational changes to achieve DNA translocation is not well understood (5–7). Understanding the fundamental mechanism of the helicase motor protein may aid in understanding the functional mechanism of other ATPase motor proteins. Furthermore, as the malfunctioning of helicases is linked to cancer and premature aging (8,9), it is biomedically important to understand how they function at the molecular level.
The E1 helicase of papillomavirus and Simian Virus 40 helicase (SV40) are two structurally well-characterized helicases that belong to the same family (1,10,11). In their functional form, these two helicases assemble to form a ring-shaped structure with six identical protein subunits encircling the DNA in the channel (Fig. 1 a). Based on the static crystal structures and kinetic data, different mechanisms that couple ATP cycling to DNA translocation have been proposed for these two systems (10,11). Gai et al. (10) suggested a concerted nucleotide binding and hydrolysis mechanism for the SV40 helicase. Alternatively, based on the crystal structure of E1 hexameric helicase bound to a single-stranded DNA, a sequential ATP-binding mechanism was proposed (11). According to the sequential mechanism (Fig. 1 c), after binding ATP in the empty pocket, the DNA-binding loop of the protein that makes close contact with the DNA backbone moves forward along the ssDNA in the direction of motion (from the 3′ → 5′ end of the DNA (12,13)). In the next step, another ATP binds in the adjacent pocket, which moves the DNA-binding loop of that subunit forward in the direction of motion. Thus, it is believed that the sequential ATP binding from one pocket to the next coordinates the motion of DNA binding loops, and thereby leads to a processive, unidirectional motion of the hexamer along the ssDNA (Fig. 1 d). The question whether the DNA translocation by helicases occurs via concerted or sequential ATP binding mechanism remains unresolved.
Figure 1.
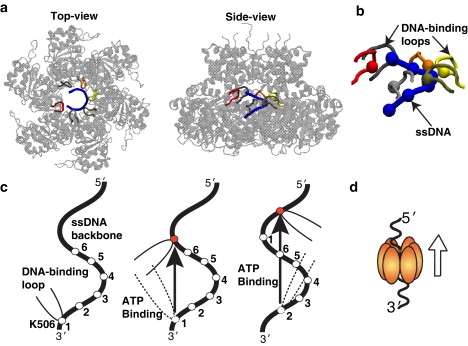
Sequential ATP-binding mechanism proposed for a hexameric helicase, the E1 protein of papillomavirus (11). (a) Crystal structure of the E1 hexameric structure in complex with ssDNA (PDB ID: 2GXA). The ssDNA (blue), which consists of six nucleotides, is encircled in the hexamer channel (cartoon representation), making contacts with the DNA-binding loops (tube representation) of E1 hexamer. (b) Positively-charged lysine residue (K506) at the tip of the DNA binding loop closely interacting with the negatively-charged phosphates of the ssDNA backbone (blue). The position of the lysine residues follow the helical coordinates of the DNA backbone. (c) Schematic of the sequential ATP-binding mechanism: i), ATP binding occurs in the empty pocket, which induces a large conformational change in the associated protein subunit (see Fig. 2d); ii), the DNA-binding loop of the interacting subunit moves in the direction; iii), the ATP binding occurs in the empty pocket adjacent to the ATP-bound pocket and associated conformational change further translocates the DNA-binding loop of that subunit in the direction; and iv), the ATP hydrolysis products are released, emptying the binding pockets for the next cycle (d).
The DNA translocation by the helicase motor proteins involves motions that occur over a vast range of spatial and temporal timescales. Based on the structure of the E1 helicase of papillomavirus, it has been proposed that a single cycle of ATP hydrolysis leads to the movement of ssDNA through the hexamer channel, with a step of one nucleotide in the 3′ → 5′ direction. However, the large nucleotide movements per ATP consumed have been suggested for several other helicases, and they range from one-nucleotide movement per ATP to 10-nucleotide movement per ATP or even higher in certain helicases (14,15). Moreover, the rate of single nucleotide translocation is approximately millisecond-to-second timescales as measured via kinetic experiments for T7 DNA helicase (16,17). Thus, testing hypothesized ATP binding mechanisms is a challenging task for both experimental and theoretical methods. In the experiments, the major difficulties are related to precisely controlling the sequence of ATP binding around the hexameric ring of the helicase and capturing the hexamer's motion (including the DNA) at the molecular level. In contrast, employing computational methods to different ATP binding mechanisms can easily be examined by ensuring that ATP binding occurs in a specific order (e.g., sequential, concerted, etc.) and directly observing the dynamics of the hexameric helicase along the ssDNA. Computer simulations are a powerful tool to investigate such mechanisms at the molecular level (18–22). However, for a protein of the size of hexameric helicase (∼17,600 heavy atoms), performing all-atom simulations over biologically relevant timescales is still prohibitive (see (23) and references therein). Instead, in this case, dynamics of the system can be effectively described by using the coarse-grained representation of the system. In the past, coarse-grained models have been successfully used to capture the dynamics of several large proteins and their complexes at the molecular level (24–29).
To date, two computational studies aimed at understanding the vectorial translocation of helicases have also been performed (30,31). Earlier, Yu et al. (30) investigated the two-domain PcrA helicase translocation. These authors used all-atom simulations on the nanosecond timescale to obtain the mean friction force between the ssDNA and monomeric helicase, with and without ATP binding. Then, a stochastic model with the effective potentials obtained from nanosecond-timescale simulations was built to predict a unidirectional motion of the monomeric helicase. However, this approach may not be feasible for investigating the dynamics of the hexameric helicase, as evaluating the mean forces for multiple ATP-binding and ssDNA-binding states may lead to convergence issues. More recently, Liu et al. (31) explored the vectorial translocation mechanism of LTag hexameric helicase via structure-energy studies of the helicase. These authors built an effective electrostatic free energy surface of the of protein/DNA complex using the linear-response approximation version of the semimacroscopic protein dipoles, i.e., via the Langevin dipoles method, and subsequently performed Langevin dynamics simulations on this surface. This allowed them to capture the unidirectional motion of the helicase. Taken together, both of these computational studies have provided insights into the translocation mechanism in DNA helicases. However, several key conformational dynamics questions regarding the function of the hexameric helicase, such as the cooperativity of the ATP binding and DNA translocation and how the binding of ATP in a specific order may affect the motion of the helicase, remain largely unexplored.
In this study, we investigate the dynamics of a hexameric helicase employing a reduced representation of the hexameric protein and ssDNA. In our coarse-grained model of the hexamer and ssDNA system, although atomic-level details are removed, the most important molecular features for the hexamer motion (i.e., ATP binding, ATP-driven conformational changes in the protein subunit, and DNA-helicase interactions) are included. We also incorporate prior information available from structural and mutation studies of helicases into our initial model. Specifically, to mimic the binding of ligands, distance restraints were imposed between the key residues in the ATP-binding pockets based on the crystal structures of helicase in the different nucleotide binding states (Table S1 in the Supporting Material). Further, experimental mutation studies suggest that the lysine residue on the DNA binding motif of the E1 helicase is essential for the translocase activity (32). This information was incorporated into our model by introducing a weak interaction between the conserved lysine residues in the DNA binding loops and the DNA backbone phosphates of the ssDNA. Including these two essential features in the model allowed us to capture the ligand-dependent, large-scale conformational changes of the helicase and test the hypothesized ligand-binding mechanisms (10,11).
Our results show that the attractive interactions between the DNA-binding domain of the helicase and the DNA backbone along with the conformational changes are equally essential in generating unidirectional motion along the ssDNA. Upon testing hypothesized alternate ATP binding mechanisms, we conclude that DNA translocation preferentially occurs via sequential ATP binding. Notably, the reverse order of ATP binding moves the helicase unidirectionally in the opposite direction. Overall, these results highlight a potential recognition mechanism employed by ATP molecules to distinguish between ATP binding pockets. Further detailed analysis shows that ATP-binding at one pocket induces an opening of the next ATP binding pocket that may be crucial for sequential ATP binding and for generating an unidirectional motion along DNA. We also discuss the implications of these findings for other motor proteins.
Methods
Overall strategy
The hexameric protein structure was modeled from the x-ray structure of the SV40 helicase solved in the absence of ATP, ADP, and DNA. Our coarse-grained hexamer model consists of spherical interaction sites (i.e., residues) connected by a bonded potential and residue-specific Lennard-Jones (LJ) potentials (33) (Fig. 2 a). Similar residue-based models have been successfully used to study protein folding (27–29), where the model parameters were chosen to stabilize the native protein structure and to reproduce the backbone flexibility. We note at this point that there are important differences between our model and similar models used to study multiconfigurational state processes. Our model requires the relaxed form of the protein only. Transition to other functional states occurs as a result of the strain introduced by binding of different substrates, e.g., ATP versus ADP, etc.
Figure 2.
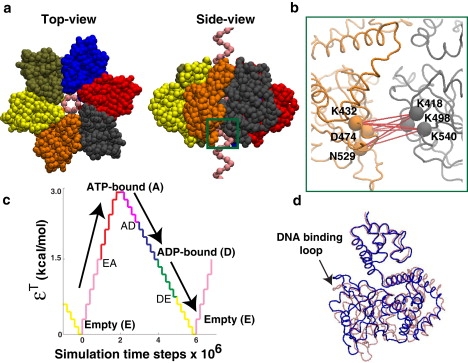
Simulation model for SV40 hexameric helicase. (a) Different views of the hexamer and ssDNA model. The hexamer model was constructed from the crystal structure of SV40 hexameric helicase in the apo-state (PDB ID: 1SVO), and the ssDNA model was built from the crystal structure solved with the channel of E1 hexameric helicase (PDB ID: 2GXA) (see Fig. 1a). Colored spheres represent the C′α values of the hexamer and the phosphates of the ssDNA. The six protein subunits shown in different colors are structurally identical to one another. The ATP-binding pockets are located at the interfaces between the neighboring subunits. (For example, the ATP-binding pocket between the protein subunits colored gray and orange are located within the green box.) (b) Amplified view of ATP-binding pocket. Several key residues that are involved in ATP binding and hydrolysis are labeled. The pairwise distances (red lines) between the key residues undergo large changes from the empty to ATP-bound state, or from the ATP-bound, through ADP-bound, to empty state (Table S1). In the simulations, distance changes between these key residues were mimicked by manipulating the LJ potential with parameter, εT. (c) ATP cycle. At the start of the simulation there are no distance restraints between the key residues (εT = 0). As the simulation progresses, the pairwise distances are slowly forced to be in the crystallographic ATP-bound state by increasing the value of εT in a stepwise manner via the weak ATP binding state (EA). After the tight ATP-bound state (A) is reached, εT is gradually reduced via the weak ADP-bound state (AD) to simulate the ADP-bound (D) and eventually the empty state (E) via the weak ADP release state (DE). (d) Conformational change of the model hexamer via ATP binding (red and blue represent the structure before and after the ATP binding event (RMSD = 4.3 Å)). The ATP-bound distance restraints were applied to all six binding pockets of the apo-hexamer. The resulting conformational change in all the protein subunits was similar. For clarity, only one protein subunit is shown.
The ssDNA was represented as a helical strand that consisted of spherical interaction site, coincident with the phosphates. The ssDNA was inserted into the hexamer channel (Fig. 2 a). The repeating unit of this helical strand consisted of six bases with a diameter and pitch of 14 Å and 3.3 Å, respectively, as in the x-ray structure of the ssDNA bound to the channel of E1 hexameric helicase (see Fig. 1 a). Attractive interactions were introduced between the lysine residues (K512) at the tip of the DNA-binding loops and the phosphates of the ssDNA, to mimic the close contacts between the DNA binding loops and the DNA backbone observed in the channel of E1 hexamer. During the simulations, the helical structure of the ssDNA was fixed, whereas the hexamer was allowed to move freely along the DNA strand. The assumptions and limitations of our coarse-grained model are discussed in Scheme S1 (Supporting Material). The modeling and simulation details of the hexameric helicase are as follows.
Construction of the coarse-grained hexamer model
We followed the procedures described in Hoang and Cieplak (27) to build a coarse-grained model of SV40 hexameric helicase. The Cα coordinates were taken from the nucleotide-free crystal structure of the hexamer (Protein DataBank (PDB) ID: 1SVO). Adjacent pairs of Cα atoms were bonded with a bonded potential,
| (1) |
where rij is the distance between the Cα atoms i and j, d0 is the equilibrium distance (= 3.8 Å), and k1 and k2 are the spring constants (k1 = 3.0 kcal/Å2, k2 = 300.0 kcal/Å4). Nonadjacent pairs of Cα atoms were classified into two types: a native contact pair if rij < 8.0 Å in the crystal structure, or nonnative contact pair if otherwise. The native contact pairs were interconnected through a LJ potential,
| (2) |
where is the corresponding interatomic distance in the apo-crystal structure (PDBID: 1SVO). The LJ energy parameter ε was set at 3.0 kcal/mol. This potential was truncated and shifted to zero at rij = 18.0 Å. The nonnative contact interactions were modeled by a repulsive potential,
| (3) |
if rij < a0, and 0 if otherwise. The distance a0 is the average of over all native-contact pairs (= 5.5 Å).
Idealized and reduced representation of ssDNA
The DNA model was constructed from the crystal structure of ssDNA solved with E1 hexameric helicase (Fig. 1). Because the ssDNA segment was short (i.e., six bases) and made only one helical turn, first we fitted a perfect helix to the backbone (i.e., phosphates) of that ssDNA, and then repeated the structure. The following formula was used for the fitting:
| (4) |
Here n is the phosphate index (n = 1, 2, …, 6) and θ is the turn per phosphate (= 2π/6). The two parameters, radius R and rise per phosphate Z, were varied between 6.0 Å and 8.0 Å and between 2.5 Å and 4.5 Å, respectively. The best fit to the crystallographic ssDNA was obtained for R = 6.4 Å and Z = 3.3 Å, with root mean-square deviations (RMSDs) for phosphate atoms of 0.46 Å. We note that the ssDNA conformation in the channel may adopt a coiled or linear conformation. However, due to the intrinsic helical nature of ssDNA, and the strong electrostatic interactions between the positively charged DNA-binding loops and the negatively-charged DNA backbone, it is expected that a similar helical structure will be continuously formed near the DNA-binding loops during the translocation process.
DNA-hexamer interactions
Close contacts between the DNA-binding loops and the ssDNA backbone are observed in the channel of the E1 hexameric helicase (Fig. 1). In our model, these attractive interactions were mimicked by an LJ potential introduced between the lysine residues (K512) at the tip of the loops and the phosphates on the backbone as given by the following equation:
| (5) |
This potential was truncated and shifted to zero at 18.0 Å. The equilibrium distance was obtained from the corresponding distance in the crystal structure of the E1 complexed with the ssDNA (= 4.6 Å). The other residues were set to be repulsive to the ssDNA backbone, only if they overlapped with one another. The repulsive potential was given by
| (6) |
if , and 0 if otherwise.
ATP cycle: modeling ATP binding and ADP release
During the ATP cycle, the helicase motor goes through three major conformational states: 1), ATP binding; 2), ATP hydrolysis; and 3), ADP release. These three major states are labeled as A, D, and E, respectively, in the schematic illustrating the ATP cycle (Fig. 2 c). In addition, there are three intermediate states corresponding to weak ATP binding, weak ADP binding, and a weak ADP release between the major conformational states. These states are labeled as EA, AD, and DE, respectively. The existence of intermediate states was indicated by crystallographically resolved ADP-like states with active site interactions between those of ADP-bound and ATP-bound structures (11). In the simulations, these states represent a gradual change in the substrate-binding site conformation transitioning to the next major conformational state. For example, the ATP binding procedure can be divided into an initial ATP docking state from empty or Apo (E) to weak binding state (EA), and a binding transition state leading to the tight bound state (A) (Fig. 2 c). Throughout the text, we refer to tightly bound ATP or ADP states as simply ATP- or ADP-bound states.
Incorporating ATP-binding events in the coarse-grained model
In a coarse-grained model, the ability to account for the explicit local interactions with a ligand (i.e., ATP or ADP) in the active site pocket is a challenging task. In this work, to incorporate the ATP binding events (i.e., binding, hydrolysis, and product release) into the model of the hexameric helicase, distance restraints were imposed between key residues in the binding pockets (Fig. 2 b). The key residues were selected from the conserved sequence motifs for ATP binding in superfamily III helicases (5,11): lysine in the Walker A motif (K432), aspartic acid in Walker B (D474), asparagine in Sensor I (N529), lysine in Sensor II (K418), arginine in Sensor III (R498), and arginine in Arginine Finger (R540). As shown in Fig. 2 b, the first three residues belong to one subunit, and the last three to the adjacent subunit. In the ATP-bound and ADP-bound x-ray structures (PDB ID: 1SVM and 1SVL), intersubunit distances between these key residues become significantly smaller than those observed in the apo x-ray structure (PDB ID: 1SVO; and see Table S1). In our simulations, these distances were modulated by changing the parameter εT of the LJ potential,
| (7) |
where is the crystallographic distance obtained from the ATP-bound crystal structure (PDB ID: 1SVM). The ATP binding event was mimicked by gradually increasing the parameter εT from 0.0 to 3.0 kcal/mol (see Fig. 2 c). As εT was increased, the distances between the key residues, rij, were switched from the apo- to the ATP-bound state. During the release of the ATP-bound distance restraints, rij became very similar to the ADP-bound distances at εT = 1.5 kcal/mol and eventually became similar to the distances found in the apo-conformation at εT = 0.0 kcal/mol. The change in distances between the key residues in the ATP-binding pocket resulted in a large conformational change of the entire protein structure (Fig. 2 d). The RMSD between the model hexamers for the empty and ATP-bound state was 4.3 Å, which is consistent with the RMSD between the apo and ATP-bound x-ray structures of SV40 hexamer (10).
Molecular dynamics protocol
The ssDNA helical strand was inserted in the central channel of the apo hexamer and this protein-DNA complex was relaxed by molecular dynamics (MD) simulations. All MD simulations were performed at a constant temperature of 300 K using a Nosé-Hoover thermostat. Depending on the ATP-binding mechanisms (i.e., sequential, concerted, etc.), the same or different initial values of εT were assigned to the six ATP binding pockets. During the simulations, the εT was increased or decreased in a gradual manner, following the same ATP cycle (described above) for the different ATP-binding mechanisms (see Fig. 2 c). The time interval of transition from one major conformational state to the other was set at 2,000,000 time steps (i.e., apo (E) (εT = 0.0 kcal/mol) → ATP-bound (A) (εT = 3.0 kcal/mol); ATP-bound (A) (εT = 3.0 kcal/mol) → ADP-bound (D) (εT = 1.5 kcal/mol); and ADP-bound (D) (εT = 1.5 kcal/mol) → apo (E) (εT = 0.0 kcal/mol)).
Results and Discussions
Coupling between the conformational changes and DNA-hexamer interactions is required for unidirectional motion of the helicase
We simulated hexamer motion using the most probable sequential ATP-binding mechanism proposed for the E1 hexameric helicase based on the structural studies of Enemark and Joshua-Tor (11). The MD simulations were performed at a constant temperature of 300 K over the course of 40,000,000 time steps. As shown in the schematic of the sequential ATP binding mechanism (Fig. 3 a), ATP was allowed to bind sequentially in one empty ATP binding pocket (E) and then the next adjacent empty pocket at every 1,000,000 time steps. Following the ATP cycle (Fig. 2 c), the state of each ATP-binding pocket was gradually turned over to the next major conformational state (e.g., from the empty (E) to the tight-bound ATP-bound state (A) via the weak-binding state (EA)) at every 2,000,000 time steps. In addition, in our simulations the salt-bridge interactions between the lysine residue on the DNA binding motif and DNA phosphate backbone were introduced via a weak interaction potential (see Methods).
Figure 3.
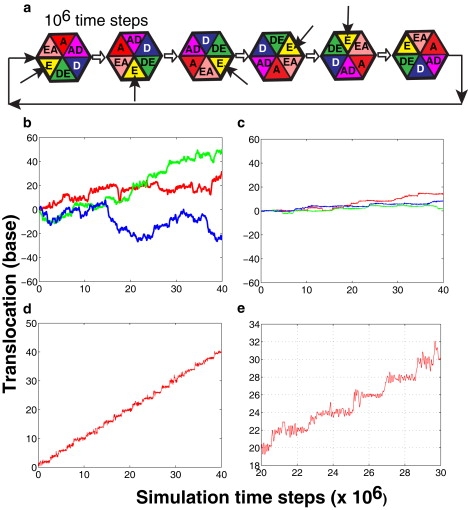
Motion of the hexamer along the ssDNA with the sequential ATP binding mechanism. (a) Schematic diagram of the sequential ATP-binding mechanism used in the simulations. Color represents the state of the ATP binding pocket (see Fig. 2c). Each binding pocket follows the same ATP cycle but starts in a different state. Consequently, ATP binding occurs sequentially from one pocket to the next, at every 106 time steps. The arrow indicates the empty pocket where the ATP binding event starts. (b–e) Trajectory of the center-of-mass of the hexamer, captured over the course of 40 ATP binding events (40 × 106 simulation time steps). All trajectories are scaled by the base pitch of the ssDNA model (3.3 Å). The upward direction corresponds to the forward direction of motion (3′ → 5′ end of the ssDNA). (b) Weakly attractive interactions between the DNA-binding loops and the DNA backbone (εDNA = 1.5 kcal/mol) result in a random motion of the hexamer. The three trajectories shown in different colors were obtained from the simulations started with different initial conditions (i.e., hexamer's position along the ssDNA). (c) Interestingly, strong attractive interactions between the hexamer and ssDNA (εDNA = 7.5 kcal/mol) eliminate all motions along the ssDNA. (d) In a moderately attractive case (εDNA = 4.5 kcal/mol), unidirectional motion of the hexamer is observed. (e) Magnified view of the trajectory undergoing unidirectional motion.
We tested different scenarios where the strength of DNA-hexamer interactions was varied by modulating a parameter, εDNA (Eq. 5), in the interaction potential. Fig. 3 b shows the results of a simulation for the case of a weakly-attractive interaction between the ssDNA and the hexamer (εDNA = 1.5 kcal/mol). As shown, the hexamer moves forward and backward, but no unidirectional motion is observed. The absence of directionality in the helicase motion is consistent with the experimental observations which showed that the mutation of the lysine residue at the tip of the DNA-binding loop (K512 for SV40; K506 for E1) to a nonpolar residue significantly reduces the helicase activity (32,34,35). On the other hand, strongly attractive interactions between K512 and the DNA phosphates (εDNA = 7.5 kcal/mol) immobilize the hexamer in its initial position (Fig. 3 c). This result is equivalent to cross-linking experiments of the DNA-binding loop to the DNA backbone (36).
Interestingly, for the case of moderately attractive interactions between the DNA-binding loop and the DNA backbone (εDNA = 4.5 kcal/mol), the hexamer exhibits a unidirectional motion along the ssDNA (Fig. 3 d). Over the course of 40 ATP-binding events, the hexamer moves forward by ∼40 bases (i.e., ∼13 nm). The step-size of one base per ATP observed in our simulations for SV40 helicase is consistent with the step-size of one basepair per ATP measured for DnaB helicase that unwinds dsDNA (37), and also hypothesized based on the structural studies (11). At a finer resolution, a stepwise motion of the hexamer is observed (Fig. 3 e). These stepwise jumps are directly related to the conformational changes in a protein subunit, particularly the change in the DNA-binding loop as it ratchets from one ssDNA contact to the next (38). A simulation snapshot in Fig. S1 (Supporting Material) shows the movement of one protein subunit along the ssDNA. Clearly, upon ATP binding, both the DNA-binding loop and the protein subunit move forward by one helical turn. After the ATP-binding event, the protein structure gradually relaxes back to the initial apo state. During the release process, the position of the protein subunit along the ssDNA remains the same due to the interaction between the lysine at the tip of the loop and the DNA backbone. Upon the next ATP binding at the same pocket, the interaction between the loop and the ssDNA breaks and the protein subunit shifts further up. Overall, these studies show that the coupling between conformational changes and optimal DNA-hexamer interactions is essential to capture unidirectional motion of the helicase.
Sequential versus other ATP-binding mechanisms
One of the critical factors to capture the unidirectional hexamer motion is the sequence of ATP binding at the six binding pockets. For example, we find that the concerted mechanism, which was proposed based on the structural studies for the SV40 helicase (10), does not translocate the hexamer along the ssDNA (Fig. 4, a and b). In our simulations, we modeled the concerted mechanism by mimicking a simultaneous ATP binding event in all six ATP binding pockets at every 6,000,000 time steps. The resulting conformational change of the entire hexamer generated a large inertia along the ssDNA. As a result of this inertia, the loops bound to the ssDNA were easily detached even during the transition from the ATP-bound to the empty state, unlike the jump mechanism observed in the sequential-binding mechanism (Fig. S1).
Figure 4.
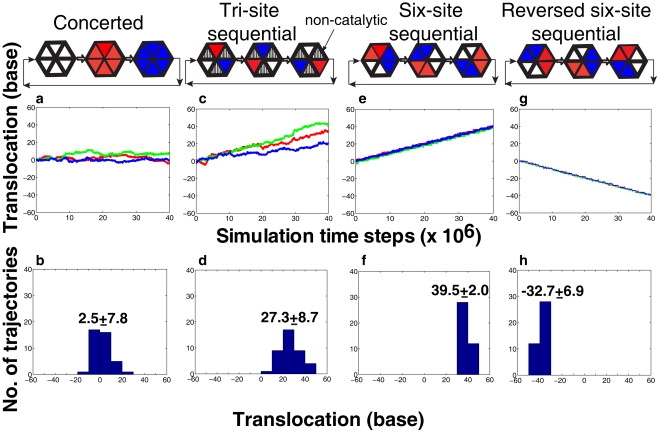
Hexamer's motion with different ATP-binding mechanisms: (a and b) concerted, (c and d) tri-site sequential, and (g and h) reversed six-site sequential mechanism. The six-site sequential mechanism in panels e and f is same as that used in Fig. 3. (Middle row) Three independent trajectories of the hexamer. (Bottom row) Distribution of total bases translocated from 40 independent molecular dynamics trajectories started with the different initial conditions. The rate of ATP binding is 6:6 (number of ATP/106 time steps) in the case of concerted ATP binding mechanism; 1:2 (number of ATP/106 time steps) in the case of tri-site sequential mechanism; and 1:1 (number of ATP/106 time steps) in the case of six-site sequential mechanism.
Fig. 4, c and d, shows the hexamer motion for a three-site sequential ATP-binding model proposed for gp4 protein, a hexameric helicase of bacteriophage T7 (39). In this case, the ATP molecule is only allowed to bind at every alternate binding pocket. Thus, the ATP binding occurs sequentially from one active pocket to the next active pocket at every 2,000,000 time steps. This mechanism is quite similar to the sequential ATP-binding mechanism of the F1-ATPase molecular motor where the catalytic and the noncatalytic binding pockets are alternatively located around the hexamer ring (40).
As shown in Fig. 4 c, a unidirectional motion of the hexamer along the DNA strand is observed for each trajectory in the three-site sequential mechanism. After averaging over 40 independent trajectories, the total bases translocated at the expense of 20 ATP (over 40,000,000 time steps) is ∼27 nucleotide bases. This result agrees with the experimentally measured step-size of one base per ATP hydrolyzed, also seen in the six-site sequential ATP mechanism (37). However, the trajectories are not as robust as those observed in the six-site sequential case (Fig. 4 e). Interestingly, ∼10% of the trajectories reach 40 base translocation just after 20 ATP-binding events (Fig. 4 d). This finding suggests that the step-size larger than the ideal limit may occur in some helicases as suggested by experiments (16,17), which would be unity for the six-site sequential case.
In the three-site case, the hexamer utilizes the same driving force as described in Fig. 3 d. Upon ATP binding, the two neighboring loops from the catalytic and the noncatalytic binding pockets move forward in a concerted manner through a conformational change of the catalytic protein subunit. However, the loop associated with noncatalytic binding pocket (i.e., empty state) tends to revert to its previous position. Therefore, those loops are not stabilized at the new forward positions, and often fall down to the initial backward positions before the next ATP-binding event occurs (in Fig. 4 c, note the points where the red and blue lines depart from the green line).
We also examined the reversed (or clockwise) sequential pathway (Fig. 4, g and h). Interestingly, the hexamer continuously moves backward along the ssDNA with the same step-size observed in the counterclockwise case (Fig. 4, e and f), i.e., one base per ATP. In this reverse motion, the conformational change of the subunit corresponds to the transition of the ATP-bound state (A) to the empty state (E).
How do ATP molecules distinguish between different binding pockets?
The strong dependence of the hexamer motion on the sequence of ATP binding (e.g., clockwise or counterclockwise sequential) implies that in nature, ATP molecules must be able to distinguish between different ATP binding pockets. We hypothesize that, in the case where there are two or more empty binding pockets available in the hexamer, the deformation of the pockets (i.e., opening or closing) could be a key criterion to differentiate one binding pocket from the others. To test this hypothesis, MD simulations with two different starting hexameric helicase models were performed: 1), only ssDNA was inserted into the channel of the apo hexameric helicase; and 2), ATP was bound in one of the pockets and its influence on the size of other ATP binding pockets was determined in the absence of the ssDNA. In the first case, as shown in Fig. 5, top, the largest closing is observed in the pocket 1, in which the associated loop is bound to the most forward position (5′ direction) of the ssDNA, while binding pocket 4, whose associated loop is bound to the backward position (3′ direction) of the ssDNA, is opened. The DNA binding loops of pockets 5 and 6 are not tightly bound to the ssDNA backbone. If a closing of the empty binding pockets increases the ATP-binding affinity, this asymmetric deformation of six empty pockets may naturally lead to an initial conformation of the sequential ATP-binding mechanism (see Fig. 3 a).
Figure 5.
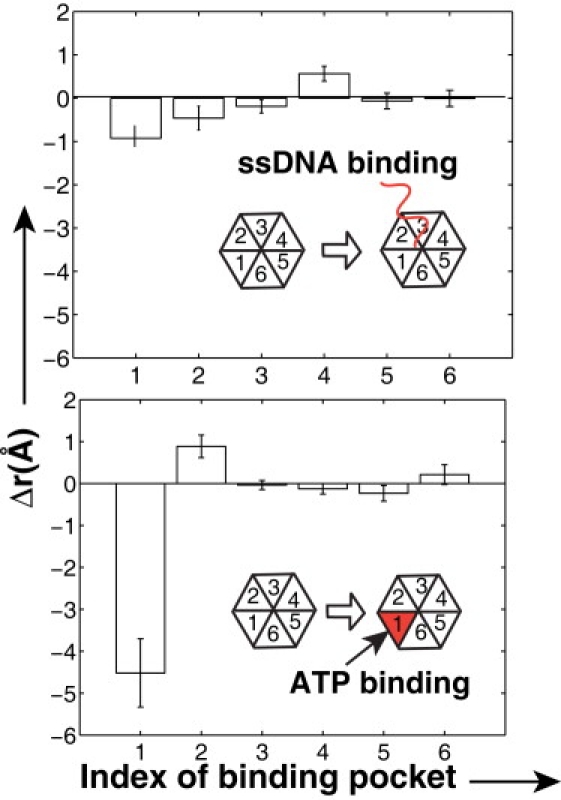
Asymmetric deformation of the ATP-binding pockets on binding either ssDNA or only ATP in one of the pockets. The deformation Δr was calculated from a change in distances between the key residues in the binding pocket before and after the ATP (or DNA) binding. A positive value of Δr represents an opening of the binding pocket. The error bar was obtained from 10 independent simulation runs. (Top) DNA binding in the channel of apo-hexamer. (Inset) Schematic of DNA-binding loop and ssDNA. The pocket number 1 corresponds to the protein subunit whose DNA-binding loop is nearest to the 5′ end of DNA in the 3′ → 5′ direction. (Bottom) ATP binding in the one of the pocket without DNA. Here, ATP was bound to the empty pocket number 1 of the apo-hexamer structure.
In the second case, where ATP-bound distance restraints were applied on the pocket 1 of the apo-hexamer structure in the absence of the ssDNA (Fig. 5, bottom), it is observed that the ATP binding at pocket 1 induces a relatively large opening of the empty pocket 2 (clockwise adjacent to pocket 1). On the other hand, pocket 6 (counterclockwise to pocket 1) and the other three empty pockets 3–5, exhibit little deformation. The direction of a pocket opening is opposite to the desired direction for the (counterclockwise) sequential ATP-binding mechanism in Fig. 3 a. When the counterclockwise pocket is occupied by ATP, this unidirectional transfer of mechanical energy may be used to trigger ATP hydrolysis for enhancing the product release.
Conclusions
Our dynamic representation of the hexameric helicase translocation along ssDNA fills a gap between the static information obtained from the atomistic crystal structures and the bulk experimental data (e.g., step-size) obtained from dynamic helicase activity studies. Using a coarse-grained model that includes the ATP-binding events and the DNA-hexamer interactions at the molecular level, we demonstrate that the sequential ATP-binding mechanism can generate a unidirectional motion of the hexameric helicase along ssDNA with the step-size of one base per ATP, consistent with experimental data for the translocation rates. Importantly, our results show that the hexamer may have intrinsic structural properties to coordinate ATP binding in a sequential order, which can be affected by either binding the ssDNA in the channel or upon binding ATP in the active site pocket. These novel suggestions from our computational studies can be tested experimentally using single molecule and rapid kinetics techniques that can directly monitor motor function. For example, the two state-of-the-art experimental techniques to our knowledge that may be able to test our findings in solution are 1), the fluorescence stopped-quench-flow method (41) and 2), single molecule studies that use magnetic tweezers (42). Specifically, using the fluorescence-quench-flow method, one can explore kinetics of complex helicase interactions with nucleic acids and ligands in solution, and the examine the mechanism of the functioning of the hexameric helicase.
Recently, coarse-grained models have had some success in studying conformational changes of large biomolecular complexes (24,43,44). Notably, the rotational motion of the shaft protein for F1-ATPase was investigated using the switching Gō model (24). In this model, the change between different nucleotide binding states was modeled as a vertical excitation, allowing for switching between two single basin models of the endpoints. Recently, Okazaki et al. suggested an improved multiple-basin model to account for the probability that proteins sample multiple conformations during conformational transitions (43). However, the important ingredient that is still missing in these models is the ability to account for the local interactions with a ligand in the active site pocket. Comparatively, in our coarse-grained model, DNA-helicase interactions with ATP were included by imposing distance restraints between the key residues in the ATP-binding pockets. This enhancement in the model allowed the system to evolve naturally between different conformational states by simply turning on and off the distance restraints, which very closely mimics the binding and release of ATP.
Hexameric helicases discussed here belong to the AAA+ family of proteins and thus the detailed sequential ATP-binding mechanism discovered for the hexameric helicase may have implications for other AAA+ proteins. In particular, interesting comparison can be drawn with proteasomes that are involved in protein degradation by unfolding and translocating proteins into the degradation chamber for proteolysis (34). The ATP hydrolysis is required for the protein denaturation and translocation steps of degradation. Compared to the helicases, the number of ATPs consumed per residue translocated or degraded is high and substrate-dependent for the proteasomes. As our studies show, the successful translocation of the hexameric helicases requires relatively strong interactions between charged DNA backbone phosphates and polar residues of DNA binding loops. We hypothesize that the poor efficiency of the proteasomes may arise due to the lack of a regular shape and properties of translocating polypeptides resulting in weak coupling between the substrate and the hexameric subunits. Interestingly, it has been suggested that ATP hydrolysis of some proteases may occur in a probabilistic rather than strictly-ordered sequential manner (34). We are currently exploring the detailed dynamics of other AAA+ family members in search of a unifying mechanism of these fascinating molecular motors. In future work, we will perform quantitative analysis of the ATP binding and hydrolysis steps, and make a thorough comparison with experimentally measurable quantities such as ATP energy efficiency, to gain better understanding of these fascinating molecular motors.
Supporting Material
One table, one figure, one scheme, and two movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00053-6.
Supporting Material
Acknowledgments
This work was supported by the National Institutes of Health through the Center for Multi-Scale Modeling Tools for Structural Biology (grant No. RR012255) and the National Science Foundation through the Center for Theoretical Biological Physics (grant No. PHY0216576).
Footnotes
Kenji Yoshimoto's present address is Globalfoundries c/o IBM, 2070 Route 52, Mailstop 10A, Hopewell Junction, NY 12533.
References
- 1.Enemark E.J., Joshua-Tor L. On helicases and other motor proteins. Curr. Opin. Struct. Biol. 2008;18:243–257. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohman T.M., Bjornson K.P. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 3.Patel S.S., Picha K.M. Structure and function of hexameric helicases. Annu. Rev. Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 4.Hickman A.B., Dyda F. Binding and unwinding: SF3 viral helicases. Curr. Opin. Struct. Biol. 2005;15:77–85. doi: 10.1016/j.sbi.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Erzberger J.P., Berger J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 6.Wessel R., Schweizer J., Stahl H. Simian virus 40 T-antigen DNA helicase is a hexamer which forms a binary complex during bidirectional unwinding from the viral origin of DNA replication. J. Virol. 1992;66:804–815. doi: 10.1128/jvi.66.2.804-815.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouts E.T., Yu X., Botchan M.R. Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J. Biol. Chem. 1999;274:4447–4458. doi: 10.1074/jbc.274.7.4447. [DOI] [PubMed] [Google Scholar]
- 8.van Brabant A.J., Stan R., Ellis N.A. DNA helicases, genomic instability, and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2000;1:409–459. doi: 10.1146/annurev.genom.1.1.409. [DOI] [PubMed] [Google Scholar]
- 9.Mohaghegh P., Hickson I.D. DNA helicase deficiencies associated with cancer predisposition and premature aging disorders. Hum. Mol. Genet. 2001;10:741–746. doi: 10.1093/hmg/10.7.741. [DOI] [PubMed] [Google Scholar]
- 10.Gai D., Zhao R., Chen X.S. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell. 2004;119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 11.Enemark E.J., Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 12.Goetz G.S., Dean F.B., Matson S.W. The unwinding of duplex regions in DNA by the simian virus 40 large tumor antigen-associated DNA helicase activity. J. Biol. Chem. 1988;263:383–392. [PubMed] [Google Scholar]
- 13.Seo Y.S., Müller F., Hurwitz J. Bovine papilloma virus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc. Natl. Acad. Sci. USA. 1993;90:2865–2869. doi: 10.1073/pnas.90.7.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D.E., Narayan M., Patel S.S. T7 DNA helicase: a molecular motor that processively and unidirectionally translocates along single-stranded DNA. J. Mol. Biol. 2002;321:807–819. doi: 10.1016/s0022-2836(02)00733-7. [DOI] [PubMed] [Google Scholar]
- 15.Johnson D.S., Bai L., Wang M.D. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell. 2007;129:1299–1309. doi: 10.1016/j.cell.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucius A.L., Vindigni A., Lohman T.M. DNA unwinding step-size of E. coli RecBCD helicase determined from single turnover chemical quenched-flow kinetic studies. J. Mol. Biol. 2002;324:409–428. doi: 10.1016/s0022-2836(02)01067-7. [DOI] [PubMed] [Google Scholar]
- 17.Serebrov V., Pyle A.M. Periodic cycles of RNA unwinding and pausing by hepatitis C virus NS3 helicase. Nature. 2004;430:476–480. doi: 10.1038/nature02704. [DOI] [PubMed] [Google Scholar]
- 18.Brooks C.L. Protein and peptide folding explored with molecular simulations. Acc. Chem. Res. 2002;35:447–454. doi: 10.1021/ar0100172. [DOI] [PubMed] [Google Scholar]
- 19.Karplus M., McCammon J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 20.Karplus M., Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arora K., Brooks C.L. Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arora K., Brooks C.L. Functionally important conformations of the Met20 loop in dihydrofolate reductase are populated by rapid thermal fluctuations. J. Am. Chem. Soc. 2009;131:5642–5647. doi: 10.1021/ja9000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlick T. Springer-Verlag; New York: 2002. Molecular Modeling and Simulation: An Interdisciplinary Guide. [Google Scholar]
- 24.Koga N., Takada S. Folding-based molecular simulations reveal mechanisms of the rotary motor F1-ATPase. Proc. Natl. Acad. Sci. USA. 2006;103:5367–5372. doi: 10.1073/pnas.0509642103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elcock A.H. Molecular simulations of cotranslational protein folding: fragment stabilities, folding cooperativity, and trapping in the ribosome. PLOS Comput. Biol. 2006;2:824–841. doi: 10.1371/journal.pcbi.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.West D.K., Olmsted P.D., Paci E. Mechanical unfolding revisited through a simple but realistic model. J. Chem. Phys. 2006;124:154909. doi: 10.1063/1.2185100. [DOI] [PubMed] [Google Scholar]
- 27.Hoang T.X., Cieplak M. Molecular dynamics of folding of secondary structures in Gō-type models of proteins. J. Chem. Phys. 2000;112:6851–6862. [Google Scholar]
- 28.Shea J.E., Brooks C.L. From folding theories to folding proteins: a review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- 29.Onuchic J.N., Wolynes P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Yu J., Ha T., Schulten K. How directional translocation is regulated in a DNA helicase motor. Biophys. J. 2007;93:3783–3797. doi: 10.1529/biophysj.107.109546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H., Shi Y., Warshel A. Simulating the electrostatic guidance of the vectorial translocations in hexameric helicases and translocases. Proc. Natl. Acad. Sci. USA. 2009;106:7449–7454. doi: 10.1073/pnas.0900532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen J., Gai D., Chen X.S. The roles of the residues on the channel β-hairpin and loop structures of Simian virus 40 hexameric helicase. Proc. Natl. Acad. Sci. USA. 2005;102:11248–11253. doi: 10.1073/pnas.0409646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen M.P., Tildesley D.J. Oxford University Press; New York: 1989. Computer Simulation of Liquids. [Google Scholar]
- 34.Martin A., Baker T.A., Sauer R.T. Rebuilt AAA+motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 35.Liu X., Schuck S., Stenlund A. Adjacent residues in the E1 initiator β-hairpin define different roles of the β-hairpin in Ori melting, helicase loading, and helicase activity. Mol. Cell. 2007;25:825–837. doi: 10.1016/j.molcel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Wong I., Lohman T.M. ATPase activity of Escherichia coli Rep helicase crosslinked to single-stranded DNA: implications for ATP driven helicase translocation. Proc. Natl. Acad. Sci. USA. 1996;93:10051–10056. doi: 10.1073/pnas.93.19.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galletto R., Jezewska M.J., Bujalowski W. Unzipping mechanism of the double-stranded DNA unwinding by a hexameric helicase: quantitative analysis of the rate of the dsDNA unwinding, processivity and kinetic step-size of the Escherichia coli DnaB helicase using rapid quench-flow method. J. Mol. Biol. 2004;343:83–99. doi: 10.1016/j.jmb.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 38.Wang H., Oster G. Ratchets, power strokes, and molecular motors. App. Phys. A. 2002;75:315–323. [Google Scholar]
- 39.Hingorani M.M., Washington M.T., Patel S.S. The dTTPase mechanism of T7 DNA helicase resembles the binding change mechanism of the F1-ATPase. Proc. Natl. Acad. Sci. USA. 1997;94:5012–5017. doi: 10.1073/pnas.94.10.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyer P.D. The ATP synthase—a splendid molecular machine. Annu. Rev. Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 41.Adreeva I.E., Roychowdhury A., Bujalowski W. Mechanisms of nucleotide cofactor interactions with the RepA protein of plasmid RSF1010. Binding dynamics studied using fluorescence stopped-flow method. Biochemistry. 2009;48:10620–10636. doi: 10.1021/bi900940q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lionnet T., Spiering M.M., Croquette V. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:19790–19795. doi: 10.1073/pnas.0709793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okazaki K., Koga N., Wolynes P.G. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2006;103:11844–11849. doi: 10.1073/pnas.0604375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pu J., Karplus M. How subunit coupling produces the γ-subunit rotary motion in F1-ATPase. Proc. Natl. Acad. Sci. USA. 2008;105:1192–1197. doi: 10.1073/pnas.0708746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.