

Abstract
The serotonin transporter (SERT) terminates neurotransmission by removing serotonin from the synaptic cleft. In addition, it is the site of action of antidepressants (which block the transporter) and of amphetamines (which induce substrate efflux). We explored the functional importance of the N terminus in mediating the action of amphetamines by focusing initially on the highly conserved threonine residue at position 81, a candidate site for phosphorylation by protein kinase C. Molecular dynamics simulations of the wild type SERT, compared with its mutations SERTT81A and SERTT81D, suggested structural changes in the inner vestibule indicative of an opening of the inner vestibule. Predictions from this model (e.g. the preferential accumulation of SERTT81A in the inward conformation, its reduced turnover number, and a larger distance between its N and C termini) were verified. Most importantly, SERTT81A (and the homologous mutations in noradrenaline and dopamine) failed to support amphetamine-induced efflux, and this was not remedied by aspartate at this position. Amphetamine-induced currents through SERTT81A were comparable with those through the wild type transporter. Both abundant Na+ entry and accumulation of SERTT81A in the inward facing conformation ought to favor amphetamine-induced efflux. Thus, we surmised that the N terminus must play a direct role in driving the transporter into a state that supports amphetamine-induced efflux. This hypothesis was verified by truncating the first 64 amino acids and by tethering the N terminus to an additional transmembrane helix. Either modification abolished amphetamine-induced efflux. We therefore conclude that the N terminus of monoamine transporters acts as a lever that sustains reverse transport.
Keywords: Phosphorylation, Transport/Neurotransmitter, Oocyte, Transport Drugs, Zinc, Amphetamine, Carrier-mediated Efflux, Fluorescence Resonance Energy Transfer Microscopy, Serotonin
Introduction
The transporters for the monoamines noradrenaline (NET),4 dopamine (DAT), and serotonin (5-hydroxytryptamine (5HT); SERT) mediate reuptake of previously released substrates from the synaptic cleft to rapidly terminate neurotransmission (1). The monoamine transporters belong to the neurotransmitter:sodium symporter family (2). The structure of their hydrophobic core is well understood, and educated guesses can be made based on several crystal structures that have been solved for the bacterial homolog LeuTAa (3) as follows: the hydrophobic core is comprised of 12 transmembrane-spanning segments (TM) that are arranged in two pseudo-symmetrical helical repeats of TM1–TM5 and TM6–TM10, whereas TM11 and TM12 are thought to represent the dimer interface. Movements within the inverted repeats can readily account for a conformational cycle predicted by the alternating access model (4) as follows: substrate and co-transported ion enter via an outer vestibule created by the outward facing conformation; upon binding to the substrate-binding site, access from the outer vestibule is occluded. Subsequently, the substrate and co-transported ions are released into the interior of the cell when the transporter adopts the inward facing conformation (that is pseudo-symmetrical to the outward facing conformation). LeuTAa only has short extensions at the N and C termini (5). Hence, the structures of the N and C termini of monoamine transporters cannot be inferred from LeuTAa. The intracellular termini, however, are important and have been implicated in two functionally important areas as follows: (i) in transporter trafficking (6–10) and (ii) transporter phosphorylation by different kinases (protein kinase C (PKC); see Ref. 11) and calmodulin-dependent kinase IIα (12, 13).
Stimulation of PKC by phorbol esters (14), the diacylglycerol analog 1-oleoyl-2-acetyl-sn-glycerol (15) or by receptor-mediated activation (16), results in increased N-terminal phosphorylation of DAT. This may regulate the availability of transporters at the cell surface, because phosphorylated transporters are thought to be subject to dynamin-dependent internalization (14). This interpretation has been questioned; elimination of all consensus phosphorylation sites by mutagenesis both in SERT and in DAT did not lead to any appreciable deficit in internalization (16, 17). In addition, transporter phosphorylation is thought to be relevant for the action of amphetamine and its congeners; amphetamines elicit virtually all their biological actions by inducing reverse transport, i.e. by switching monoamine transporters into a mode where the transporters mediate the efflux of dopamine (DAT), norepinephrine (NET), and SERT; inhibition of protein kinases or their genetic ablation blunt the ability of amphetamines to induce reverse transport (13, 18, 19). Several conceptual problems arise if amphetamine-induced reverse transport is treated as a special situation within the framework of the alternating access model (20). Most importantly, in the current context it is not clear how phosphorylation of the N terminus can be translated into a shift in the transport cycle that promotes outward rather than inward movement of substrate.
Here, we addressed the role of the N terminus by three approaches: (i) disruption of a consensus PKC phosphorylation (78XRXT81) site in the juxtamembrane region of the SERT N terminus; (ii) truncation of the N terminus (while preserving the putative phosphorylation site); and (iii) constraining the mobility of the N terminus by tethering it to an additional transmembrane segment. The observations are consistent with a model in which the N-terminal segment must be freely mobile to regulate the conformational cycle of the transporter and transmit the actions of amphetamines.
EXPERIMENTAL PROCEDURES
Materials
[3H]CFT (85.9 Ci/mmol), [3H]imipramine (47.5 Ci/mmol), [3H]dopamine (40 Ci/mmol), and [3H]serotonin (28.1 Ci/mmol) were supplied by PerkinElmer Life Sciences and [3H]MPP+ (85 Ci/mmol) by American Radiolabeled Chemicals (St. Louis). JHC1-64 (2-(3-s-butylimino-6-diethylamino-3H-xanthen-9-yl)-5-(5-{2-[3β-(3,4-dichloro-phenyl)-2β-methoxycarbonyl-8-aza-bicyclo[3.2.1]oct-8-yl]-ethylcarbamoyl}-pentylsulfamoyl)-benzenesulfonic acid anion) was synthesized as reported previously (21). The QuikChange II site-directed mutagenesis kit was from Stratagene Cloning Systems (La Jolla, CA); oligonucleotides were from Operon Biotechnologies (Cologne, Germany), and the M1 FLAG antibody was from Sigma. Chemicals at analytical grade were obtained from standard suppliers with the exception of ibogaine, which was donated by Marko Resinovic (Slovenia). Cell culture media, supplements, and antibiotics were from Invitrogen.
Modification of Plasmids, Cell Culture, and Transfection
Mutageneses were performed with the QuikChange II site-directed mutagenesis kit with cDNA of human isoforms of NET, DAT, or SERT as template (cloned into the pECFP and pEYFP vectors (Clontech) or into the pOTV oocyte vector) and Pfu Turbo DNA polymerase (Stratagene) to produce mutant cDNAs that code for fluorescence protein-tagged transporters (for the sake of brevity, CFP and YFP as prefix or suffix C and Y will only be used in an abbreviated form for fluorescence resonance energy transfer microscopy); all transporters were fluorescently tagged, except the transporters expressed in Xenopus laevis oocytes and the TACSERT construct.
Sense and antisense custom oligonucleotides were designed to contain the mutations of interest. The sequences of the primer sense strands (5′ to 3′ direction shown below, with mutations indicated in boldface underlined font) were as follows: SERTS62A, CGGGAGATGACACACGGCACGCTATCCCAGCGACC; SERTR79A, CTTCATCAAGGGGAAGCGGAGACCTGGGGC; SERTT81A, GGGGAACGGGAGGCCTGGGGCAAGAAGG; SERTT81D, GGGGAACGGGAGGACTGGGGCAAGAAGG; NETT58A, GCGCAGCCCCGGGAGGCCTGGGGCAAGAAGATCG; NETT58D, GCGCAGCCCCGGGAGGACTGGGGCAAGAAGATCG; DATT62A, GGCCCAGGATCGGGAGGCCTGGGGCAAGAAGATCG; and DATT62D, GGCCCAGGATCGGGAGGACTGGGGCAAGAAGATCG.
The T81A mutant in the double fluorescence-tagged C-SERT-Y background, used in intramolecular FRET measurements, was created by site-directed mutagenesis as described above. The N-terminal truncation mutant SERT-YFP with the first 64 amino acids removed, therefore termed Δ64SERT, was produced by PCR using the primers (5′ to 3′ direction) GCGCGGTACCGTCACAGCATTCAAGCGG (reverse, KpnI site) and GCGCGAATTCGATGGCGACCACC (forward, EcoRI site).
A FLAG sequence (DYKDDDDK) was inserted into the single membrane-spanning interleukin 2α receptor subunit, Tac (T-cell activation), sequence downstream of the N-terminal signal sequence (22) by a two-step PCR. The signal sequence affords the use of the M1 FLAG antibody (Sigma), because M1 only binds FLAG epitopes positioned at the extreme N terminus (23). The FLAG-tagged Tac fragment was subsequently fused to the N terminus of human SERT using PCR, and the TACSERT was inserted into the expression vector pcDNA3.1(−) (Invitrogen). All mutations were confirmed by sequencing.
HEK293 and CAD cells were grown at 37 °C in a 5% CO2-humidified atmosphere, on standard plastic cultureware, as desired. HEK293 cells were grown in Dulbecco's modified Eagle's medium, supplemented with 10% fetal calf serum and 1% kanamycin. CAD cells were grown in Dulbecco's modified Eagle's medium/Ham's F-12 (1:1) medium, supplemented with 8% fetal calf serum and 1% penicillin/streptomycin. All experiments were also carried out in HEK293 cells and showed similar results to those obtained in CAD cells (data not shown). Cells were seeded onto poly-d-lysine-coated 48-well culture plates (for uptake assays) and 13- or 16-mm glass coverslips (for confocal and FRET microscopy, respectively) or 5-mm glass coverslips (for release studies). For most experiments, the cells were transiently transfected with wild type or mutant plasmid cDNA (2–20 μg) using the calcium phosphate co-precipitation method. The experiments were carried out 48 h following the transfection.
TACSERT was transiently transfected in HEK293 cells (number CRL-1573, ATCC) and grown in Dulbecco's modified Eagles medium with Glutamax-I supplemented with 10% fetal bovine serum, 5 mm sodium pyruvate, and penicillin/streptomycin (100 μg/ml) at 37 °C in a humidified incubator with 5% CO2. Transfection was carried out using Lipofectamine 2000 (Invitrogen); 4 μg of plasmid encoding TACSERT or wild type SERT was used for transient transfection of cells in a 75-cm2 culture flask. Cells were assayed 48–72 h after transfection. For uptake and binding saturation experiments, carried out to measure transporter turnover, stable HEK293 lines were created using Geneticin (G418) for selection of positive clones, as indicated in the respective figure legends.
Uptake and Release Assays
Fluorescence tags do not introduce any functional deficit to monoamine transporters (24). Uptake experiments were performed as described previously (25). In brief, for the determination of nonspecific uptake by SERT, DAT, and NET, we used 10 μm paroxetine, mazindole, or nisoxetine, respectively, and 0.1–60 μm [3H]dopamine or [3H]5HT was added for 1 min as indicated. For TacSERT, transfected cells were plated in 96-well plates (ViewPlate; PerkinElmer Life Science). Prior to the experiment, the cells were washed once in uptake buffer (25 mm HEPES, 120 mm NaCl, 5 mm KCl, 1.2 mm CaCl2, and 1.2 mm MgSO4 supplemented with 5 mm d-glucose, 0.1 mm ascorbic acid, and 0.1 mm pargyline) and equilibrated in uptake buffer for 30 min before starting the assay. Nonlabeled 5HT was added at increasing concentrations followed by 30–50 nm [3H]5HT to a final volume of 150 μl. After incubation for 3 min at room temperature, cells were washed twice in ice-cold uptake buffer. Scintillation fluid (0.15 ml) was added, and the radioactivity was measured in a Wallac microplate liquid scintillation counter (PerkinElmer Life Sciences).
Substrate efflux assays were performed as described previously (26, 27). In brief, culture medium was removed from transiently transfected CAD or HEK293 cells (4·105 cells per well grown on coverslips in 96-well plates), and the cells were preincubated with 0.4 μm [3H]5HT or with 0.1 μm 1-[3H]methyl-4-phenylpyridinium (MPP+) for 20 min at 37 °C in a final volume of 0.1 ml of Krebs/HEPES buffer per well. The coverslips were transferred into chambers, and excess radioactivity was subsequently washed out with buffer at 25 °C for 45 min at a perfusion rate of 0.7 ml/min. Once stable efflux of radioactivity was achieved, following the initial wash, 2-min fractions were collected, and samples were counted in a β-counter.
Binding Assays
Membranes were prepared from HEK293 cells transfected with wild type or mutant transporters. The cells were washed twice in phosphate-buffered saline (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.5 mm KH2PO4, pH adjusted to 7.4), harvested, and centrifuged at 13,000 rpm for 10 min. The pellets were dissolved in HME buffer (20 mm HEPES, 2 mm MgCl2, 1 mm EDTA, pH adjusted to 7.4), centrifuged, and freeze-thawed twice using liquid nitrogen. Following another centrifugation step, the pellets were redissolved in HME buffer and sonicated three times for 7 s with 5-s intervals. The membranes were incubated with [3H]CFT for DAT or [3H]imipramine for SERT in reactions of 0.1 to 0.25 ml, respectively, in the presence or absence of other drugs in the desired concentration range. The buffer contained 20 mm Tris-HCl, 2 mm MgCl2, 120 mm NaCl, and 3 mm KCl. For experiments involving DAT, ZnCl2 was added to the buffer at a final concentration of 10 μm. Nonspecific binding was determined in the presence of 10 μm methylphenidate or paroxetine (for DAT and SERT assays, respectively). The reactions, lasting between 0.5 and 60 min, were terminated by rapid washing with ice-cold buffer and collected onto glass fiber filters (Whatman GF/B) that were dissolved in scintillation medium and counted for [3H] content.
Electrophysiology
The cDNA of the wild type SERT and various SERT mutants were cloned into the oocyte expression vector pOTV. The plasmids were linearized and in vitro transcribed with T7 RNA polymerase using the mMessage mMachine T7 RNA kit (Ambion). The resulting cRNA was injected into defolliculated, stage VI X. laevis oocytes (25 ng/oocyte; X. laevis frogs were from Nasco, Fort Atkinson, WI). Electrophysiological recordings were obtained after incubation of the injected oocytes for 5–9 days at 18 °C in standard oocyte solution (100 mm NaCl, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH adjusted to 7.6 with NaOH). Whole cell currents and voltages were recorded from oocytes by the two-electrode voltage clamp and the current clamp method as described previously (28) using the CA-1B high performance oocyte clamp (Dagan Corp.), interfaced to an IBM-compatible PC and pCLAMP 9.2 (Axon Instruments), to control membrane potentials and other parameters. Electrodes were pulled from borosilicate glass capillaries to a resistance of 0.2–1 megohm and filled with 3 m KCl. For experiments, the oocyte was impaled with two electrodes in a chamber continuously perfused with buffer (100 mm NaCl, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, pH adjusted to 7.4 with NaOH) until a stable base-line current was obtained. Currents were subjected to low pass-filtering at 20 Hz. During voltage clamp the membrane potential was held at the indicated voltage (Vhold) and the applied current (I) was recorded. In the current clamp mode, the membrane potential was monitored and the voltage recorded.
For efflux measurements, injected X. laevis oocytes were incubated with 0.4 μm [3H]5HT at 18 °C for 10 min. They were then washed three times in cold buffer and transferred to the experimental chamber. The oocytes were superfused first with ND100 for 5 min (to guarantee a constant basal efflux) and during the experiment either with PCA or serotonin, along with the addition of ND100. All experiments were carried out at room temperature with a superfusion rate of 1 ml/min. Fractions were collected in scintillation vials and counted for radioactivity. At the end of the experiment, the remaining radioactivity was recovered by dissolving the oocyte with 10% SDS. Drug-induced release of radioactivity was calculated as percentage of radioactivity present in the cell at any given time point.
Confocal Laser Scanning Microscopy
Confocal microscopy was done essentially as published (29) using a Zeiss LSM 510 confocal microscope (argon laser, 30 milliwatts; helium/neon laser, 1 milliwatt) equipped with an oil immersion objective (Zeiss Plan-NeoFluar ×40/1.3). The expression of transporters was quantified by calculating the ratios of plasma membrane intensity to cellular interior intensity, using the method described by Wuller et al. (30). For immunocytochemistry, transiently transfected HEK293 cells grown on glass coverslips were fixed in 4% paraformaldehyde for 10 min, washed twice in phosphate-buffered saline, and incubated in blocking buffer (1% bovine serum albumin in phosphate-buffered saline) for 30 min. Subsequently, cells were incubated with mouse anti-FLAG M1 primary antibody diluted 1:1000 for 1 h in blocking buffer. Following five washes in blocking buffer, secondary Alexa488-conjugated goat anti-mouse antibody (Invitrogen) diluted 1:200 was applied for another 45 min before the cells were washed twice in blocking buffer, twice in phosphate-buffered saline, and mounted using Vectashield (Vectashield Laboratories, Burlingame, CA). Coverslips were sealed with nail polish. All steps were performed at room temperature. Confocal microscopy was performed on a Zeiss 510 confocal laser scanning microscope using a 63× 1.4 NA oil immersion objective. Alexa488 was excited with the 488 nm laser line from an argon-krypton laser, and the fluorescent signal was collected with a 505–550-nm bandpass filter.
X. laevis oocytes were transferred into an experimental chamber (1μ-slide 18-well ibiTreat, Ibidi, Germany) for confocal microscopy experiments to determine SERT expression levels, by visualizing the binding of the Rhodamine-labeled 2β-carbomethoxy-3β-(3,4-dichlorophenyl)tropane, JHC1-64 (21). The compound was added in a cumulative manner (30 nm to 2 μm), and fluorescence elicited by the helium/neon laser at 542 nm (intensity: 100%; 560–615 nm broad pass filter) was recorded over 200 s in 10-s intervals.
FRET microscopy was carried out on an epifluorescence Zeiss Axiovert M200 microscope using the “three filter method” as described previously (31). Cells were transiently transfected (2 × 105 cells/22-mm coverslip) and placed into a chamber containing buffer. The images were taken using a 63× oil immersion objective and Ludl filter wheels to allow for rapid switching between the fluorescence excitation and emission filters for CFP (ICFP; excitation, 436 nm; emission, 480 nm; and dichroic mirror, 455 nm), YFP (IYFP; excitation, 500 nm; emission, 535 nm; and dichroic mirror, 515 nm), and FRET (IFRET; excitation, 436 nm; emission, 535 nm; and dichroic mirror, 455 nm). The images were captured by a CCD camera and analyzed using Metamorph (Meta Imaging, Universal Imaging Corp., version 4.6). Background fluorescence was subtracted from all images, and fluorescence intensity was measured at the plasma membrane and in cytosolic regions in all images. To calculate a normalized FRET signal (NFRET), we used Equation 1,
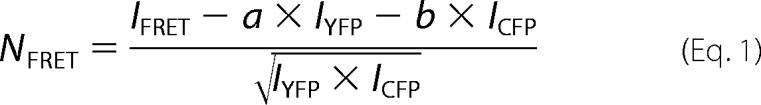 |
where a and b represent the bleed through values for YFP and CFP.
Molecular Modeling
The conformational changes in the wild type protein compared with the T81A and T81D mutants were investigated by a series of molecular dynamics (MD) simulations using a three-dimensional homology model of SERT based on the crystal structure of the leucine transporter from Aquifex aeolicus (LeuTAa), Protein Data Bank code 2A65 (5), with its natural substrate 5HT bound. The underlying alignment was chosen from the comprehensive study published by Beuming et al. (32), and generation of the homology model and subsequent docking of 5HT were performed with the software package MOE (Molecular Operating Environment, version 2007.09). Two Na+ ions were positioned into the binding site equivalently to those in LeuTAa, and one Cl− ion was placed on the basis of the description of its coordinating residues (33). It is worth pointing out that the model is truncated at both C- and N-terminal ends (C terminus, 23 residues; N terminus, 78 residues are missing) because there is no corresponding sequence in the LeuTAa template within those regions; it should be noted explicitly that the mutated Thr81 is only three amino acid side chains away from the N-terminal truncation point. Thus, MD simulations performed have to be interpreted cautiously as this missing fragment might influence the simulation; furthermore, it should be noted that the MD simulations have not been performed down to the millisecond time frame that would be necessary to compute the actual conformational changes on a biological time scale. MD simulations were performed with the software package DESMOND (Desmond Molecular Dynamics System, version 2.0, D. E. Shaw Research, New York), and the resulting trajectories were analyzed using DESMOND and VMD (34). The three-dimensional model of wild type SERT with bound 5HT was placed into a 1-palmitoyl-2-oleoyl-l-phosphatidylcholine membrane and solvated into an orthorhombic box filled with water. Na+ and Cl− ions were added (excluding the region within a radius of 7 Å around 5HT) to provide a final salt concentration of 0.15 m. The OPLS all-atom force field was used throughout. Constant temperature (310 K) and pressure (1 atm) were achieved by using the Berendsen algorithm. The whole system, consisting of about 55,000 atoms, underwent several short energy minimization and relaxation steps (constrained MD). After equilibration of the wild type protein, data were collected from 3-ns runs. In the end point structure from the wild type MD simulation, Thr81 was mutated into alanine and aspartate. Consequently, two individual trajectories of 6 ns each were calculated for each mutant.
Statistical Analysis
Parameter estimates were obtained by fitting data from uptake and binding experiments to the appropriate equations (i.e. rectangular hyperbola, monophasic inhibition, and monoexponential association) using the Marquardt-Levenberg algorithm. IC50 values for inhibition of [3H]imipramine binding by PCA and ibogaine were converted to Ki values using the Cheng-Prusoff equation (35). Data are reported as arithmetic means ± S.E. or geometric means with 95% confidence limits.
Confocal laser scanning microscopy images were analyzed either by Image J 1.35 (National Institutes of Health, Bethesda) or by Zeiss LSM Image Browser (version 4,2,2,121; Carl Zeiss Microimaging Gmbh). For FRET experiments, background fluorescence was subtracted, and bleed through values for YFP and CFP were determined in each experiment, and normalized FRET (NFRET) signals were calculated using Metamorph and Image J PixFRET Plugin (36).
Appropriate statistical analyses of the data were performed, as indicated in figure legends. In short, to calculate the statistical significance of differences between wild type and mutant transporters and/or different experimental conditions, one-way analysis of variance (ANOVA) was followed by Tukey post hoc t tests. Paired or unpaired t tests were used in those instances where two samples were being compared.
RESULTS
Inspection of the SERT N terminus with NetPhos, a software that recognizes serine, threonine, and tyrosine phosphorylation sites in eukaryotic proteins predicted that Thr81 and Ser62 were candidate sites for phosphorylation by PKC (via the X(R/K)X(S/T) recognition sequence). Ser62 is located in an amino acid stretch present only in SERT (and conserved throughout all species examined with the notable exception of Drosophila melanogaster) but not in other monoamine transporters (Table 1). In contrast, Thr81 is located within the absolutely conserved RETWGKK motif proximal to the first TM of the N termini of DAT (Thr62), NET (Thr58), and SERT (Table 1). Previously reported mutations of the adjacent tryptophan residue in DAT led to dramatic reductions in dopamine uptake (37, 38). In DAT, an N-terminal deletion mutant of 65 amino acids that included the RETWGK motif was unable to afford substrate uptake (39), although the deletion of the first 59 amino acids did not affect dopamine uptake (40). Importantly, both truncation mutants were expressed to an appreciable extent at the cell surface. This suggests that this region plays a significant role in monoamine transporter function. Taking other possible recognition sequences into consideration, Thr81 and Ser62 may be liable to phosphorylation by cAMP-dependent protein kinases (e.g. PKA or cAPK) at their characteristic recognition motif RX(S/T).
TABLE 1.
Sequence alignment of the N-terminal region of monoamine transporters
The conserved RETWGKK motif adjacent to TM1 is illustrated in shaded gray. The conserved threonine residues (T), at position 81 of human (h) SERT, 62 of human (h) DAT, and 58 of human (h) NET are indicated in underlined boldface font.

We replaced both sites in YFP-tagged SERT by alanine residues, creating the SERTS62A and SERTT81A mutants. Both constructs showed surface expression comparable with that of wild type SERT (Fig. 1). Table 2 displays the ratios of plasma membrane fluorescence intensity to cell interior fluorescence intensity and thus serves as a quantitative measure of transporter surface expression; no significant differences between expression levels of wild type SERT and the mutants were observed (p > 0.05, one-way ANOVA followed by Tukey's post hoc t-tests). Subsequent uptake assays showed that the mutants were all functional and comparable with wild type SERT (Fig. 1B; p > 0.05, one-way ANOVA followed by Tukey's post hoc t-tests, n = 3–6, performed in triplicate). The Km values (μm) were as follows: wild type SERT = 1.9 (0.9–2.9), SERTS62A = 1.1 (0.2–2.0), and SERTT81A = 1.7 (0.1–3.3); the Vmax values (pmol/106 cells/min) were as follows: wild type SERT = 151 ± 11, SERTS62A = 72 ± 7, and SERTT81A = 56 ± 7. Thus, the mutated transporters were functional and afforded substrate influx. Accordingly, it was possible to examine transporter-mediated efflux.
FIGURE 1.

Expression and function of wild type SERT and of mutated versions that lack candidate N-terminal phosphorylation sites. A, representative confocal laser scanning microscopy images illustrating that the cellular expression of SERTS62A and SERTT81A is comparable with that of wild type SERT. All constructs are almost exclusively targeted to the plasma membrane (also see for the ratio staining of plasma membrane to cell interior in Table 2). HEK293 cells were transiently transfected with CFP-tagged transporter constructs; the cell membrane was visualized by trypan blue staining (26). B, saturation of [3H]5HT uptake by SERT and the mutants expressed in transiently transfected CAD cells. Nonspecific uptake was determined in the presence of 10 μm paroxetine (n = 3–6, each experiment performed in triplicate).
TABLE 2.
Levels of cell surface expression of the wild type SERT and N-terminal mutants
The transporters were transiently expressed in HEK293 cells in at least three individual experiments and visualized by confocal laser scanning microscopy. Images (at least eight) were analyzed by ImageJ to quantify plasma membrane and cell interior fluorescence intensity.
| Transporter | Plasma membrane fluorescence intensity/cell interior fluorescence intensity ± S.E. |
|---|---|
| Wild type SERT | 4.03 ± 0.48 |
| SERTS62A | 4.53 ± 0.58a |
| SERTT81A | 3.80 ± 0.79a |
| Δ64SERT | 5.13 ± 0.86a |
a p > 0.05 compared with wild type SERT.
SERTThr81 Mutants Impair Amphetamine-induced Release without Affecting Oligomerization of the Mutated Transporters
Addition of PCA to the superfusion buffer did not alter [3H]5HT efflux in CAD cells expressing SERTS62A in comparison with wild type SERT (Fig. 2, A and B). In contrast, cells transfected with SERTT81A failed to show any detectable [3H]5HT release when challenged with PCA (Fig. 2C). Under the assumption that Thr81 is part of a canonical PKC phosphorylation consensus site, we (i) disrupted the site by replacing Arg79 by alanine (SERTR79A) and (ii) created an aspartate mutant (SERTT81D) to mimic phosphorylation at position 81. The resulting SERT mutants reached the cell surface (data not shown) and were functional (Fig. 1B). The Km values (μm) were as follows: SERTT81D = 2.7 (0.5–4.8) and SERTR79A = 3.6 (1.3–5.9); the Vmax values (pmol/106 cells/min) were as follows: SERTT81D = 33 ± 4 and SERTR79A = 13 ± 2. The SERTR79A mutation abrogated efflux, and higher PCA concentrations even abolished basal efflux (Fig. 2D). However, the phosphate-mimicking mutation SERTT81D did not result in recovery of amphetamine-induced efflux; SERTT81D (Fig. 2E) did not differ from SERTT81A (Fig. 2C). Interestingly, we found subtle but significant differences in the magnitude of basal efflux between wild type SERT (0.42 ± 0.01%·min−1; n = 129 observations) and the mutants, SERTS62A (0.65 ± 0.04%·min−1; p < 0.01; n = 67 observations), SERTT81A (0.89 ± 0.07%·min−1; p < 0.001; n = 99 observations), and SERTR79A (1.28 ± 0.05%· min−1; p < 0.001; n = 41 observations) but not to SERTT81D (0.53 ± 0.02%·min−1; p > 0.05, ANOVA followed by Tukey's post hoc t test; n = 72 observations).
FIGURE 2.

Amphetamine-stimulated release of [3H]5HT by wild type SERT (A), SERTS62A (B), SERTT81A (C), SERTR79A (D), and SERTT81D (E). Transiently transfected CAD cells expressing wild type and mutant SERTs were preloaded with [3H]5HT for 30 min at 37 °C and subsequently superfused (as described under “Experimental Procedures”). Upon reaching a stable base line, three 2-min fractions (6 min) were collected to define basal efflux (basal efflux, mean of the three fractions before PCA addition: wild type SERT, 0.42 ± 0.01%·min−1, i.e. 296 ± 10 dpm·min−1, n = 99 observations of randomly chosen experiments performed on different days; SERTS62A; 0.65 ± 0.04%·min−1, i.e. 480 ± 38 dpm·min−1, n = 67; SERTT81A, 1.77 ± 0.14%· min−1, i.e. 517 ± 22 dpm·min−1, n = 99; SERTR79A, 1.28 ± 0.05%·min−1, i.e. 563 ± 29 dpm·min−1, n = 41; and SERTT81D, 1.06 ± 0.04%·min−1, i.e. 318 ± 21 dpm·min−1, n = 104). Thereafter, cells were exposed to PCA (10 μm).
These observations naturally led us to question whether this PKC phosphorylation consensus site, and in particular Thr81, plays a role in determining amphetamine-induced efflux in other monoamine transporters. Thus, we generated both alanine and aspartate mutants in NET and DAT, corresponding to SERTThr81. The mutants were all capable of inward transport (data not shown); and as expected, their outward transport mode was clearly defective (Fig. 3, B and C), comparable with the SERT mutants (Fig. 3A).
FIGURE 3.

Concentration-response curve for amphetamine-induced release in wild type monoamine transporters and juxtamembrane threonine mutants. A, PCA-stimulated [3H]5HT release in wild type and mutant SERT. B and C, d-amphetamine (D-AMPH)-stimulated release of [3H]MPP+ by wild type DAT, DATT62A, and DATT62D (B) and wild type NET, NETT58A, and NETT58D (C). The experiments were performed as described in the legend to Fig. 2 and under “Experimental Procedures” (basal efflux for wild type DAT, 0.25 ± 0.01%·min−1, i.e. 255 ± 8 dpm·min−1, n = 148; DATT62A, 0.29 ± 0.02%·min−1, i.e. 226 ± 4 dpm·min−1, n = 132; DATT62D, 0.17 ± 0.01%·min−1, i.e. 105 ± 4 dpm·min−1, n = 144. Basal efflux for wild type NET, 0.039 ± 0.002%·min−1, i.e. 47 ± 2 dpm·min−1, n = 48; NETT58A, 0.074 ± 0.002%·min−1, i.e. 73 ± 2 dpm·min−1, n = 47; NETT58D, 0.065 ± 0.009%· min−1, i.e. 69 ± 15 dpm·min−1, n = 39).
A reduction in the affinity of the mutated transporters for amphetamine might explain the loss of amphetamine-induced efflux. However, when we determined the Ki values of PCA for wild type SERT and SERTT81A, calculated from inhibition of [3H]imipramine binding, we did not detect any significant differences (n = 3, p > 0.05, Mann Whitney U test). The Ki values were 2.5 μm (1.5–4.1) for the wild type SERT and 2.6 μm (2.1–3.3) for SERTT81A. Therefore, the substantial decline we observed in amphetamine-induced efflux of the threonine mutant cannot be accounted for by a decrease in its affinity for PCA. Furthermore, this finding is supported by the lack of amphetamine-induced efflux at higher concentrations in CAD cells expressing wild type or mutant DAT, NET, and SERT (Fig. 3). In contrast to this typical bell-shaped concentration-response relationship for the wild type transporters, which was also reported previously (19, 41), the mutant monoamine transporters collectively present as being incapable of supporting amphetamine-induced efflux over the entire PCA concentration range (Fig. 3). Our earlier experiments showed that transporter oligomerization was a prerequisite for amphetamine-induced release (19). Hence, we examined whether the mutation of Thr81 altered the quaternary structure of SERT by performing FRET experiments with fluorescently labeled SERTT81A and wild type SERT. However, whether we co-expressed the SERTT81A with itself (CFP- and YFP-tagged proteins) or with the wild type transporter, we could not detect any significant differences in NFRET values (Fig. 4; p > 0.05, one-way ANOVA followed by Tukey's post hoc t test, n = 3–4). Thus, SERTT81A assembles into oligomeric structures in a manner indistinguishable from wild type SERT. This ruled out the possibility that impaired efflux by Thr81 mutants of SERT resulted from defective oligomerization.
FIGURE 4.

FRET in wild type SERT and SERT-T81A. Transiently transfected CAD cells co-expressing wild type C-SERT and Y-SERT (labeled SERT/SERT), C-SERT and Y-SERTT81A (SERTT81A/SERT), or C-SERTT81A and Y-SERTT81A (SERTT81A/SERTT81A) were grown on glass coverslips. FRET was recorded as outlined under “Experimental Procedures.” The C-SERT-Y construct was used as positive control, whereas CFP co-expressed with YFP represented the negative control in each experiment. The NFRET values from at least three individual experiments (with 5–10 visible fields) were calculated, and there were no statistical differences (p > 0.05, one-way ANOVA followed by Tukey's post hoc t tests) between NFRET values for the following conditions: SERT/SERT, SERTT81A/SERT, and SERTT81A/SERTT81A.
Amphetamine-induced Currents through SERTT81A
Transporter-mediated currents play a major role in amphetamine-induced efflux (42, 43); uncoupled conductances (i.e. ionic currents in excess of that associated with substrate translocation) have been observed in all monoamine transporters investigated (25, 43–47). They give rise to elevated intracellular Na+ concentrations (48). The increase in intracellular Na+ is thought to facilitate outward transport because it allows for accumulation of the transporter in the inward facing conformations. It was conceivable that mutation of Thr81 altered the conducting state of the transporter; if this was the case, it could account for the blunted amphetamine-induced release. Accordingly, we examined the electrophysiological properties of SERTT81A in comparison with wild type SERT after expression of the proteins in X. laevis oocytes.
Control (i.e. water-injected) X. laevis oocytes did not reveal any detectable current upon application of PCA regardless of which protocol was employed (data not shown). The transporter-mediated leak conductance was defined by application of 10 μm cocaine and was identical in wild type SERT and SERTT81A (data not shown). In both wild type SERT and SERTT81A-expressing oocytes, PCA induced a current. To examine the current-voltage (I-V) relation, cells were clamped at a holding potential of −40 mV, which was subsequently shifted to −100 mV followed by a ramp to +40 mV. As reported previously (44), the inward current triggered by PCA showed inward rectification (Fig. 5A). The I-V relationships of wild type SERT and SERTT81A were similar in shape, but at each voltage examined, the current through SERTT81A was larger than that through the wild type transporter (Fig. 5A). The amphetamine-induced current is large enough to afford membrane depolarization (28), and this may change the driving force for transport of substrate. We therefore also employed the current clamp mode to estimate the extent of depolarization resulting from the PCA-triggered current through wild type and mutant SERT (Fig. 5B). Consistent with the recordings shown in Fig. 5A, the depolarizing effect of PCA was significantly greater in SERTT81A than in wild type SERT (Mann-Whitney U test; n = 3).
FIGURE 5.

Amphetamine-induced currents (A), membrane depolarization (B), quantification by binding of the fluorescent inhibitor JHC1-64 (C), and [3H]5HT release (D) by the wild type and mutant SERT expressed in X. laevis oocytes. A, current/voltage (I-V) relations were obtained by jumping from a holding potential (Vhold) of −40 mV to −100 mV followed by a ramp to +40 mV over a total time of 10 s (n = 9). The PCA-induced current was defined as the current measured in the presence of PCA (10 μm) after subtraction of the current measured in the corresponding control buffer (IPCA − Icontrol). B, membrane potentials recorded during release experiments under current clamp conditions for wild type SERT and SERTT81A. The error bars represent the standard deviation (p < 0.05; Mann-Whitney U test, two-tailed; n = 3). C, saturation curves for JHC1-64 binding to X. laevis oocytes expressing wild type SERT, SERTT81A, and Δ64SERT. Oocytes (that had been used for voltage clamp experiments) were subsequently incubated in a cumulative manner with increasing concentrations of JHC1-64 for 200 s. Images were captured using the same configuration for all cells, with the indicated concentrations of JHC1-64. D, PCA-induced release of [3H]5HT in cRNA-injected X. laevis oocytes under current clamp conditions (n = 4).
It was surprising to find that amphetamine-stimulated currents elicited in X. laevis oocytes were higher for the SERTT81A compared with those by the wild type SERT (Fig. 5). Hence, we reasoned that this could be accounted for by a difference in expression levels. We used a rhodamine-labeled 2β-carbomethoxy-3β-(3,4-dichlorophenyl)tropane, JHC1-64 (21), to examine SERT surface expression levels in oocytes. This compound was used recently to visualize the cell surface of DAT-expressing neurons in dopaminergic explant cultures (49). We added increasing concentrations of JHC1-64 and examined SERT-expressing oocytes by confocal laser scanning microscopy; images were captured every 10 s following an incubation period of at least 200 s, which sufficed to reach binding equilibrium even at the lowest concentration employed (supplemental Fig. A). Water-injected oocytes served as negative control; JHC1-64 did not bind to a measurable extent to the surface of these oocytes (data not presented). The fluorescence intensity was analyzed on a pixel-by-pixel basis using ImageJ and expressed as fluorescence arbitrary units against the JHC1-64 concentration; JHC1-64 bound in a saturable manner to the cell surface-expressed transporters (Fig. 5C); affinity estimates (KD = 262.2 ± 37.2, 315.2 ± 67.6, and 378.1 ± 136.8 nm for wild type, SERTT81A, and SERTΔ64, respectively) were comparable with those reported previously (21). SERTT81A (and SERTΔ64) displayed a slightly higher maximum binding to wild type SERT (Fig. 5C; Bmax = 272 ± 24, 432 ± 34, and 398 ± 57 arbitrary units for wild type, SERTT81A, and SERTΔ64, respectively).
The higher currents detected in oocytes expressing SERTT81A can be correlated to its higher expression levels as revealed by the JHC1-64 binding experiments. We also confirmed that PCA did not trigger an efflux in oocytes expressing the mutant SERT, although it promoted ion flux through the protein. SERTT81A failed to support PCA-induced efflux under current clamp conditions (Fig. 5D). Taken together, these data show that PCA stimulates similar inward currents through the mutant SERT as in the wild type. Hence, neither a change in the oligomeric state nor an impaired Na+ influx can account for the inability of PCA to trigger monoamine efflux through SERTThr81.
Molecular Dynamics Simulations Point toward the Importance of Thr81 for the Conformational Equilibrium of SERT
We searched for structural changes in the juxtamembrane region by studying the formation of H-bonds along the molecular dynamics trajectories of wild type SERT compared with mutant SERTs. One H-bond in particular, which is present at 78% of the last 3-ns period of simulation time in the wild type SERT, is absent if Thr81 is replaced by alanine or aspartate (Fig. 6, A–C). The hydroxyl group of Thr81 forms a stable H-bond with the backbone carbonyl group of Tyr350 of the third internal loop (IL3) in the SERT (Fig. 6A). In the DAT, the corresponding residue (Tyr335) has been recognized to be part of a network of interactions on the intracellular side of the transporter (50, 51). According to Fig. 6, mutation of Thr81 to alanine (Fig. 6B) or to aspartate (Fig. 6C) disrupts the interaction between Thr81 and Tyr350. Hence, the decay of this particular interaction leads to subsequent conformational changes. Importantly, the cytoplasmic region of TM1 moves apart from TM6-IL3 and IL2 (shown for SERTT81D in Fig. 6D; analogous results were obtained for SERTT81A, data not shown). Thus, mutation of Thr81 to Ala or Asp alters the conformational equilibrium of the SERT transport cycle toward the inward facing conformation; and more specifically, it does so by interacting with IL3 (and also IL2). Furthermore, we observed an increase in the distance between the C terminus (i.e. the most distal point of TM12) and the N terminus after mutation (indicated by the arrows in Fig. 6, E and F). Therefore, mutation of Thr81 to alanine or aspartate changes the conformation of the mutant SERTs to assume a predominantly inward facing state. The supplemental Fig. B depicts the course of the root mean square deviations of the protein backbones during the above mentioned MD simulations.
FIGURE 6.

Molecular dynamics simulations of SERTThr-81 mutants reveal models favoring inward facing states. A, snapshot of wild type SERT after 16 ns of MD simulation. The Thr81 side chain forms a stable H-bond with the backbone carbonyl of Tyr350 in IL3. B, snapshot of SERTT81A after 6 ns of MD simulation; the H-bond is not formed between Ala81 and Tyr350 during the course of the simulation. C, snapshot of SERTT81D after 6 ns of MD simulation; no H-bond is formed between Asp81 and Tyr350 during the course of the simulation. D, snapshots of wild type SERT and SERTT81D, following an MD simulation (backbones superimposed). In the in silico mutant (displayed in orange), the cytoplasmic region of TM1 is moved apart from TMD6 and IL3 (indicated by magenta arrows) in comparison with wild type SERT (green). E and F, snapshots of wild type SERT and SERTT81D during MD production simulation (backbones superimposed); TM12 moves away from TM1 (N terminus) after mutation of Thr81 to alanine or aspartate (wild type SERT, green; SERTT81A, yellow; SERTT81D, orange; Cα atoms of Thr81 are displayed in sphere representation).
Based on this model, the following predictions can be made. (i) SERTT81A ought to have a higher affinity for ibogaine, a compound that binds preferentially to the inward facing conformation (52). (ii) Destabilizing the interaction of Thr81 with IL3 ought to increase the distance between the N and the C termini, which in the wild type are in close proximity (53). (iii) There should be a decline in the affinity for inhibitors such as imipramine, which bind to the outward facing conformation (54). These three predictions have been verified. As illustrated in Fig. 7A, the affinity of ibogaine for SERTT81A (IC50 = 0.53 ± 0.04 μm) was significantly higher than that for the wild type SERT (IC50 = 0.95 ± 0.10 μm; p < 0.05, Mann Whitney U test, n = 3). We measured the distance between the C and N termini by recording intramolecular FRET in transporters tagged on both the N terminus (with CFP) and the C terminus (with YFP, to yield C-SERT-Y). Resonance energy transfer was significantly lower in C-SERT-YT81A than in wild type C-SERT-Y (Fig. 7B). Using the Förster equation (55), we estimated the distances to be 44.8 Å for the wild type C-SERT-Y and 48.0 Å for C-SERT-YT81A. Finally, we determined that the affinity of [3H]imipramine was reduced by ∼4-fold for the SERTT81A mutant (Fig. 8A; the KD values (nm) were 4.7 (2.4–7.0) for the wild type SERT and 20 (5–35) for SERTT81A). The same effect can also be seen for SERTT81D and SERTR79A (data not shown).
FIGURE 7.

Probing the conformational state of SERTT81A by measuring the affinity of ibogaine (A) and by using intramolecular FRET as a molecular ruler (B). A, inhibition by ibogaine of [3H]imipramine binding to membranes (10–15 μg) prepared from HEK293 cells expressing wild type SERT or SERTT81A. The incubation was done in a final volume of 0.25 ml containing 20 mm Tris-HCl, pH 7.4, 1 mm MgCl2, 120 mm NaCl, and 5 and 1 nm [3H]imipramine for 20 min at 25 °C. Specific binding in individual experiments amounted to 1500–2000 cpm and was set at 100% to normalize for inter-assay variation; nonspecific binding (determined in the presence of 10 μm paroxetine) was ≤200 cpm. Data are means ± S.D. of three independent experiments carried out in duplicate. B, Thr81 was mutated in a SERT construct tagged with CFP and YFP at the N and C termini, respectively. FRET was recorded microscopically in HEK293 cells transiently expressing C-SERT-Y and C-SERT-YT81A as outlined in the legend to Fig. 4. A significant reduction in the NFRET value (determined with Image J PixFRET software as outlined under “Experimental Procedures”) was observed for the mutant in comparison with the wild type SERT (p = 0.0052, from three individual transfections comprising a total of 22 (wild type) and 26 (mutant SERT) cells, unpaired t test).
FIGURE 8.

Binding of [3H]imipramine to wild type SERT and SERTT81A (A, C, and D) and of [3H]CFT to wild type DAT and DATT62A (B). A and B, saturation binding experiments were carried out in a final volume of 0.25 ml containing membranes expressing wild type (5 μg) and mutant SERT (7 μg) or wild type and mutant DAT (20 μg). The reaction was carried out in 20 mm Tris-HCl, pH 7.4, 1 mm MgCl2, 120 mm NaCl, 3 mm KCl and the indicated concentrations of [3H]imipramine for 10 min at 25 °C. For binding of [3H]CFT, the assay indications were similar except that the buffer did not contain KCl and it was supplemented with 10 μm ZnCl2 as indicated (empty symbols in B). Shown are representative experiments carried out in duplicate that were replicated at least three times. C, reaction was done at a constant protein concentration (40 μg/ml), but the reaction volume was adjusted to 0.15 and 0.5 ml for 10 and 3 nm [3H]imipramine, respectively. At the time point indicated, the reaction was terminated by rapid filtration over glass fiber filters precoated with 1% polyethyleneimine. D, pseudo-first order rate constant (kobs) calculated from experiments carried out as outlined for 3 and 10 nm in C was plotted as a function of ligand concentrations. Shown are data obtained in three independent experiments that were carried out in parallel for wild type and mutant SERT; error bars represent the standard error of the estimate calculated for each individual curve.
We replicated the experiment for wild type DAT and DATT62A to test whether this effect is exclusively seen in SERT or extends to other members of the monoamine transporter family. As expected, DATT62A (Fig. 8B, closed triangle) bound [3H]CFT with lower affinity than wild type DAT (Fig. 8B, closed circle). Hence, it is evident that this highly conserved threonine residue is involved in the conformational switch between inward and outward facing conformation of presumably all monoamine transporters. DAT carries a high affinity Zn2+-binding site at its extracellular side, spanning from EL2 to the top of TM7 (56, 57). Zn2+ enhances binding of inhibitors (28, 56) by stabilizing the outward facing conformation, presumably because Zn2+ restricts the helical movements (58). Therefore, we tested if Zn2+ could stabilize DATT62A in an outward facing conformation to an extent that overcame the effect of the mutation. The presence of Zn2+ increased the affinity of DATT62A for [3H]CFT, although the KD values still exceeded that of wild type DAT (Fig. 8D, open symbols). The KD values for the wild type DAT were 35 ± 4 and 12 ± 1 nm in the absence and presence of Zn2+, respectively. For DATT62A, the KD value was 124 ± 9 nm in the absence and 27 ± 4 nm in the presence of Zn2+.
These findings can be interpreted to indicate that the switch of the transporter into the outward facing conformation is promoted by interactions of the N terminus (specifically Thr81 in SERT or Thr62 in DAT) with the inner vestibule. Inhibitors bind to neurotransmitter transporters with rate constants that are slower than the diffusion controlled rate, indicating that there is a rate-limiting conformational step preceding high affinity binding. Accordingly, kinetic experiments can be used to explore conformational cycling of the transporters (29, 59). We determined the association rate of [3H]imipramine at different concentrations for binding to wild type SERT and SERTT81A (representative curves are shown in Fig. 8C). A replot of the data (with kobs as a function of ligand concentration, see Fig. 8D) allowed us to estimate the bimolecular association rate constant from the slope and the dissociation rate from the intercept; for wild type SERT, koff = 0.56 min−1 and kon = 0.34 nm−1 min−1; for SERT-T81A, koff = 0.96 min−1 and kon = 0.12 nm−1 min−1. The kinetic KD values (calculated from the ratio koff/kon) were 1.7 nm (wild type SERT) and 7.7 nm (SERTT81A), and thus in reasonable agreement with the KD value estimated from the saturation experiments (Fig. 8A). More importantly, the drop in kon and the increase (albeit less pronounced) in koff suggest that the mutation impairs both the conformational switch that allows the transporter to enter into outward facing conformation (kon) and, to a lesser extent, the capacity of the transporter to maintain this outward facing conformation (koff).
Based on these observations, we predicted that the mutation ought to affect the turnover number of the transporter. To calculate turnover numbers, we created stable HEK293 lines of wild type SERT and SERTT81A and carried out the pertinent uptake and saturation binding experiments. The turnover numbers (calculated from the ratio of Vmax of [3H]5HT uptake/Bmax of [3H]imipramine binding) were 187 and 62 min−1, for the wild type SERT and SERTT81A, respectively. These numbers imply that the conformational cycle underlying the translocation process is three times slower in the mutant than in the wild type SERT.
N-terminal Truncation Mutant Operates in a Fashion Analogous to SERTT81A
It is likely that Thr81 coordinates a network of interactions that are stabilized by additional contacts. We used a two-pronged approach to test this conjecture. (i) We truncated the first 64 residues of the N terminus, and the resulting Δ64SERT leaves Thr81 intact but is devoid of all other possible phosphorylation sites. (ii) We constrained the flexible movements of the N terminus by attaching a membrane-spanning single Tac helix that tethers the N terminus to the plasma membrane (see Fig. 9 for a schematic representation of the constructs). The Δ64SERT was targeted to the cell surface (Fig. 9B and Table 2) and was functional (Fig. 10A). The Km values were not significantly different from each other, i.e. wild type SERT = 1.8 (1.2–2.8) μm and Δ64SERT = 1.7 (0.5–5.6) μm, whereas the Vmax of Δ64SERT (84 ± 20 pmol/106 cells/min) was significantly lower than that obtained with cells transiently transfected in parallel to express wild type SERT (166 ± 22 pmol/106 cells/min; p < 0.05, unpaired t test, n = 3–5). Subsequently, we examined the ability of HEK293 cells expressing Δ64SERT to respond to amphetamine and release [3H]5HT. We found that transporter-mediated release was dramatically reduced (Fig. 10B). Furthermore, the KD value for [3H]imipramine binding (Fig. 10C) was increased to an extent comparable with SERTT81A (i.e. ∼4-fold; KD Δ64SERT, 22.2 (3.9–40.4); wild type SERT, 4.5 (3.5–5.5); Bmax (pmol/mg protein) Δ64SERT, 9.5 ± 1.8; wild type SERT, 15 ± 0.5; n = 2). Along with electrophysiological data showing that Δ64SERT induces a marked decrease in amphetamine-stimulated release while maintaining normal currents (supplemental Fig. C), it reinforces even further that Δ64SERT operates in much the same fashion as SERTT81A. Finally, we suspected that the fluorescent tag may introduce steric hindrance to helical movements of the Δ64SERT and thus affect amphetamine-induced release. Therefore, we created an additional Δ64SERT mutant, where the tag is located at the C terminus. This mutant was indistinguishable from the one employed in Figs. 5, 9, and 10, which was true for all other assays examined (i.e. cell surface localization, substrate uptake, inhibitor binding, and PCA-induced release; data not shown).
FIGURE 9.

Expression of A64SERT and TACSERT in comparison with wild type SERT. The schematic representation illustrates the nature of the constructs employed in our study. The star indicates the position of Thr81. HEK293 cells were transiently transfected with cDNA encoding wild type or the Δ64SERT truncation mutant tagged with CFP (A and B) or the TACSERT (C). The cell surface expression of CFP-tagged transporters (A and B) was visualized by confocal microscopy by sequentially capturing images for CFP (green) and trypan blue (red; to delineate the cell surface). Overlays (in yellow) highlight the cell surface expression of the transporters. C, expression of TACSERT was visualized by staining fixed, nonpermeabilized cells with the M1 antibody directed against the N-terminal FLAG epitope followed by a secondary Alexa488-conjugated goat anti-mouse antibody using a Zeiss LSM510 confocal microscope (left image; the scale bar indicates 10 μm) as outlined under “Experimental Procedures”; the cell surface is shown in a bright field image (right image).
FIGURE 10.

Specific [3H]5HT uptake (A), abolition of amphetamine-stimulated [3H]5HT release (B), and marked reduction in the affinity of [3H]imipramine (C), resulting from the truncation of the SERT N terminus. A, uptake of [3H]5HT was determined in HEK293 cells transiently expressing wild type SERT and Δ64SERT as outlined in the legend to Fig. 1. B, PCA-induced release was determined as in Fig. 2, and the concentration-response curves generated as in Fig. 3. C, saturation of [3H]imipramine binding was performed with membranes (20 μg) prepared from HEK293 cells transiently expressing wild type SERT and Δ64SERT as outlined in the legends to Figs. 7 and 8.
Tethering the N Terminus to the Membrane Impairs Amphetamine-induced Efflux
The presence of the intact Thr81 did not influence the efflux impairment of Δ64SERT; hence, we inferred that the N terminus itself plays a structurally greater role than originally thought. Previously, it has been proposed that the interaction of the N terminus of the γ-aminobutyric acid transporter GAT1 with IL4 plays an essential role in the control of transport (60). Therefore, we rationalized that the conformational flexibility of the SERT-N terminus might be of relevance to amphetamine-induced efflux. Hence, we created an additional SERT construct, termed TACSERT, with a FLAG-tagged Tac fragment fused to the N terminus; because the Tac fragment is inserted into the plasma membrane due to its hydrophobic core, it therefore tethers the N terminus to the plasma membrane and restricts its movement(s) (refer to schematic in Fig. 9).
First, we employed immunocytochemistry to establish that TACSERT is targeted to the cell surface (Fig. 9C). We subsequently examined whether TACSERT exhibits normal functional activity, and we thus showed that its substrate uptake was not significantly different from that of the wild type SERT (Km (μm), 3.19 ± 1.16 versus 4.50 ± 0.10; and Vmax (pmol/min/106 cells), 36.7 ± 3.2 versus 36.5 ± 1.6, respectively; see Fig. 11A). Therefore, the TACSERT construct displayed no functional impairments, in clear contrast to the mutants described above. Further uptake and binding assays, testing whether the affinities of two different SERT inhibitors, citalopram and imipramine, were altered upon the appendage of the Tac fragment to SERT also failed to reveal any noticeable effects compared with the wild type transporter (data not shown). Surprisingly, however, when PCA (10 μm) was applied to HEK293 cells expressing TACSERT, we found that the construct was incapable of eliciting serotonin release (Fig. 11B).
FIGURE 11.

Constraining the flexibility of the amino tail in TACSERT does not impair substrate uptake (A) but does preclude PCA-induced efflux (B). A, uptake of [3H]5HT was determined in HEK293 cells expressing wild type SERT and TACSERT as outlined in the legend to Fig. 1. B, comparison of [3H]5HT efflux induced by PCA in TACSERT and wild type SERT. HEK293 cells transiently expressing the indicated versions of SERT were preloaded with [3H]5HT as described in Fig. 2. Release was induced by superfusing the cells with 10 μm PCA. The results are presented as bar graphs showing specific uptake (A, n = 3 from 3 individual experiments) or base-line-subtracted efflux (B, n = 8 from two independent experiments).
DISCUSSION
Three aspects have been argued as important in accounting for the ability of amphetamines to promote substrate efflux via monoamine transporters (20, 61). (i) Accumulation of Na+ in the vicinity of the inner vestibule promotes the accumulation of transporters in the inward facing conformation and thus makes more transporter moieties available for outward transport (48). (ii) Amphetamines have been observed to activate protein kinase C isoforms by an unknown mechanism; it has been argued that the N terminus of monoamine transporters is subject to phosphorylation by isoforms of protein kinase C and/or calmodulin-dependent kinase-II and that this modification is required to facilitate transport reversal by amphetamines (13). (iii) In the oligomeric counter-transport model (19), a dimeric arrangement of monoamine transporters is a prerequisite for the action of amphetamines, because one transporter moiety is bound to amphetamine, which in turn triggers the outward movement of the substrate by the second moiety. These models are not mutually exclusive; both intracellular Na+ and N-terminal phosphorylation may, for instance, be important in triggering the counter-transport mode in the transporter oligomer (19).
Our experiments were originally designed to examine the role of Thr81 of SERT as a potential site of phosphorylation by protein kinase C isoforms. Related experiments were recently reported for the homologous position Thr62 in DAT (62). Although we agree with Guptaroy et al. (62) that Thr81 (and the homologous residues in NET and DAT) is crucial for amphetamine-induced release, we find little evidence for a major difference between substituting alanine and the phosphate mimicking aspartate mutation. We stress that this conclusion is based on experiments carried out on all three amphetamine-sensitive monoamine transporters, i.e. SERT, NET, and DAT, and in three different cellular backgrounds, including CAD cells, which are of neuronal origin and express syntaxin-1A (which is thought to bind to the N terminus of SLC6 family transporters; Refs. 63, 64). Instead, our experiments uncovered a previously unappreciated role of the N terminus in regulating the transport cycle and in mediating the action of amphetamines. The latter finding is remarkable; the loss of amphetamine-induced release in SERTT81A (and in all other SERT mutants studied) provides incontrovertible evidence that permeation of Na+ does not suffice to trigger reverse transport. SERTT81A supported amphetamine-induced currents that were at the very least not smaller than those flowing through the wild type transporter.
In our molecular dynamics simulation, Thr81 was found to participate in a network of interactions that can be readily rationalized to stabilize the outward facing conformation. The network of interactions observed here are consistent with earlier calculations and with their interpretation (33, 50, 51, 62). Following these lines of evidence, destabilization of the interaction network can therefore readily be envisaged to shift the conformational equilibrium of the transporter toward an inward facing conformation; we provide several arguments that support this interpretation as follows: (i) increase in affinity of ibogaine, which binds preferentially to the inward facing conformation (54); (ii) drop in affinity of imipramine, which binds to the outward facing conformation; and (iii) increased distance between the N and C termini, which was detected by FRET. These observations allow us to rationalize the lower turnover number for serotonin uptake. However, they again do not explain why the action of amphetamine should be abolished. In fact, it is generally assumed that allowing the transporter to accumulate in the inward facing conformation is a prerequisite for amphetamine-induced efflux (61).
Similarly, the models emerging from molecular dynamics calculations do not explain why truncation of the N terminus (Δ64SERT) results in the same phenotype as substituting alanine for Thr81. Several residues are known to shift the conformational equilibrium between inward and outward facing conformation, e.g. mutations in the GQXXRXG motif in IL1 (between TM2 and TM3) of NET (65, 66) and replacement of Glu136 at the bottom of TM2 of SERT, Tyr335 (50), and Asp345 (67) in IL3 of DAT. These effects can be explained based on these residues clustering at the inner vestibule. Similarly, the present and earlier modeling studies (62, 68) suggest that the juxtamembrane portion of the N terminus also interacts with the internal loops lining the inner vestibule. In fact, mutation of the Trp63 residue following Thr62 in DAT (i.e. the residue homologous to Thr81 of SERT) abolishes inhibitor binding and dopamine inward transport (69). Moreover, when we substituted Arg79 of SERT by alanine, to disrupt the RETWGKK motif (and thereby the recognition sequence for PKC) at a site other than Thr81, we found that this mutation also leads to a phenotype comparable with SERTT81A. These findings highlight the importance of this particular region of the N terminus in the conformational switch between the inward and outward facing state. Nevertheless, in additional experiments to explore this notion, we illustrate that this fragment of the N terminus per se does not suffice to support amphetamine-induced substrate efflux, i.e. preserving the RETWGKK motif (and a preceding segment) while truncating the first 64 amino acids also abolishes amphetamine-induced efflux. It is worth pointing out that earlier work (18) reported that truncation of the first 22 amino acids of DAT sufficed to eliminate amphetamine-induced reverse transport. This was ascribed to the removal of phosphorylation sites. Although we do not intend to discard activation of protein kinases as an important step in facilitating amphetamine-induced monoamine efflux, we propose an alternative role of the N-terminal extension (20). This is based on the recently published structure of the bacterial Na+/betain symporter BetP, which is arranged in a trimer (70). In this trimeric arrangement, the C terminus of one BetP moiety (which is topologically equivalent to the N terminus of monoamine transporters) is thought to trigger the activation of the adjacent transporter moiety. In our oligomeric counter-transport model, the amphetamine-liganded transporter activates its adjacent oligomeric partner. The communication is achieved by the N terminus of the amphetamine-liganded moiety that acts as a lever to trigger outward movement of the substrate through the second moiety. This model predicts that the releasing action of amphetamine is abolished if the mobility of the lever is restricted. It is evident that this was the case; tethering the N terminus to an additional transmembrane domain, as shown for the TACSERT construct, abolished amphetamine-induced reverse transport. Remarkably, although the addition of the Tac fragment to and the truncation of the amino tail both abolish amphetamine-stimulated substrate release by SERT, they appear not to cause matching effects on other transporter functions, in particular inhibitor binding (i.e. TACSERT remaining unchanged, whereas that of the Δ64SERT becomes markedly reduced). These data, being the first observations of monoamine transporters behaving in this manner, tempt us to suggest that uptake, binding (core-protein-function) channel, and efflux modes of transporter action may exist as independent events, with the latter relying immensely on the flexibility of the N terminus.
At the very least, the model has the merit to explain observations that are otherwise difficult to reconcile, most importantly, the dissociation between amphetamine-induced currents, which are preserved in SERTT81A (and Δ64SERT), and reverse transport. Additional implications of the model are currently being explored to understand the underlying mechanisms of amphetamine action. This may allow us to understand the paradoxical actions of amphetamines on transporter variants associated with psychological disorders (71).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant P01 DA 12408 (to U. G.) and the National Institute on Drug Abuse Intramural Research Program (to A. H. N.). This work was also supported by FWF Grants P18706 and SFB3506 (to H. H. S.), SFB3502 (to G. F. E.), and SFB3510 (to M. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. A–C.
- NET
- noradrenaline transporter
- CFP
- cyan fluorescent protein
- CFT
- 2-β-carbomethoxy-3-β-(4-fluorophenyl)-N-methyltropane
- DAT
- dopamine transporter
- FRET
- fluorescence resonance energy transfer
- HEK
- human embryonic kidney
- 5HT
- 5-hydroxy tryptamine (serotonin)
- IL
- intracellular loop
- MD
- molecular dynamics
- MPP+
- 1-methyl-4-phenylpyridinium
- PCA
- para-chloroamphetamine
- PKC
- protein kinase C
- SERT
- serotonin transporter
- TM
- transmembrane domain
- YFP
- yellow fluorescent protein
- ANOVA
- analysis of variance.
REFERENCES
- 1.Rudnick G., Clark J. (1993) Biochim. Biophys. Acta 1144, 249–263 [DOI] [PubMed] [Google Scholar]
- 2.Saier M. H., Jr., Tran C. V., Barabote R. D. (2006) Nucleic Acids Res. 34, D181–D186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh S. K. (2008) Channels 2, 380–389 [DOI] [PubMed] [Google Scholar]
- 4.Jardetzky O. (1966) Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 5.Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 6.Torres G. E., Carneiro A., Seamans K., Fiorentini C., Sweeney A., Yao W. D., Caron M. G. (2003) J. Biol. Chem. 278, 2731–2739 [DOI] [PubMed] [Google Scholar]
- 7.Reiterer V., Maier S., Sitte H. H., Kriz A., Rüegg M. A., Hauri H. P., Freissmuth M., Farhan H. (2008) J. Neurosci. 28, 12453–12464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farhan H., Reiterer V., Kriz A., Hauri H. P., Pavelka M., Sitte H. H., Freissmuth M. (2008) J. Cell Sci. 121, 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farhan H., Korkhov V. M., Paulitschke V., Dorostkar M. M., Scholze P., Kudlacek O., Freissmuth M., Sitte H. H. (2004) J. Biol. Chem. 279, 28553–28563 [DOI] [PubMed] [Google Scholar]
- 10.Farhan H., Reiterer V., Korkhov V. M., Schmid J. A., Freissmuth M., Sitte H. H. (2007) J. Biol. Chem. 282, 7679–7689 [DOI] [PubMed] [Google Scholar]
- 11.Foster J. D., Cervinski M. A., Gorentla B. K., Vaughan R. A. (2006) Handb. Exp. Pharmacol. 175, 197–214 [DOI] [PubMed] [Google Scholar]
- 12.Gnegy M. E. (2003) Eur. J. Pharmacol. 479, 83–91 [DOI] [PubMed] [Google Scholar]
- 13.Fog J. U., Khoshbouei H., Holy M., Owens W. A., Vaegter C. B., Sen N., Nikandrova Y., Bowton E., McMahon D. G., Colbran R. J., Daws L. C., Sitte H. H., Javitch J. A., Galli A., Gether U. (2006) Neuron 51, 417–429 [DOI] [PubMed] [Google Scholar]
- 14.Daniels G. M., Amara S. G. (1999) J. Biol. Chem. 274, 35794–35801 [DOI] [PubMed] [Google Scholar]
- 15.Foster J. D., Pananusorn B., Vaughan R. A. (2002) J. Biol. Chem. 277, 25178–25186 [DOI] [PubMed] [Google Scholar]
- 16.Granas C., Ferrer J., Loland C. J., Javitch J. A., Gether U. (2003) J. Biol. Chem. 278, 4990–5000 [DOI] [PubMed] [Google Scholar]
- 17.Sakai N., Sasaki K., Nakashita M., Honda S., Ikegaki N., Saito N. (1997) J. Neurochem. 68, 2618–2624 [DOI] [PubMed] [Google Scholar]
- 18.Khoshbouei H., Sen N., Guptaroy B., Johnson L., Lund D., Gnegy M. E., Galli A., Javitch J. A. (2004) PLoS Biol. 2, E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seidel S., Singer E. A., Just H., Farhan H., Scholze P., Kudlacek O., Holy M., Koppatz K., Krivanek P., Freissmuth M., Sitte H. H. (2005) Mol. Pharmacol. 67, 140–151 [DOI] [PubMed] [Google Scholar]
- 20.Sitte H. H., Freissmuth M. (2010) J. Neurochem. 112, 340–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha J. H., Zou M. F., Adkins E. M., Rasmussen S. G., Loland C. J., Schoenenberger B., Gether U., Newman A. H. (2005) J. Med. Chem. 48, 7513–7516 [DOI] [PubMed] [Google Scholar]
- 22.Waldmann T. A. (1991) J. Biol. Chem. 266, 2681–2684 [PubMed] [Google Scholar]
- 23.Einhauer A., Jungbauer A. (2001) J. Biochem. Biophys. Methods 49, 455–465 [DOI] [PubMed] [Google Scholar]
- 24.Schmid J. A., Scholze P., Kudlacek O., Freissmuth M., Singer E. A., Sitte H. H. (2001) J. Biol. Chem. 276, 3805–3810 [DOI] [PubMed] [Google Scholar]
- 25.Hilber B., Scholze P., Dorostkar M. M., Sandtner W., Holy M., Boehm S., Singer E. A., Sitte H. H. (2005) Neuropharmacology 49, 811–819 [DOI] [PubMed] [Google Scholar]
- 26.Sitte H. H., Hiptmair B., Zwach J., Pifl C., Singer E. A., Scholze P. (2001) Mol. Pharmacol. 59, 1129–1137 [DOI] [PubMed] [Google Scholar]
- 27.Scholze P., Nørregaard L., Singer E. A., Freissmuth M., Gether U., Sitte H. H. (2002) J. Biol. Chem. 277, 21505–21513 [DOI] [PubMed] [Google Scholar]
- 28.Meinild A. K., Sitte H. H., Gether U. (2004) J. Biol. Chem. 279, 49671–49679 [DOI] [PubMed] [Google Scholar]
- 29.Korkhov V. M., Holy M., Freissmuth M., Sitte H. H. (2006) J. Biol. Chem. 281, 13439–13448 [DOI] [PubMed] [Google Scholar]
- 30.Wüller S., Wiesner B., Löffler A., Furkert J., Krause G., Hermosilla R., Schaefer M., Schülein R., Rosenthal W., Oksche A. (2004) J. Biol. Chem. 279, 47254–47263 [DOI] [PubMed] [Google Scholar]
- 31.Bartholomäus I., Milan-Lobo L., Nicke A., Dutertre S., Hastrup H., Jha A., Gether U., Sitte H. H., Betz H., Eulenburg V. (2008) J. Biol. Chem. 283, 10978–10991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 33.Forrest L. R., Tavoulari S., Zhang Y. W., Rudnick G., Honig B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y., Prusoff W. H. (1973) Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 36.Feige J. N., Sage D., Wahli W., Desvergne B., Gelman L. (2005) Microsc. Res. Tech. 68, 51–58 [DOI] [PubMed] [Google Scholar]
- 37.Chen N., Vaughan R. A., Reith M. E. (2001) J. Neurochem. 77, 1116–1127 [DOI] [PubMed] [Google Scholar]
- 38.Sorkina T., Richards T. L., Rao A., Zahniser N. R., Sorkin A. (2009) J. Neurosci. 29, 1361–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorkina T., Doolen S., Galperin E., Zahniser N. R., Sorkin A. (2003) J. Biol. Chem. 278, 28274–28283 [DOI] [PubMed] [Google Scholar]
- 40.Gu H. H., Wu X., Giros B., Caron M. G., Caplan M. J., Rudnick G. (2001) Mol. Biol. Cell 12, 3797–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gobbi M., Funicello M., Gerstbrein K., Holy M., Moya P. R., Sotomayor R., Forray M. I., Gysling K., Paluzzi S., Bonanno G., Reyes-Parada M., Sitte H. H., Mennini T. (2008) J. Neurochem. 105, 1770–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerstbrein K., Sitte H. H. (2006) Handb. Exp. Pharmacol. 175, 95–111 [DOI] [PubMed] [Google Scholar]
- 43.Sitte H. H., Huck S., Reither H., Boehm S., Singer E. A., Pifl C. (1998) J. Neurochem. 71, 1289–1297 [DOI] [PubMed] [Google Scholar]
- 44.Mager S., Min C., Henry D. J., Chavkin C., Hoffman B. J., Davidson N., Lester H. A. (1994) Neuron 12, 845–859 [DOI] [PubMed] [Google Scholar]
- 45.Mager S., Kleinberger-Doron N., Keshet G. I., Davidson N., Kanner B. I., Lester H. A. (1996) J. Neurosci. 16, 5405–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galli A., Blakely R. D., DeFelice L. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 8671–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galli A., DeFelice L. J., Duke B. J., Moore K. R., Blakely R. D. (1995) J. Exp. Biol. 198, 2197–2212 [DOI] [PubMed] [Google Scholar]
- 48.Saunders C., Ferrer J. V., Shi L., Chen J., Merrill G., Lamb M. E., Leeb-Lundberg L. M., Carvelli L., Javitch J. A., Galli A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6850–6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eriksen J., Rasmussen S. G., Rasmussen T. N., Vaegter C. B., Cha J. H., Zou M. F., Newman A. H., Gether U. (2009) J. Neurosci. 29, 6794–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loland C. J., Norregaard L., Litman T., Gether U. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kniazeff J., Shi L., Loland C. J., Javitch J. A., Weinstein H., Gether U. (2008) J. Biol. Chem. 283, 17691–17701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacobs M. T., Zhang Y. W., Campbell S. D., Rudnick G. (2007) J. Biol. Chem. 282, 29441–29447 [DOI] [PubMed] [Google Scholar]
- 53.Just H., Sitte H. H., Schmid J. A., Freissmuth M., Kudlacek O. (2004) J. Biol. Chem. 279, 6650–6657 [DOI] [PubMed] [Google Scholar]
- 54.Rudnick G. (2006) in Neurotransmitter Transporters (Sitte H. H., Freissmuth M. eds) Vol. 175, pp. 59–73, Springer-Verlag, Berlin [Google Scholar]
- 55.Patterson G. H., Piston D. W., Barisas B. G. (2000) Anal. Biochem. 284, 438–440 [DOI] [PubMed] [Google Scholar]
- 56.Norregaard L., Frederiksen D., Nielsen E. O., Gether U. (1998) EMBO J. 17, 4266–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loland C. J., Norregaard L., Gether U. (1999) J. Biol. Chem. 274, 36928–36934 [DOI] [PubMed] [Google Scholar]
- 58.Nørgaard-Nielsen K., Gether U. (2006) Handb. Exp. Pharmacol. 175, 1–22 [DOI] [PubMed] [Google Scholar]
- 59.Rudnick G. (1998) Methods Enzymol. 296, 233–247 [DOI] [PubMed] [Google Scholar]
- 60.Hansra N., Arya S., Quick M. W. (2004) J. Neurosci. 24, 4082–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robertson S. D., Matthies H. J., Galli A. (2009) Mol. Neurobiol. 39, 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guptaroy B., Zhang M., Bowton E., Binda F., Shi L., Weinstein H., Galli A., Javitch J. A., Neubig R. R., Gnegy M. E. (2009) Mol. Pharmacol. 75, 514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beckman M. L., Bernstein E. M., Quick M. W. (1998) J. Neurosci. 18, 6103–6112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haase J., Killian A. M., Magnani F., Williams C. (2001) Biochem. Soc. Trans. 29, 722–728 [DOI] [PubMed] [Google Scholar]
- 65.Sucic S., Bryan-Lluka L. J. (2002) Brain Res. Mol. Brain Res. 108, 40–50 [DOI] [PubMed] [Google Scholar]
- 66.Sucic S., Bryan-Lluka L. J. (2007) Eur. J. Pharmacol. 556, 27–35 [DOI] [PubMed] [Google Scholar]
- 67.Chen N., Rickey J., Berfield J. L., Reith M. E. (2004) J. Biol. Chem. 279, 5508–5519 [DOI] [PubMed] [Google Scholar]
- 68.Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., Rudnick G. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen N. H., Reith M. E. (2002) in Neurotransmitter Transporters: Structure, Function, and Regulation (Reith M. E. ed) 2nd Ed., pp. 53–110, Humana Press Inc., Totowa, NJ [Google Scholar]
- 70.Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., Ziegler C. (2009) Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 71.Mazei-Robison M. S., Bowton E., Holy M., Schmudermaier M., Freissmuth M., Sitte H. H., Galli A., Blakely R. D. (2008) J. Neurosci. 28, 7040–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.