

Abstract
This paper describes a model system for studying the auto-catalytic phosphorylation of an immobilized substrate by a kinase enzyme. This work uses self-assembled monolayers (SAMs) of alkanethiolates on gold to present the peptide substrate on a planar surface. Treatment of the monolayer with Abl kinase results in phosphorylation of the substrate. The phosphorylated peptide then serves as a ligand for the SH2 adaptor domain of the kinase and thereby directs the kinase activity to nearby peptide substrates. This directed reaction is intramolecular and proceeds with a faster rate than does the initial, intermolecular reaction, making this an auto-catalytic process. The kinetic non-linearity gives rise to properties that have no counterpart in the corresponding homogeneous phase reaction: in one example, the rate for phosphorylation of a mixture of two peptides is faster than the sum of the rates for phosphorylation of each peptide when presented alone. This work highlights the use of an adaptor domain in modulating the activity of a kinase enzyme for an immobilized substrate and offers a new approach for studying biochemical reactions in spatially inhomogeneous settings.
Keywords: Adaptor domain, autocatalysis, interfacial, SAMDI, phosphorylation
Introduction
It is now recognized that biochemical reactions within the cell exploit kinetic non-linearities to regulate the many cellular functions. For example, allosteric regulation of enzymes by metabolites serves to control biosynthetic pathways with spatial and temporal resolution in the cell.[1] Likewise, the extensive connectivity in gene regulatory networks—where the expression of regulatory genes are regulated by their protein products—is vital to coordinating the global expression programs.[2] Enzymes are also equipped with adaptor domains that can direct their locations in the cell and therefore sequester the enzyme from active substrates and redirect the activity towards less active substrates.[3,4] This mechanism is important in the B-cell receptor complex, where an SH2 adaptor domain localizes a kinase to the membrane where it can rapidly initiate signaling.[5] There remains a need for model systems that can be used to identify and understand the mechanisms by which enzyme function can be regulated spatially and temporally. In this paper we describe a model for the auto-catalytic phosphorylation of an immobilized substrate by a kinase having an adaptor domain that recognizes the product it generates, and we demonstrate kinetic features of this reaction that have no counterpart in the corresponding solution phase reaction.
We designed a model system to characterize the amplification in rate that can occur when a kinase acts on an immobilized substrate and also includes an adaptor domain that binds to the resulting phosphorylated peptide (Figure 1A). In this system, phosphorylation of a peptide on the surface is followed by association of the SH2 domain with the phosphopeptide (with a dissociation constant of Kd). The kinase is then present at the surface at an effective concentration that is higher than that in the bulk,[6] where it is expected to rapidly phosphorylate those peptides that are within reach of the tethered kinase (with a rate constant of k2). We refer to these two pathways for reaction as the ‘intermolecular’ and ‘intramolecular’ pathways, respectively. We show below that this interfacial reaction displays a non-linear kinetic profile—that is, the reaction displays an initial lag phase followed by an increasing rate—that is consistent with the auto-catalytic model. We also show that the duration of the lag phase and the magnitude of the rate acceleration depend on the density of the substrate and that the activity of a poor substrate is enhanced when it is presented as a mixture with a good substrate. This work highlights the use of a modular adaptor domain in regulating the activity of a kinase enzyme for an immobilized substrate and offers a new approach for studying biochemical reactions in spatially inhomogeneous settings.
Figure 1.
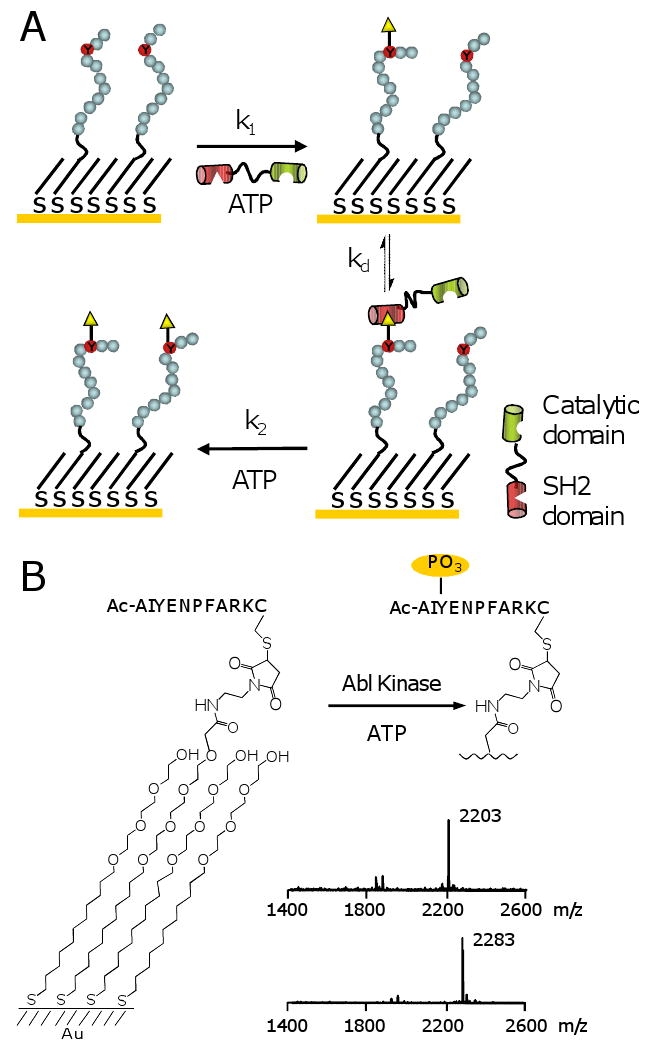
(A) Model system for the auto-catalytic phosphorylation of an immobilized peptide substrate by a kinase that includes an adaptor domain that recognizes its phosphopeptide product. The kinase can phosphorylate the peptide substrate by way of an ‘intermolecular’ pathway (with rate constant k1) or an ‘intramolecular’ pathway (with rate constant k2). (B) SAMDI mass spectrometry was used to characterize the phosphorylation of a peptide immobilized to a monolayer by Abl kinase. A spectrum of the monolayer prior to reaction showed a peak at m/z of 2203, corresponding to the peptide-terminated disulfide. Following treatment with Abl kinase, a mass spectrum of themonolayer revealed a new peak at m/z of 2283, resulting from phosphorylation of the peptide.
Results and Discussion
Experimental Design
To prepare the substrate, we used a self-assembled monolayer that presents a peptide substrate for Abl kinase at a density of 10% mixed with tri(ethylene glycol) groups.[7,8,9] The latter prevent non-specific interactions of the enzyme with the surface and maintain the activity of immobilized peptide. An important benefit of the monolayer is that it can be characterized using matrix-assisted laser desorption-ionization mass spectrometry (i.e., the SAMDI method) to monitor progress of the kinase reaction. [10,11,12] A mass spectrum of a monolayer presenting the substrate Ac-AIYENPFARKC-NH2 revealed a peak at m/z of 2203, corresponding to the peptide-terminated disulfide (Figure 1B). The monolayer was then treated with Abl kinase and ATP for 30 minutes and characterized again to reveal a new peak at m/z of 2283, as expected for the phosphorylation reaction.
Kinase Reaction is Auto-Catalytic
We determined the time course of the reaction by treating separate monolayers with Abl kinase for times ranging from 0 to 45 minutes, and then analyzing the monolayers by SAMDI (Figure 2A). We determined the yield at each time point by dividing the intensity of the peak for the phosphopeptide by the sum of the intensities for the phosphopeptide and the peptide substrate. To confirm that the peak intensities can be used to accurately determine yields for the interfacial reactions, we prepared several monolayers having defined densities of the phosphorylated and parent peptides. We acquired SAMDI mass spectra of these monolayers and found that the determined yields were within 10% of the actual yields. We found that the kinetic profile of the reaction is initially sluggish but then accelerates after a minor amount of product has accumulated. This profile is consistent with the model in Figure 1 and requires that the velocity for the SH2-mediated reaction (the ‘intramolecular’ pathway) is greater than that for the initial reaction (the ‘intermolecular’ pathway). When we repeated the experiment with a monolayer presenting a mixture of the peptide substrate and a phosphorylated peptide (Ac-PVpYENVAKKKC-NH2),[13] at a ratio of 9:1, we found that the lag phase was shortened (Figure 2A). We could also inhibit the intramolecular pathway by adding the soluble phosphopeptide (Ac-PVpYENVAKKKC-NH2) to the reaction mixture (Figure 2B). This same inhibitor had no effect on the phosphorylation of a soluble peptide substrate. These observations are consistent with a model wherein the initial phosphorylation of the immobilized peptide is followed by association of the kinase with the product and an increased rate for the phosphorylation of proximal peptides.
Figure 2.

(A) A time course of the reaction shown in Figure 1 revealed that the phosphorylation reaction was initially slow and then accelerated after accumulation of a minor amount of product (◆). A corresponding time course for a reaction on a monolayer that presents a mixture of peptide and phosphopeptide showed a much shorter lag phase (▲). The data were fit with equation 1 as described in the text. (B) The reaction was repeated in the presence of a soluble inhibitor of the SH2 adaptor domain. The interfacial reaction showed a dose-dependent inhibition in rate (◆) while an analogous reaction performed in solution was unaffected by the inhibitor (▲). The data for the interfacial reaction were fit to a sigmoidal curve described by y=min + (max-min)EC50/(EC50 + 10x) with the minimum (min) and maximum (max) values of y and the concentration of ligand at half-maximum response (EC50) as adjustable parameters. The data for the homogeneous phase reaction were fit to a straight line.
Kinetic Model
Because the ‘intermolecular’ and ‘intramolecular’ pathways operate simultaneously, the observed rate of the phosphorylation reaction is the sum of the rates for the two pathways. In the analysis that follows, we ignore the effect that binding of the kinase to a phosphorylated peptide has in sterically blocking access of a second enzyme to nearby peptide substrates. Equation 1 expresses the rate for the kinase reaction and includes terms for the intermolecular process and the intramolecular process, where the latter explicitly treats inhibition of the SH2 domain by a phosphopeptide I (Ac-AIpYENPFARK-NH2) with a dissociation constant Kd. Γo and Γs are the initial and time-dependent densities of substrate on the monolayer, respectively (where the difference between these two quantities represents the density of the product phosphopeptide Γp). The statistical factor Γs/Γo in the second term reflects the decreasing number of peptide substrates available to a tethered kinase as the reaction progresses. The rate constants k1 and k2 are defined in Figure 1 and are aggregate constants that include the kcat and Km terms; note that k2 is an average rate constant because the actual value depends on the distance that separates the phosphorylated peptide that binds the SH2 domain and the peptide substrate. Of course, the statistical arrangement of peptides on the monolayer results in a range of distances separating each pair of peptides. The first pathway depends only on the density of the substrate and the second pathway depends on the density of both the substrate and the product. This second term has an explicit dependence on the square of the substrate density and gives rise to the auto-catalytic properties of the reaction. In this way, the flux through the second pathway increases as the reaction proceeds and then decreases as the substrate is consumed. Differentiation of equation 1 reveals that the maximum rate is expected to occur when the densities of substrate and product are equal.
We determined the time-course of the reaction and fit the experimental data to equation 1 (with [I]=0) to obtain best-fit values for k1 and k2 of 0.0079 min-1 and 0.525 min-1, respectively. Hence, at the time of maximum rate (that is, when Γs and Γp are equal) the flux through the intramolecular pathway (9.8 pmolcm-2 min-1) was approximately 30-fold greater than that through the intermolecular pathway (0.30 pmol cm-2 min-1). We then repeated the reaction in the presence of increasing concentrations of a soluble phosphopeptide inhibitor (I) and found that the magnitude of the rate amplification decreased with higher concentrations of inhibitor. Figure 3 shows that the experimental data are consistent with the model described above. We used equation 1 to simulate kinetic curves, using the values of k1 and k2 reported above and a dissociation constant for the binding of the phosphopeptide to the Abl SH2 domain of 40 μM. This correlation supports the reasonableness of the model and again establishes the role of the adaptor domain in the intramolecular pathway.
Figure 3.

The kinase reaction was performed in the presence of a soluble phosphopeptide (I) for concentrations ranging from 0 to 1 mM. The solid curves are simulations derived from equation (1) with k1=0.0079 min-1, k2=0.525 min-1 and Kd=40 μM.
| (1) |
Substrate Specificity
We next asked how the relative activities of two different peptide substrates would compare when presented in solution and on a surface. To address this question, we compared the phosphorylation rates of a ‘good’ and a ‘poor’ substrate. Our ‘good’ substrate is that used above (substrate I), Ac-AIYAAPFKKGC-NH2,[4b] and for the ‘poor’ substrate we choose a peptide that still has, in its phosphorylated form, a reasonable affinity for the Abl SH2 domain (substrate II, Ac-AIYENPFARKC-NH2). We first characterized the values of kcat and Km for the two peptides in a homogeneous phase reaction using a ‘pull-down’ assay wherein the reaction proceeds in a homogeneous solution, is stopped, and then the reaction mixture is applied to a monolayer to immobilize the peptides.[14] With the assumption that the peptide substrate and phosphorylated product immobilize with the same rate constant—a reasonable assumption, since the immobilization is mediated by a terminal cysteine residue—the monolayer can be analyzed by SAMDI to determine the extent of reaction in solution. We used a saturating concentration of ATP (1 mM)[15] and found that the phosphorylation of both peptides followed Michaelis-Menten kinetics. We measured the initial velocities of the reaction for multiple concentrations of the substrates and fit these data to a Lineweaver-Burke plot to obtain kcat and kcat/Km values for each peptide. These values are shown in figure 4A and reveal that substrate I is eight-fold more active than is substrate II. We then characterized the activities of the two substrates separately immobilized to monolayers at a density of 10%. We again performed a best fit of the kinetic data to equation 1 (with [I]=0) to obtain k1 and k2 for each substrate (Figure 4B). The values of k1 differed by a factor of 6, in approximate agreement with the relative activities of the soluble peptides. This result is reasonable, since enzyme reactions at surfaces proceed with kinetics similar to that in solution, provided that the surface does not play a steric role in the reaction.[16] The values of k2 differed by only a factor of 1.5, which we suggest is consistent with the relative activity of the kinase for the two substrates in solution in the following sense. In solution, the two substrates have kcat values that differ by a factor of 1.2. On the surface, we believe that the tethered kinase should act on the two substrates with a relative activity that is determined by the kcat values. The Km values, by contrast, are less relevant because the ‘intramolecular’ reaction proceeds with a first order rate constant. The relative values we determine for the interfacial and the homogenous phase reactions are the same within experimental error and suggest that the activities of the peptides on the surface are consistent with a characterization of the kinetic constants for the reactions in solution.
Figure 4.

(A) Phosphorylation of two peptide substrates in solution follows Michaelis-Menten kinetics. Peptide I (◆) is a more active substrate for Abl kinase than is peptide II (▲). (B) The two peptides were separately immobilized to monolayers and treated with Abl. Kinetic profiles for the reactions were fit with equation 1 and gave values of k1 and k2.
Mixture of Peptide Substrates
We next show that the activity of the poor substrate can be substantially enhanced when it is present on the monolayer as a mixture with the good substrate. We prepared a monolayer that had the poor substrate (peptide II) immobilized at a density of 5% and a second monolayer that had a mixture of the two peptide substrates, each at a density of 5%. We treated each monolayer with the kinase (as described above) for times ranging from 0 to 60 minutes and analyzed the extent of reaction using SAMDI mass spectrometry. Because the two peptides had distinct masses, we could independently monitor the rates of phosphorylation of each peptide on the same monolayer (Figure 5). These data show that phosphorylation of the bad substrate is accelerated when the good substrate is present on the monolayer. In the presence of the good substrate, we find that the lag phase for phosphorylation of the poor substrate is shortened and the maximum rate—the slope of the kinetic profile at an extent of reaction of 50%—is increased. These data are consistent with a model wherein rapid phosphorylation of the good substrate generates a phosphorylated peptide that recruits kinase to the monolayer, where it can then phosphorylate the poor substrate (in addition to the good substrate). This model is consistent with the kinetic characterization of the kinase for each peptide alone. As shown above, the kinase acts on the two substrates with a six-fold difference in k1, which governs the length of the lag phase in the reaction. But the values of k2 are comparable, meaning that once tethered, the kinase efficiently phosphorylates both the good and the bad substrate. Hence, phosphorylation of the good substrate initiates the intramolecular pathway for both the good and the poor substrate, thereby circumventing the longer lag phase for the latter. This result again demonstrates a unique contrast between reactions at surfaces and in solution. The activities of two peptides in solution are independent of one another, whereas the role of the adaptor domain connects the activities of the two substrates at the surface.
Figure 5.

A comparison of the rates for phosphorylation of substrate II when present alone (◆) or as a mixture with substrate I (▲). The phosphorylation of substrate II displays a shorter lag phase and a greater maximum rate when it is present as a mixture with substrate I. For the mixed monolayer, the rate for phosphorylation of substrate I is also shown (●).
Intramolecular Reaction Depends on Density
A remaining question concerns the molecularity of the auto-catalytic process, and whether it does indeed proceed through an ‘intramolecular’ pathway wherein the kinase is tethered to the surface at the time the peptide substrate binds the active site or whether the kinase dissociates prior to the next reaction. We reasoned that the intramolecular reaction would show a strong dependence on the density of substrate, and therefore repeated the reaction on monolayers presenting peptide at densities of 1, 2.5, 5, and 10% relative to the total alkanethiolate (Figure 6A). The kinetic profiles reflect two effects of density on rate. First, the lag phase is shorter for higher densities of peptide, as would be expected from the first order dependence of rate on the density of ligand in the intermolecular pathway.[16] Second, the magnitude of the rate acceleration decreases with lower densities of the substrate, and is essentially absent for a monolayer presenting the substrate at a density of 1%. We use a simplified model to suggest that this trend is consistent with the intramolecular pathway (Figure 6B). In the model, the adaptor domain is bound to the monolayer and allows the kinase domain to explore an area of the monolayer approximating a circular ring. We estimate from structural data [17] that the mean separation of the SH2 binding site and the kinase active site is 6 nm and assume that flexibility of the protein allows this distance to vary by ±0.5 nm. With these assumptions, the kinase active site can sweep a perimeter of monolayer of approximately 38 nm2, or 175 alkanethiolates (note that the kinase cannot sample the entire area of this circle, which is sterically blocked by the adaptor domain or conformationally inaccessible to the kinase). Hence, for a substrate density of 1%, there are, on average, one to two peptides available for the tethered kinase, placing this density near the threshold value required for a significant rate enhancement with the intramolecular pathway. For monolayers presenting higher densities of substrate, a tethered kinase might access of order ten substrates, enabling a high degree of rate acceleration.
Figure 6.
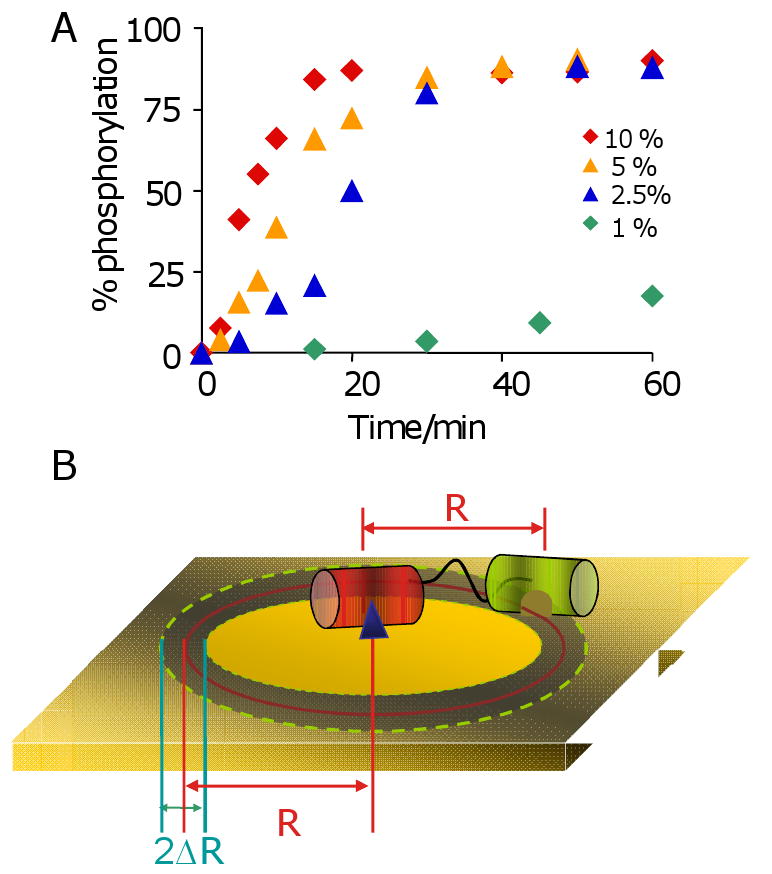
(A) A series of monolayers presenting peptide substrate at densities of 1, 2.5, 5 and 10% were treated with kinase for times ranging from 0 to 60 minutes. The kinetic profiles depend on the density of the substrate and show little auto-amplification at low density. (B) A model to approximate the number of peptide substrates that are available to a kinase that is tethered to the monolayer.
Discussion
The regulation of enzyme activities to give non-linear activities is a well-established feature of biochemical reaction networks. An early set of examples are found in DNA-binding proteins, which bind DNA with low affinity in their monomeric states, but can dimerize at higher concentrations to give potent activation of gene expression. The cooperativity underlying binding of the dimer to the DNA gives rise to a sensitive dependence of activity (here, DNA binding and initiation of gene expression) on the concentration of protein, leading to a switch-like control over gene expression.[18] But the proteins are otherwise present at a uniform concentration in the sample and therefore have a uniform activity; that is the activity of an enzyme does not vary because of its location in the reaction mixture.
The example reported here belongs to a smaller class of enzymes that have rate profiles that vary with position in a sample; that is, the activity of the enzyme is spatially inhomogeneous. In the present example, the immobilization of a substrate for Abl kinase to a planar surface allows the kinase to be recruited to substrates that are proximal to phosphopeptide products. In this way, an initial phosphorylation reaction by the kinase is followed by subsequent phosphorylation reactions that proceed with an increased rate, but only in the immediate vicinity of the initial product. The reaction is auto-catalytic because the product generated by the kinase participates in the molecular mechanism for the subsequent reaction but is itself unchanged during the second reaction. The apparent activity of a substrate is increased if it is found within a critical distance of a product phosphopeptide. This dependence of enzyme activity on location has no analogy in the corresponding homogeneous phase reaction where both the enzyme and peptide are soluble and free to diffuse within the sample to maintain spatially uniform concentrations of all solutes. Further, unlike the examples of auto-catalytic enzyme function that derive from allosteric control over the active site, the example of rate acceleration reported here derives from a co-localization of the enzyme and substrates and not a direct modulation of the catalytic activity of the enzyme at its active site.
We compare this system with two other examples of biochemical reactions whose rates have a dependence on position. Ismagilov and coworkers used a microfluidic device to mimic the threshold response of the blood clotting cascade.[19] They patterned surfaces with tissue factor, a protein that stimulates initiation of blood clotting, and delivered plasma across the surface to probe the formation of a fibrin clot. This work found that the size, pattern and orientation (relative to flow) of the patterned shape had a significant influence on clotting. Simulations supported a reaction-diffusion model wherein a balance of generation and diffusion of the initiator was required to achieve a threshold concentration that then led to auto-catalytic production of the clot. Significantly, this example demonstrates the emergent properties possible with a simple biochemical network and shows how the clever design of model systems can be used to access these properties in the laboratory. In another example, Kuriyan and coworkers characterized the role of membrane localization in the nucleotide exchange of Ras by Son of Sevenless (SOS).[20] SOS has two binding sites for Ras that are allosterically linked. A nucleotide-loaded Ras binds to SOS and enhances the activity of the enzyme for subsequent nucleotide exchange reactions.[21] In this way, the product of the SOS-mediated reaction serves to increase the activity of SOS for further reactions. As with our example, there is a spatial profile to the increased activity, as the reaction is accelerated in the vicinity of products. This kinetic feature is also consistent with the localization of Ras in nanoclustered domains in the cell membrane, which can further accelerate the reaction in signaling processes.[22]
The localization of the kinase to the surface by way of the adaptor domain has an analogy to the role of exosites in enzyme-catalyzed processes. An exosite is a region of the substrate that interacts with an enzyme at a site removed from the active site and that increases the binding affinity of the substrate for the enzyme.[23] The deacetylase HDAC8, for example, interacts with a peptide from the N-terminal tail of histone 4 at a distal site and is required for activity.[24] In the present example, the interaction of the kinase SH2 domain with a phosphorylated peptide serves to increase the concentration of the kinase near its peptide substrate—that is, the combination of the monolayer and the phosphorylated and unmodified peptides represent a ‘substrate’—but with two important distinctions. First, not all peptides have uniform activity since there is a distribution of distances that separate each peptide from its nearest phosphorylated neighbor and those that are not at the optimal distance will have a reduced activity. Second, enzymes that recognize substrates having exosites display an enhanced activity, but this activity is uniform and applies to all of the substrate molecules. For the kinase substrates immobilized to a monolayer, the activities of the peptide substrates are dependent both on location and on time.
This work also has an analogy to studies of processive phosphorylation of substrates having multiple tyrosine residues that are recognized by the kinase. Mayer and coworkers reported studies of the phosphorylation of Crk-associated substrate, a model substrate that has multiple phosphorylation sites for Abl kinase.[3a] When the SH2 domain was active, Abl efficiently hyperphosphorylated the substrate whereas inactivation of the adaptor domain resulted in a lower level of phosphorylation. Control experiments show that the processive activity requires that the adaptor domain of the kinase binds to the phosphopeptide and is consistent with a model wherein initial phosphorylation is followed by association of the kinase with the substrate which then can efficiently act on other sites within the bound complex. This work also showed that the specificity of the adaptor domain could override the specificity of the kinase domain; that is, the adaptor domain could direct kinase activity to substrates that were otherwise poorly active. This collaboration between the intrinsic activity of a substrate and its recruitment to an enzyme gives another dimension to control substrate specificity.[25] Finally, Cantley and coworkers have reported that the kinase substrates of several non-receptor kinases have specificities that match those of the SH2 domains for the phosphorylated product, raising the possibility that auto-catalytic phosphorylation of substrates may be a common motif in signaling pathways.[4b]
Another feature of this system that has no analogy in the corresponding solution-phase reaction is the increased activity of a poor substrate when it is present on the monolayer together with a good substrate. The reaction again begins with phosphorylation of the better substrate, but then yields a product that can catalyze phosphorylation of the poor substrate by recruiting a kinase to the surface for a subsequent ‘intramolecular’ reaction. In the corresponding homogeneous phase reaction, the rate of phosphorylation of each peptide is constant whether it is present alone or in a mixture, though under certain conditions—at low concentrations of the kinase—the presence of a good substrate can suppress the phosphorylation of the poor substrate through competition for access to the enzyme, which would lead to the opposite effect—an increase in the discrimination of the two substrates.[26]
Finally, we note one more aspect in which interfacial and homogenous reactions differ. We reason that the interfacial reaction must proceed to give ‘islands’ of phosphorylated peptide on the surface, though we have not characterized these structures directly. With studies of enzyme function in solution, rapid diffusion of the peptide substrate relative to the enzyme serves to maintain equilibrium concentrations of the substrate and product throughout the sample. When the peptides are immobilized to a surface, however, not all peptides are equal in their activity. Those that are neighbors to a phosphopeptide have a higher activity and will be converted to product by way of the intramolecular pathway. It is tempting to speculate that this type of effect could play a role in the generation of lipid rafts, or domains of enzymatically-modified phosopholipid in the cell membrane. The present work does not address these possibilities, but related work points to the modularity inherent to engineering adaptor domains to rewire cellular pathways.[27]
Conclusion
In summary, this paper describes a model system for studying the reactions of enzymes whose activities towards immobilized substrates vary with position and time. The system is attractive because it is experimentally simple and well-suited to mechanistic studies of the factors that give rise to emergent kinetic features of enzyme-mediated reactions. The extension of this model system to include monolayers that are initially patterned with domains of phosphorylated and unmodified peptides, and the inclusion of an opposing phosphatase activity represent directions for further exploring the features of this system.
Experimental Section
Synthesis of Peptides
Peptides were synthesized at 0.1mmol scale each on Fmoc-Rink amide 4-methylbenzhydrylamine resin (AnaSpec. Inc., San Jose, CA). All peptides were purified by reverse phase HPLC on a C18 column (Waters).
Preparation of SAMs Presenting Maleimide Groups
The maleimide-presenting SAMs were prepared as previously reported.[7] Briefly, gold-coated coverslips (4 nm Ti, 22 nm Au) were immersed in an ethanolic solution containing a symmetric disulfide presenting tri(ethylene glycol) groups and an asymmetric disulfide presenting one maleimide group and one tri(ethylene glycol) group at different ratio(in case of 10% maleimide density, a molar ratio of 4:1 was used) overnight with a total concentration of disulfide of 0.2mM.
Kinase Assay
Phosphate buffer (pH 7.4) containing cysteine-terminated peptides (0.2mM) were applied to SAMs and incubated at 30 °C for 1 hour to immobilize peptides on monolayers. The monolayers were rinsed with distilled water and ethanol and dried under nitrogen flow. Abl kinase (New England Biolabs, Ipswich, MA) was diluted to 1∼2 U μL-1 in kinase buffer(50 mM Tris-HCl, 10 mM MgCl2, 1 mM EGTA, 2 mM DTT, 0.01% Brij 35, pH 7.5) with ATP (0.2∼1 mM), and 1-2 μL was applied to each monolayer (∼4 mm2) and incubated at 30 °C for different time periods.
Mass Spectrometry
Following the peptide phosphorylation by kinase, monolayers were rinsed as described above and then treated with matrix solution (2,4,6-trihydroxyacetophenone, 5 mg mL-1 in acetonitrile), dried, and loaded in a Voyager DE-PRO Biospectrometry mass spectrometer (Applied Biosystems, Framingham, MA). A 337 nm nitrogen laser was used as a desorption/ionization source with accelerating voltage of 20 kV and extraction delay time of 50 ns. All spectra were acquired using positive reflector mode. All spectra were Gaussian-smoothed and baseline-corrected.
Semi-Quantitative Analysis and Ionization Efficiency of Peptides
For quantitation, the extent of phosphorylation was determined based on the relative peak intensity of product and substrate on SAMDI spectra: Y= Ip/(Ip+Is), where p refers to the phosphorylated peak and s refers to the parent peak. To calibrate the ionization efficiency of parent peptides and phosphorylated peptides, we prepared a series of monolayers with defined densities of maleimide-conjugated phosphorylated and parent peptides. SAMDI mass spectra of these monolayers showed that the determined yields were within 10% of the actual yields.
Acknowledgments
This work was supported by the NSF.
References
- 1.(a) Umbarger HE. Science. 1956;123:848. doi: 10.1126/science.123.3202.848. [DOI] [PubMed] [Google Scholar]; (b) Madsen NB, Kasvinsky PJ, Fletterick RJ. J Biol Chem. 1978;253:9097–9101. [PubMed] [Google Scholar]
- 2.Shen-Orr SS, Milo R, Mangan S, Alon U. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mayer BJ, Hirai H, Sakai R. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]; (b) Pellicena P, Stowell KR, Miller TW. J Biol Chem. 1998;273:15325–15328. doi: 10.1074/jbc.273.25.15325. [DOI] [PubMed] [Google Scholar]; (c) Lewis LA, Chung CD, Chen Jy, Parnes JR, Moran M, Patel VP, Miceli MC. J Immunol. 1997;159:2292–2300. [PubMed] [Google Scholar]; (d) Duyster J, Baskaran R, Wang JYJ. Proc Natl Acad Sci U S A. 1995;92:1555–1559. doi: 10.1073/pnas.92.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]; (b) Zhou S, Carraway KL, III, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley LC. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 5.(a) Reth M, Brummer T. Nat Rev Immunol. 2004;4:269–277. doi: 10.1038/nri1335. [DOI] [PubMed] [Google Scholar]; (b) Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WWA, Zurn C, Reth M. Mol Cell. 2002;10:1057–1069. doi: 10.1016/s1097-2765(02)00739-6. [DOI] [PubMed] [Google Scholar]
- 6.Kholodenko BN, Hoek JB, Westerhoff HV. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 7.Houseman TB, Gawalt SE, Mrksich M. Langmuir. 2003;19:1522–1531. [Google Scholar]
- 8.Su J, Mrksich M. Langmuir. 2003;19:4867–4870. [Google Scholar]
- 9.Mrksich M. ACS Nano. 2008;2:7–18. doi: 10.1021/nn7004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su J, Mrksich M. Angew Chem, Int Ed. 2002;41:4715–4719. doi: 10.1002/anie.200290026. [DOI] [PubMed] [Google Scholar]
- 11.Min DH, Su J, Mrksich M. Angew Chem, Int Ed. 2004;43:5973–5977. doi: 10.1002/anie.200461061. [DOI] [PubMed] [Google Scholar]
- 12.Su J, Bringer MR, Ismagilov RF, Mrksich M. J Am Chem Soc. 2005;127:7280–7281. doi: 10.1021/ja051371o. [DOI] [PubMed] [Google Scholar]
- 13.Cowburn D, Zheng J, Xu Q, Barany G. J Biol Chem. 1995;270:26738–26741. doi: 10.1074/jbc.270.45.26738. [DOI] [PubMed] [Google Scholar]
- 14.Min DH, Yeo WS, Mrksich M. Anal Chem. 2004;76:3923–3929. doi: 10.1021/ac049816z. [DOI] [PubMed] [Google Scholar]
- 15.Foulkes GJ, Chow M, Gorka C, Frackelton RA, Jr, Baltimore D. J Biol Chem. 1985;260:8070–8077. [PubMed] [Google Scholar]
- 16.Nayak S, Yeo WS, Mrksich M. Langmuir. 2007;23:5578–5583. doi: 10.1021/la062860k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]; (b) Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, Kuriyan J. Mol Cell. 2006;21:787–798. doi: 10.1016/j.molcel.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 18.(a) Pabo CO, Sauer RT. Annu Rev Biochem. 1992;61:1053–1095. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]; (b) Ackers GK, Johnson AD, Shea MA. Proc Natl Acad Sci USA. 1982;79:1129–1133. doi: 10.1073/pnas.79.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Kastrup CJ, Runyon MK, Shen F, Ismagilov RF. Proc Natl Acad Sci U S A. 2006;103:15747–15752. doi: 10.1073/pnas.0605560103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kastrup CJ, Shen F, Ismagilov RF. Angew Chem, Int Ed. 2007;46:3660–3662. doi: 10.1002/anie.200604995. [DOI] [PubMed] [Google Scholar]
- 20.Gureasko J, Galush WJ, Boykevisch S, Sondermann H, Bar-Sagi D, Groves JT, Kuriyan J. Nat Struct Mol Biol. 2008;15:452–461. doi: 10.1038/nsmb.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi Dafna, Kuriyan J. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 22.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Nat Cell Biology. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 23.(a) Krishnaswamy S. J Thromb Haemostasis. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]; (b) Overall CM. Methods Mol Biol. 2001;151:79–120. [PubMed] [Google Scholar]
- 24.Gurard-Levin ZA, Mrksich M. Biochemistry. 2008;47:6242–6250. doi: 10.1021/bi800053v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu G, Liu Y, Shaw S. Cell Cycle. 2005;4:52–56. doi: 10.4161/cc.4.1.1353. [DOI] [PubMed] [Google Scholar]
- 26.Ubersax JA, Ferrell JE., Jr Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 27.(a) Yeh BJ, Rutigliano RJ, Deb A, Bar-Sagi D, Lim WA. Nature. 2007;447:596–600. doi: 10.1038/nature05851. [DOI] [PubMed] [Google Scholar]; (b) Haword PL, Chia MC, Rizzo SD, Liu FF, Pawson T. Proc Natl Acad Sci U S A. 2003;100:11267–11272. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]