

Abstract
Nicotinic acetylcholine receptor (nAChR) genes form a highly conserved gene cluster at the lung cancer susceptibility locus 15q25.1. In this study, we show that the CHRN α 3 gene encoding nAChR α 3 subunit is a frequent target of aberrant DNA hypermethylation and silencing in lung cancer, whereas the adjacent CHRN β 4 and CHRN α 5 genes exhibit moderate and no methylation, respectively. Treatment of cancer cells exhibiting CHRN α 3 hypermethylation with DNA methylation inhibitors caused demethylation of the CHRN α 3 promoter and gene re-activation. Restoring CHRN α 3 levels through ectopic expression induced apoptotic cell death. shRNA-mediated depletion of nAChR α 3 in CHRN α 3-expressing lung cancer cells elicited a dramatic Ca2+ influx response in the presence of nicotine, followed by activation of the Akt survival pathway. CHRN α 3-depleted cells were resistant to apoptosis-inducing agents, underscoring the importance of epigenetic silencing of the CHRN α 3 gene in human cancer. In defining a mechanism of epigenetic control of nAChR expression in non-neuronal tissues, our findings offer a functional link between susceptibility locus 15q25.1 and lung cancer, and suggest nAChRs as theranostic targets for cancer detection and chemoprevention.
Keywords: epigenetic silencing, DNA methylation, nicotine acetylcholine receptor genes, apoptosis, lung cancer
INTRODUCTION
Nicotinic acetylcholine receptors (nAChRs) encoded by the nicotinic acetylcholine receptor (CHRN) genes clustered at the lung cancer susceptibility locus 15q25 (1-4), are prototypic ligand-gated ion channels that are activated by the endogenous agonists (acetylcholine and choline) and the exogenous compound nicotine. Activation of nAChR induces the opening of nonselective cation channels and voltage-gated Ca2+ channels (5) and the principles of nAChR function have been preserved in evolution (6). CHRN genes are expressed in both neuronal and non-neuronal tissues, suggesting that nAChRs may play an important role in processes other than synaptic transmission. Indeed, apart from their classical role at neuromuscular junctions, nAChRs have also been implicated in the regulation of cellular processes such as proliferation, cell-cell interaction, and cell death (7-10), although underlying mechanisms remain poorly understood.
Different nAChR-subunits are expressed in normal lung tissues, and nicotine exposure has been theorized as influencing the expression of nicotinic acetylcholine receptor subunit genes (9, 10). The nAChR subunit composition in-turn further regulates function and pharmacology of nAChR; however, the exact mechanisms governing expression and assembly of nAChRs in normal lung epithelium and lung cancer tissues is largely unknown (7, 8, 11). nAChRs are thought to be hetero-pentamers composed of combinations of different α and β subunits, encoded by a conserved family of at least 12 CHRN genes (CHRNα2-α10 and CHRNβ2-β4). Although several single nucleotide polymorphisms (SNPs) spanning the CHRNα3-CHRNβ4-CHRNα5 gene cluster have been associated with lung cancer incidence and susceptibility, only SNP RS16969968 has been identified to result in the frequent amino acid substitution Asn398Asp in the CHRNα5 gene (12).
Interestingly, it was found that lung cancer cells may express a distinct pattern of nAChR subunits (13), and activation of nAChR receptors and nAChR subunit composition may regulate critical cellular processes in non-neuronal tissues (Schuller, 2009). For example, nicotine, at concentrations found in active smokers, was shown to inhibit apoptosis in lung cancer cells (14), whereas the activation of nAChRs in lung epithelial cells triggered stimulation of cell proliferation (14, 15). These results suggest that deregulation of CHRN gene expression and changes in nAChR functional states may lead to disruption of normal cell proliferation and cell death in normal lung tissues. Watanabe et al found that the nAChRα4 gene promoter exhibit differential patterns of DNA methylation in murine non-neuronal tissues (liver, muscle and brain), suggesting that epigenetic mechanisms may be responsible for the tissue-specific expression of the nAChR genes (16). However, little is known on the extent of deregulation of nAChR-encoding genes in human cancer and possible mechanisms underlying the disruption of nAChR function in lung tissues.
In this study, we tested the hypothesis that expression of nAChR encoding genes clustered at the 15q25.1 locus may be under epigenetic regulation and that epigenetic silencing of CHRN genes may contribute to lung cancer. We present evidence indicating that the CHRNα3 gene exhibits frequent DNA hypermethylation in lung tumours, and that these epigenetic changes are associated with unscheduled gene silencing and abrogation of cell death.
MATERIALS AND METHODS
Tumour samples
Lung cancer samples and blood control samples were obtained from a case-control study on lung cancer conducted at Cancer Research Centre, Moscow (Russia), as a part of a larger multicenter case-control study coordinated by the International Agency for Research on Cancer (2, 17). Both lung cancer samples and blood control samples used were described elsewhere (2, 17). Informed consent was obtained from all patients, and the study was approved by the relevant Institutional Review Committee.
Cell lines, culture conditions and transfections
Human lung cancer cell lines used were maintained in standard medium under conditions recommended by the American Type Culture Collection. Transient transfections for these cells were carried out using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Mammalian expression constructs containing full-length cDNAs of the CHRNα3, CHRNβ4 and CHRNα5 genes, under control of CMV promoter, were kindly provided as a gift from Dr. Jon Lindstrom (University of Pennsylvania, Philadelphia). For inducible depletion of the CHRNα3 gene, the H1299 and H1650 lung cancer cells were transfected with pTRIPZ plasmid encoding shRNA against the CHRNα3 gene (V2LHS_112345; Thermo Fisher Scientific), followed by TET-ON induction of shRNA for 24–48 hours with 1.0 μg/ml doxycycline. In this plasmid, RFP is induced as part of the same transcript as the shRNA, and hence was used as a reporter for morphometric scoring.
DNA methylation analysis
DNA methylation analysis was performed by Pyrosequencing following DNA extraction and bisulfite conversion as previously described (17, 18). For this analysis, we used 142 primary tumours of the lung and 164 blood samples (Supplementary Table 1). Adjacent nonmalignant lung tissues were available from 60 cases and were also used for the analysis. Bisulfite treatment was carried out with EZ Methylation-Gold kit (Proteigene) following the manufacturer’s recommendations. The bisulfite-converted DNA was pyrosequenced using a pyrosequencing system (PSQ 96MA, Biotage). Pyrosequencing assays were designed for quantitative measurement of DNA methylation levels in the promoter region of the CHRNα3, CHRNβ4 and CHRNα5 genes (Supplementary Table 2).
Gene expression analysis
Total RNA was extracted from different cell cultures using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. The reverse transcriptase reaction was subjected to semi-quantitative or quantitative PCR analysis (Supplementary Table 3). For quantitative PCR analysis, the real-time quantitative PCR were performed in quadruplicate with the MXP3000 real-time PCR system (Stratagene) using the Universal Probe Library System (Roche) and TaqMan Universal PCR Master Mix according to the manufacturer’s instructions. Primers and probes were designed using Universal Probe Library Assay Design Center (Roche) (Supplementary Table 3).
Demethylating drug treatment
Cells were incubated in culture medium containing 5 μM of 5-Aza-2’-deoxycytidine (Sigma, St. Louis, MO) or 50 μM RG108 (a kind gift from Deutsches Krebsforschungszentrum, Heidelberg) for 6 days with daily medium change. The effects of demethylating agents on the methylation and expression levels of the candidate CHRN genes were monitored by pyrosequencing and RT-qPCR, respectively.
Annexin V-FITC Assay
Cells in early stages of apoptosis were detected using the ApoAlert Annexin V-FITC apoptosis kit (Clontech), by following the manufacturers protocol.
Western Blot Analysis
Western blotting was carried out essentially as described previously (19). Blots were probed with Anti-CHRNα3 antibody (ab55773; Abcam) at a 1:1000 dilution. Antibodies against native and phospho- Akt,-p42/p44, -p38, NFkb and PARP were all from Cell Signaling Technology. Mouse monoclonal antibody (Clone C4; MP Biomedicals, France) to β-actin was used for loading control.
Immunohistochemical analysis
The avidin-biotin-peroxidase complex method was used for immunohistochemical analysis of α3 nAChR. After deparaffinization, unstained slides were treated with microwave heating in antigen retrieval solution for 10 minutes. Anti-CHRNα3 antibody (a gift from Prof. Lindstrom; University of Pennsylvania, Philadelphia) was applied at a 1:50 dilution.
Ca2+ influx microscopy
The effect of nicotine on Ca2+ influx was monitored essentially as previously described (20). Briefly, the cells were seeded at concentrations of 1×105/ml into a 4-chamber glass (Labtek). Cells positive for induction were tracked by RGFP reporter gene expression. Dye-loaded lung cancer cells were treated with 10mM nicotine and examined using a confocal argon ion laser scanning unit (LSM 410, class 3B; Carl Zeiss, Oberkochen, Germany).
Statistical Analysis
Statistical analysis was carried out essentially as described previously (17), using SPSS software, version 16.1. Correlation between immunohistochemical staining intensity scores and DNA methylation level was determined using Spearman’s nonparametric correlation analysis. ANOVA, followed by Newman-Keuls’ test, was used to determine mean differences between different groups of replicates, to find differences within the methylation and expression levels. Mean differences for apoptotic responses between mock-transfected and transfected with different CHRN gene constructs were calculated using Students ‘t’ test. P < 0.05 or P < 0.01 was considered statistically significant.
RESULTS
CHRNα3 and CHRNβ4 genes exhibit lung tumour specific hypermethylation
To test the hypothesis that nAChR encoding genes may be regulated epigenetically, we quantitatively determined DNA methylation levels at multiple CpG sites in the promoter of three nAChR encoding genes (CHRNα3, CHRNβ4 and CHRNα5) clustered at locus in chromosome region 15q25.1 (Figure 1A). All the CpG sites included in the analysis are in the bona fide CpG islands and located at 377-403 bp, 3-49 bp and 277-316 bp upstream of the transcription start sites of the α3,β4 and α5 subunit encoding CHRN genes, respectively. We combined pyrosequencing, the technique allowing quantitative analysis of multiple CpG sites, with a series of lung tumours (n=142) and control blood samples (n=164) (Figure 1B). Results of the pyrosequencing analysis of CHRNα3, CHRNβ4 and CHRNα5 genes in lung tumours and blood samples are shown in Figure 1C and Supplementary Figure 1. The results revealed that CHRNα3 exhibited significantly higher levels of DNA methylation in lung tumours (Figure 1C, Supplementary Figure 1). CHRNα5 was virtually unmethylated (mean methylation levels <5%) (Figure 1), whereas CHRNβ4 exhibited intermediate methylation levels (Figure 1C). These results indicate that CHRNα3 and CHRNβ4 exhibit a highly significant and moderate hypermethylation, respectively, whereas CHRNα5 remains unmethylated in lung tumours.
Figure 1. DNA Methylation analysis of the nicotine acetylcholine receptor genes clustered at the 15q25.1 locus in lung cancer.
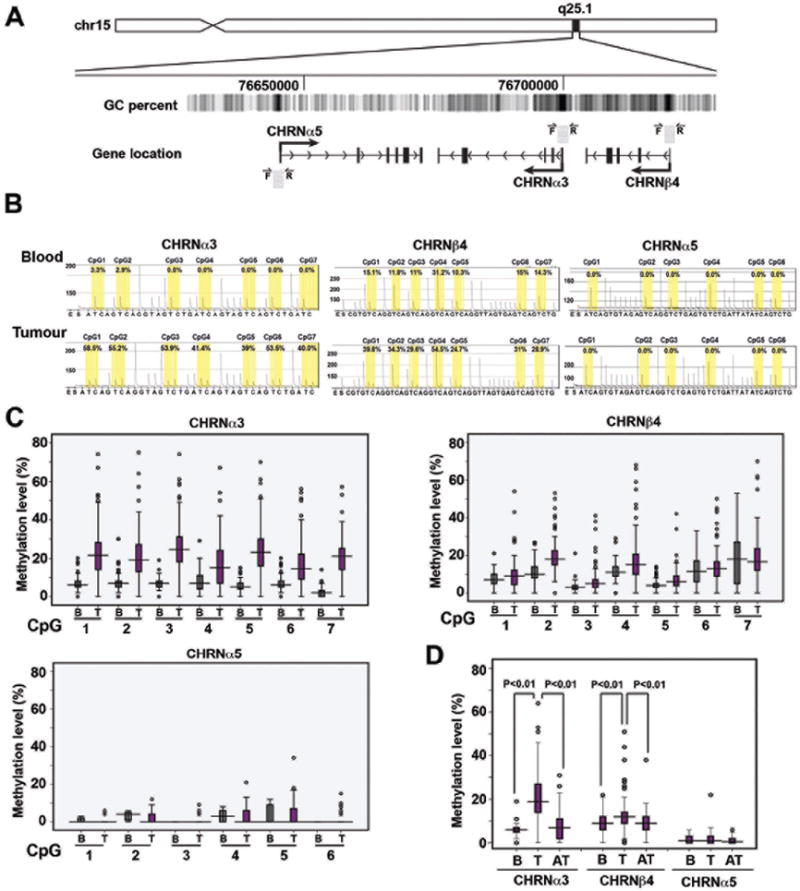
A) Schematic representation of the chromosomal arrangement of the 15q25.1 loci containing three nicotine acetylcholine receptor genes. Location of the analyzed sequence and sets of primers for each gene analyzed are indicated by grey boxes and arrows, respectively. B) Representative pyrograms of all three CHRN genes obtained from the pyrosequencing assays used to quantitatively determine the DNA methylation status across multiple CpG sites. C) Boxplots of the results obtained by the analysis of individual genes and CpG sites. DNA methylation levels obtained by analyzing the mean methylation levels of all the CpG sites for CHRNα3, CHRNβ4 and CHRNα5 gene in tumour tissues (T) and blood samples (B). (D) Analysis of methylation of CHRN genes in lung tumours and normal appearing adjacent tissues. Levels of methylation obtained for CHRNα3, CHRNβ4 and CHRNα5 genes in blood (B), lung tumours (T), and normal-appearing adjacent tissues (AT). The statistical significance for differential methylation in tissues analyzed was calculated using Newman-Keuls’ test.
We next analyzed methylation levels of CHRNα3, CHRNβ4 and CHRNα5 in normal appearing adjacent lung tissues and found that levels of DNA methylation in CHRNα3 and CHRNβ4 in adjacent tissues were comparable to those in blood samples, but significantly lower in comparison to the methylation levels seen in lung cancer (Figure 1D). These results confirm the notion that CHRNα3 and CHRNβ4 exhibit tumour-specific hypermethylation.
Analysis of DNA methylation frequency (defined as the percentage of tumour samples with methylation levels >90% quantile levels in non-tumour adjacent lung tissue samples), showed that CHRNα3 and CHRNβ4 genes were frequently hypermethylated (in 37% and 24% patient samples, respectively), while hypermethylation of the CHRNα5 gene was relatively infrequent (7%) in lung tumors (Table 1; Supplementary Figure 1).
Table 1.
Frequency of CpG hypermethylation in lung tumours
| Gene | Methylation levels Maximum/Minimum/Average | Methylation level cut-offˆ | Hypermethylated tumor samples* (%) |
|---|---|---|---|
| CHRNα3 | 64.83% / 0% / 21.14% | 22.67% | 52/142 (36.6%) |
| CHRNβ4 | 51.14% / 0% / 13.36% | 15.03% | 30/124 (24.2%) |
| CHRNα5 | 7.51% / 0% / 1.57% | 5.72% | 7/103 (6.8%) |
The 90% percentile value for surrounding non-tumour lung tissue samples.
Samples with methylation levels above the quantile representing the upper 90% of methylation in surrounding non-tumour lung tissue samples.
Hypermethylation of CHRNα3 is associated with its silencing in lung cancer
Because aberrant DNA hypermethylation of the gene promoter can lead to downregulation of gene expression, we next determined whether levels of CHRN methylation correlate with expression of the gene. Immunohistochemical analysis using antibody against CHRNα3 protein revealed that the tumour samples with CHRNα3 hypermethylation showed lower staining intensities in comparison to those with less methylation (significant correlation; r=-0.66), indicating that samples having hypermethylation of CHRNα3 gene have less expression levels of the nAChRα3 protein (Figure 2A, B). These results suggest that hypermethylation of CHRNα3 gene may result in partial or complete silencing of the gene. Interestingly, a similar association between staining intensities of nAChRβ4 protein and the CHRNβ4 methylation levels in lung cancer was not observed (data not shown).
Figure 2. Expression of CHRN genes and its association with DNA hypermethylation.
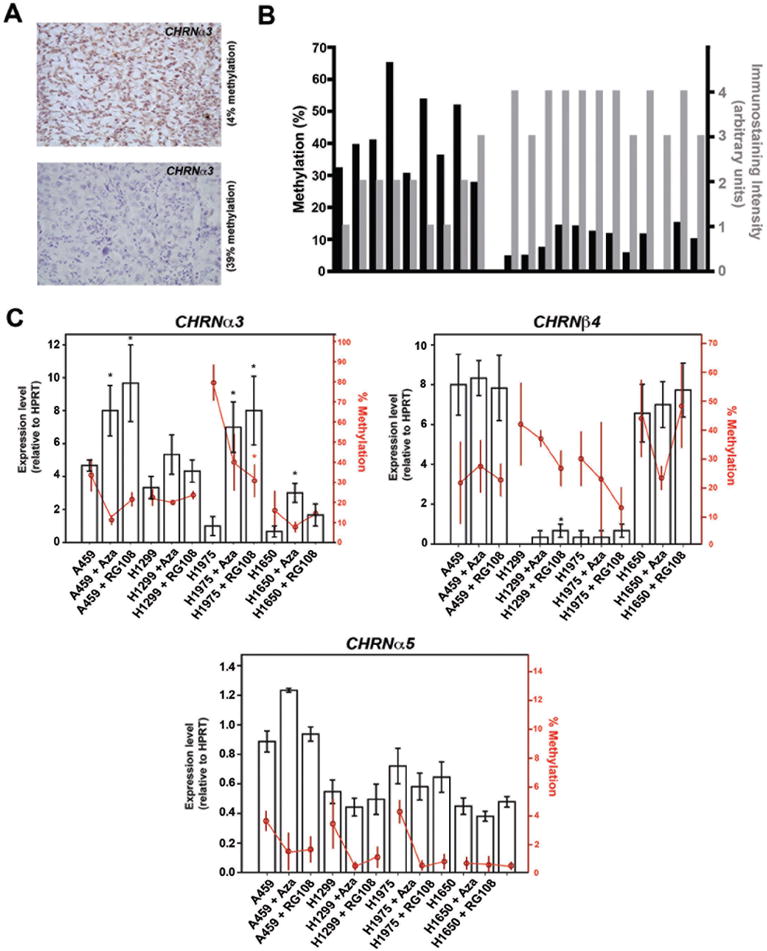
A) Immunohistochemical analysis showing methylation level-dependent expression of nAChRα3 among different lung tumour samples. Representative micrographs of immunohistochemical analysis show high (upper panel) and low (lower panel) expression of nAChRα3 in the two representative lung cancer samples. Dark brown staining is indicative of the presence of nAChRα3 protein. Tissue is counterstained to indicate cell nuclei (blue staining). B) Comparison of DNA methylation and expression levels of nAChRα3 in individual lung tumour samples. Intensity of staining was scored from 0 to 4 as described in Materials and Methods. A significant inverse correlation between CHRNα3 methylation and nAChRα3 expression levels was observed (Pearson Correlation coefficient, r = -0.66). C) The effect of treatment with DNA demethylating drugs on CHRN gene expression in lung cancer cells. Four different lung cancer cell lines were treated with indicated doses of two different demethylating drugs (5-aza-2’deoxycytidine, Aza; and RG108) for 5 days and mRNA and methylation levels of CHRNα3, CHRNβ4, and CHRNα5 were analyzed. The gene expression levels were determined by quantitative-RT-PCR assays specific for each CHRN gene and DNA methylation levels were measured quantitatively by pyrosequencing. Black bars and red circles represent mean expression and methylation values, respectively. The error bars represent standard error. For a given cell line, statistically significant (P<0.05) methylation or expression levels were calculated by comparing the treated groups with the vehicle treated group (control). Statistically significant expression or methylation values (P<0.05) are indicated by asterisks.
We next treated four lung cancer cell lines (A459, H1299, H1975 and H1650) with two different demethylating drugs, 5-Aza-2’-Deoxycytidine (AzaC) and RG108 (21), and examined both the expression and methylation levels of the CHRN genes. Although there were notable differences in DNA methylation levels between cell lines, the observed methylation levels in the cell lines were comparable to those observed for the individual genes in different lung tumour samples (Figure 2C and Figure 1C).
Treatment with either of two different DNA methylation inhibitors resulted in a modest but consistent demethylation of CHRNα3 gene in cancer cell lines. Importantly, CHRNα3 responded with higher levels of expression following treatment with a demethylating agent (Figure 2C), consistent with previous reports that DNA methylation inhibition can effectively reactivate transcriptionally silent genes associated with unscheduled hypermethylation (22). In contrast, demethylation treatment failed to significantly increase expression levels of CHRNβ4 and CHRNα5 despite a noticeable reduction in methylation levels in these genes (Figure 2C), suggesting that methylation levels of these genes found in these cancer cells do not interfere with their expression. Therefore, DNA hypermethylation appears to specifically target the expression of the CHRNα3 gene, whereas the CHRNβ4 and CHRNα5 genes may not be deregulated through aberrant DNA methylation in lung cancer cells.
Restoration of expression or overexpression of CHRNα3 induces apoptotic cell death
To understand the functional impact of deregulated expression of the CHRNα3 gene on cellular processes, we sought to restore expression of CHRNα3 gene in cancer cell lines. To this end, CMV promoter-driven CHRNα3, CHRNβ4 or CHRNα5 expression vectors or an empty vector (control) were transfected into H1975 cells, wherein high levels of the CHRNα3 promoter methylation were found to result in lower expression of the gene (Figure 2C), and examined their expression. RT-PCR analysis of transfected cells revealed high levels of expression of the corresponding CHRN genes in comparison to the mock-transfected control (Figure 3A). In addition, Western blot analysis of cell lysates from CHRNα3-transfected cells showed significantly higher levels of nAChRα3 protein levels in comparison to the mock-transfected cells (Figure 3A). To test the effect of increased expression of CHRN genes on cell death, H1975 cells were mock-transfected or transfected with increasing concentrations of plasmids expressing CHRNα3, CHRNβ4, or CHRNα5, and cell death fraction after staining with FITC-conjugated Annexin V (whose binding to externalized phosphatidyl-serine on cell membrane is an early indicator of apoptosis) was analyzed. As shown in Figure 3B, C, CHRNα3 transfection resulted in a dose-dependent increase in apoptotic fraction, whereas transfection of cells with the vector expressing CHRNβ4 and CHRNα5 genes failed to induce appreciable levels of apoptotic cell death (Figure 3B, and data not shown). Increased apoptotic cell death in cells transfected with CHRNα3 was further verified by flow cytometric analysis of cell cycle profiles following PI staining, which revealed significantly higher sub-G1 population in CHRNα3-transfected cells (76%) in comparison to mock transfected cells (8.5%) (Figure 3C), and the induction of proteolytic cleavage of PARP (Figure 3C), a hallmark of apoptosis. These results indicate that restoration of nAChRα3 expression in cancer cells induces cell death, and that cell death triggered by ectopic expression of CHRNα3 gene is mediated by the apoptotic pathway. Induction of apoptosis following restoration of CHRNα3 expression was further found to be associated with activated MAPK signaling pathway (as judged by a dose-dependent increase in phosphorylation levels of both p44/p42 MAP kinase and p38 MAP kinase proteins), whereas activation of the NFκB signalling pathway was not observed in these cells (Figure 3D). These results suggest that epigenetic silencing of CHRNα3 may impair apoptotic cell death during tumour development, and that the restoration of its expression may sensitize cancer cells to apoptotic death through activation of MAPK stress signalling.
Figure 3. Restoration of expression of CHRNα3 in cancer cells induces apoptosis.
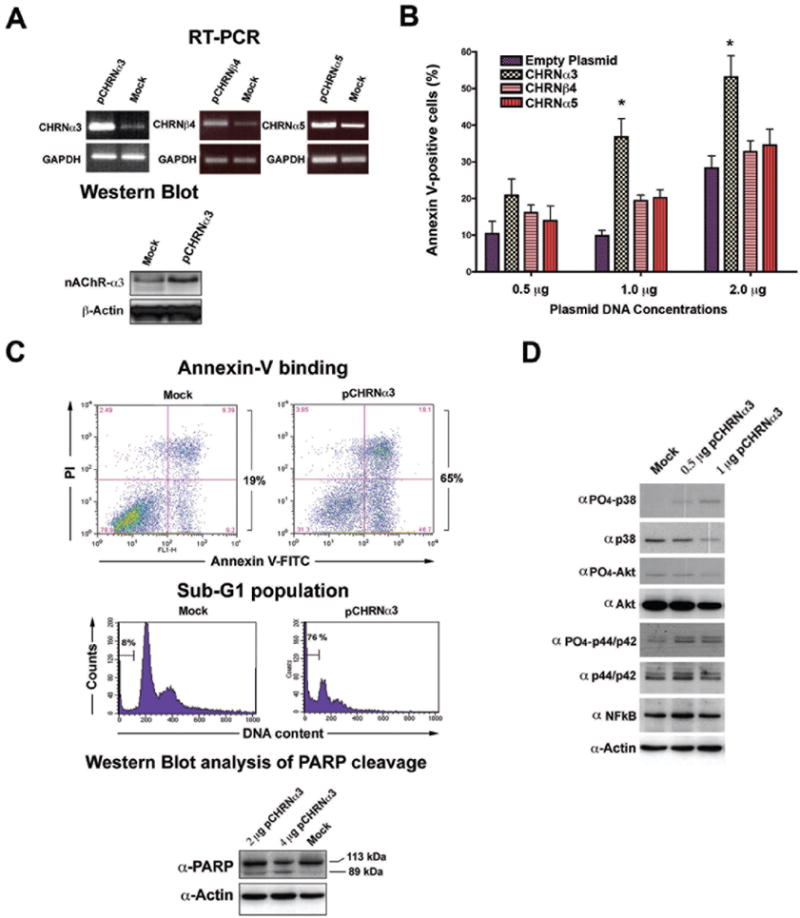
A) H1975 cells were transfected with empty vector (mock) or the vector containing the cDNA of CHRNα3, β4 and α5 genes and expression levels of individual genes were determined by RT-PCR analysis (upper panel). GAPDH expression levels are used as a loading control. Western blot analysis of CHRNα3 expression (lower panel). H1975 cells were transfected with empty vector (mock) or the indicated concentrations of the CHRNα3 expressing vector, and the nAChRα3 protein levels were determined by Western blotting. Equal loading was verified by anti-actin antibody. B) H1975 cells were transfected with indicated concentrations of the CHRNα3, β4 and α5 expression vector, and the apoptotic fraction was determined 36 hours post-transfection by flow cytometry following staining with Annexin V. The results are given as the means +/- standard deviations for triplicate samples. Data are from one of two independent experiments. Statistical significance (P<0.05) was calculated by comparing mock transfected samples with all other sample types in the same group, and is indicated by an asterisk. C) H1975 cells were mock transfected (empty vector) or transfected with the vector containing the cDNA of CHRNα3, and apoptotic fraction (Annexin V staining) (upper panel) and sub-G1 population (middle panel) were determined by flow cytometry. Western blot analysis of PARP cleavage after the re-expression of CHRNα3 in H1975 cells. Full-length PARP (113 kDa) and cleaved fragments (89 kDa) are indicated. D) Restoration of CHRNα3 expression is associated with activated MAPK/p38 stress signalling. H1975 cells were mock-transfected (empty vector) or transfected with indicated concentrations of the nAChRα3 expression vector, and 36 hours post-transfection cells were harvested and analysed by Western blotting using different antibodies.
Knockdown of CHRNα3 gene deregulates nAChR-associated signalling and impairs cell death response
To examine the physiological significance of the methylation-mediated silencing of CHRNα3 gene, we used small hairpin RNAs (shRNAs) to inhibit the expression of the CHRNα3 gene in human cancer cells, and tested the function of nAChR receptors and apoptotic proficiency. Two lung cancer cell lines, H1650 and H1299, both of which exhibit low methylation levels and express appreciable levels of nAChRα3 (Figure 4A and Figure 2C), were stably transfected with a doxycycline-inducible shRNA expression construct targeting CHRNα3. Successful silencing of the target gene in the presence of doxycycline (+Dox) was confirmed at both transcript (data not shown) and protein levels (Figure 4A). We next tested the effect of nAChRα3 knockdown on the function of nAChRs, activation of which is known to induce opening of non-selective cation and voltage-gated Ca2+ channels leading to the downstream cellular signalling events (8, 9). As shown in Figure 4B, nAChRα3 depleted cells (+Dox) exhibited a significantly higher Ca2+ influx response upon nicotine exposure, in comparison to the nAChRα3-expressing control cells (-Dox), indicating that CHRNα3 depletion triggers hyperactivation and dramatic opening of Ca2+ channels in the presence of nicotine.
Figure 4. Effect of the CHRNα3 gene knockdown on nAChR signalling and apoptotic proficiency of lung cancer cells.
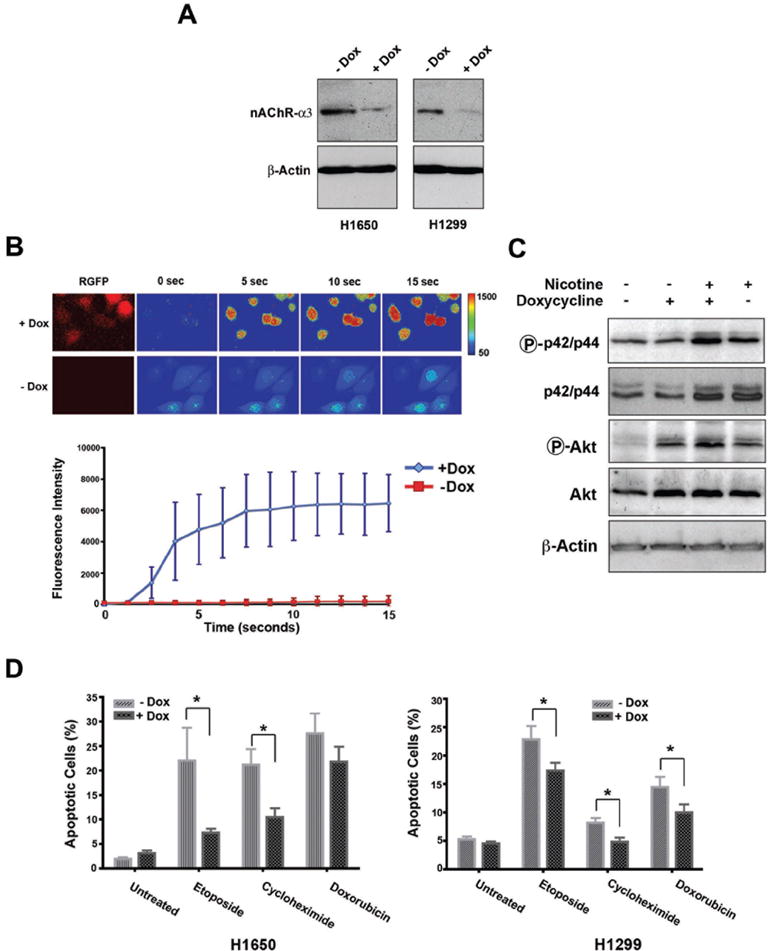
A) H1650 and H1299 cells were stably transfected with an inducible shRNA expression vector targeting CHRNα3 gene, and the efficiency of shRNA-mediated silencing was revealed by Western blots using anti-nAChRα3 antibody. To silence the CHRNα3 gene, expression of shRNA was induced with 1μM doxicycline (+Dox) for 48 hours. B) Analysis of nicotine-induced Ca2+ influx in H1299 cells expressing different levels of nAChRα3. H1299 cells were grown in the presence (+Dox) or absence (-Dox) of doxycycline, and following addition of 10 mM nictone (time point, 0 sec), the imaging of intracellular Ca2+ was performed using the Ca2+ indicator fluo-4 (10μM) and confocal laser microscopy (upper panel). RGFP expression is under the control of the same doxycyclin-inducible promoter as the α3-shRNA and thus is used as a reporter to shRNA expression. Plot comparing the quantified fluorescent intensity changes in CHRNα3-depleted (+Dox) and CHRNα3-expressing (-Dox) cells (lower panel). C) Knockdown of CHRNα3 expression triggers activation of the Akt-signalling pathway. H1299 cells were treated with nicotine (10 mM) for 48 hours following prior incubation in the presence (+Dox) or absence (-Dox) of doxycycline for 48 hours. Phosphorylation of Akt (PO4-Akt) and p42/44 (PO4-p42/44), and native Akt and p42/44 protein levels in total protein lysates were monitored by Western blot analysis. Equal loading was verified by anti-actin antibody. D) Knockdown of CHRNα3 gene impairs cell death response. H1650 and H1299 cells stably transfected with inducible CHRNα3 shRNA were incubated in the presence or absence of doxycycline for 24 hours and treated with etoposide, cyclohexamide or dexamethasone (100μM) for a further 24 hours. Cells were then stained with Annexin V/propidium iodide, and apoptotic fraction was analysed by FACS. Shown are mean values (+/-SD) from three different experiments. Statistical significance was calculated using Student’s unpaired ‘t’ test (P < 0.05; two tailed) and significant values are indicated by an asterisk.
To test whether cholinergic signalling by nicotine triggers a survival signalling pathway in nAChRα3-deficient cells we investigated MEK and Akt signalling, two pathways known to be activated by nicotine/cholinergic signalling through AChRα7 receptors (9, 15, 23-26). Incubation of cells in the presence of nicotine resulted in a higher levels of phosphorylation of Akt and p42/p44 proteins in nAChRα3 knockdown cells compared to nAChRα3-expressing cells (Figure 4C), indicating that a higher Ca2+ influx in nAChRα3 depleted cells is followed by activation of the MEK and Akt signalling cascade. Together, these results show that knockdown of CHRNα3 results in an activation of cholinergic signalling by nicotine and triggers the MEK/Akt survival signalling pathway.
To further explore the effects of CHRNα3 inactivation on cell survival and apoptosis proficiency, H1299 and H1650 cells expressing inducible shRNA were treated with different apoptosis-inducing agents in the absence or presence of doxycycline, and apoptotic fraction (Annexin V-positive cells) was determined by flow cytometry. While both CHRNα3-expressing and CHRNα3-knockdown cells in the absence of apoptosis-inducing agents exhibited comparable levels of annexin-V positive cells, suggesting that loss of CHRNα3 does not lead to spontaneous cell death, CHRNα3-depleted cells treated with apoptosis-inducing agents exhibited significantly lower apoptotic responses to etoposide, cycloheximide and doxorubicine (Figure 4D). These findings support the notion that downregulation of CHRNα3 gene expression impairs apoptotic proficiency.
DISCUSSION
The present study revealed that the 15q25.1 locus may be under epigenetic regulation and that its deregulation may contribute to the development of lung cancer. Although CHRNα3, CHRNβ4 and CHRNα5 are clustered together at the same locus, they exhibit differential susceptibility to aberrant methylation. Interestingly, we found that elevated CHRNβ4 methylation, in contrast to hypermethylation of the CHRNα3 gene, failed to inhibit expression of the gene. However, as levels of CHRNβ4 methylation in lung tumours were markedly lower than those of CHRNα3, it is therefore possible that methylation levels of CHRNβ4, although increased, were insufficient to induce significant silencing of the gene. Alternatively, lack of transcriptional repression may be due to lower CpG density in the CHRNβ4 promoter (57 CpG sites in the island) compared to the CHRNα3 promoter (137 CpG sites in the island) (http://genome.ucsc.edu/cgi-bin/hgGateway), consistent with the notion that the density of methylated CpG sites is a critical factor for transcriptional silencing and that repression by DNA methylation requires a high density of methylated cytosines (27).
The finding that restoration of CHRNα3 expression in lung cancer cells harbouring silenced endogenous CHRNα3 gene induces apoptosis suggests that silencing of CHRNα3 expression may abrogate susceptibility to cell death and contribute to cancer development. Consistent with this notion, CHRNα3 downregulation using an shRNA approach in CHRNα3-expressing cells leads to apoptotic defects in response to different apoptosis-inducing agents. These results argue that silencing of CHRNα3 expression in non-neuronal tissues (such as normal lung epithelium) may abrogate physiological cell death and promote cell survival. Transfection experiments using varying concentrations of CHRNα3-expression vector resulted in a dose-dependent increase in apoptotic fraction, suggesting that CHRNα3 gene dosage and its transcriptional control play an important role in cellular susceptibility to apoptosis. The mechanism by which CHRNα3 silencing contributes to cellular evasion of apoptosis appears to involve nAChR receptors whose activation induces opening of voltage-gated Ca2+ channels and the downstream pro-survival signalling events (8, 9). This notion is supported by our results showing a significantly higher Ca2+ influx in CHRNα3-depleted cells followed by activation of MEK and Akt survival signalling pathways, inhibition of apoptosis and promotion of survival. However, it remains unclear how inactivation of CHRNα3 gene expression and consequent depletion of nAChRα3 subunit may result in aberrant function of the nAChR receptors. The genes encoding nAChR subunits are thought to be under a coordinated transcriptional regulation (28, 29), and nAChRα3, α5 and β4 subunits assemble together to form functional nAChR receptor complexes. Loss of one subunit of heteromeric nAChRs may affect activation properties of the receptors and cellular response to different receptor agonists/antagonists including nicotine, therefore higher Ca2+-influx in nAChRα3 subunit depleted cells may result from altered nicotine binding affinity and activation properties of nAChRs lacking the nAChRα3 subunit. Alternately, these altered properties of nAChRs in nAChRα3-depleted cells may also result from the overrepresentation of other subtype-containing nAChRs. Because nAChRα7 receptors are the primary receptor types that mediate the proliferative effects of nicotine in the tumour cells (15) and are also considered to be the major stimulator of cancer development and progression (30, 31), silencing of the nAChRα3-encoding gene may result in over-representation of other nAChR subunits (notably nAChRα7 and nAChRα5), which may stimulate cell survival and provide a proliferation advantage to tumour cells. Based on our results we propose a model (Supplementary Figure 2) in which the CHRNα3 gene, apart from its classical regulatory role in neurotransmission, plays an important role in the regulation of cell survival/cell death of non-neuronal cells (i.e., lung epithelial cells).
In summary, our results provide mechanistic insights into the events through which silencing of the CHRN gene may deregulate cellular response to apoptotic stimuli in non-neuronal tissue and promote tumour development, providing a functional link between susceptibility locus 15q25.1 and lung cancer. However, it should be noted that our results indicate that in lung cancer tumours (and not in surrounding non-tumour lung tissue of same patients) the 15q25.1 locus is aberrantly methylated resulting in silencing of the CHRNα3 gene, suggesting that irrespective of the known risk factor exposure, hypermethylation of CHRNα3 gene is a common and tumour tissue-specific phenomenon. While further studies are required to discover possible causes of selective hypermethylation at 15q25.1 locus, the information provided may open new research avenues to investigate different epigenetic mechanisms by which nicotinic deregulation of acetylcholine receptors in non-neuronal tissues may contribute to cancer development and progression. Our study also reinforces the interest in nicotinic acetylcholine receptors as markers for cancer detection, clinical predictors and potential chemopreventive targets.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr Jon Lindstrom (University of Pennsylvania, Philadelphia) for kindly providing the anti-CHRNα3 antibody and expression constructs containing cDNAs of the CHRNα3, CHRNβ4 and CHRNα5 genes and Deutsches Krebsforschungszentrum (Heidelberg) for providing RG108. We also thank Dr Hector Hernandez Vargas for critical reading of the manuscript and helpful discussions. Further thanks are due to John Daniel for editing the manuscript. T. Vaissière is supported by a PhD fellowship from la Ligue National (Française) Contre le Cancer. A. Krais received supported from the Bio-Silc programme of the French National Cancer Institute (INCa). This work was supported by la Ligue Nationale (Française) Contre le Cancer (Comité Loire, France). The work in IARC Epigenetics Group is supported by grants from the National Institutes of Health/National Cancer Institute (NIH/NCI), United States; L’Association pour la Recherche sur le Cancer (ARC), France; the European Network of Excellence Environmental Cancer Risk, Nutrition and Individual Susceptibility (ECNIS), and the Swiss Bridge Award (to Z.H.). This work was also supported in part by the European Commission, DG-E1, Inco-Copernicus grant.
References
- 1.Schwartz AG, Prysak GM, Bock CH, Cote ML. The molecular epidemiology of lung cancer. Carcinogenesis. 2007;28:507–18. doi: 10.1093/carcin/bgl253. [DOI] [PubMed] [Google Scholar]
- 2.Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–7. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 3.Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindstrom JM. Nicotinic acetylcholine receptors of muscles and nerves: comparison of their structures, functional roles, and vulnerability to pathology. Ann N Y Acad Sci. 2003;998:41–52. doi: 10.1196/annals.1254.007. [DOI] [PubMed] [Google Scholar]
- 6.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–71. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- 8.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catassi A, Servent D, Paleari L, Cesario A, Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–31. doi: 10.1016/j.mrrev.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Dasgupta P, Chellappan SP. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5:2324–8. doi: 10.4161/cc.5.20.3366. [DOI] [PubMed] [Google Scholar]
- 11.Gahring LC, Rogers SW. Neuronal nicotinic acetylcholine receptor expression and function on nonneuronal cells. AAPS J. 2005;7:E885–94. doi: 10.1208/aapsj070486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JC, Cruchaga C, Saccone NL, et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet. 2009;18:3125–35. doi: 10.1093/hmg/ddp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam DC, Girard L, Ramirez R, et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007;67:4638–47. doi: 10.1158/0008-5472.CAN-06-4628. [DOI] [PubMed] [Google Scholar]
- 14.Maneckjee R, Minna JD. Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 1994;5:1033–40. [PubMed] [Google Scholar]
- 15.West KA, Brognard J, Clark AS, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111:81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe H, Zoli M, Changeux JP. Promoter analysis of the neuronal nicotinic acetylcholine receptor alpha4 gene: methylation and expression of the transgene. Eur J Neurosci. 1998;10:2244–53. doi: 10.1046/j.1460-9568.1998.00235.x. [DOI] [PubMed] [Google Scholar]
- 17.Vaissiere T, Hung RJ, Zaridze D, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243–52. doi: 10.1158/0008-5472.CAN-08-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaissière Thomas, C C, Paliwal Anupam, Vineis Paolo, Genair-EPIC Investigators. Hainaut Pierre, Herceg Z. Quantitative analysis of DNA methylation after whole bisulfitome amplification of a minute amount of DNA from body fluids. Epigenetics. 2009;4:1–10. doi: 10.4161/epi.8833. [DOI] [PubMed] [Google Scholar]
- 19.Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 20.Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–98. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- 21.Kangaspeska S, Stride B, Metivier R, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–5. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 22.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 23.Dasgupta P, Rizwani W, Pillai S, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marrero MB, Bencherif M. Convergence of alpha 7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: central role for JAK2 activation of STAT3 and NF-kappaB. Brain Res. 2009;1256:1–7. doi: 10.1016/j.brainres.2008.11.053. [DOI] [PubMed] [Google Scholar]
- 25.Paleari L, Catassi A, Ciarlo M, et al. Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif. 2008;41:936–59. doi: 10.1111/j.1365-2184.2008.00566.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Paleari L, Sessa F, Catassi A, et al. Inhibition of non-neuronal alpha7-nicotinic receptor reduces tumorigenicity in A549 NSCLC xenografts. Int J Cancer. 2009;125:199–211. doi: 10.1002/ijc.24299. [DOI] [PubMed] [Google Scholar]
- 27.Weber M, Schubeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–80. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Scott MM, Deneris ES. Shared long-range regulatory elements coordinate expression of a gene cluster encoding nicotinic receptor heteromeric subtypes. Mol Cell Biol. 2006;26:5636–49. doi: 10.1128/MCB.00456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benfante R, Flora A, Di Lascio S, et al. Transcription factor PHOX2A regulates the human alpha3 nicotinic receptor subunit gene promoter. J Biol Chem. 2007;282:13290–302. doi: 10.1074/jbc.M608616200. [DOI] [PubMed] [Google Scholar]
- 30.Egleton RD, Brown KC, Dasgupta P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci. 2008;29:151–8. doi: 10.1016/j.tips.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Arias HR, Richards VE, Ng D, Ghafoori ME, Le V, Mousa SA. Role of non-neuronal nicotinic acetylcholine receptors in angiogenesis. Int J Biochem Cell Biol. 2009;41:1441–51. doi: 10.1016/j.biocel.2009.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.