

Abstract
Recent studies have revealed that the TAM receptor protein tyrosine kinases — TYRO3, AXL and MER — have pivotal roles in innate immunity. They inhibit inflammation in dendritic cells and macrophages, promote the phagocytosis of apoptotic cells and membranous organelles, and stimulate the maturation of natural killer cells. Each of these phenomena may depend on a cooperative interaction between TAM receptor and cytokine receptor signalling systems. Although its importance was previously unrecognized, TAM signalling promises to have an increasingly prominent role in studies of innate immune regulation.
Receptor protein tyrosine kinases (PTKs) are cell-surface transmembrane receptors that contain a regulated PTK activity within their cytoplasmic domains. They function as sensors for extracellular ligands, the binding of which triggers receptor dimerization and activation of the receptor’s kinase. This leads to the recruitment, phosphorylation and activation of multiple downstream signalling proteins, which ultimately change the physiology of cells. Although there are only 58 receptor PTK genes in the human genome1 (see the human kinome website), the signal transduction cascades initiated by receptor PTK activation control diverse cellular processes — from cell differentiation to cell death. Well-known receptor PTK subfamilies include the ERBB receptors, which have essential roles in cardiac and neural development and the progression of some forms of breast cancer2; and the ephrin receptors, which are required for tissue morphogenesis and the patterning of neuronal connections in the developing brain3.
The focus of this Review, the TAM group, was among the last receptor PTK subfamilies to be identified, and the biological roles of its three members — TYRO3, AXL and MER — remained uncharacterized for several years. Largely through the analysis of engineered loss-of-function mutants in mice, these roles have become increasingly apparent. They reflect a specific requirement for TAM signalling in settings in which fully differentiated cells, tissues and organs must be maintained in the face of continuous challenge, turnover and renewal. In humans, ongoing homeostatic regulation of this sort must be carried out, frequently on a daily basis, for decades. Although an essential role for TAM regulation of tissue homeostasis is evident in the adult nervous, reproductive and vascular systems, it is in the regulation of the innate immune response where the TAM receptors have especially profound effects.
In this Review, we highlight the central roles that TAM signalling has in the intrinsic inhibition of the inflammatory response to pathogens by dendritic cells (DCs) and macrophages; during phagocytosis of apoptotic cells by these same cells; and in the maturation and killing activity of natural killer (NK) cells. We also discuss how, in many or all of these settings, TAM receptors depend on and interact with cytokine receptors.
TAM receptors and ligands
The three TAM receptors, TYRO3, AXL and MER, were identified as a distinct receptor PTK subfamily in 1991 (REFS 4,5). Subsequent cloning of full-length cDNAs by multiple laboratories resulted in a profusion of different names for the receptors, but TYRO3, AXL and MER are now the official NCBI designations. Similar to class 1 and class 2 cytokine receptors (such as the interleukin-2 and type I interferon receptors), the TAM receptors are specific to chordates, and this receptor PTK subfamily was among the last to appear in evolution6. The extracellular, ligand-binding regions of the TAMs have a defining arrangement of two immunoglobulin-related domains and two fibronectin type III repeats, each in tandem. These are followed by a single-pass transmembrane domain, and a catalytically competent, cytoplasmic PTK (FIG.1). The TAM receptors are most closely related to RON (also known as CD136, MST1R), the PTK receptor for macrophage-stimulating protein, and to MET, the hepatocyte growth-factor receptor1. Similar to all other receptor PTKs, the TAMs seem to signal as dimers7 (FIG. 1).
Figure 1. TAM receptors and their ligands.

TYRO3 (also known as BRT, DTK, RSE, SKY and TIF), AXL (also known as ARK, TYRO7 and UFO) and MER (also known as EYK, NYM and TYRO12) are receptor protein tyrosine kinases (PTKs) that are expressed by dendritic cells, macrophages and immature natural killer (NK) cells of the immune system, Sertoli cells of the testis, retinal pigment epithelial (RPE) cells of the eye, and several other cell types. TAM receptor dimers bind to their two ligands, growth-arrest-specific 6 (GAS6) and protein S, through interaction between the two N-terminal immunoglobulin-like domains of the receptors and the two C-terminal laminin G (LG) regions, which together make up the SHBG (sex hormone binding globulin) domain, of the ligands. (The solved X-ray crystal structure of the GAS6 SHBG domain bound to the immunoglobulin domains of AXL7 reveals that both ligand and receptor crystallize as dimers.) Via their N-terminal Gla domains, GAS6 and protein S then bind to phosphatidylserine that is displayed on the extracellular surface of the plasma membranes of apoptotic cells or on the outer segments of photoreceptors. EGF, epidermal growth factor; FNIII, fibronectin type III.
The two ligands that bind to and activate the TAM receptors remain the subject of investigation. When originally cloned, TYRO3, AXL and MER were orphans, in that their ligands were unknown and could not be predicted. A series of biochemical and cell-culture experiments identified two closely related proteins — growth-arrest-specific 6 (GAS6) and protein S — as TAM agonists8 (FIG. 1). GAS6 was found to bind and activate — that is, stimulate the tyrosine autophosphorylation of — all three receptors, albeit with markedly different affinities (AXL≥TYRO3≫MER)9. Although protein S was initially reported to be a specific agonist for TYRO3 (REF. 8), more recent studies have demonstrated that in cells in which TYRO3 is co-expressed with MER, protein S is also a potent MER agonist10. An important issue with regard to published ligand–receptor interaction studies is that most have involved the assay of single agonists against single receptors. However, both TAM ligands and TAM receptors heterodimerize (C.V.R. and G.L., unpublished observations), and heterodimeric ligand–receptor binding and activation profiles are, based on what is known for other receptor PTKs, almost certain to differ from those of homodimers. A recent analysis has demonstrated that the ability of protein S to stimulate both the phagocytosis of apoptotic cells by macrophages (see later) and the autophosphorylation of MER in macrophages, requires protein S dimerization11.
In this Review, we generally discuss GAS6 and protein S interchangeably. It is important to emphasize, however, that these proteins have different patterns of expression in mammalian tissues (Supplementary information S1 (table)) and different bioactivities. In addition to its role as a TAM ligand, for example, protein S has an important, TAM-independent activity as a blood anticoagulant — an activity that is not exhibited by GAS6. Protein S, which is present at relatively high levels (~300 nM) in the serum, serves as an essential cofactor for activated protein C, a protease that degrades factor Va and factor VIIIa, and thereby inhibits blood coagulation12.
GAS6 and protein S share the same distinctive arrangement of structural motifs. Each protein has a ~60 amino-acid Gla domain at its amino terminus, a region rich in glutamic acid residues that are γ-carboxylated in a vitamin-K-dependent reaction. (Gla-domain-containing proteins are prominent components of the blood coagulation cascade.) These Gla domains bind the phospholipid phosphatidylserine13, and this is an important feature of the in vivo function of GAS6 and protein S14-17. The Gla domain is followed by four epidermal growth factor (EGF)-like modules, and then by two tandem laminin G domains that are related to those of the sex hormone binding globulin (SHBG) (FIG. 1). This SHBG-like module is both necessary and sufficient for TAM receptor binding and activation, whereas the Gla domain is dispensable for these activities7,18. Overall, GAS6 and protein S share ~42% amino-acid identity.
Biological roles of TAM signalling
TAM receptors are broadly expressed in cells of the mature immune, nervous, reproductive and vascular systems (BOX 1; Supplementary information S1 (table)). Usually, there is expression of more than one TAM receptor in a given cell type, such as in DCs; and co-expression of all three receptors, as occurs in Sertoli cells of the testis, is not uncommon. Many TAM-positive cells also express one or both TAM ligands. In contrast to most other receptor PTKs, expression of the TAMs is substantially upregulated postnatally, and is maintained at relatively high levels in adult tissues. Although cDNAs encoding the TAM receptors have been cloned repeatedly from tumour cells, and TAM receptors, particularly AXL, have been implicated in cancer progression19, genuine insights into the biological roles of these receptors were only obtained when mice lacking TYRO3, AXL, MER or GAS6 were generated and analysed20-25.
Box 1 ∣ TAM receptor signalling in other tissues and organs.
In addition to the roles discussed in this Review, TAM receptor signalling has been shown have important regulatory roles in vascular smooth-muscle homeostasis92-94, in platelet function and thrombus stabilization23,95,96, in erythropoiesis97 and in cancer development and progression19,98,99. TAM receptors are also implicated in the regulation of osteoclast function100,101, in the control of oligodendrocyte cell survival102,103, and in the infection of dendritic cells and other cells by Ebola and Marburg viruses104,105. In many of these instances, the primary downstream TAM signalling pathway appears to be the phosphoinositide 3-kinase–AKT pathway95,97,99,102,103, rather than the Janus kinase–STAT (signal transducer and activator of transcription) pathway that is highlighted in this Review.
Because knockout of each of the TAM genes still produced viable and fertile mice, and because the Tyro3 and Mer genes are linked in the mouse genome, it was possible to generate knockout mice that were deficient for all three TAM receptors20. Remarkably, even these triple knockout mice were found to be viable at birth, and to be superficially normal for the first several weeks thereafter. This notwithstanding, mice lacking all three TAM receptors were found to develop a plethora of debilitating phenotypes, all of which appeared to be degenerative in nature20,21.
Beginning at about 3 weeks after birth, for example, germ cells in the testis were observed to die in increasing numbers, such that by three months after birth, the seminiferous tubules in the TAM-deficient mutants were depleted of spermatogonia, spermatocytes, spermatids and mature sperm20. Another dramatic phenotype was observed in the retinae of TAM-deficient mice, where, beginning at about the same time, photoreceptors were seen to die by apoptosis. By 2–3 months of age, essentially all photoreceptors were lost from these mice20. As discussed below, these two apparently unrelated degenerative phenotypes are in fact directly linked, in that they both result from the inability of TAM-deficient cells to properly phagocytose apoptotic cells and membranes.
Autoimmunity in TAM mutant mice
Although the TAMs have important roles in the nervous, reproductive and vascular systems (see BOX 1), perhaps the most serious consequences of mutations of the TAM genes are seen in the immune system. Also at around 3 weeks after birth, the peripheral lymphoid organs of the TAM triple mutant mice begin to grow at elevated rates, such that by 6 months of age, the spleens and lymph nodes of these mice are often 10 times the weight of those from wild-type mice. This is due to the expansion of both myeloid- and lymphoid-cell populations, and colonies of lymphocytes are eventually observed in essentially all tissues of the triple mutant animals21. T and B cells, as well as macrophages and DCs, are constitutively activated in these mice: they express elevated levels of both acute and chronic activation markers, such as CD95 and CD44 (in B cells), CD25 (in T cells), and MHC class I and II, CD86 and IL-12 (in peritoneal macrophages and CD11c+ splenic DCs)21,25. The TAM receptors are not expressed by most lymphocytes, and multiple lines of evidence demonstrate that the lymphocyte activation seen in the TAM-deficient mice is due primarily to the loss of TAM receptor function in antigen-presenting cells (APCs).
Not surprisingly, the TAM-deficient mice eventually develop broad-spectrum autoimmune disease21. In addition to the enlargement of peripheral lymphoid organs, clinical manifestations of autoimmunity include swollen joints and footpads, skin lesions, blood vessel haemorrhages and IgG deposits in kidney glomeruli. Humoral manifestations include high titres of circulating antibody to multiple autoantigens, including double-stranded DNA (dsDNA), a variety of plasma membrane phospholipids and collagen21. As for most other features of TAM-deficient mice, a clear gene dosage effect is seen, with mice deficient in a single TAM receptor exhibiting milder phenotypes, such as lower dsDNA-specific autoantibody titres, than mice deficient in two TAM receptors, and with mice deficient in all three receptors exhibiting the most severe phenotypes21. These gene dosage effects are in keeping with the co-expression of more than one TAM receptor in most, if not all, TAM-positive cells. In addition to developing autoimmunity, TAM-deficient mice and APCs are hyper-responsive to endotoxins, as monitored by the production of pro-inflammatory cytokines25,26. Most Mer−/− mice, for example, die from a dose of lipopolysaccharide (LPS) that is otherwise non-lethal in wild-type mice26.
TAM inhibition of inflammation
The autoimmune disease that develops in the TAM mutant mice is likely to result from the loss of TAM regulation of two related phenomena: the innate inflammatory response to pathogens by DCs and macrophages, and the phagocytosis of apoptotic cells by these APCs. Recent analyses of signal transduction events have provided important insights into these two phenomena.
As for any dynamic system, the innate immune response must be carefully regulated — a mechanism that turns it off must be tied to a mechanism that turns it on. A rapid inflammatory response to bacteria, viruses and other disease-causing pathogens, for example, is crucial to their defeat. Yet unrestrained signalling by Toll-like receptors (TLRs) and cytokine receptors in DCs and macrophages generates a chronic inflammatory milieu that can lead to disease and even death. A recent study has shown that the TAM receptors function as pivotal inhibitors that prevent this dysregulation from occurring25.
Consistent with the observations in TAM-deficient mice that are outlined earlier, TLR activation of wild-type DCs has been found to be potently inhibited by prior incubation with either GAS6 or protein S25,27. TAM-mediated inhibition is seen irrespective of whether the TLR activated is TLR3, TLR4 or TLR9; and multiple points in TLR signal transduction cascades — including the activation of the p38 mitogen-activated protein kinase (MAPK), extracellular-signal-regulated kinase 1 (ERK1)/ERK2, nuclear factor-κB (NF-κB), tumour-necrosis factor (TNF)-receptor-associated factor 3 (TRAF3) and TRAF6 — are inhibited. TAM receptor signalling has also been found to inhibit TLR-induced production of pro-inflammatory cytokines — including TNF, interleukin-6 (IL-6), IL-12 and type I interferons (IFNs)25.
This inhibition requires new gene expression, and among the most important of the inhibitory genes that are induced by the TAMs are the suppressor of cytokine signalling (sOCs) proteins SOCS1 and SOCS3 (REF. 25). SOCS proteins have been studied extensively, and have for many years been known to be induced through the activation of cytokine receptors, most notably by type I IFN receptor (IFNAR) activation28,29. This induction was thought to represent a classic negative-feedback loop, as SOCS1 and SOCS3 inhibit JAK (Janus kinase)–STAT (signal transducer and activator of transcription) signalling downstream of cytokine receptors. However, the induction of SOCS proteins by IFNAR activation has recently been shown to proceed through and be dependent on TAM receptors25.
As the transcription of SOCS genes is known to be driven by STAT transcription factors, the activation (that is, the tyrosine phosphorylation) of these transcription factors in response to TAM signalling was also examined25. It was found that STAT1, but not STAT2 or STAT3, is activated directly by TAM activation in DCs. Moreover, studies in Stat1−/− DCs demonstrated that TAM-mediated inhibition of TLR-activated inflammatory responses requires STAT1. That STAT1 is normally activated downstream of cytokine receptors30, notably IFNAR, prompted an examination of the role that this receptor might have in TAM signalling. Using Ifnar1−/− DCs, it was found that all aspects of TAM-mediated inhibition of inflammation also require IFNAR25. Consistent with this, the R1 subunit of IFNAR and AXL can be reciprocally co-immunoprecipitated from wild-type DCs, demonstrating a physical association between cytokine and TAM receptors that parallels their physiological association. Together, these results indicate that TAM receptors bind to and usurp the IFNAR–STAT1 complex, and thereby switch it from a signalling complex that initiates and amplifies inflammation to one that inhibits inflammation25.
That the TAM system inhibits inflammation in DCs suggests that, in principle, this system should not be fully engaged at the onset of a TLR-initiated immune response, and further, that some feature of the system — for example, TAM receptor or ligand expression — should be upregulated subsequent to TLR and cytokine receptor engagement. This is indeed the case — AXL mRNA and protein levels are markedly elevated upon treatment of DCs with either TLR agonists (LPS, CpG-containing oligonucleotides or polyI:C (polyinosinic–polycytidylic acid)) or with IFNα (REFS 25,31). Both of these forms of AXL upregulation depend on the presence of both STAT1 and IFNAR25.
Taken together, these results illuminate a tri-partite cycle of inflammation, for which the ultimate, previously unrecognized regulatory arm consists of TAM-mediated inhibition (FIG. 2). The integration of TAM signalling into such a regulatory cycle has significant implications for both the understanding and treatment of human immune system disorders (see later).
Figure 2. An inflammation cycle regulated by TAM signalling.
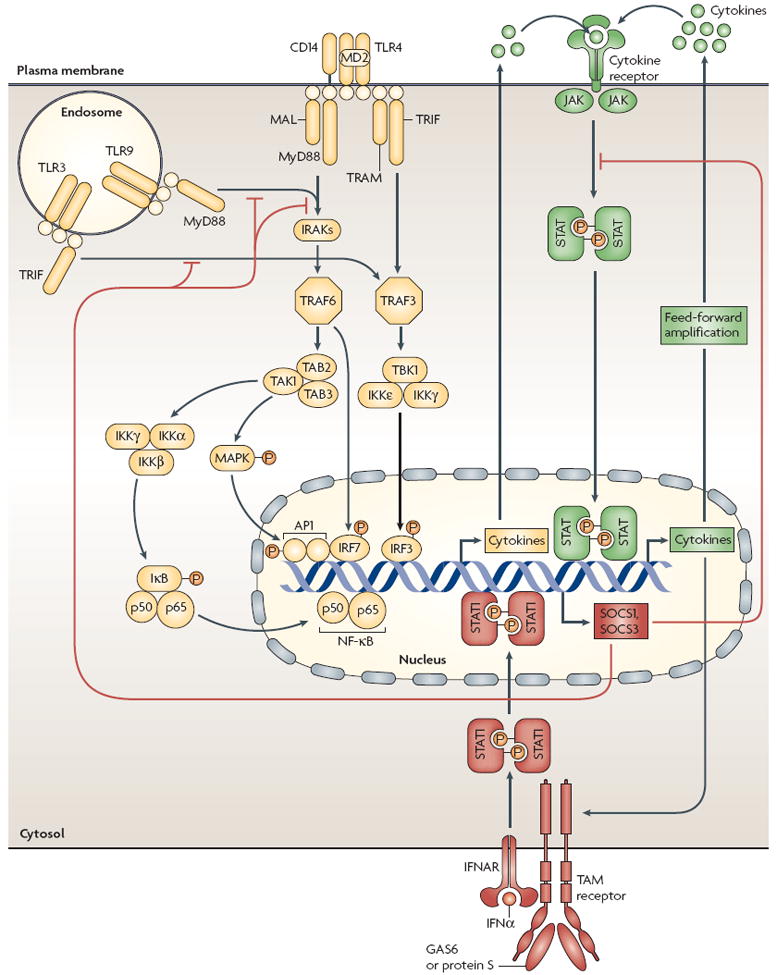
Quiescent macrophages and dendritic cells (DCs) are stimulated by pathogen encounter, which activates Toll-like receptor (TLR) signalling pathways (yellow). This results in an initial burst of pro-inflammatory cytokines, the levels of which are then greatly amplified in a feed-forward loop through cytokine receptor signalling pathways (green). Cytokine signalling also drives the upregulation of expression of the TAM receptor AXL, which engages TAM receptor signalling pathways (red). These result in the induction of expression of suppressor of cytokine signalling 1 (SOCS1) and SOCS3, which broadly inhibit both TLR and cytokine receptor cascades, thereby ending the inflammatory response. TAM receptor signalling requires coordinate interaction with both the type I interferon receptor (IFNAR) and the transcription factor STAT1 (signal transducer and activator of transcription 1), which is also used for both cytokine amplification and the upregulation of AXL. AP1, activator protein 1; GAS6, growth-arrest-specific 6; IKK, inhibitor of NF-κB kinase; IFNα, interferon-α; IRAK, interleukin-1-receptor-associated kinase; IRF, interferon-regulatory factor; JAK, Janus kinase; MAL, MyD88-adaptor-like protein; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; TAB, TAK1-binding protein; TAK1, transforming-growth-factor-β-activated kinase 1; TBK1, TANK-binding kinase 1; TRAF, tumour-necrosis-factor-receptor-associated factor; TRAM, TRIF-related adaptor molecule; TRIF, Toll/interleukin-1-receptor-domain-containing adaptor protein inducing interferon-β.
TAM regulation of phagocytosis
In addition to a failure in the inhibition of TLR-induced inflammation, the development of blindness, sterility and autoimmunity in the TAM mutant mice also reflects dysregulation of a second TAM-dependent process — the phagocytic clearance of apoptotic cells and membranes. TAM receptor signalling is not required for this process during development, as the large number of apoptotic-cell corpses that are generated during embryogenesis appear to be cleared normally. Instead, it is essential for the phagocytosis that occurs continuously in adult organs as part of normal tissue homeostasis (FIG. 3). These organs include, but are not restricted to, those of the immune system.
Figure 3. TAM signalling and the ‘homeostatic phagocytosis’ of apoptotic cells and membranes.

TAM receptor signalling is required for the clearance of apoptotic cells in fully differentiated adult tissues and organs that undergo constant challenge, renewal and remodelling. These include the testis, where Sertoli cells clear the large number of apoptotic cells that are generated during spermatogenesis (a); the retina, where retinal pigment epithelial (RPE) cells pinch off the distal ends of photoreceptor outer segments (b); and the lymphoid organs, where macrophages and dendritic cells remove apoptotic cells generated by infection (c). Each of these events is dramatically impaired in mice that lack TAM receptors. A significant fraction of TAM receptor signalling in macrophages, RPE cells and Sertoli cells appears to be autocrine and/or paracrine, in that each of these TAM+ cells also express the TAM receptor ligands growth-arrest-specific 6 (GAS6) and/or protein S.
Indeed, results from studies of the retina and testis — the two organs in which TAM-mediated regulation of what might be called ‘homeostatic phagocytosis’ was first appreciated — highlight similarities with the roles of TAM receptors in the immune system. As noted above, male mice that lack all three TAM receptors are sterile owing to the postnatal death of essentially all germ cells, a degeneration that results from the compromised function of Sertoli cells20 (FIG. 3a). These cells, but not germ cells, express all three TAM receptors and both ligands20,32. Sertoli cells are highly phagocytic, binding and ingesting both apoptotic germ cells and cellular contents that are extruded as these cells mature into sperm33. This phagocytic activity is particularly important, as in mammals more than half the population of differentiating spermatogenic cells dies by apoptosis34. The absence of phagocytic clearance in the TAM-deficient testis leads to the toxic accumulation of dead cells and debris. Phagocytosis by Sertoli cells increases and decreases as a function of stage in spermatogenesis, with ingestion of apoptotic germ cells being minimal between spermatogenic stages IV–VIII (REF. 35). The levels of GAS6 and protein S expression also cycle in Sertoli cells as a function of stage in spermatogenesis, with ligand levels being lowest at stage VIII (REF. 20). This fluctuation in ligand expression levels may therefore provide a mechanism for the cyclic regulation of phagocytosis. It is interesting to note that rhythmic or cyclic variation in the intensity of TAM signalling — in Sertoli cells during spermatogenesis20, in DCs and macrophages during inflammation25 and in cells of the retinal pigment epithelium (RPE) during the circadian phagocytosis of photoreceptor membranes10 (see later) — is a general feature of TAM action.
In the retina, MER and TYRO3 are expressed by a specialized set of large epithelial cells that line the back of the eye10. These RPE cells have several functions, among the most important of which is the phagocytosis of the distal ends of photoreceptor outer segments36, which are the opsin-containing membranous organelles in which visual input is first transduced. New membrane stacks are added at the proximal base of each photoreceptor outer segment each day, and to maintain a constant steady-state length of the organelle, the distal ends of the outer segments must be pinched off and internalized by RPEs (FIG. 3b). This process does not occur in the TAM-deficient mice. As noted above, degeneration of photoreceptors was originally noted in mice lacking all three TAM receptors20. However, this was subsequently found — in rats, mice and humans — to be due to mutations of the Mer gene alone37-41. A long-studied inherited form of retinitis pigmentosa in rats37-39, and a more recently described inherited form of the disease in humans40,41, are both due to Mer gene mutations. MER and TYRO3 have been localized to the apical tips of the RPE cell processes that penetrate the photoreceptor outer segment layer and pinch off the distal ends of the outer segments10. The failure of RPE cells to carry out this process in the absence of TAM signalling results in the death of essentially all photoreceptors by 3 months of age, thereby resulting in blindness.
These defects in the clearance of apoptotic cells in TAM-deficient testes and eyes have direct parallels in the mutant immune system. Bacterial and viral infections generate a large number of apoptotic-cell corpses, which must be cleared by macrophages and DCs, and this form of homeostatic phagocytosis is also impaired in the TAM-deficient mice42-44 (FIG. 3c). The presence of an elevated steady-state number of apoptotic cells in these mutant mice is likely to contribute to the development of autoimmune disease45, as unremoved apoptotic-cell debris presents danger signals and autoantigens46,47, and elevated numbers of apoptotic cells are often seen in the germinal centres of lymph nodes and other lymphoid organs in human autoimmune syndromes48,49. At the same time, apoptotic cells are known to have multiple immunomodulatory — but generally immunosuppressive — effects on APCs50-52.
Most of the analyses of the role of TAM receptors in the phagocytosis of apoptotic cells, and of the ability of apoptotic cells to engage TAM receptor signalling, have been carried out with the MER receptor and with macrophages and DCs isolated from Mer−/− mice42,43. Recent studies, however, suggest that all three receptors contribute to these events, albeit to different degrees in different APC populations44. When challenged with a large number of experimentally induced apoptotic thymocytes, Mer−/− macrophages are inefficient in clearing the dead cells42. This defect is specific to the phagocytosis and clearance of apoptotic cells, and does not reflect diminished general phagocytic activity, as assayed by the uptake of labelled bacteria, yeast or latex spheres42. Particularly intriguing, with regard to the action of TAM receptors in macrophages, is the demonstration that the long-recognized ability of serum to stimulate cultured human macrophages to phagocytose apoptotic cells is due to the presence of protein S, and more specifically to its ability to bind phosphatidylserine17. An interaction between TAM and cytokine receptors during homeostatic phagocytosis has not been clearly demonstrated. However, IL-10 has been found to stimulate the phagocytosis of apoptotic cells by monocytes and macrophages53,54, and GAS6 and MER are among the monocyte genes that are upregulated by IL-10 (REF. 55).
Apoptotic cells appear to exert their immunosuppressive effect on DCs through a TAM signal transduction pathway that is similar to that outlined earlier for the inhibition of inflammation. The prior addition of apoptotic cells to cultured DCs, for example, has been shown to inhibit both LPS-induced NF-κB activation and secretion of TNF and IL-12, and this inhibition has been found to depend on and be transduced through MER56,57. Although apoptotic cells are immunosuppressive and can induce tolerogenic populations of DCs, how they actually do this biochemically remains to be determined. Whereas some apoptotic cells might carry GAS6 and/or protein S, most cells in the body that die by apoptosis do not express these TAM ligands. We suggest an alternative possibility: that the primary, and perhaps only, stimulant that apoptotic cells provide to activate TAM receptors on macrophages and DCs is phosphatidylserine58. In this model, the relevant TAM ligands are the GAS6 and protein S that are produced by macrophages and DCs themselves. That is, TAM signalling induced by apoptotic cells is autocrine, similar to TAM signalling during the inhibition of inflammation. Phosphatidylserine, tethered to a large structure (the extracellular membrane face of the apoptotic cell (FIG. 1)) would serve to stabilize the interaction between TAM receptors and APC-produced TAM ligands, through binding to the Gla domains of the ligands. This would in theory greatly increase the binding affinity and effective potency of these ligands for the TAM receptors by slowing the rate at which GAS6 and protein S dissociate from the receptors. This form of TAM activation by phosphatidylserine is supported by recent findings on the evolution of the TAM receptors (BOX 2).
Box 2 ∣ Primordial TAM receptors and ligands.
A single TAM-like receptor and a single GAS6 (growth-arrest-specific 6)- and protein-S-like ligand first appeared in evolution in the genomes of urochordates such as Ciona intestinalis and Ciona savignyi89. This was also the case for class 1 and class 2 cytokines and their receptors90,91. Given their physiological and physical interaction, it will be interesting to assess whether cytokine and TAM receptors may have co-evolved.
In addition to a vertebrate-like TAM receptor–ligand pair, the genomes of Ciona also encode a hybrid receptor molecule in which a TAM-like protein tyrosine kinase (PTK) and transmembrane domain are fused directly to a TAM-ligand-like Gla domain89. The Ciona TAM agonists GAS6 and protein S are able to physically bridge two cells: their C-terminal SHBG (sex hormone binding globulin) domains bind TAM receptors on one cell, and their N-terminal Gla domains bind to phosphatidylserine on the plasma membrane of an apposed apoptotic cell (see figure). However, the Ciona receptor–ligand hybrid fusion streamlines this signalling arrangement and may function as a direct phosphatidylserine receptor. This domain-shuffling evolutionary experiment was apparently abandoned with the emergence of true vertebrates — so far, it has not been observed in other genomes. EGF, epidermal growth factor; FNIII, fibronectin type III.

Given the dependence on phosphatidylserine of TAM receptor action during phagocytosis, it will be interesting to examine potential physical interaction between TAM receptors and the recently described direct phosphatidylserine receptors TIM4 (T-cell immunoglobulin domain and mucin domain protein 4)59 and BAI1 (brain-specific angiogenesis factor 1)60, particularly as a clear physiological interaction between MER and αvβ5-integrin, which is also required for the phagocytosis of both apoptotic cells58 and photoreceptor outer segment membranes61, has been demonstrated. Similarly, it will be important to assess downstream TAM signalling pathways that mediate phagocytosis62 versus those that are required for immunosuppression25,27,56. A recent report suggests that these pathways may be dissociable27.
TAM control of natural-killer-cell differentiation
In addition to recognizing pathogens directly, the innate immune system must also recognize and destroy cells that are infected with pathogens. The effectors of this arm of the innate response are NK cells63, and the TAM signalling system also has an important role in regulating the activity of these cells22. This regulation again appears to be carried out in concert with cytokine receptors.
On activation, NK cells kill their target cells through the exocytosis of perforin- and granzyme-containing granules, and the secretion of CD95 ligand and TNF-related apoptosis-inducing ligand (TRAIL)64-68. NK cells also produce a variety of cytokines, including IFNγ, TNF and granulocyte/macrophage colony-stimulating factor (GM-CSF)69-71. NK-cell target recognition and killing activity rely on the expression of a set of so-called inhibitory receptors (such as members of the Ly49 and CD94 families) and activating receptors (such as NK1.1, DX5 and CD69), which recognize ligands on target cells72-74. The acquisition of expression of these receptors has been found to require TAM signalling in the bone marrow22 (FIG. 4).
Figure 4. TAM signalling and the maturation of NK cells.
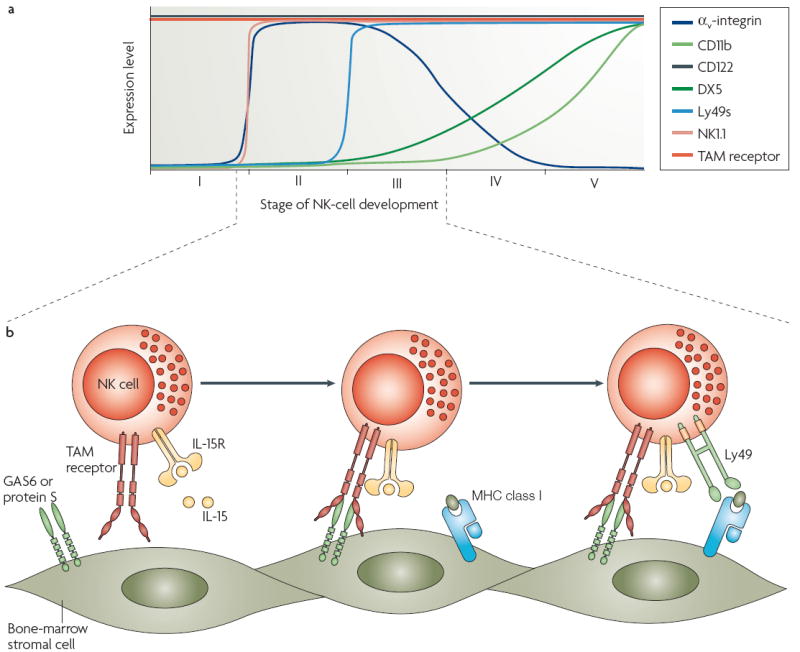
Natural killer (NK)-cell differentiation proceeds through five stages, which are delimited by the expression of markers such as CD122, NK1.1 and Ly49s (a). TAM receptor signalling is required for NK-cell maturation during stage III. In this instance, growth-arrest-specific 6 (GAS6) and protein S are produced by stromal cells of the bone-marrow niche, and activate the TAM receptors, TYRO3, AXL and MER, that are expressed by immature NK cells (b). In the absence of TAM receptor signalling, these immature NK cells do not acquire expression of the inhibitory and activating receptors (for example, Ly49) required for target-cell recognition and killing. TAM receptor signalling may require interaction with a cytokine receptor signalling system — in this case, the interleukin-15 receptor (IL-15R), as physical and physiological interactions between the α-subunit of the IL-15R and AXL have been demonstrated.
The molecular mechanics of this process have been recently reviewed75. The basic finding is that NK cells isolated from TAM-deficient mice have very poor cytotoxic activity. As for the TAM-deficient phenotypes outlined above, the impairment of killing activity increases as more TAM genes are inactivated, consistent with the fact that all three receptors are expressed by immature NK cells in the bone marrow22,76. The acquisition of expression of inhibiting and activating receptors by these immature cells is driven by the stromal cells of the bone marrow, and these cells have been found to express both GAS6 and protein S22 (FIG. 4). Furthermore, the ability of stromal cells to drive NK-cell maturation in vitro can be fully recapitulated if immature cells are grown on NIH3T3 fibroblasts that express either recombinant GAS6 or protein S. Although NK cells from mice lacking all three TAM receptors have normal levels of perforin and granzyme B, these cells lack the full complement of activating and inhibitory receptors that are expressed by cytotoxic NK cells, are unable to secrete IFNγ in response to stimulation, and most importantly, exhibit a 10-fold lower killing ability against target cells than wild-type NK cells22. Therefore, TAM signalling drives the end-stage differentiation of NK cells (FIG. 4).
Another factor that has a critical role in NK-cell maturation is IL-15. Analyses of both IL-15-deficient and IL-15 receptor (IL-15R)-deficient mice have demonstrated that this cytokine promotes the differentiation of NK-cell precursors (CD122+NK1.1− cells) into immature NK cells (CD122+NK1.1+ cells), and have additionally suggested that IL-15 also operates at the slightly later stages of NK-cell maturation during which TAM signalling is required77-81. It is therefore interesting that a physical and physiological association between the TAM receptor AXL and the α-subunit of the IL-15R has been demonstrated82. This demonstration has been made in L929 fibroblasts and DCs rather than NK cells, but the AXL–IL-15Rα association is of clear functional consequence to these cells. IL-15 protects L929 cells from TNF-induced cell death, and this IL-15 effect is entirely dependent on the presence of AXL. IL-15Rα and AXL co-localize on the L929 cell plasma membrane and can be co-immunoprecipitated. (This is also the case in DCs.) In addition, treatment of L929 cells with IL-15 mimics treatment with GAS6, and results in the rapid tyrosine phosphorylation of both IL-15Rα and AXL. Thus, an intimate interaction between a TAM receptor and a cytokine receptor, which is structurally distinct from IFNAR, is again required for both TAM receptor and cytokine receptor function. In this example, and also with respect to the interaction of TAM receptors and the IFNAR signalling complex in DCs, it will be of interest to determine if the demonstrated physical association between cytokine receptors and AXL extends to TYRO3 and MER.
Prospects and implications
Recent insights into TAM receptor function have many implications for our understanding of innate immune regulation and the treatment of immune system disorders. These include a possible role for TAM-receptor-mediated inhibition of endotoxin tolerance and immune suppression — phenomena in which hypo-responsiveness to TLR engagement is induced by prior TLR activation83,84. In macrophages, components of the TAM pathway that are upregulated during inflammation must turn over with a half-life that allows responding cells to return to their baseline levels. If a secondary encounter with pathogen occurs before this happens, then the TLR response to the secondary pathogen will be blunted. Immunosuppression also occurs in end-stage sepsis. This compromises the ability of patients to eradicate their primary infection and also predisposes them to nosocomial infections. In this context, it is interesting to note that a recent clinical study found that the levels of circulating GAS6 are consistently elevated in patients with severe sepsis, and are correlated with the occurrence of septic shock85. These observations suggest that small molecule inhibitors of the TAM receptors may be reasonable candidates both for potential therapies in acute sepsis and for new-generation vaccine adjuvants for immunization. On the flip side, diminished or interrupted TAM receptor signalling may lead to the development of chronic inflammatory and autoimmune diseases, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis. This clearly happens in TAM-deficient mice21,25; and in humans, reduced levels of free circulating protein S, which should result in reduced TAM receptor signalling, are evident in patients with SLE86-88. Therefore, the analysis of TAM receptor signalling is likely to have an increasingly prominent role in our understanding of both the normal innate immune response and its perturbation in disease.
Supplementary Material
Acknowledgments
Work in the authors’ laboratory is supported by grants from the Lupus Research Institute and the US National Institutes of Health (G.L.), by the Salk Institute (G.L. and C.V.R.) and by the Pew Latin American Fellows Program (C.V.R.).
- Natural killer (NK) cells
NK cells are lymphoid cells capable of lysing bacteria- and virus-infected cells as well as many tumour cells, without prior sensitization. They have important roles in combating infections, in the immune surveillance of cancer and in host-versus-graft rejection.
- Toll-like receptors (TLRs)
Pattern-recognition receptors that recognize molecules — such as the lipopolysaccharide of bacterial cell walls, the unmethylated CpG-containing deoxynucleotides of bacterial DNA and the double-stranded RNAs of viruses — that are broadly shared by pathogens but not by host cells. Pathogen activation of TLRs initiates the innate immune response in dendritic cells and macrophages.
- SOCS proteins (suppressor of cytokine signalling proteins)
SOCS proteins inhibit STAT (signal-transducer and activator of transcription) phosphorylation by binding and inhibiting JAKs (Janus-family kinases) and/or competing with STATs for phosphotyrosine binding sites on cytokine receptors. They also inhibit signal transducers downstream of Toll-like receptor activation.
- STATs (signal transducers and activators of transcription)
STATs are latent cytoplasmic transcription factors that upon phosphorylation, typically by Janus-family kinases (JAKs), are activated. They then translocate to the nucleus, where they drive the transcription of their target genes. The signal transduction pathways downstream of many cytokine receptors depend on STAT protein activation
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
AXL ∣ GAS6 ∣ IFNAR ∣ IL-15R ∣ MER ∣ protein S ∣ STAT1 ∣ TYRO3
FURTHER INFORMATION
Greg Lemke’s homepage: http://www.salk.edu/faculty/faculty_details.php?id=35
The human kinome: http://www.kinase.com/human/kinome
See online article: S1 (table)
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol. 2007;19:124–134. doi: 10.1016/j.ceb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nature Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 4.Lai C, Lemke G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. 1991;6:691–704. doi: 10.1016/0896-6273(91)90167-x.. This study first identified the TAM receptors as a distinct receptor PTK subfamily.
- 5.O’Bryan JP, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11:5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapraz F, et al. RTK and TGF-β signaling pathways genes in the sea urchin genome. Dev Biol. 2006;300:132–152. doi: 10.1016/j.ydbio.2006.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki T, et al. Structural basis for Gas6–Axl signalling. EMBO J. 2006;25:80–87. doi: 10.1038/sj.emboj.7600912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stitt TN, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0.. This study reports the identification of two ligands for TAM receptors.
- 9.Nagata K, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- 10.Prasad D, et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. 2006;33:96–108. doi: 10.1016/j.mcn.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 11.Uehara H, Shacter E. Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinase-mediated phagocytosis of apoptotic cells. J Immunol. 2008;180:2522–2530. doi: 10.4049/jimmunol.180.4.2522. [DOI] [PubMed] [Google Scholar]
- 12.Rezende SM, Simmonds RE, Lane DA. Coagulation, inflammation, and apoptosis: different roles for protein S and the protein S–C4b binding protein complex. Blood. 2004;103:1192–1201. doi: 10.1182/blood-2003-05-1551. [DOI] [PubMed] [Google Scholar]
- 13.Huang M, et al. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nature Struct Biol. 2003;10:751–756. doi: 10.1038/nsb971. [DOI] [PubMed] [Google Scholar]
- 14.Nakano T, et al. Requirement of γ-carboxyglutamic acid residues for the biological activity of Gas6: contribution of endogenous Gas6 to the proliferation of vascular smooth muscle cells. Biochem J. 1997;323:387–392. doi: 10.1042/bj3230387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasanbasic I, Rajotte I, Blostein M. The role of γ-carboxylation in the anti-apoptotic function of Gas6. J Thromb Haemost. 2005;3:2790–2797. doi: 10.1111/j.1538-7836.2005.01662.x. [DOI] [PubMed] [Google Scholar]
- 16.Benzakour O, Kanthou C. The anticoagulant factor, protein S, is produced by cultured human vascular smooth muscle cells and its expression is up-regulated by thrombin. Blood. 2000;95:2008–2014. [PubMed] [Google Scholar]
- 17.Anderson HA, et al. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nature Immunol. 2003;4:87–91. doi: 10.1038/ni871.. This work identified the TAM ligand protein S as the factor responsible for serum-stimulated phagocytosis of apoptotic cells.
- 18.Sasaki T, et al. Crystal structure of a C-terminal fragment of growth arrest-specific protein Gas6. Receptor tyrosine kinase activation by laminin G-like domains. J Biol Chem. 2002;277:44164–44170. doi: 10.1074/jbc.M207340200. [DOI] [PubMed] [Google Scholar]
- 19.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–728. doi: 10.1038/19554.. This study documented the progressive degeneration of germ cells in the TAM triple mutant mice.
- 21.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663.. This study demonstrated that TAM-deficient mice develop severe lymphoproliferation and systemic autoimmunity.
- 22.Caraux A, et al. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nature Immunol. 2006;7:747–754. doi: 10.1038/ni1353.. This work showed that the TAM receptors are required for NK-cell differentiation and maturation.
- 23.Angelillo-Scherrer A, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nature Med. 2001;7:215–221. doi: 10.1038/84667. [DOI] [PubMed] [Google Scholar]
- 24.Yanagita M, et al. Essential role of Gas6 for glomerular injury in nephrotoxic nephritis. J Clin Invest. 2002;110:239–246. doi: 10.1172/JCI14861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034.. This study identified a new negative regulatory pathway driven by the TAM receptor family, and established its role as an inhibitor of both TLR-and cytokine-driven immune responses in APCs.
- 26.Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-α production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999;162:3498–3503. [PubMed] [Google Scholar]
- 27.Tibrewal N, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-κB transcriptional activation. J Biol Chem. 2008;283:3618–3627. doi: 10.1074/jbc.M706906200. [DOI] [PubMed] [Google Scholar]
- 28.Wormald S, Hilton DJ. The negative regulatory roles of suppressor of cytokine signaling proteins in myeloid signaling pathways. Curr Opin Hematol. 2007;14:9–15. doi: 10.1097/00062752-200701000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005;7:100–110. doi: 10.1186/ar1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 31.Sharif MN, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203:1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, et al. Immunoexpression of Tyro 3 family receptors — Tyro 3, Axl, and Mer — and their ligand Gas6 in postnatal developing mouse testis. J Histochem Cytochem. 2005;53:1355–1364. doi: 10.1369/jhc.5A6637.2005. [DOI] [PubMed] [Google Scholar]
- 33.Nakanishi Y, Shiratsuchi A. Phagocytic removal of apoptotic spermatogenic cells by Sertoli cells: mechanisms and consequences. Biol Pharm Bull. 2004;27:13–16. doi: 10.1248/bpb.27.13. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa A, Shiratsuchi A, Tsuda K, Nakanishi Y. In vivo analysis of phagocytosis of apoptotic cells by testicular Sertoli cells. Mol Reprod Dev. 2005;71:166–177. doi: 10.1002/mrd.20278. [DOI] [PubMed] [Google Scholar]
- 35.Ueno H, Mori H. Morphometrical analysis of Sertoli cell ultrastructure during the seminiferous epithelial cycle in rats. Biol Reprod. 1990;43:769–776. doi: 10.1095/biolreprod43.5.769. [DOI] [PubMed] [Google Scholar]
- 36.Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403. doi: 10.1083/jcb.42.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Cruz PM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 38.Nandrot E, et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mechanisms of physiological cell adhesion and apoptosis. Neurobiol Dis. 2000;7:586–599. doi: 10.1006/nbdi.2000.0328. [DOI] [PubMed] [Google Scholar]
- 39.Duncan JL, et al. An RCS-like retinal dystrophy phenotype in mer knockout mice. Invest Ophthalmol Vis Sci. 2003;44:826–838. doi: 10.1167/iovs.02-0438. [DOI] [PubMed] [Google Scholar]
- 40.Gal A, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nature Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 41.Tschernutter M, et al. Clinical characterisation of a family with retinal dystrophy caused by mutation in the Mertk gene. Br J Ophthalmol. 2006;90:718–723. doi: 10.1136/bjo.2005.084897.. References 37 to 41 identified the essential role of MER in retinal homeostasis and phagocytosis in mice, rats and humans.
- 42.Scott RS, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603.. This is the first study that demonstrated the importance of the TAM receptor MER in the clearance of apoptotic cells by macrophages.
- 43.Cohen PL, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–5642. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 45.Mahoney JA, Rosen A. Apoptosis and autoimmunity. Curr Opin Immunol. 2005;17:583–588. doi: 10.1016/j.coi.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 46.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nature Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nature Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 48.Baumann I, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 49.Gaipl US, et al. Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun. 2006;9:173–187. doi: 10.1159/000090781. [DOI] [PubMed] [Google Scholar]
- 50.Stuart LM, et al. Inhibitory effects of apoptotic cell ingestion upon endotoxin-driven myeloid dendritic cell maturation. J Immunol. 2002;168:1627–1635. doi: 10.4049/jimmunol.168.4.1627. [DOI] [PubMed] [Google Scholar]
- 51.Sauter B, et al. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ravichandran KS. “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell. 2003;113:817–820. doi: 10.1016/s0092-8674(03)00471-9. [DOI] [PubMed] [Google Scholar]
- 53.Ogden CA, et al. Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt’s lymphoma. J Immunol. 2005;174:3015–3023. doi: 10.4049/jimmunol.174.5.3015. [DOI] [PubMed] [Google Scholar]
- 54.Lingnau M, Hoflich C, Volk HD, Sabat R, Docke WD. Interleukin-10 enhances the CD14-dependent phagocytosis of bacteria and apoptotic cells by human monocytes. Hum Immunol. 2007;68:730–738. doi: 10.1016/j.humimm.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Jung M, et al. Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur J Immunol. 2004;34:481–493. doi: 10.1002/eji.200324323. [DOI] [PubMed] [Google Scholar]
- 56.Sen P, et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood. 2007;109:653–660. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallet MA, et al. MerTK is required for apoptotic cell-induced T cell tolerance. J Exp Med. 2008;205:219–232. doi: 10.1084/jem.20062293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Miyanishi M, et al. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 60.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 61.Finnemann SC, Nandrot EF. MerTK activation during RPE phagocytosis in vivo requires αVβ5 integrin. Adv Exp Med Biol. 2006;572:499–503. doi: 10.1007/0-387-32442-9_69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahajan NP, Earp HS. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: a mechanism for localizing guanine nucleotide-exchange factor action. J Biol Chem. 2003;278:42596–42603. doi: 10.1074/jbc.M305817200. [DOI] [PubMed] [Google Scholar]
- 63.Lodoen MB, Lanier LL. Natural killer cells as an initial defense against pathogens. Curr Opin Immunol. 2006;18:391–398. doi: 10.1016/j.coi.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smyth MJ, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–510. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 65.Screpanti V, Wallin RP, Grandien A, Ljunggren HG. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol. 2005;42:495–499. doi: 10.1016/j.molimm.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 66.Screpanti V, Wallin RP, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol. 2001;167:2068–2073. doi: 10.4049/jimmunol.167.4.2068. [DOI] [PubMed] [Google Scholar]
- 67.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nature Rev Immunol. 2003;3:361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 68.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nature Rev Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 69.Arase H, Arase N, Saito T. Interferon γ production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. J Exp Med. 1996;183:2391–2396. doi: 10.1084/jem.183.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gosselin P, et al. Induction of DAP12 phosphorylation, calcium mobilization, and cytokine secretion by Ly49H. J Leukoc Biol. 1999;66:165–171. doi: 10.1002/jlb.66.1.165. [DOI] [PubMed] [Google Scholar]
- 71.Ortaldo JR, Young HA. Expression of IFN-γ upon triggering of activating Ly49D NK receptors in vitro and in vivo: costimulation with IL-12 or IL-18 overrides inhibitory receptors. J Immunol. 2003;170:1763–1769. doi: 10.4049/jimmunol.170.4.1763. [DOI] [PubMed] [Google Scholar]
- 72.Raulet DH, Vance RE, McMahon CW. Regulation of the natural killer cell receptor repertoire. Annu Rev Immunol. 2001;19:291–330. doi: 10.1146/annurev.immunol.19.1.291. [DOI] [PubMed] [Google Scholar]
- 73.Vivier E, Nunès JA, Vély F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 74.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 75.Roth C, Rothlin C, Riou S, Raulet DH, Lemke G. Stromal-cell regulation of natural killer cell differentiation. J Mol Med. 2007;85:1047–1056. doi: 10.1007/s00109-007-0195-0. [DOI] [PubMed] [Google Scholar]
- 76.Behrens EM, et al. The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol. 2003;33:2160–2167. doi: 10.1002/eji.200324076. [DOI] [PubMed] [Google Scholar]
- 77.Kennedy MK, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cooper MA, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100:3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- 79.Prlic M, Blazar BR, Farrar MA, Jameson SC. In vivo survival and homeostatic proliferation of natural killer cells. J Exp Med. 2003;197:967–976. doi: 10.1084/jem.20021847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koka R, et al. Interleukin (IL)-15Rα-deficient natural killer cells survive in normal but not IL-15Rα-deficient mice. J Exp Med. 2003;197:977–984. doi: 10.1084/jem.20021836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 82.Budagian V, et al. A promiscuous liaison between IL-15 receptor and Axl receptor tyrosine kinase in cell death control. EMBO J. 2005;24:4260–4270. doi: 10.1038/sj.emboj.7600874.. This study documented the IL-15-induced transactivation of the TAM receptor AXL and showed both physical and functional crosstalk between the IL-15 receptor and AXL.
- 83.Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 84.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116:203–212. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Borgel D, et al. Elevated growth-arrest-specific protein 6 plasma levels in patients with severe sepsis. Crit Care Med. 2006;34:219–222. doi: 10.1097/01.ccm.0000195014.56254.8a. [DOI] [PubMed] [Google Scholar]
- 86.Meesters EW, et al. The inflammation and coagulation cross-talk in patients with systemic lupus erythematosus. Blood Coagul Fibrinolysis. 2007;18:21–28. doi: 10.1097/01.mbc.0000256022.01900.c2. [DOI] [PubMed] [Google Scholar]
- 87.Brouwer JL, Bijl M, Veeger NJ, Kluin-Nelemans HC, van der Meer J. The contribution of inherited and acquired thrombophilic defects, alone or combined with antiphospholipid antibodies, to venous and arterial thromboembolism in patients with systemic lupus erythematosus. Blood. 2004;104:143–148. doi: 10.1182/blood-2003-11-4085. [DOI] [PubMed] [Google Scholar]
- 88.Song KS, Park YS, Kim HK. Prevalence of anti-protein S antibodies in patients with systemic lupus erythematosus. Arthritis Rheum. 2000;43:557–560. doi: 10.1002/1529-0131(200003)43:3<557::AID-ANR11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 89.Kulman JD, et al. Vitamin K-dependent proteins in Ciona intestinalis, a basal chordate lacking a blood coagulation cascade. Proc Natl Acad Sci USA. 2006;103:15794–15799. doi: 10.1073/pnas.0607543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liongue C, Ward AC. Evolution of Class I cytokine receptors. BMC Evol Biol. 2007;7:120. doi: 10.1186/1471-2148-7-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krause CD, Pestka S. Evolution of the Class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther. 2005;106:299–346. doi: 10.1016/j.pharmthera.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 92.Melaragno MG, et al. Gas6 inhibits apoptosis in vascular smooth muscle: role of Axl kinase and Akt. J Mol Cell Cardiol. 2004;37:881–887. doi: 10.1016/j.yjmcc.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 93.Korshunov VA, Daul M, Massett MP, Berk BC. Axl mediates vascular remodeling induced by deoxycorticosterone acetate-salt hypertension. Hypertension. 2007;50:1057–1062. doi: 10.1161/HYPERTENSIONAHA.107.096289. [DOI] [PubMed] [Google Scholar]
- 94.Korshunov VA, Mohan AM, Georger MA, Berk BC. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res. 2006;98:1446–1452. doi: 10.1161/01.RES.0000223322.16149.9a. [DOI] [PubMed] [Google Scholar]
- 95.Angelillo-Scherrer A, et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J Clin Invest. 2005;115:237–246. doi: 10.1172/JCI22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gould WR, et al. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J Thromb Haemost. 2005;3:733–741. doi: 10.1111/j.1538-7836.2005.01186.x. [DOI] [PubMed] [Google Scholar]
- 97.Angelillo-Scherrer A, et al. Role of Gas6 in erythropoiesis and anemia in mice. J Clin Invest. 2008;118:583–596. doi: 10.1172/JCI30375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Graham DK, et al. Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2006;12:2662–2669. doi: 10.1158/1078-0432.CCR-05-2208. [DOI] [PubMed] [Google Scholar]
- 99.Keating AK, et al. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene. 2006;25:6092–6100. doi: 10.1038/sj.onc.1209633. [DOI] [PubMed] [Google Scholar]
- 100.Nakamura YS, et al. Tyro 3 receptor tyrosine kinase and its ligand, Gas6, stimulate the function of osteoclasts. Stem Cells. 1998;16:229–238. doi: 10.1002/stem.160229. [DOI] [PubMed] [Google Scholar]
- 101.Katagiri M, et al. Mechanism of stimulation of osteoclastic bone resorption through Gas6/Tyro 3, a receptor tyrosine kinase signaling, in mouse osteoclasts. J Biol Chem. 2001;276:7376–7382. doi: 10.1074/jbc.M007393200. [DOI] [PubMed] [Google Scholar]
- 102.Shankar SL, et al. The growth arrest-specific gene product Gas6 promotes the survival of human oligodendrocytes via a phosphatidylinositol 3-kinase-dependent pathway. J Neurosci. 2003;23:4208–4218. doi: 10.1523/JNEUROSCI.23-10-04208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shankar SL, et al. Gas6/Axl signaling activates the phosphatidylinositol 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factor α-induced apoptosis. J Neurosci. 2006;26:5638–5648. doi: 10.1523/JNEUROSCI.5063-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shimojima M, Ikeda Y, Kawaoka Y. The mechanism of Axl-mediated Ebola virus infection. J Infect Dis. 2007;196(Suppl. 2):259–263. doi: 10.1086/520594. [DOI] [PubMed] [Google Scholar]
- 105.Shimojima M, et al. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol. 2006;80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.