

Abstract
The first example of a computationally de novo designed protein that binds an emissive abiological chromophore is presented, in which a sophisticated level of cofactor discrimination is pre-engineered. This heterotetrameric, C2-symmetric bundle, AHis:BThr, uniquely binds (5,15-di[(4-carboxymethyleneoxy)phenyl]porphinato)zinc [(DPP)Zn] via histidine coordination and complementary noncovalent interactions. The A2B2 heterotetrameric protein reflects ligand-directed elements of both positive and negative design, including hydrogen-bonds to second-shell ligands. Experimental support for the appropriate formulation of [(DPP)Zn:AHis:BThr]2 is provided by UV/visible and circular dichroism spectroscopies, size exclusion chromatography, and analytical ultracentrifugation. Time-resolved transient absorption and fluorescence spectroscopic data reveal classic excited-state singlet and triplet PZn photophysics for the AHis:BThr:(DPP)Zn protein (kfluorescence = 4 × 108 s−1; τriplet = 5 ms). The A2B2 apoprotein has immeasurably low binding affinities for related [porphinato]metal chromophores that include a (DPP)Fe(III) cofactor and the zinc metal ion hemin derivative [(PPIX)Zn], underscoring the exquisite active site binding discrimination realized in this computationally designed protein. Importantly, elements of design in the AHis:Bthr protein ensure that interactions within the tetra-α-helical bundle are such that only the heterotetramer is stable in solution; corresponding homomeric bundles present unfavorable ligand-binding environments, and thus preclude protein structural rearrangements that could lead to binding of (porphinato)iron cofactors.
Introduction
The design and synthesis of de novo proteins that bind abiological cofactors provide a general strategy to interrogate important protein structure-function relationships1–5 and build proteins that possess functions not seen in nature. Although de novo designed proteins capable of binding hemin [(PPIX)Fe] have been realized,6–9 such systems typically mimic the structures, functions and spectral features of naturally occurring hemoproteins. While advances in protein design have been used to craft tetra-α-helical peptides that bind abiological (porphinato)iron (PFe) cofactors,10–12 elaborating entirely new metalloprotein functions requires the ability to engineer proteins that uniquely accommodate targeted ligand frameworks as well as specific metal ions. Designed protein complexes containing abiological Zn-porphyrin cofactors are of particular interest due to the fact that electron and energy transfer reactions can be activated by pulsed laser excitation of such chromophores,13–17 and the fact that these moieties are key components in systems exhibiting a variety of novel nonlinear optical and photophysical properties.18–23 Herein, a helical bundle protein that selectively encapsulates an emissive zinc porphyrin chromophore is computationally designed and experimentally characterized.
(Porphinato)zinc (PZn) chromophores have been utilized to provide fundamental insights into electron and energy transfer reactions13,16 and develop high performance electronic and optical materials.18–23 Synthetic multiporphyrin arrays13,24–27 yield supramolecular architectures inspired in part by the naturally occurring multiporphyrin array found in the light harvesting complex of photosystem II.28–30 While PZn may be dispersed in polymer-based systems, controlling the cofactor’s orientation and local environment can be difficult or limited.20,31,32
Though subtle, the folding and self-assembly properties of proteins can be leveraged to arrive at well defined local environments for porphyrin-based cofactors. Bundles of helical peptides can encapsulate and coordinate natural and abiotic Zn porphyrins.33,34 Inspection of fiducial structures have guided the design of such systems, where Zn is coordinated with appropriate side-chain ligands (e.g., histidine) and hydrophobic residues are patterned so as to stabilize a helical bundle. In addition, such designed α-helical proteins can be displayed in oriented molecular arrays at interfaces.34–36 Many of these systems examined thus far have fluctuating tertiary structures, however, and likely do not have the geometric complementarity and highly structured physico-chemical protein-porphyrin interactions observed in natural proteins.33,35 Alternatively, non-natural (porphinato)metal derivatives have been incorporated into myoglobin and related proteins.37–42 Complete control and creation of a protein environment that is complementary to an abiological cofactor, however, might best be accomplished through automated de novo design. Such approaches explicitly consider the atomic details of the backbone structure as well sidechain-sidechain and sidechain-cofactor packing.
The de novo design of soluble, proteins selective for PZn is complicated by the preference of zinc porphyrins for a five-coordinate environment.43–45 In contrast, the symmetry in six-coordinate Fe3+ ligation can be leveraged in the design of proteins that encapsulate iron porphyrins.10–12 Such preferred local asymmetry in the metal coordination of PZn suggests use of helical bundles having reduced symmetry, e.g., heterotetrameric bundles.46,47 Along these lines, heterotetramers have been designed that present a dinuclear metal ion site48,49 including an A2B2 protein.50 Heterotetrameric systems of this type have not been identified for the much larger PZn cofactors. Many previous bundle proteins resulted from coarse-grained design approaches, including the manipulation of complementary solvent-exposed electrostatic interactions and empirical matching of interior hydrophobic side chains.50
Computational design methods provide powerful tools for engineering structure and sequence.3,10–12,51–55 Herein, these methods are applied to address specific protein-protein and protein-PZn interactions in an atomically detailed manner. Elements of both positive and negative design are used to arrive at a de novo heterotetrameric, α-helical peptide bundle that selectively incorporates an abiotic PZn choromophore that can be electronically excited to produce a long-lived fluorescent state. We exploit the rigidity of tetra-α-helical peptides to organize a (5,15-diarylporphinato)Zn cofactor [(DPP)Zn] at specified locations within an α-helical bundle. Previous designs of helical peptides that bind abiological cofactors provided for selective binding of (DPP)FeIII over iron(III) protoporphyrin IX [(PPIX)FeIII].10–12 In contrast to the bis-His ligation environment of (DPP)FeIII, the (DPP)Zn chromophore requires mono-His coordination,33 which demands a less symmetric protein environment relative to previous homotetrameric D2-symmetric four-helix bundles. An appropriate A2B2 heterotetrameric protein is computationally designed and experimentally characterized.
Experimental Methods
Synthesis of (DPP)Zn
5,15-Di[((4-ethyl ester)methylene-oxy)phenyl]porphyrin was synthesized according to a previously published procedure.10
5,15-Di[((4-ethyl ester)methylene-oxy)phenyl]porphinato zinc [(DPPESTER)Zn]. In a round bottom flask equipped with a condenser, the free base porphyrin (126 mg, 0.19mmol) was dissolved in 150 mL of chloroform. Zn(OAc)2 (340 mg, 2.1 mmol) was added to the stirring solution and the mixture was refluxed for 3 h. The color changed from a dark purple to an intense violet. The crude material was obtained by removing the chloroform via rotary evaporation. The product was purified by silica gel column chromatography (40:60 THF:hexanes), which eluted as an intense violet band. The solvent was removed under reduced pressure yielding a purple solid. Isolated yield = 130 mg (95% based on 0.19 mmol of the free-base starting material).
5,15-Di[(4-carboxymethylene-oxy)phenyl]porphinato zinc [(DPP)Zn]
(DPPESTER)Zn (130 mg) was saponified by refluxing with KOH (90 mg, 1.6 mmol) in 80 mL of CHCl3:EtOH (3:1) for 3 h. The solvent was removed under reduced pressure and redissolved in water where the solution was acidified with 1N HCl. The precipitate was filtered, washed with 1N HCl, dissolved in THF, and then reprecipitated with hexanes. The supernatant was decanted and the remainder of the solvent evaporated under reduced pressure yielding additional (DPP)Zn product. Isolated yield = 118 mg (97% yield based on 0.18 mmol of the ethyl-ester starting material). _1H-NMR (CDCl3/1% pyridine-d5, 500 MHz): δ 10.5 (s, 2H), 9.6 (d, 4H), 9.3 (d, 4H), 8.3 (d, 4H), 7.7 (d, 4H), 5.4 (s, 4H). MS(MALDI-TOF): m/z = 672.52 (M+) (Calcd for C36H24N4O6: 672.10).
Peptide Purification
Two synthesized peptides AHis: Ac-SLEEALQEIQ QAAQEAQQAL QKHQQALQKF QKYG-CONH2 and BThr: Ac-SLEEALQEAQ QTAQEFQQAL QKIQQAFQKF QKYG-CONH2, were purchased from Biopeptide. The impure material was further purified by reversed phase HPLC (Varian ProStar) over a C4 column (Vydac). Analytical reversed phase HPLC (Hewlett Packard 1100) indicated successful purification of the peptides. MALDI-TOF (PerSeptive Biosystems Voyager DE) indicated correct mass spectral data (AHis Calcd, 3953.4; found, 3953.2. BThrCalcd, 4027.5; found 4027.9).
General Sample Preparation
Samples were prepared by introducing equimolar amounts of AHis and BThr to a 50 mM phosphate buffer, 150 mM sodium chloride, pH 7.5. A twofold excess of the cofactor (DPP)Zn was added from either a 5mM DMSO or 0.5 M NaOH stock solution. (Note: final DMSO concentration was kept below 1%). The sample was heated to >50°C were 15 min, followed by cooling to room temperature. Any unbound cofactor precipitates under these conditions, which was removed via spin filtration (Millipore Ultrafree MC 0.22 μm).
UV-Visible and Circular Dichroism (CD) Spectroscopy
To verify binding of the cofactor, complex formation was monitored at the Soret band at 420 nm and Q-band at 551 nm. Electronic absorption spectra were obtained on a Hewlett Packard 8453 UV-visible spectrophotometer at room temperature in 1-cm quartz cells. To evaluate the backbone conformation and stability of the protein-cofactor complex, CD spectroscopy was carried out at 25 °C using a Jasco 810 Spectropolarimeter in 0.1-cm cells. Buffer conditions for CD experiments were 50 mM phosphate buffer, 150 mM sodium chloride, pH 7.5; the protein concentration was 50 μM for the component peptides and 100 μM for the apo- and holo-heterotetramers. (DPP)Zn was added from a 0.5 M NaOH stock solution, as DMSO absorbs light in the 190–260 nm region of the spectrum. All samples were filtered through a 0.22 μm spin filter. For thermal denaturation experiments, the ellipticity at 222 nm was monitored as a function of temperature. Data were collected every 2 °C; a heating rate of 1 °C/min was followed by a 4-min equilibration time.
Stoichiometry
A solution of 2.5 μM (DPP)Zn was prepared in 50 mM phosphate buffer, 150 mM sodium chloride, pH 7.5, 1% octyl glucopyranoside (OG). (The surfactant, OG, aids in cofactor solubilization). Aliquots of AHis:BThr (0.50 μM) were introduced into the solution and allowed to equilibrate for 5 min before a spectrum was obtained. Spectrophotometric titrations were monitored at 421 nm.
Job Analysis
Ten separate samples were prepared that varied the ratio of the two peptides from 0:1 → 1:1 → 1:0 AHis:BThr in 50 mM phosphate buffer, 150 mM sodium chloride, pH 7.5, (total peptide] = 150 μM). A twofold excess of the cofactor (DPP)Zn was added from a concentrated DMSO stock solution, such that the final DMSO concentration was kept below 1%. The sample was heated to >50°C for 15 min and then cooled to room temperature. Any unbound cofactor formed visible aggregates that were removed via spin filtration (0.2 μm). Each sample was analyzed for cofactor binding via electronic absorption spectroscopy. A Job plot monitoring peptide ratio versus cofactor absorbance at 550 nm was generated from the UV/visible spectral data.
Size Exclusion Chromatography
Gel filtration profiles were obtained using a Superdex 75 10/300GL column on an FPLC system (GE Healthcare AKTA FPLC System). To evaluate the nature of the oligomeric state, 100 μL of a 250 μM sample was injected onto the column and eluted with a flow rate of 0.5 mL/min, and a 50 mM phosphate buffer, 100 mM NaCl, pH 7.5 mobile phase. MWapp was calculated from a standard curve generated from mass standards blue dextran (Vo), aprotinin (6500 Da), cytochrome c (12 400 Da), carbonic anhydrase (29 000 Da), and albumin (66 000 Da) mass standards. Results are provided in the Supporting Information.
Analytical Ultracentrifugation
For analytical ultracentrifugation, 125 μL samples of 600 μM AHis, BThr, AHis:BThr (1:1) and 25 μM (DPP)Zn:AHis:BThr in 50 mM phosphate buffer (100 mM NaCl, pH 7.5) were prepared. The samples were filtered prior to analysis using a 0.2 μm spin filter. The analysis was performed at 25 °C using a Beckman XL-I analytical ultracentrifuge. The absorbance was monitored at 280 nm for peptide-only samples and 555 nm for samples containing the cofactor, and the sample was centrifuged at 35 000, 40 000, and 48 000 rpm. The data were analyzed using a modified global fitting routine in IGOR Pro (Wavemetric). The data were well-described by assuming a single molecular weight species. The partial specific volume (v) of 0.735 mL/g was calculated using SEDNTERP (http://www.rasmb.bbri.org/).
Nanosecond Transient Absorption Spectroscopy
Transient absorption spectra of 3 μM (DPP)Zn:AHis:BThr in 50 mM phosphate buffer (100 mM NaCl, pH 7.5) were recorded with the previously described Q-switched Nd:YAG laser (DCR-1A, Quanta Ray, Mountain View, CA).56
Picosecond Fluorescence Lifetime Measurements (Streakscope)
Time-resolved emission spectra of 3 μM (DPP)Zn:AHis:BThr, in 50 mM phosphate buffer (100 mM NaCl, pH 7.5) were recorded using a Hamamatsu C4780 picosecond fluorescence lifetime measurement system. This system employs a Hamamatsu Streakscope C4334 as its photon-counting detector and a Hamamatsu C4792-01 synchronous delay generator. A Hamamatsu 405 nm diode laser served as the excitation light source. All fluorescence data were acquired in single-photon-counting mode using Hamamatsu HPD-TA software. The data were analyzed using a single exponential kinetic fit in OriginLab (7.5).
Computational Methods
Requirements for efficient cofactor binding dictated the protein backbone geometry as described previously in the design of an iron-porphyrin binding protein.10 Zn2+ prefers a pentacoordinate environment, and only one axial His ligand was specified to coordinate each cofactor, yielding a C2 symmetric structure (Figure 1). The resulting A2B2 tetramer contains two distinct peptides, labeled AHis and BThr; the two AHis helices are antiparallel to one another as are the two BThr helices. AHis contributes a Zn-coordinating His, and BThr contributes a Thr that is poised to interhelically hydrogen bond with the His of AHis. The identities of these two residues (His and Thr) are constrained within the tetrahelical structure, and those of the remaining 62 variable positions are determined using computational sequence design. In Zn-porphyrins, as opposed to ferric-porphyrins, the Zn ion is slightly domed out of the plane defined by the porphyrin macrocycle (see Table S1 of supporting material). The sequence calculations described below were found to be insensitive to dome heights in the range 0.0–0.3 Å.
Figure 1.
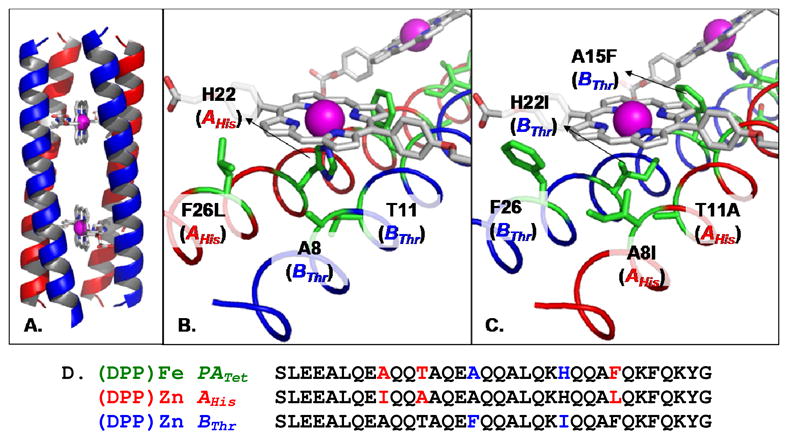
Model of the designed protein illustrating: (A) the AHis:BThr:(DPP)Zn structure; (B) residues near the coordinating H22; (C) details of four residue modifications on distal side; and (D) the final sequences compared to that of PATet.10–12
Protein Sequence Design
The identities of the 62 variable positions were determined by recursive computational design calculations of the probabilities of the amino acids at variable positions.3,57 This statistical, computationally-assisted design strategy (SCADS) has been used previously.3,10,58–60 Seventeen natural amino acids were allowed at the variable positions. Cysteine, histidine, and proline were excluded, as was methionine at positions facing the protein interior; proline has a low helix propensity, and histidine, cysteine and methionine could introduce nontargeted metal-coordination. Positions 11 in BThr and 22 in AHis were constrained to be Thr and His, respectively. Three rounds of calculations were performed. In the first round, hydrophobic residues A, F, G, I, L, V, and W were allowed in the interior positions 5, 8, 15, 19, 26, and 29. The site-specific probabilities of the amino acids were determined as described previously.10,11 After the first round, identities L1, E3, A4, E7, I8(AHis), A8(BThr), E14, A15(AHis), F15 (BThr), A18, F26(BThr), and F29 were specified. Selections were made from the probable amino acids at each site, and unless indicated, the computationally determined amino acid identity applies to both peptides. At the underlined positions the most probable amino acid identities were selected. E7 and E14 form complementary electrostatic interactions with positions 21 and 28, positions that were subsequently constrained to allow only positively charged amino acids (K, R). Subject to these constraints on sequence, a second calculation was performed. The resulting probabilities were used to specify E2, L5, A12, L19, K21, A25 (AHis), K28, K31, and Y32. A12 was the second-most probable identity in position 12, which was selected over S since it resides at the interhelical interface. K was the third-most probable identity in position 31, an exterior position at which many amino acids have comparable probabilities. Before the third calculation, at all remaining variable exterior positions, ionizable amino acids and proline (D, E, H, K, P, and R) were precluded so as to not frustrate predetermined interhelical electrostatic interactions. After this third calculation, the remaining identities were fixed: Q6, Q9, Q10, A11 (AHis), Q13, Q16, Q17, Q20, I22 (BThr), Q23, Q24, A25 (BThr), L26 (AHis), Q27, and Q30. All of these selections, except A25, were the most probable choices at these respective positions. The third-most probable A25 (BThr), was chosen over the most probable G and the second-most probable S, to support an interhelical hydrophobic zipper motif involving I8 (AHis).
Figure 1 provides a comparison of the newly designed sequences (PZn peptide) with an earlier (DPP)Fe design (parent tetramer PATet) of Cochran et al.10,11 Among the 64 unique residue positions, the (DPP)Zn design differs from the previous (DPP)Fe protein at five positions. Two mutations, T11A (AHis) and H22I (BThr), result from changing the metal environment from six-point to five-point coordination; these residues are distal to the coordinating His, effectively removing the second coordination site while maintaining a hydrophobic interior (Figure 1). Mutations A8I (AHis) and F26L (AHis) are sterically linked to the mutation H22I (BThr). Mutation A15F (BThr) results from the reduced symmetry compared to the previous homotetramer. A8I (AHis), F26L(AHis), and A15F(BThr) yield a protein interior that is both more well-packed and more hydrophobic than the previously designed (DPP)Fe protein.10,11
Results
Protein Design
Requirements for efficient cofactor binding dictated the protein backbone geometry as described previously in the design of a homotetrameric precursor bundle (PAtet) that bound ferrous and ferric DPP cofactor in a bis-His geometry.10 Zn2+ prefers a pentacoordinate environment, and only one axial His ligand was specified to coordinate each cofactor, yielding a C2 symmetric structure (Figure 1). Following a minimalist strategy, the sequences of the two helices, designated AHis and BThr, were designed starting with the aim of making the least number of changes required to introduce the desired specificity; the peptides should bind DPP(Zn) only when both the AHis and BThr peptides were present as a binary mixture. In the original homotetrameric PAtet bundle, each helix contains a single His and Thr residue. Thr and His residues on adjoining helices form a network of inter-helical second-shell hydrogen bonds that create two symmetry-related bis-His binding sites appropriate for interaction with ferrous and ferric porphyrin derivatives. In contrast, the AHis and BThr helices of the current design have a single His and Thr, respectively. The resulting (AHis: BThr)2 heterotetamer retains the inter-helical second-shell hydrogen bonds of PAtet, but due to the lack of a His residue in BThr, two single-His sites are formed to provide specificity for pentacoordinate Zn(II) porphyrins. Moreover, the side-chain packing algorithm showed that specific buried positions had a clear preference for large apolar sidechains that could not be accommodated in the starting PAtet sequence, due to steric overlap when all four chains of the protein were forced to have the same sequence. The relaxed symmetry of the heterotetrameric bundle allowed positioning of these larger apolar sidechains in a geometrically complementary manner, potentially increasing the stability of the complex, as well as the specificity for binding to the hetero-tetrameric bundle, versus potential homo-oligomers of the constituent individual peptides. In summary, the AHis:BThr heterotetramer should bind specifically to two (DPP)Zn cofactors only if the two peptides were present at a 1:1 stoichiometry, and with the helices in an antiparallel arrangement (a parallel bundle would position the two His residues on the same side leading to a bis-His site for a single cofactor, or two side-to-side sites leading to negative cooperativity, with the second binding porphyrin more weakly than the first).4,33
Cofactor
(DPP)Zn (Chart 1) shows two pyrrole peaks that appear at 9.6 and 9.3 ppm in the 1H-NMR data (d6-DMSO) suggesting D2-symmetry. This cofactor in buffer (50 mM phosphate, 150 mM NaCl, pH 7.5) is not readily soluble and aggregates appreciably, resulting in broad UV/visible bands in the Soret and Q-band regions of the spectrum (vide infra). For clarity, Chart 1 shows the zinc protoporphyrin IX ((PPIX)Zn) structure; note that the nature and position of the (PPIX)Zn peripheral macrocycle substituents (β-CH3, CHCH2, and -CH2COO−) differ markedly from those of (DPP)Zn (meso-4′-carboxymethyleneoxyphenyl).
Chart 1.
Electronic Absorption Spectroscopy
In the absence of peptide, the cofactor partially aggregates, resulting in broad, red-shifted B and Q-band manifolds (Figure 2) having low intensities; upon addition of surfactant (1 wt% OG), B (λmax(B-state) = 411 nm) and Q-state absorption (λmax(Q-state) =545 nm) maxima are evident. When a 1:1 AHis:BThr peptide mixture is added, both bands red-shift (λ max(B-state) = 421 nm, λ max(Q-state) = 551 nm), consistent with His ligation to the (DPP)Zn cofactor. Electronic absorption spectroscopy was utilized to monitor titrations of protein into (DPP)Zn solutions, which demonstrates a 1:1:1 [(DPP)Zn:AHis:BThr] cofactor:protein complex stoichiometry, as expected for mono-His coordination of (DPP)Zn (Figure 3). Addition of protein in excess of the stoichiometric amount does not further augment the B band absorbance. The titration data of Figure 3 may be fit with a two-site binding model having values of the dissociation constants for the first and second equivalents of the cofactor of K1 = 10 μM and K2 = 8 × 10−3 μM, respectively (see Supporting Information). Such estimates are approximate and nonrigorous thermodynamically due to the poor solubility of the cofactor and the presence of detergent in the samples. Notwithstanding, this analysis provides an empirical description of the binding process that demonstrates: 1) a high cooperativity to the assembly process (the first cofactor is bound weakly, while the second is bound tightly); 2) the mean overall dissociation constant, (K1K2)1/2 = 0.28, is in the sub-micromolar range (and consistent with values found for a five-coordinate heme-protein maquette).7,61 Ratiometric titrations (Job plot analysis) involving the two peptides (AHis:BThr: 1:0 → 1:1 → 0:1) evince a maximal (DPP)Zn binding efficiency at 1:1 AHis:BThr (Figure 4, inset), consistent with the targeted structure of the complex. Homogeneous peptide solutions of either AHis or BThr do not bind (DPP)Zn as indicated by the overall low absorption intensity in the visible spectrum consistent with aggregated, unbound cofactor (Figure 4).
Figure 2.

UV/Visible spectrum of (DPP)Zn, 2.5 μM in 50 mM phosphate buffer, 150 mM NaCl, pH 7.5 (solid black line), with 1% w/v octylglucopyranoside (dashed black line), and in the presence of 1:1 AHis:BThr (solid red line) with 1% w/v octylglucopyranoside.
Figure 3.

Titration of 1:1 AHis:BThr mixture into a 2.5 μM (DPP)Zn solution in 50 mM Pi, 150 mM NaCl, pH 7.5 and 1% OG surfactant (w/w). Inset: binding curve monitored at 420 nm fit with a dissociation curve. Dashed lines are included in the inset plot of absorbance vs. [AHis:BThr]/2[(DPP)Zn] to indicate a stoichiometry of 1:1:1 AHis:BThr:(DPP)Zn.
Figure 4.

Job plot (inset, λprobe = 550 nm) and corresponding (DPP)Zn-based electronic absorption spectral dependence upon the AHis:BThr molar ratio. Experimental conditions: [(DPP)Zn] = 75 μM, [total peptide] = 150 μM, 50 mM Pi, 150 mM NaCl, pH = 7.5.
The designed heterotetrameric PZn-binding protein is porphyrin macrocycle selective. UV-visible spectroscopic studies probing cofactor specificity show that 1:1 solutions of AHis:BThr do not bind (PPIX)Zn as indicated by the absence of any significant change in the absorption spectrum, particularly the intensity of the B-band at 420 nm (see Figure 2), upon exposure to the designed peptides (Figure 5). In addition, B-band (423 nm) and Q-band maxima (549–550 nm and 585–587 nm) were not observed, as has been reported for bound (PPIX)Zn analogs of hemoglobin62 and cytochrome c.63 Interestingly, similar studies64 as well as Job analysis (Figure 6) indicate that (DPP)FeIII is not bound at any AHis:BThr molar ratio, including homogeneous peptide solutions where a 2:1 AHis:(DPP)Fe stoichiometry is maintained (Figure S1), congruent with the homomeric bundles of AHis being underpacked and unstable. This sophisticated level of cofactor differentiation contrasts sharply the properties of a ligation site in a helix-turn-helix peptide dimer that bound either two (PPIX)Zn species with mono-His coordination or one (PPIX)Fe cofactor in a classic bis-His mode.33
Figure 5.
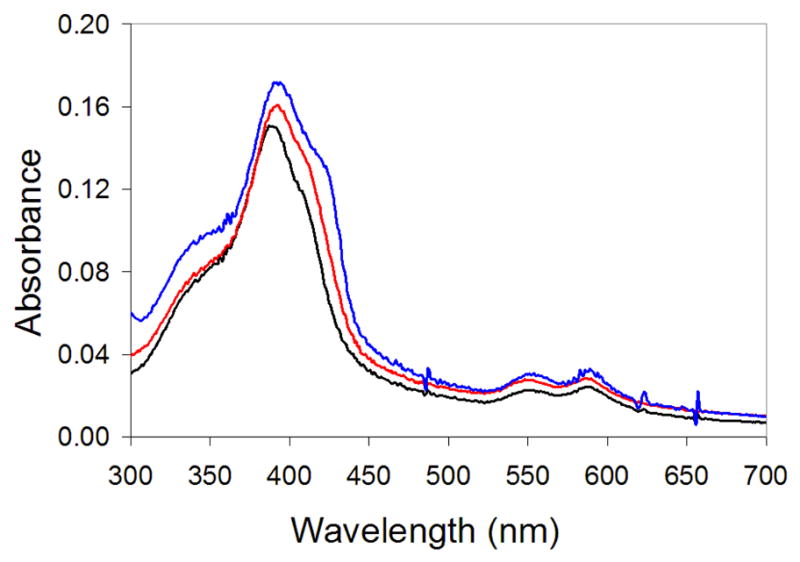
UV/vis of 5 μM (PPIX)Zn in 50 mM Pi, 150 mM NaCl, pH 7.5 (black), 5 μM (PPIX)Zn: AHis:BThr (red), and 5 μM (PPIX)Zn with 25 μM AHis and 25 μM BThr(blue).
Figure 6.

UV/vis of (DPP)FeIII (5 μM in 50 mM Pi, 150 mM NaCl, pH 7.5) in the presence of AHis, (red), 1:1 AHis:BThr (purple), and BThr (blue) (Total peptide concentration, 10 μM). Inset: Job plot monitored at 408 nm indicating no binding of the cofactor to the peptide mixture.
CD Spectroscopy
CD spectroscopy confirms varying degrees of α-helical secondary structure for the individual units of AHis, and BThr, as well as the 1:1 mixture AHis:BThr and AHis:BThr:(DPP)Zn (Figure 7A) as indicated by the minima at 208 and 222 nm.65 A binary 1:1 mixture of AHis:BThr showed a CD spectrum typical of a helical protein. No change was observed upon addition of stoichiometric (DPP)Zn cofactor, again supporting the conclusion that the apo-protein was already folded in the absence of cofactor. The enhanced stability of apo-AHis:BThr is likely due in part to its superior hydrophobic interior packing.
Figure 7.
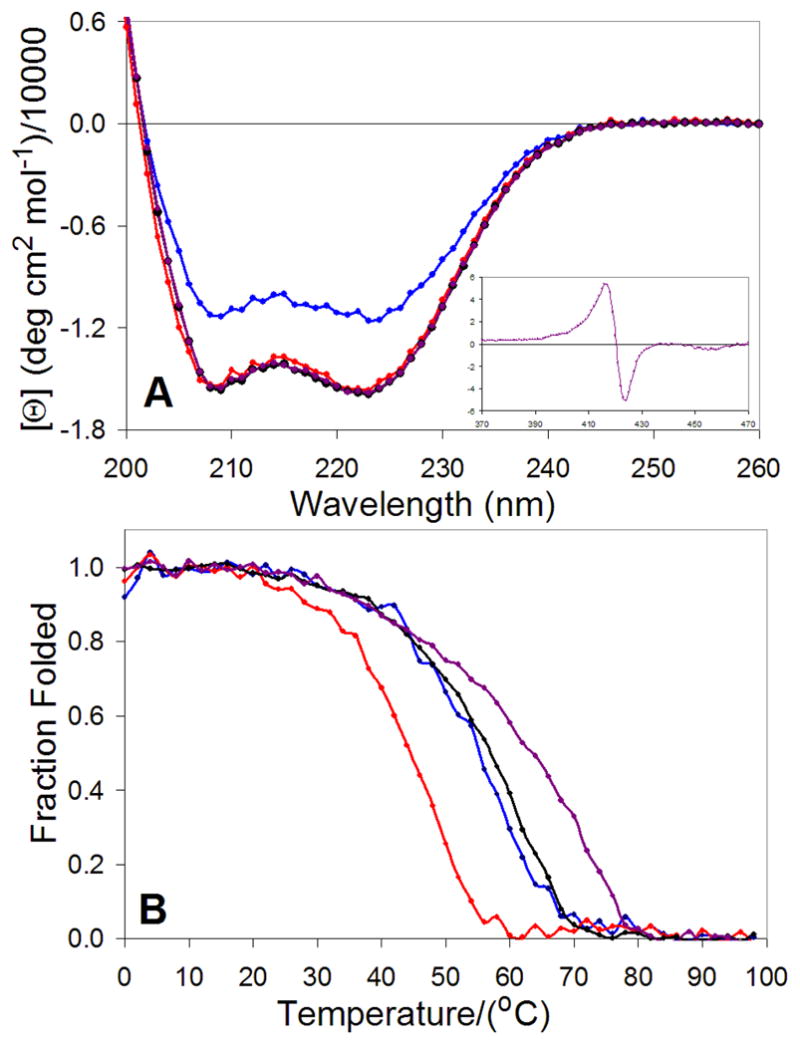
A) Normalized circular dichroism spectra (25 °C) of AHis (red), BThr (blue), AHis:BThr (1:1 mixture black), and (DPP)Zn:AHis:BThr (1:1:1 mixture; purple); the inset shows the corresponding visible/Soret spectrum). B) Thermal unfolding profiles (λprobe = 222 nm) of AHis (red, TM = 44 °C), BThr (blue, TM = 54 °C), AHis:BThr (black, TM = 56 °C), and complexed (DPP)Zn:AHis:BThr (purple, TM = 63 °C). The fraction folded was estimated using the ellipticity relative to folded and unfolded baselines.
Although the (DPP)Zn cofactor is achiral, it becomes optically active when bound in the chiral environment of the AHis:BThr tetramer. The visible region of the spectrum indicates a positive Cotton effect for the two PZn chromophores with λmin = 423 nm and λmax = 416 nm. This observation suggests that the two (DPP)Zn chromophores are aligned in a specific rigid conformation along the superhelical axis of the bundle; in the absence of peptide, no (DPP)Zn CD signal is observed.65,66
The stabilities of the component peptides, AHis:BThr and AHis:BThr:(DPP)Zn were further examined by monitoring their thermal unfolding profiles (Figure 7B). Although the unfolding curves were not fully reversible, precluding a thermodynamic analysis, they provide qualitative data concerning the stabilities of the various assemblies. The BThr peptide had greater stability than the AHis peptide, and when mixed, the sample unfolded in a single transition with a midpoint near that of the BThr sample. Thus, addition of BThr leads to the formation of a complex in which AHis is substantially stabilized towards denaturation. Furthermore, the addition of stoichiometric (DPP)Zn further increased the midpoint by approximately 7°C, indicative of specific binding to the hetero-tetrameric protein.
Equilibrium Analytical Ultracentrifugation (AUC)
AUC was employed to determine the oligomerization states of: the individual peptides, AHis and BThr; the 1:1 mixture of the peptides, AHis:BThr; and the fully formed bundle, (DPP)Zn:AHis:BThr (Figure 8). AUC for AHis suggests trimer formation: MWcalc [AHis]3, 11860.2; MWfound [AHis]3, 11731 ± 153. Similarly, AUC analysis for BThr is consistent with tetramer formation: MWcalc [BThr]4, 16110; MWfound [BThr]4, 16462 ± 188. For a 1:1 mixture of AHis:BIle (apo form), the computed molecular weight was slightly higher than that expected for the heterotetrameric bundle (MWcalc ([AHis]:[BThr])2 = 15960; MWfound ([AHis]:[BThr])2, 18797 ± 322), suggestive of a small amount of higher-order aggregation or lack of accuracy in the computed partial specific volumes used to convert the buoyant MW to the observed MW. In contrast, upon incorporation of the cofactor, a homogeneous tetra-α-helical holoprotein sample was formed, MWcalc (DPP)Zn:AHis:BThr, 17309.6; MWfound (DPP)Zn:AHis:BThr, 17265±200 Da. Size exclusion chromatography confirmed the association states of the peptides in the absence of the cofactor, but the chromatography matrix interacted with (DPP)Zn, leading to problems with reproducibility and difficulties in the interpretation of the results for the holo-protein.
Figure 8.
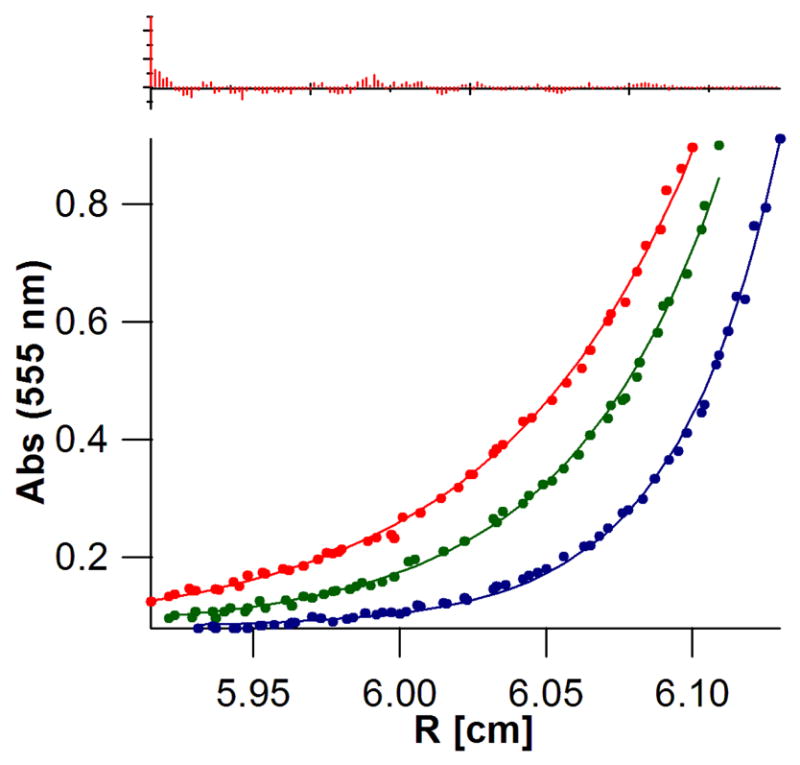
Sedimentation equilibrium of (DPP)Zn:AHis:BThr (30 μM, RT) at various angular velocities (35, red; 40, green; and 48 kRPM, blue) monitoring the absorbance at 555 nm (Q-band of (DPP)Zn) yields an appropriate MW for the bundle, 17265 ± 200. MWcalc = 17309.8, (χ2 = 3.06). The residuals are shown in the top panel; they were random within the experimental error of the measurement and showed no systematic errors.
Photophysical Characterization
Steady-state emission, as well as time-resolved transient absorption and fluorescence spectroscopic data, demonstrate classic excited-state singlet and triplet PZn photophysics for the AHis:BThr:(DPP)Zn protein. AHis:BThr:(DPP)Zn displays S1→S0 fluorescence emission (λem(max) = 641 nm) typical of its protein-bound chromophore, (Figure S2). Time resolved experiments (λex = 405 nm) yield a fluorescence decay time constant (kf = 4 × 108 s−1) similar to that exhibited by Zn cytochrome c (S1→S0, λem = 595, 650 nm; kf = 3 × 108 s−1), Figure 9.67 Pump-probe transient absorption spectroscopy (λex = 532 nm) indicates high quantum yield for relaxation of AHis:BThr:1(DPP)Zn* to its corresponding triplet state, AHis:BThr:3(DPP)Zn *, Figure 10. The measured triplet lifetime of the electronically excited holoprotein, τtriplet = 5 ms, is consistent with similar lifetime determinations of the photoexcited triplet states of PZn compounds in organic solvents (τtriplet = 1.2 ms),68 as well as with that evaluated for Zn cytochrome c (τtriplet = 14 ms).67 The triplet lifetime of PZn compounds fully exposed to water is τtriplet = 0.1–0.4 ms, suggesting that the protein sequesters the cofactor from solvent exposure.69
Figure 9.
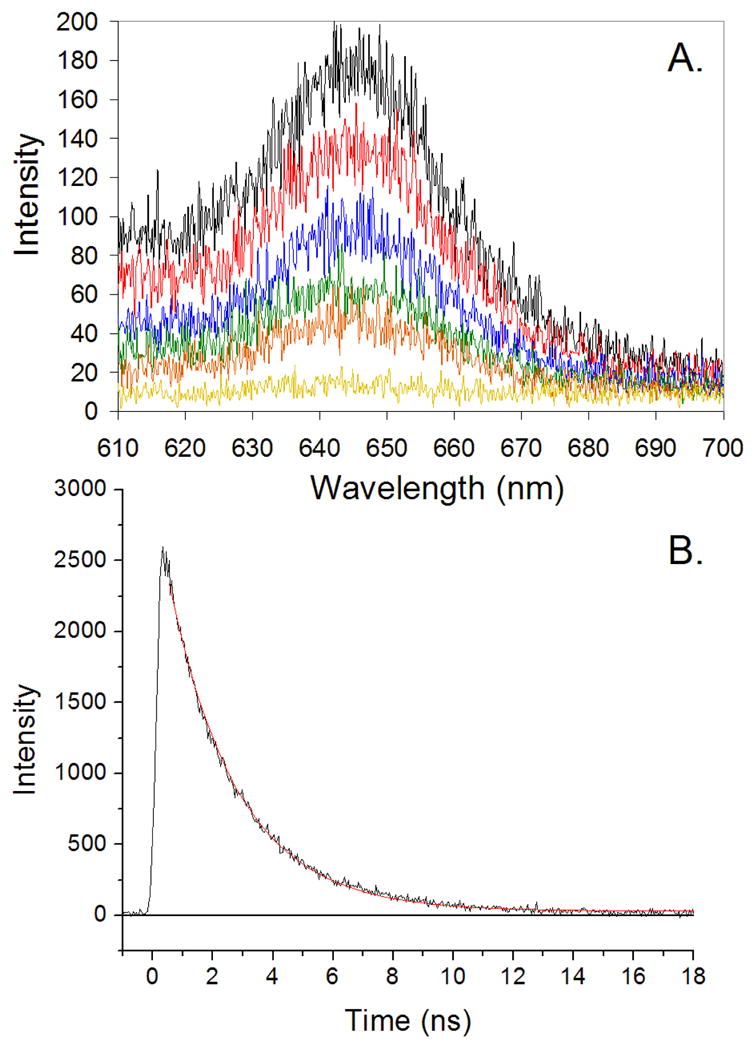
Time resolved fluorescence of AHis:BThr:(DPP)Zn in 50 mM Pi, 150 mM NaCl, pH 7.5 buffer, λex = 405 nm. (A) Fluorescence spectra obtained at various time delays (black, <1 ns; red, 1 ns; blue, 2 ns; green, 3ns; orange, 4 ns; yellow, 10 ns. (B) The kinetic profile obtained from the time-dependent integrated emission over the 600 – 700 nm spectral region; the red line is a first-order fit yielding a time constant of 4 × 108 s−1.
Figure 10.

(A) Transient absorption difference spectra of AHis:BThr:(DPP)Zn in 50 mM Pi, 150 mM NaCl, pH 7.5 buffer, λex = 532 nm. (B) The kinetic profile determined at 480 nm of the corresponding Figure 9A data (black dots); a fit to two exponential processes (red line) gives τ1 = 175 μs and τ2 = 4.5 ms.
Discussion
A tetrameric alpha-helical bundle capable of binding an abiological (porphinato)zinc cofactor has been successfully designed and characterized. This protein binds the cofactor via a combination of designed shape complementarity, electrostatic interactions, and a single histidine to coordinate the Zn. The computationally designed protein assembly forms the predicted bundle when all three components (AHis, BThr, and (DPP)Zn) are present, and the targeted peptide stoichiometry has the highest affinity for the cofactor. The protein selectively incorporates the targeted (DPP)Zn chromophore and binds neither (DPP)Fe nor (PPIX)Zn (Zn-hemin). In this work, computational protein design has been used to organize two synthetic emissive PZn chromophores, in a targeted spatial arrangement and molecular environment, that is selective for both metal ion and the nature of the peripheral porphyrin macrocycle substituents.
Five-Coordinate Geometry
(Porphinato)zinc compounds are known to bind a single axial ligand.70–75 The heterotetrameric design A2B2 was engineered such that each histidine would be associated with a single (DPP)Zn chromophore. The design departed from the previously reported PATet, which bound two equivalents of a (DPP)Fe cofactor using a bis-His coordination site.10 The calculations identified isoleucine as an appropriate amino acid to position opposite the coordinating histidine. Red shifts in the B- and Q-bands of the UV/visible spectrum provide evidence of (DPP)Zn binding to a histidine side chain, which is consistent with the 1:1:1 AHis:BThr:(DPP)Zn stoichiometry of association observed in the spectrophotometric titrations. Finally, the ratiometric titration suggests a 1:1 mixture is required to bind the (DPP)Zn cofactor. We are aware of only one other designed peptide capable of binding a (porphinato)zinc cofactor - a homo-dimeric tetra-α-helical bundle that is known to undergo a structural rearrangement when it binds a (porphinato)iron compound.33 The tetra-α-helical system reported herein does not only preferentially bind (DPP)Zn with respect to its corresponding (porphinato) iron derivative; the AHis:BThr apo-protein is designed in a manner that bars structural rearrangement that would produce a stable bis-His coordination environment for the equivalent Fe-containing cofactor analog, (DPP)Fe (vide infra).
Controlled Organization of Bundle
We employed the Statistical Computationally Assisted Design Strategy (SCADS) algorithm57,76 to identify an appropriate heterotetrameric (A2B2) bundle similar to the work with the homotetrameric (DPP)FeII/III-binding protein10 (Figure 1). The final sequences reflect ligand-directed elements of both positive and negative design and a mix of interactions that include hydrogen-bonds to second-shell ligands. One peptide chain, AHis contains the His ligand while chain BThr contains its complementary secondary ligand, Thr. An implicit element of negative design in this arrangement is that this interaction can form only in the heterotetramer; homomeric bundles thus present an unstable ligand-binding geometry. A second element of positive design is the enhanced interior packing associated with A16F mutation on BThr and F26L on AHis; these side chain replacements also serve as a negative design element in that the homotetramer formed from the AHis chains is underpacked in these regions, while the corresponding homogeneous BThr bundle shows repulsive steric interactions at Phe16. A third element of positive design is the identification of interior side chains that form a largely hydrophobic cavity tailored to the (DPP)Zn cofactor.
All three elements of positive and negative design were checked by a Job analysis with the (DPP)FeIII synthetic cofactor. The ferric center prefers a six coordinate geometry, therefore 4 AHis peptides should provide a bis-His ligation environment in a homotetrameric bundle. However, binding of (DPP)FeIII was not observed, presumably due to the lack of the hydrogen-bonding residue, Thr8. In addition, the lack of bundle formation when mixing 4 AHis + (DPP)FeIII can be directly related to the lack of interhelical and core Phe residues present in the previous homotetramer;10 the absence results in significant underpacking of the 4 AHis homotetramer. Furthermore, a 1:1 ratio of AHis:BThr provides an appropriate ligand environment for (DPP)Zn, but a single histidine for (DPP)FeIII renders the d5 transition metal coordinatively unsaturated. These design features present in a 1:1 AHis:BThr ratio preclude rearrangements to accommodate a ferric porphyrin cofactor.
Interestingly, bundle formation occurs in the absence of the cofactor. This is in contrast to the previous work with (DPP)FeIII, where the (porphinato)iron unit played a prominent role in the formation of the bundle as shown by enhanced helical formation in the presence of the cofactor.10,12 CD spectra of AHis, BThr, and AHis:BThr are consistent well folded α-helical bundles. In addition, size exclusion chromatography suggests partial formation of multimeric bundles of AHis, BThr, and tetrameric bundles of AHis:BThr. Where AUC of the peptides alone suggest bundle formations of a trimeric AHis, a tetrameric BThr, and a mixture of oligomeric states exists when both AHis and BThr peptides are present. These data are consistent with the fact that the trimer forming AHis peptide is underpacked due to the lack of interior hydrophobic groups. The BThr peptide has bulky Phe groups that complement the AHis peptide. In the presence of the (DPP)Zn chromophore, the preformed bundles/oligomeric states reorganize to form the targeted hetero-tetrameric bundle.
We have shown here through computational design and experimental characterization, a high level of control over the formation of complex metalloporphyrin-peptide bundles. The primary coordination sphere is illustrated in electronic absorption spectroscopy experiments which highlight a red-shift of the (DPP)Zn B- and Q-bands with binding of a nitrogenous base (histidine) to the cofactor’s axial position. Circular dichroism spectroscopic data highlight the secondary structure of the bundle; AHis, BThr, and AHis:BThr, as well as the complexed peptide AHis:BThr:(DPP)Zn form well-folded, stable α-helices. Tertiary structure and predicted bundle formation were confirmed by spectrophotometric titrations and analytical ultracentrifugation. The correct stoichiometry of 1:1:1 AHis:BThr:(DPP)Zn was clearly evinced in the titration data, and tetrameric bundle formation was supported by AUC experiments.
Controlled Organization of Cofactors
Directed-assembly of multiple chromophores in a single protein via computational design is necessary when protein function requires vectorially organized pigments. Such control is manifest in biological light-harvesting and energy-transducing proteins; a similar ability to structurally organize light-addressable cofactors is required in order to fabricate de novo proteins that possess opto-electronic functionality. The positive exciton coupling evident in CD spectra is consistent with the positioning of the chromophores in proximity to one another. This ability to organize (porphinato)zinc units and maintain important photophysical properties in the protein bundle such as a ns singlet excited-state lifetime and a fluorescence rate constant that resembles that determined in organic solvents (kF = 4 × 108 s−1), confirms de novo protein design as a useful approach to organize emissive moieties in a unidirectional manner (i.e., along the superhelical axis of the designed bundle), without alteration of chromophore properties.
This work offers an alternative and important complement to synthetic multiporphyrin arrays that utilize covalent and/or coordinative interactions between metalloporphyrin units to drive modest degrees of chromophore-chromophore electronic coupling.13,24–26 Protein design offers an approach to achieve specific spatial arrangements of cofactors in a completely water soluble system; furthermore, protein surface functionality can in principle be exploited to direct higher levels of organization of these electro-optically addressable components. As peptide design strategies become increasingly more sophisticated, covalent connections between cofactors that enable strong coupling will yield systems that offer entirely new electro-optic functionality in protein environments.18–23
Conclusion
In summary, a de novo light-emissive protein has been computationally designed and engineered. The protein organizes two synthetic PZn chromophores in a targeted spatial arrangement and molecular environment. The binding site of this protein is selective for both metal ion and the nature of the peripheral porphyrin macrocycle substituents. The protein achieves a sophisticated level of cofactor discrimination: the heterotetrameric, C2-symmetric bundle, AHis:BThr, uniquely binds the abiological (DPP)Zn chromophore with only single histidine coordination. The immeasurably low binding affinity of this protein for analogous (DPP)Fe cofactors or for zinc hemin derivatives [e.g., (PPIX)Zn], confirms the utility of probabilistic computational design77,78 in arriving at proteins complementary to a chosen cofactor, and lays the groundwork for the design of proteins that incorporate more complex light-addressable elements.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM-071628). The authors gratefully acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant DE-FG02-04ER46156 for supporting the protein design elements of this study. The authors thank the MRSEC (DMR-00-79909) and NSEC (DMR-0425780) programs of the National Science Foundation for infrastructural support. MJT is grateful to the Francqui Foundation (Belgium), and VLAC (Vlaams Academisch Centrum), Centre for Advanced Studies of the Royal Flemish Academy of Belgium for Science and the Arts, for research fellowships.
Footnotes
Supporting Information Available. Compiled benchmark (porphinato)zinc X-ray crystallographic data, detailed binding model, size exclusion chromatography, additional absorption and emission spectra.
References
- 1.Koder RL, Dutton PL. Dalton Trans. 2006. pp. 3045–3051. [DOI] [PubMed] [Google Scholar]
- 2.Case MA, McLendon GL. Accounts Chem Res. 2004;37:754–762. doi: 10.1021/ar960245+. [DOI] [PubMed] [Google Scholar]
- 3.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Sharp RE, Moser CC, Rabanal F, Dutton PL. P Nat Acad Sci. 1998;95:10465–10470. doi: 10.1073/pnas.95.18.10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei YN, Hecht MH. Prot Eng Des Sel. 2004;17:67–75. doi: 10.1093/protein/gzh007. [DOI] [PubMed] [Google Scholar]
- 6.Reedy CJ, Gibney BR. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 7.Zhuang JY, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Inorg Chem. 2004;43:8218–8220. doi: 10.1021/ic048502r. [DOI] [PubMed] [Google Scholar]
- 8.Rosenblatt MM, Wang JY, Suslick KS. P Nat Acad Sci. 2003;100:13140–13145. doi: 10.1073/pnas.2231273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghirlanda G, Osyczka A, Liu WX, Antolovich M, Smith KM, Dutton PL, Wand AJ, DeGrado WF. J Am Chem Soc. 2004;126:8141–8147. doi: 10.1021/ja039935g. [DOI] [PubMed] [Google Scholar]
- 10.Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. J Am Chem Soc. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 11.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. J Am Chem Soc. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAllister KA, Zou HL, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, Therien MJ, Blasie JK, DeGrado WF. J Am Chem Soc. 2008;130:11921–11927. doi: 10.1021/ja800697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wasielewski MR. Chem Rev. 1992;92:435–461. [Google Scholar]
- 14.Hoffman BM, Natan MJ, Nocek JM, Wallin SA. Struct Bond. 1991;75:85–108. [Google Scholar]
- 15.Hoffman BM, Celis LM, Cull DA, Patel AD, Seifert JL, Wheeler KE, Wang JY, Yao J, Kurnikov IV, Nocek JM. P Nat Acad Sci. 2005;102:3564–3569. doi: 10.1073/pnas.0408767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winkler JR, Gray HB. Chem Rev. 1992;92:369–379. [Google Scholar]
- 17.Redmore NP, Rubtsov IV, Therien MJ. J Am Chem Soc. 2003;125:8769–8778. doi: 10.1021/ja021278p. [DOI] [PubMed] [Google Scholar]
- 18.Uyeda HT, Zhao YX, Wostyn K, Asselberghs I, Clays K, Persoons A, Therien MJ. J Am Chem Soc. 2002;124:13806–13813. doi: 10.1021/ja020651q. [DOI] [PubMed] [Google Scholar]
- 19.Duncan TV, Rubtsov IV, Uyeda HT, Therien MJ. J Am Chem Soc. 2004;126:9474–9475. doi: 10.1021/ja0400638. [DOI] [PubMed] [Google Scholar]
- 20.Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ. P Nat Acad Sci. 2005;102:2922–2927. doi: 10.1073/pnas.0409394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Susumu K, Duncan TV, Therien MJ. J Am Chem Soc. 2005;127:5186–5195. doi: 10.1021/ja040243h. [DOI] [PubMed] [Google Scholar]
- 22.Duncan TV, Susumu K, Sinks LE, Therien MJ. J Am Chem Soc. 2006;128:9000–9001. doi: 10.1021/ja061897o. [DOI] [PubMed] [Google Scholar]
- 23.Frail PR, Susumu K, Huynh M, Fong J, Kikkawa JM, Therien MJ. Chem Mat. 2007;19:6062–6064. [Google Scholar]
- 24.Wagner RW, Johnson TE, Lindsey JS. J Am Chem Soc. 1996;118:11166–11180. [Google Scholar]
- 25.Burrell AK, Officer DL, Plieger PG, Reid DCW. Chem Rev. 2001;101:2751–2796. doi: 10.1021/cr0000426. [DOI] [PubMed] [Google Scholar]
- 26.Holten D, Bocian DF, Lindsey JS. Accounts Chem Res. 2002;35:57–69. doi: 10.1021/ar970264z. [DOI] [PubMed] [Google Scholar]
- 27.Fletcher JT, Therien MJ. Inorg Chem. 2002;41:331–341. doi: 10.1021/ic010871p. [DOI] [PubMed] [Google Scholar]
- 28.Cogdell RJ, Isaacs NW, Howard TD, McLuskey K, Fraser NJ, Prince SM. J Bacteriol. 1999;181:3869–3879. doi: 10.1128/jb.181.13.3869-3879.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasil’ev S, Orth P, Zouni A, Owens TG, Bruce D. P Nat Acad Sci. 2001;98:8602–8607. doi: 10.1073/pnas.141239598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guskov A, Kern J, Gabdulkhakov A, Brose M, Zouni A, Saenger W. Nat Struct Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto M, Ueno A, Mihara H. Chem-Eur J. 2001;7:2449–2458. doi: 10.1002/1521-3765(20010601)7:11<2449::aid-chem24490>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 32.Aubert N, Troiani V, Gross M, Solladie N. Tetrahedron Lett. 2002;43:8405–8408. [Google Scholar]
- 33.Sharp RE, Diers JR, Bocian DF, Dutton PL. J Am Chem Soc. 1998;120:7103–7104. [Google Scholar]
- 34.Xu T, Wu SP, Miloradovic I, Therien MJ, Blasie JK. Nano Lett. 2006;6:2387–2394. doi: 10.1021/nl062091p. [DOI] [PubMed] [Google Scholar]
- 35.Ye SX, Discher BM, Strzalka J, Xu T, Wu SP, Noy D, Kuzmenko I, Gog T, Therien MJ, Dutton PL, Blasie JK. Nano Lett. 2005;5:1658–1667. doi: 10.1021/nl050542k. [DOI] [PubMed] [Google Scholar]
- 36.Strzalka J, Xu T, Tronin A, Wu SP, Miloradovic I, Kuzmenko I, Gog T, Therien MJ, Blasie JK. Nano Lett. 2006;6:2395–2405. doi: 10.1021/nl062092h. [DOI] [PubMed] [Google Scholar]
- 37.Neya S, Suzuki M, Ode H, Hoshino T, Furutani Y, Kandori H, Hori H, Imai K, Komatsu T. Inorg Chem. 2008;47:10771–10778. doi: 10.1021/ic801406x. [DOI] [PubMed] [Google Scholar]
- 38.Neya S, Imai K, Hori H, Ishikawa H, Ishimori K, Okuno D, Nagatomo S, Hoshino T, Hata M, Funasaki N. Inorg Chem. 2003;42:1456–1461. doi: 10.1021/ic020504t. [DOI] [PubMed] [Google Scholar]
- 39.Neya S, Kaku T, Funasaki N, Shiro Y, Iizuka T, Imai K, Hori H. J Biol Chem. 1995;270:13118–13123. doi: 10.1074/jbc.270.22.13118. [DOI] [PubMed] [Google Scholar]
- 40.Sato T, Tanaka N, Neya S, Funasaki N, Iizuka T, Shiro Y. Biochim Biophys Acta. 1992;1121:1–7. doi: 10.1016/0167-4838(92)90329-c. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi T, Hitomi Y, Ogoshi H. J Am Chem Soc. 1998;120:4910–4915. [Google Scholar]
- 42.Monzani E, Alzuet G, Casella L, Redaelli C, Bassani C, Sanangelantoni AM, Gullotti M, De Gioia L, Santagostini L, Chillemi F. Biochemistry. 2000;39:9571–9582. doi: 10.1021/bi000784t. [DOI] [PubMed] [Google Scholar]
- 43.Nappa M, Valentine JS. J Am Chem Soc. 1978;100:5075–5080. [Google Scholar]
- 44.Feitelson J, Spiro TG. Inorg Chem. 1986;25:861–865. [Google Scholar]
- 45.Ye SY, Shen CY, Cotton TM, Kostic NM. J Inorg Biochem. 1997;65:219–226. doi: 10.1016/s0162-0134(97)00001-9. [DOI] [PubMed] [Google Scholar]
- 46.Woolfson DN. Fibrous Proteins: Coiled Coils, Collagen and Elastomers. 2005;70:79. [Google Scholar]
- 47.Root BC, Pellegrino LD, Crawford ED, Kokona B, Fairman R. Prot Sci. 2009;18:329–336. doi: 10.1002/pro.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsh ENG, DeGrado WF. P Nat Acad Sci. 2002;99:5150–5154. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan J, DeGrado WF. P Nat Acad Sci. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. J Mol Biol. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 51.Dahiyat BI, Mayo SL. Science. 1997;278:82–7. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 52.Ali MH, Taylor CM, Grigoryan G, Allen KN, Imperiali B, Keating AE. Structure. 2005;13:225–234. doi: 10.1016/j.str.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 54.Kortemme T, Joachimiak LA, Bullock AN, Schuler AD, Stoddard BL, Baker D. Nat Struct Mol Biol. 2004;11:371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 55.Butts CA, Swift J, Kang SG, Di Costanzo L, Christianson DW, Saven JG, Dmochowski IJ. Biochemistry. 2008;47:12729–12739. doi: 10.1021/bi8016735. [DOI] [PubMed] [Google Scholar]
- 56.Papp A, Vanderkooi JM, Owen CS, Holtom GR, Phillips CM. Biophys J. 1990;58:177–186. doi: 10.1016/S0006-3495(90)82363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kono H, Saven JG. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 58.Swift J, Wehbi WA, Kelly BD, Stowell XF, Saven JG, Dmochowski IJ. J Am Chem Soc. 2006;128:6611–6619. doi: 10.1021/ja057069x. [DOI] [PubMed] [Google Scholar]
- 59.North B, Cristian L, Stowell XF, Lear JD, Saven JG, DeGrado WF. J Mol Biol. 2006;359:930–939. doi: 10.1016/j.jmb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 60.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, DeGrado WF. J Am Chem Soc. 2005;127:5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 61.Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Inorg Chem. 2004;43:4793–4795. doi: 10.1021/ic0497679. [DOI] [PubMed] [Google Scholar]
- 62.Leonard JJ, Yonetani T, Callis JB. Biochemistry. 1974;13:1460–1464. doi: 10.1021/bi00704a022. [DOI] [PubMed] [Google Scholar]
- 63.Vanderkooi JM, Adar F, Erecinska M. Eur J Biochem. 1976;64:381–387. doi: 10.1111/j.1432-1033.1976.tb10312.x. [DOI] [PubMed] [Google Scholar]
- 64.This result is in contrast to the parent homotetramer which binds both Fe(DPP) and Zn(DPP) (results not shown).
- 65.Pescitelli G, Gabriel S, Wang YK, Fleischhauer J, Woody RW, Berova N. J Am Chem Soc. 2003;125:7613–7628. doi: 10.1021/ja030047v. [DOI] [PubMed] [Google Scholar]
- 66.Guryanov I, Moretto A, Campestrini S, Broxterman QB, Kaptein B, Peggion C, Formaggio F, Toniolo C. Biopolymers. 2006;82:482–490. doi: 10.1002/bip.20500. [DOI] [PubMed] [Google Scholar]
- 67.Sudha BP, Dixit N, Moy VT, Vanderkooi JM. Biochemistry. 1984;23:2103–2107. [Google Scholar]
- 68.Darwent JR, Douglas P, Harriman A, Porter G, Richoux MC. Coord Chem Rev. 1982;44:83–126. [Google Scholar]
- 69.Kim JE, Pribisko MA, Gray HB, Winkler JR. Inorg Chem. 2004;43:7953–7960. doi: 10.1021/ic048972l. [DOI] [PubMed] [Google Scholar]
- 70.Scheidt WR, Lee YJ. Struct Bonding. 64:1–70. [Google Scholar]
- 71.Collins DM, Hoard JL. J Am Chem Soc. 1970;92:3761–71. doi: 10.1021/ja00715a038. [DOI] [PubMed] [Google Scholar]
- 72.Barkigia KM, Berber MD, Fajer J, Medforth CJ, Renner MW, Smith KM. J Am Chem Soc. 1990;112:8851–8857. [Google Scholar]
- 73.DiMagno SG, Lin VSY, Therien MJ. J Org Chem. 1993;58:5983–5993. [Google Scholar]
- 74.Goll JG, Moore KT, Ghosh A, Therien MJ. J Am Chem Soc. 1996;118:8344–8354. [Google Scholar]
- 75.LeCours SM, DiMagno SG, Therien MJ. J Am Chem Soc. 1996;118:11854–11864. [Google Scholar]
- 76.Park S, Xi Y, Saven JG. Curr Op Struct Biol. 2004;14:487–494. doi: 10.1016/j.sbi.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 77.Kang SG, Saven JG. Curr Op Chem Biol. 2007;11:329–334. doi: 10.1016/j.cbpa.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 78.Saven JG. Curr Op Colloid & Interface Science. 2009. in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.