

Abstract
Monocular deprivation normally alters ocular dominance in the visual cortex only during a postnatal critical period (20 to 32 days postnatal in mice). We find that mutations in the Nogo-66 receptor (NgR) affect cessation of ocular dominance plasticity. In NgR−/− mice, plasticity during the critical period is normal, but it continues abnormally such that ocular dominance at 45 or 120 days postnatal is subject to the same plasticity as at juvenile ages. Thus, physiological NgR signaling from myelin-derived Nogo, MAG, and OMgp consolidates the neural circuitry established during experience-dependent plasticity. After pathological trauma, similar NgR signaling limits functional recovery and axonal regeneration.
Central nervous system myelin proteins limit axonal growth and regeneration after traumatic and ischemic injury in adult mammals (1-14), but a physiological role for the myelin inhibitor pathway has not been defined. Ocular dominance (OD) within visual cortex provides a paradigm to study experience-dependent plasticity. Monocular deprivation of the contralateral eye induces a relative shift in ocular dominance of cortical responses toward the nondeprived ipsilateral eye (15). Both anatomical and electrophysiological studies in cats have defined a critical period during which the cerebral cortex is sensitive to experience-dependent plasticity, but after which altered visual experience does not change visual cortex responsiveness (15-17). In mice, single-unit recordings under barbiturate anesthesia have revealed a similar critical period for OD between 19 and 32 days postnatal (P19 to P32) (18-20). Although mouse OD plasticity measured with this method ceases after P32, a level of adult OD plasticity can be detected by other methods, such as immediate early gene expression and visually evoked potential field recordings (19, 21-23). Adult plasticity is distinct from adolescent critical period plasticity. Adult plasticity relies on the slow onset of strengthened inputs from the nondeprived eye rather than a suppression of responses from the contralateral eye (23). Barbiturate anesthesia masks OD plasticity in adult but not juvenile mice (22). Plasticity achieved during the critical period is more persistent than that obtained in the adult (22). Here, we focus on the abrupt loss of OD plasticity at the end of the critical period in single-unit cortical recordings from anesthetized mice.
Previous investigations have revealed a critical role for parvalbumin-positive γ-aminobutyric acid (GABA)–ergic neurons in timing the critical period. Dark rearing impairs inhibitory circuit maturation (24) and delays the closure of the critical period (25). Genetic disruption of a GABA synthetic enzyme, glutamic acid decarboxylase 65 (GAD65), precludes OD plasticity (26). Brain-derived neurotrophic factor is thought to expedite critical period closure by maturing GABAergic neurons (27). Loss of dendritic spines correlates with OD plasticity and requires both GAD65 function and tissue plasminogen activator (tPA) (28).
Chondroitin sulfate proteoglycans (CSPGs) are astrocyte-and neuron-derived axon-outgrowth inhibitors that have also been implicated in OD plasticity. Infusion of chondroitinase ABC into spinal cord–injured animals cleaves glycosaminoglycan chains and promotes a degree of regeneration and functional recovery (29) comparable to that of Nogo/NgR antagonism (8, 11). Injecting chondroitinase into adult rat visual cortex partially reactivates OD plasticity in response to monocular deprivation (30). To consider the cellular site of CSPG action, we examined wisteria floribunda agglutinin–stained sections of visual cortex. It is remarkable that CSPG-positive perineuronal nets predominantly (>85%) surround parvalbumin-positive inhibitory neurons, leaving nearly all other neurons unencumbered (Fig. 1A). Although genetic and pharmacological manipulation of cortical inhibition supports a model in which parvalbumin-positive inhibitory neurons initiate the critical period for OD plasticity (31, 32), glutamatergic synapses also contribute substantially to OD plasticity (33). Both the incomplete extent of OD plasticity restoration by chondroitinase treatment and the GABA-restricted CSPG distribution led us to consider whether more widely distributed neurite-inhibiting mechanisms might participate in OD plasticity. As the vast majority of cortical neurons express NgR (Fig. 1B), we considered whether NgR-mediated myelin inhibition of neurite outgrowth contributes to closing the critical period.
Fig. 1.
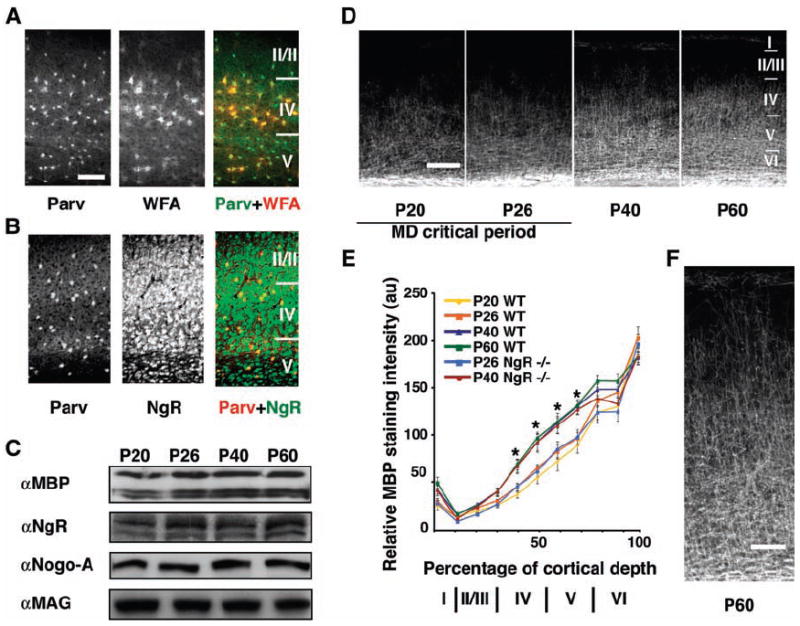
Expression of myelin, NgR, and CSPG in mouse visual cortex during the critical period for OD plasticity. (A) P40 visual cortex labeled for parvalbumin (green in merge) and wisteria floribunda agglutinin (red in merge). (B) Sections as in (A), labeled for parvalbumin (red in merge) and NgR (green in merge). (C) Homogenates of visual cortex were immunoblotted with the indicated antibodies. Microdensitometry revealed that the concentration of any one protein varied by <20% across these ages, and there were no significant changes in protein levels with age. (D) P20 to P60 visual cortex labeled with antibodies to MBP. Layers I to VI are indicated (right). (E) Distribution of relative MBP intensity within visual cortex. (F) A higher magnification image of MBP distribution at P60. Error bars reflect SEM; n = 3 mice. Asterisks denote significant differences (P < 0.05) between P20 and P26 versus P40 and P60 for both genotypes. Scale bars in [(A) and (B)] = 100 μm, in (D) = 200 μm, and in (F) = 100 μm.
Myelin-associated proteins, including ligands for NgR, are easily detected in postnatal visual cortex (Fig. 1C). The absolute abundance of the NgR ligands, Nogo-A and MAG, is essentially constant in homogenates of visual cortex over the time course of the critical period, whereas NgR tends to increase slightly (Fig. 1C). Similarly, levels of the compact myelin marker, myelin basic protein (MBP), increase only minimally during this period. A limitation of the immunoblot analysis is a mixing of all cortical layers such that selective expression changes in certain cortical layers most critical for plasticity may be obscured. Therefore, we used an immunohistochemical analysis of MBP between P26 and P60 to allow for a layer-specific assessment of expression. Although the total concentration of MBP remains nearly constant, layer-specific levels of intracortical myelin mature considerably as the critical period ends (Fig. 1D). At P20 and P26, the beginning and height of the critical period, respectively, the half-maximal staining intensity is obtained at 30% of the distance from the subcortical white matter to the pial surface, whereas after the end of the critical period, at P40 and P60, this measure of myelination extends significantly further, into 50% of the cortex (P < 0.001) (Fig. 1E). Within layers IV and V, the relative intensity of MBP staining increases by nearly 60% and 40%, respectively. The onset and distribution of cortical myelination in mice lacking NgR is indistinguishable from that of wild-type mice (Fig. 1E). Overall, the maturation of intracortical myelination correlates with the end of the critical period.
To assess OD plasticity, we first characterized the electrophysiological responsiveness of the binocular visual cortex in NgR mutant mice exposed to unmodified visual stimuli. NgR null mice have normal vision as assayed by receptive field azimuth distribution, receptive field size, response properties, and spontaneous activity (fig. S1, A to C). The weighted ocular dominance (WOD) scores for NgR mutants and wild-type mice are comparable (Fig. 2D).
Fig. 2.
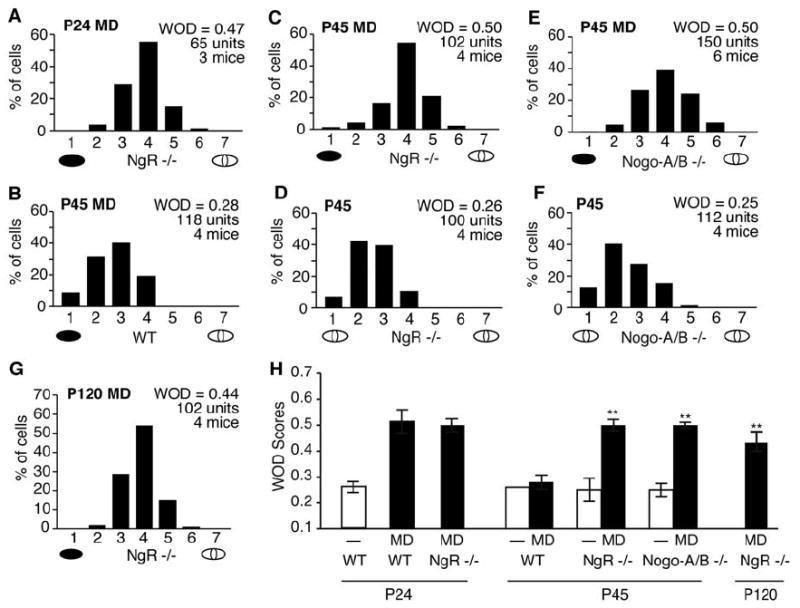
NgR and Nogo-A/B mutant mice are sensitive to monocular deprivation after the critical period. (A) OD histogram and average WOD score for NgR mutant mice receiving monocular deprivation (black ellipse beneath the abscissa) at P24. (B to G) OD histograms and WOD scores as in (A), for wild-type (WT), NgR, and Nogo-A/B mutant mice with and without monocular deprivation. (H) Mean WOD scores for monocularly deprived and nondeprived animals across genotype and age. Bars represent the WOD values in [(A) to (G)]; error bars are SEM. Double asterisks denote significant differences (P < 0.001) of P45 and P120 WOD scores from P45 WT nondeprived and monocularly deprived scores.
To explore whether NgR regulates the magnitude of OD plasticity, we tested NgR mutant mice during the critical period. OD shifts in NgR mutant mice were induced by 4 days of monocular deprivation beginning at P24. The OD histograms and calculated WOD scores (Fig. 2A) are indistinguishable from the values for wild-type mice receiving monocular deprivation at the same age. Thus, visual system development and immature cortical plasticity are normal in the absence of NgR.
To investigate whether NgR participates in restricting OD plasticity in older animals, we deprived NgR mutants and wild-type littermates at P45, after the end of the critical period. Consistent with reported findings, 4 days of monocular deprivation do not induce an OD shift in wild-type mice (Fig. 2B). However, monocular deprivation of NgR mutant mice generates an OD shift (Fig. 2C) that differs significantly from the OD of either P45 nondeprived NgR mutant mice (Fig. 2D) or wild-type controls. Longer periods of monocular deprivation (8 days in one mouse) in NgR mutant mice beginning at P45 did not increase the magnitude of the OD shift. NgR is known to mediate myelin inhibition of neurite outgrowth; a simple model for enhanced P45 plasticity is a reduction in myelin inhibition of process growth. As the NgR ligand, Nogo-A, accounts for a considerable fraction of myelin inhibition of neurite outgrowth (9, 34, 35), a similar OD phenotype is predicted for mice lacking Nogo-A. Indeed, mice homozygous for a “gene-trap” insertion that eliminates expression of Nogo-A (and the related isoform Nogo-B) (9) display OD shifts following 4 days of monocular deprivation beginning at P45 (Fig. 2, E and F). The magnitudes of these shifts are identical to those observed with NgR mutants. Although myelinated fibers are more abundant in layers IV to VI, the OD scores from more superficial and deeper recording depths for a given penetration were indistinguishable in monocularly deprived NgR mutant mice at all ages tested.
OD plasticity at P45 in NgR and Nogo-A/B mutant mice, but not in wild-type mice, might be explained either by an absence of plasticity-limiting mechanisms or by a 2-week delay in developmental maturation of the visual cortex. Nissl staining and immunohistochemistry did not reveal any general deviation in the pace of brain development for these two strains from wild-type mice (Fig. 1). To determine whether OD plasticity persists into adulthood in NgR mutant mice, we examined mice at 4 months of age, roughly three times as old as mice at the end of the critical period. Four days of monocular deprivation is sufficient to induce OD shifts comparable to those observed in mice tested during the critical period (Fig. 2G). Therefore, a slight developmental delay in OD plasticity restriction cannot explain these findings, which indicates that NgR-dependent mechanisms participate directly in restricting visual cortex experience-dependent plasticity.
NgR signaling might function upstream in a plasticity cascade to regulate GABAergic maturation, neurotrophin levels, and/or tPA signaling. Alternatively, NgR may function downstream or independently of these mediators and limit anatomical rearrangements directly. To examine whether NgR regulates OD plasticity by modifying the GABAergic system or tPA-plasmin activity, we compared the expression of GAD65 and tPA protein in visual cortex from wild-type and NgR mutant mice (Fig. 3). Absence of NgR protein does not alter GAD65, parvalbumin, or tPA immunoreactivity in the visual cortex of P60 mice (Fig. 3, A to E), which suggests that NgR functions independently or downstream of these proteins to regulate plasticity. Dark rearing delays closure of the critical period by altering GABAergic neurotransmission (24) but does not alter the maturation of intracortical myelination, as indicated by MBP staining of sections of visual cortex in P40 mice or NgR levels (fig. S2, A and B). Thus, NgR signaling serves as a necessary gate on visual cortex plasticity. Although we cannot exclude the possibility that NgR signaling is modulated by GABA neurotransmission or tPA activity, our results support a model in which myelin and NgR function independently. These pathways presumably converge to regulate anatomical rearrangements in visual cortex.
Fig. 3.
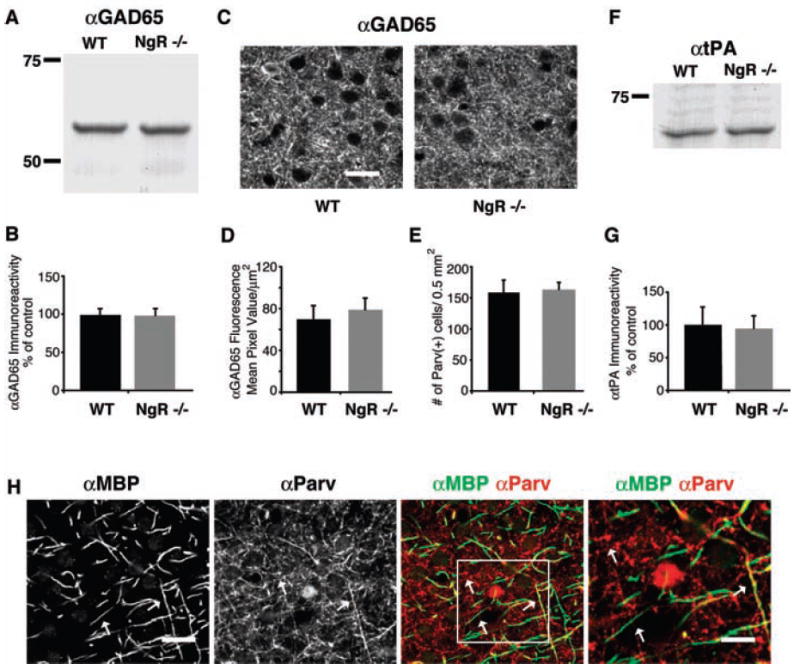
GAD65 and tPA are normal in NgR mutant mice. (A) Homogenates of visual cortex from wild-type (WT) and NgR mice were immunoblotted with antibodies to GAD65. (B) Densitometry of GAD65 immunoreactivity from blots of P60 visual cortex from WT and NgR mutants (n = 4 mice). (C) P60 visual cortex of WT and NgR−/− mice labeled with antibodies to GAD65. (D) Microdensitometric level of GAD65 immunoreactivity in the neuropil of visual cortex from WT and NgR mutants. (E) Density of parvalbumin-immunopositive interneurons in visual cortex of WT and NgR mice, per 0.5 mm2. (F) Immunoblot as in (A), with an antibody to tPA. (G) Quantification of tPA expression as in (B). (H) Confocal images of WT P60 visual cortex double-labeled with antibodies to MBP and to parvalbumin. White arrows point to immunopositive fibers in the αMBP and αParv panels, one that colocalizes in the merged image (right arrow) and others that do not (left and upper arrows). A higher magnification image, outlined by the white rectangle, is shown on the far right. Scale bar for [(C) and (H)] = 20 μm; for the higher magnification image, scale bar = 10 μm.
The distribution of intracortical myelin and NgR is widespread (Fig. 1), so multiple neuronal subtypes have the potential to be regulated by this system. Given the prominence of GABAergic systems in ocular dominance plasticity, we examined the mouse visual cortex for myelinated GABAergic processes. Consistent with electron micrographs (36, 37), roughly one-third of MBP-stained visual cortex fibers are parvalbumin-positive (Fig. 3H). Because the electrophysiological recordings presented here implicate myelin-associated inhibitors as regulators of OD plasticity, it will be interesting to examine whether the NgR pathway modulates primarily GABAergic connections, dendritic spine rearrangements, or other anatomical connections.
The current study provides genetic evidence for the hypothesis that myelination consolidates neural circuitry by suppressing plasticity in the mature brain. Specifically, NgR and Nogo-A/B are required for maturation-dependent restrictions on OD plasticity to monocular deprivation in the visual cortex. However, myelin is not the only limit on cortical plasticity, because CSPGs are also known to have a role in the visual cortex (30), and certain measures of OD persist in the adult mouse (19, 21-23). Dark rearing delays the maturation of GABAergic neurons and the deposition of CSPGs into perineuronal nets (30) but does alter the maturation of intracortical myelination, which is controlled by developmental determinants not dependent on visual experience. Thus, at least two distinct inhibitors limit OD plasticity to the critical period, and eliminating either one is sufficient to facilitate plasticity.
The Nogo/NgR pathway is not required for closure of critical period plasticity throughout the cerebral cortex. Somatosensory barrel-field anatomical plasticity to whisker ablation is not significantly altered in Nogo-A/B mutant mice (fig. S2). Nonmyelin mechanisms may be relatively important in limiting barrel-field plasticity because the relevant critical period ends earlier in development (P1 to P4), before cortical myelination matures, or because barrel-field plasticity can be mediated subcortically (38).
Recovery of motor function after pathological damage to the mature brain is facilitated by structural and synaptic plasticity. The failure of surviving neurons to reestablish functional connectivity is most obvious after spinal cord injury, but limited axon regeneration and plasticity is central to the pathophysiology of a range of neurological disorders, including stroke, head trauma, multiple sclerosis, and neurodegenerative disease. The NgR-mediated response to myelin inhibitors is known to participate in limiting recovery from spinal cord injury and stroke (8, 10-12). Thus, brain myelin proteins impede both physiological plasticity and the repair of pathologic injury by a shared NgR mechanism.
Supplementary Material
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/309/5744/2222/
DC1
Materials and Methods
Figs. S1 to S3
References
References and notes
- 1.McKerracher L, et al. Neuron. 1994;13:805. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. Neuron. 1994;13:757. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 3.Bregman BS, et al. Nature. 1995;378:498. doi: 10.1038/378498a0. [DOI] [PubMed] [Google Scholar]
- 4.Chen MS, et al. Nature. 2000;403:434. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- 5.Prinjha R, et al. Nature. 2000;403:383. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- 6.GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Nature. 2000;403:439. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- 7.Fournier AE, GrandPre T, Strittmatter SM. Nature. 2001;409:341. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- 8.GrandPre T, Li S, Strittmatter SM. Nature. 2002;417:547. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- 9.Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM. Neuron. 2003;38:187. doi: 10.1016/s0896-6273(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 10.Kim JE, Liu BP, Park JH, Strittmatter SM. Neuron. 2004;44:439. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Li S, et al. J Neurosci. 2004;24:10511. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JK, Kim JE, Sivula M, Strittmatter SM. J Neurosci. 2004;24:6209. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang KC, et al. Nature. 2002;417:941. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- 14.Schnell L, Schwab ME. Nature. 1990;343:269. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 15.Wiesel TN, Hubel DH. J Neurophysiol. 1963;26:1003. doi: 10.1152/jn.1963.26.6.1003. [DOI] [PubMed] [Google Scholar]
- 16.Hubel DH, Wiesel TN. J Physiol. 1962;160:106. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daw NW. Visual Development. Plenum; New York: 1995. [Google Scholar]
- 18.Gordon JA, Stryker MP. J Neurosci. 1996;16:3274. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawtell NB, et al. Neuron. 2003;38:977. doi: 10.1016/s0896-6273(03)00323-4. [DOI] [PubMed] [Google Scholar]
- 20.Fischer QS, et al. J Neurosci. 2004;24:9049. doi: 10.1523/JNEUROSCI.2409-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tagawa Y, Kanold PO, Majdan M, Shatz CJ. Nat Neurosci. 2005;8:380. doi: 10.1038/nn1410. [DOI] [PubMed] [Google Scholar]
- 22.Pham TA, et al. Learn Mem. 2004;11:738. doi: 10.1101/lm.75304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frenkel MY, Bear MF. Neuron. 2004;44:917. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Morales B, Choi SY, Kirkwood A. J Neurosci. 2002;22:8084. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mower GD. Brain Res Dev Brain Res. 1991;58:151. doi: 10.1016/0165-3806(91)90001-y. [DOI] [PubMed] [Google Scholar]
- 26.Hensch TK, et al. Science. 1998;282:1504. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang ZJ, et al. Cell. 1999;98:739. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 28.Mataga N, Mizuguchi Y, Hensch TK. Neuron. 2004;44:1031. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Bradbury EJ, et al. Nature. 2002;416:636. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 30.Pizzorusso T, et al. Science. 2002;298:1248. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- 31.Fagiolini M, Hensch TK. Nature. 2000;404:183. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- 32.Fagiolini M, et al. Science. 2004;303:1681. doi: 10.1126/science.1091032. [DOI] [PubMed] [Google Scholar]
- 33.Taha S, Hanover JL, Silva AJ, Stryker MP. Neuron. 2002;36:483. doi: 10.1016/s0896-6273(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 34.Simonen M, et al. Neuron. 2003;38:201. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 35.Zheng B, et al. Neuron. 2003;38:213. doi: 10.1016/s0896-6273(03)00225-3. [DOI] [PubMed] [Google Scholar]
- 36.DeFelipe J, Jones EG. J Neurosci. 1985;5:3246. doi: 10.1523/JNEUROSCI.05-12-03246.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somogyi P, Soltesz I. Neuroscience. 1986;19:1051. doi: 10.1016/0306-4522(86)90122-3. [DOI] [PubMed] [Google Scholar]
- 38.Rebsam A, Seif I, Gaspar P. J Neurosci. 2005;25:706. doi: 10.1523/JNEUROSCI.4191-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.We thank N. Tian for providing dark-reared mice. This work was supported by grants from NIH to S.M.S A.W.M. and from the McKnight Foundation to S.M.S. S.M.S. is a member of the Kavli Institute of Neuroscience at Yale.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.