

Abstract
Toll-like receptors (TLRs) are critical sensors for microbial products and are important in initiating both innate and adaptive immune defense against pathogens. Emerging evidence suggests that TLRs are also expressed by regulatory T cells (Treg) that constitute an important immune suppressive cellular mechanism to curtail TLR hyperactivity to avoid sepsis and autoimmune diseases. This review brings up to date on the expression of functional TLRs on Treg and the functional impact of TLR activation on Treg biology. We argue that the suppressive function of Treg can be augmented or attenuated depending on the nature of TLR stimulation.
Keywords: heat-shock protein, endoplasmic reticulum, gp96, grp94, HSP90b1, Toll-like receptor, TLR4, unfolded protein response, Regulatory T cells, FoxP3
Introduction
Immune system evolves primarily on the needs of combating pathogens often at the risk of inducing autoimmunity and harmful inflammation. The protective roles and the detrimental potential of the immune response to the host thus have to be delicately balanced. At the center of this balance lies the germline-coded pattern recognition receptors (PRRs) that were proposed by Charles Janeway Jr two decades ago (1) and have since been established experimentally (2). PRRs are receptors that recognize unique molecular structures of pathogens and initiate innate and immediate immune response, which also participate in optimization of the subsequent adaptive immunity. The number of PRRs is expanding which includes Toll-like receptors (TLRs), Nod and Nod-like receptors (nucleotide-binding domain and leucine-rich repeat containing molecules [NLRs]), RIG-I-like receptors (RLRs), and C-type lectin receptors (CLRs) (3, 4). TLRs, for example, are critical sensors for microbial products from bacterial, fungal, viral and protozoan origins and are important in initiating both innate and adaptive immune defenses against pathogens.
Numerous mechanisms are in place to avoid autoimmunity including deletion of self-reactive T and B cells during their ontogeny (central tolerance) and deletion/suppression/regulation of autoreactive cells in the peripheral tissues (peripheral tolerance). A key player in maintaining peripheral T cell tolerance is CD4+CD25+Foxp3+ regulator T cells (Tregs), which patrol the host organs against activation of self-reactive cells during steady state as well as in the settings of infections (5). Emerging evidence suggests that TLRs are also expressed by Tregs that constitute an important immune suppressive cellular mechanism to curtail TLR hyperactivity to avoid sepsis and autoimmune diseases. However, direct TLR activations on Treg have also been shown to block Treg function, leading to amplified immune response. In this article, we will critically examine the issues of expression and function of TLRs on Treg. We argue that the suppressive function of Treg can be augmented or attenuated depending on the nature of infection (hence, TLR stimulation) to maximize the fighting chance against pathogens and to minimize potential damage to the host.
Expression of TLRs by regulatory T cells
Up to date, there are 13 members of TLRs in mammals collectively (6). According to their subcellular localization, TLRs can be divided into intracellular and cell surface TLRs. The intracellular TLRs recognize nucleic acids in the acidic compartment, including TLR3 for dsRNA, TLR7 and TLR8 for ssRNA and TLR9 for unmethylated CpG DNA. The cell surface TLRs encompass TLR2/TLR1 for triacylated/diacylated bacterial lipopeptides, TLR2/TLR6 for lipoproteins, TLR4 for lipopolysaccharide (LPS), TLR5 for flagellin (6, 7), and TLR 11 for profilin-like protein from apicomplexan protozoa and unknown cellular component(s) from uropathogenic E. coli (8, 9). TLR10, TLR12 and TLR13 remain to be orphan receptors.
With exception of unconfirmed expression pattern of TLR12 and TLR13, most TLRs are abundantly expressed in dendritic cells (DCs), B cells and Mϕ/monocytes (6). A subset of TLRs were also detected on other immune cells, such as γδ T cells (10, 11) and conventional αβ T cells (12-19). Determined by RT-PCR, mRNA of TLR1-10 was found in freshly isolated T cells from human peripheral blood or tonsils (>95% pure) at variable levels (12, 13, 18). Further analysis revealed that both CD4+ T cells and CD8+ T cells expressed appreciable levels of mRNA for various TLRs (13, 18). Although it is difficult to directly compare TLR expression levels among different studies due to the utilizations of different internal controls (12, 13, 18), it is interesting to notice that CD4+ T cells expressed TLRs at a higher level than CD8+ T cells do (13, 18). Similarly, it has been shown that mouse CD4+ T cells expressed TLR1-9 (15, 16, 20) and rat CD4+ T cells predominantly expressed TLR3, 5, 6 and 9 (21). A concern that contaminating non-T cells could be responsible for observed TLR mRNA in these experiments could not be substantiated. Highly purified (>99.9% pure) CD4+ T cells from human PBMC and a CD4+ T cell line Jurkat were used to demonstrate the expression of TLR1, 2, 3, 4, 5, 7 and 9 (22). More convincingly, a subset of TLRs was visualized by flow cytometry at a protein level in human and murine CD4+ T cells by intracellular staining of TLR2, 3, 4, 5 and 9 (14, 23) and by surface staining of TLR2 (24, 25), 4 (26) and 5 (23).
Great attentions have been drawn to the TLR expression on CD4+CD25− effector T cells (Teff) and CD4+CD25+Foxp3+ Treg (Table I). mRNA for TLR1, 2 and 6 was ubiquitously present in CD45RBhi naïve CD4+ T cells, activated/memory CD45RBlowCD4+CD25− Teff as well as CD45RBlowCD4+CD25+ Treg (20), which is consistent with the functional heterodimeric partnership of TLR2/1 and TLR2/6. Human cord blood-derived naïve CD4+ T cells expressed significant amounts of mRNA for and intracellular proteins of TLR2 and TLR4, which were not altered by a combined anti-CD3 and IFN-α treatment. However, such a treatment induced surface expression of both TLR2 and TLR4 (25). In addition, CD45RA+ naïve CD4+ T cells from adult peripheral blood show no appreciable amount of surface TLR2, but CD45RO+ memory CD4+ T cells do. After activation with anti-CD3, CD45RA+ cells expressed a significant level of surface TLR2, which could be further enhanced by co-treatment with Pam3CSK4, a synthetic TLR2 ligand. Yet, CD45RO+ memory CD4+ T cells showed no further increase (25). Consistent with findings in human, surface expression of TLR2 was only depicted on anti-CD3 activated mouse CD4+CD25− Teff and CD4+CD25+ Treg, but not on resting T cells, though the latter expressed TLR2 intracellularly (24).
Table I.
TLR expression and direct function on Tregs (modified from (70))
| TLR | Expression | Effect on Trega | References | ||
|---|---|---|---|---|---|
| CD4+CD25− Teff |
CD4+CD25+ Treg |
Proliferation | Suppression | ||
| TLR1 | + | + | Yesb |
|
(20) |
| TLR2 | + | + | Yes |
|
(20, 24, 25, 34, 36) |
| TLR3 | + | − | (16, 21) | ||
| TLR4 | + | ++ | Yes |
|
(20, 25, 26, 49, 52) |
| TLR5 | + | ++ | no |
|
(16, 20-23, 29) |
| TLR6 | + | + | (20) | ||
| TLR7 | + | ++ | (15, 20, 22) | ||
| TLR8 | + | ++ |
|
(19, 20, 22, 29) | |
| TLR9 | + | + | Yes |
|
(14, 16, 17, 21, 30) |
| TLR10 | − | + | (29) | ||
The indirect effect of TLR via APC on Treg function is not listed here.
Based on the property of Pam3CSK4 recognized by TLR2/TLR1 heterodimeric receptor
In contrast to TLR2, TLR4 mRNA was evident in freshly isolated CD45RBlowCD4+CD25− Teff and CD4+CD25+ Treg, whereas CD45RBhi CD4+ T cells were negative for TLR4 even though they acquired CD45RBlow phenotype after in vitro activation by plate-bound anti-CD3 and anti-CD28 for 3 days (20). The mRNA signal was 3-4 fold higher in Treg compared with Teff (20). Surprisingly, surface TLR4 was readily detectable when freshly isolated human Teff and Treg were stimulated with LPS (26), which is intriguing since it is well known that TLR4 in macrophages is quickly down-regulated after engagement with LPS at mRNA and protein levels, a possible explanation for LPS tolerance (27, 28). This observation indicated that TLR4 could be differentially regulated and has unique functions, depending on its cellular expression.
Expression of TLR5 mRNA or protein has been illustrated on human peripheral blood CD4+ T cells, including CD25− Teff and CD25+ Treg (22, 23, 29), and mouse CD44low CD4+ T cells and CD45RBlow Teff and Treg (16, 20). More so, expression of TLR5 was confirmed in in vitro generated Th cell clones and Treg clones (23). It was also demonstrated that TLR5 was expressed by rat CD4+CD45RChi naïve and CD4+CD45RClow memory T cells as well as two Ag-specific CD4+ T cell clones (21). Similar to the TLR4 expression pattern, TLR5 was increased up to 4-fold higher in murine Treg when compared to Teff (20). In contrast, it was also reported that TLR5 expression was significantly down-regulated upon stimulation with anti-CD3/anti-CD8 (23).
Some reports indicated that TLR3 was not expressed by naïve or memory CD4+ T cells (17, 20). Other groups, however, found that TLR3 was detectable in murine CD4+CD25− T cells, regardless of anti-CD3 activation. Similarly, rat CD4+CD45RChi naïve and CD4+CD45RClow memory T cells as well as two Ag-specific CD4+ T cell clones expressed TLR3 (21). Furthermore, murine CD4+CD45RBhi T cells expressed TLR3 protein intracellularly (14).
In mice, TLR7 and TLR8 were detectable in CD45RBlow Teff and Treg populations, with a low-level TLR7 in CD45RBhi naïve CD4+ T cells (20). Additionally, it was also documented that mouse CD4+CD62Lhi T cells, CD4+CD45RBhi T cells or colitic CD4+ T cells from lamina propria expressed an appreciable level of TLR7 (15, 17). In human, peripheral blood CD45RA+ naïve CD4+ T cells and CD45RO+ memory CD4+ T cells as well as CD4+CD25+ Treg expressed TLR7/8 (19, 22, 29).
Similar to TLR3, TLR9 transcripts were initially reported to be absent in murine CD4+ T cells (15, 20). Accumulating evidence suggested that TLR9 is present in a subset of CD4+ T cells (14, 16, 17, 21, 30). TLR9 was expressed in CD4+CD45RBhi or CD4+CD62Lhi naïve cells and CD44low CD4+CD25− T cells (14, 16, 17). Its expression at protein level was further confirmed by intracellular staining of TLR9 (14, 17). In addition, TLR9 was found to be present in rat CD4+CD45RChi naïve T cells, CD4+CD45RClow memory T cells and CD4+CD25+ Treg (21).
Human TLR10 is an orphan receptor with unknown ligand specificity and there is yet no report of a murine homolog, although a partial, non-functional genomic sequence of the TLR10 gene is present. Genomic studies indicated that TLR10 is in a close vicinity to the locus that also contains TLR1 and TLR6, two receptors known to heterodimerize with TLR2. In addition to its homodimeric form, TLR10 can also heterodimerize with TLRs 1 and 2. Its expression has been reported on B cells and plasmacytoid DCs and associated with MyD88 (31). Most recent data elucidated that TLR10 is constitutively expressed at both mRNA and protein levels in human CD4+CD25+ Treg, but not in CD4+CD25− Teff (29).
Collectively, with the exception of missing TLR3 from Treg and lack of TLR10 expression by Teff, current data demonstrated that CD4+CD25+ Treg and CD4+CD25− Teff expressed a majority of TLRs. Moreover, Treg selectively express several TLRs at a higher level than Teff do, which include TLR2 (24), TLR4 (20, 26), TLR5 (20, 21, 23, 29), TLR7/8 (19, 20, 29) and TLR10 (29). The expression level of TLRs, namely, TLR2, 4, 5 and 7, on a particular subset of CD4+ T cells could be modulated by their activation/functional status (20, 23-25). This indicates that TLRs may serve as co-stimulatory receptors for the development and maintenance of Ag-specific effector and memory T cells (22, 25, 30).
Function of TLR stimulation on regulatory T cells
The proven expression of TLRs by both human and murine Treg indicates the physiological necessity/relevance of direct TLR signaling on modulating Treg function. A MyD88-IRAK-4-NF-κB-dependent signaling pathway is shared exclusively by all TLRs except TLR3, which utilizes TRIF-TBK-1-IRF-3 pathway. By using MyD88 knockout mice, the role of TLR signaling in CD4+ T cells has recently been investigated in vitro and in vivo. It was demonstrated in murine CD4+ T cells that CpG DNA, a TLR9 ligand, directly enhanced the proliferation, prevented anergy, promoted survival and boosted an IgG production towards an T cell-dependent Ag in a MyD88- and phosphatidylinositol 3-kinase (PI-3 kinase)-dependent fashion (30). Similarly, IRAK-4 knockout CD4+ T cells and CD8+ T cells showed a significant impairment of proliferation and effector functions in vivo and in vitro after TCR ligation, indicating a critical role for the innate immune signaling in both CD4+ and CD8+ T cell activation (32). In addition, in a T cell adoptive transfer-induced colitis model, disease severity and pathology were greatly reduced in mice when MyD88−/−CD45RBhi CD4+ T cells were transferred, compared with wild type (WT) CD45RBhi CD4+ T cells. This was contributed by decreased proliferation and production of effector cytokines, such as IL-17 (14, 15). Moreover, co-transfer of MyD88−/− Treg elicited less prevention from disease severity than WT Treg (14). Consistent with the in vivo findings, in vitro stimulation with anti-CD3 and TLR ligands (Pam3CSK4, polyI:C, LPS and CpG) promoted stronger proliferation of WT Teff than CD3 stimulation alone, which was dependent on MyD88 (except for polyI:C). On the other hand, MyD88−/− Treg had decreased suppressive function in vitro compared with WT Treg (14). Overall, these observations highly suggested the functional involvement of TLRs not only on CD4+CD25− Teff, but also on CD4+CD25+ Treg.
TLR2−/− mice are more resistant to disseminated Candida albicans infection. Analysis of TLR2−/− and MyD88−/− mice showed a significantly reduced number of CD4+CD25+ Treg in both types of mice, but not in TLR4−/− mice (33, 34). Systemic administration of TLR2 agonist Pam3CSK4 to WT mice resulted in an increase of CD4+Foxp3+ Treg (34). In vitro, Pam3CSK4, but not TLR4 agonist LPS, enhanced Treg proliferation in the presence of anti-CD3 with or without IL-2 (24, 34). It was also shown that another TLR2 ligand peptidoglycan alone was able to prolong Treg survival in vitro (33). Functionally, Pam3SCK4-treated Treg transiently reduced Foxp3 expression in vitro and also temporarily lost their suppressive activity in vitro and in vivo in models of adoptive transfer-induced colitis, leishmania or C. albican infections (24, 34). IL-2 produced by Teff played an important role in Pam3SCK4-mediated abrogation of Treg suppressor function (24). As an endogenous ligand for TLR2 and TLR4 (35), human heat shock protein 60 (HSP60) or its p277 peptide alone was able to augment a TLR2-dependent increase of human Treg function in vitro, indexed by down-regulating the proliferation of and cytokine production by both CD4+ Teff and CD8+ T cells, which was mediated by cell-cell contact and soluble factors, such as TGF-β and IL-10 (36). Mechanistically, this augmentation of Treg function required activation of PKC, PI3K and p38. Inhibition of ERK further enhanced HSP60-mediated Treg suppression (36). The discrepancy between mouse and human, exogenous or endogenous TLR ligands warrants further investigations. In contrast to previous data which demonstrated that stress-inducible HSP60, HSP70 and gp96 are also considered as endogenous ligands for TLR2 or/and TLR4 (35, 37-40), several independent groups illustrated that LPS-free HSP60, HSP70 and gp96 were not able to stimulate TLR2 or/and TLR4 by themselves (41-43). Rather, it appears that LPS chaperoned by HSP60 or by gp96 is superior to LPS alone in stimulation of cytokine production (41, 44).
A direct engagement of TLR4 by LPS on Treg has been reported to up-regulate several activation markers on Treg, induced Treg proliferation/survival without the need for TCR ligation. More importantly, LPS-treated Treg exerted enhanced function in vitro and remained suppressive in vivo (20). We recently demonstrated that the folding/assembly of most TLRs were dependent on an endoplasmic reticulum luminal HSP gp96 (grp94, HSP90b1) (45). This was revealed by both gene knockout (45, 46) and gain-of-function approaches (47). Whereas deletion of gp96 led to abrogation of all TLR responses, ectopic expression of gp96 on cell surface in transgenic mice resulted in TLR4 hyper-responsiveness (47). Interestingly, these gp96-transgenic mice developed autoimmune disease that was dependent on commensal bacteria (47), DCs and IL-12 (48). The in vivo TLR4 hyperactivity in gp96 transgenic mice allows us to examine the direct impact of TLR4 on Tregs (49). Before the experiment, we anticipated that either attenuated Treg would cause autoimmunity or potentiated Treg would curb inflammation; both of these scenarios have been reported in the literature (Table I). By intracellular staining for the Foxp3 protein, we found that the percentage and absolute number of Treg in the spleen and thymus were the same between WT and gp96 transgenic mice. We found that the suppressive function of transgenic Treg was significantly pronounced over that of WT Treg on the per cell basis. Based on the previous findings (20), we hypothesized that the enhanced Treg activity in gp96 transgenic mice is the direct effect of increased chronic triggering of TLR4 due to cell surface gp96 on Treg. Indeed, ectopic gp96 is expressed by Treg. More importantly, we found that Treg from TLR4-null gp96 transgenic mice lost their superior ability to suppress proliferation of CD4+CD25− Teff, demonstrating that the increased suppression by transgenic Treg is mediated by TLR4. This study suggested that TLR4 hyperfunction in vivo potentiates Treg function to curtail TLR4-dependent autoimmunity (49). The role of LPS on human Treg in the context of neutrophil activation has also been examined. Pre-incubation of Treg with LPS more potently inhibited production of reactive oxygen intermediates and IL-8, IL-6 and TNF-α by human neutrophils (26).
Human Treg expressed a high level of TLR5 mRNA and protein, which is equivalent to that on CD14+ monocytes (23). Interestingly, direct ligation of TLR5 agonist flagellin on Teff and Treg transiently reduced their surface expression of TLR5. In contrast, treatment of Treg with flagellin increased Foxp3 expression in Treg. Teff stimulated with flagellin and anti-CD3 demonstrated increased proliferation and IL-2 production to the level as with anti-CD28. However, anti-CD3 together with flagellin did not break unresponsiveness of Treg, rather, potentiated suppressive function of Treg in vitro in the absence of antigen-presenting cells (APCs) (23).
In a study to test if DCs have any impact on human Treg function, CpG-A but not LPS elicited a reduced suppressor function of human Treg in the presence or absence of APCs, the latter of which suggested the direct involvement of TLR in modulating human Treg activity (19). Indeed, cultured human CD4+ Treg cell lines (thus without contamination of any APCs), when pre-treated with CpG-A/B, failed to inhibit naïve CD4+ T cell proliferation in vitro in response to anti-CD3 stimulation. Surprisingly, it was the poly G tail, but not CpG motif, in CpG-A that was responsible for the abrogation of suppression activity of both Treg cell line and freshly isolated Treg. siRNA inhibition assays identified that the TLR8-MyD88-IRAK-4 pathway in Treg was required for the control of suppressive function of both Ag-specific Treg and naturally occurring Treg. More convincingly, adoptive transfer of TLR8 ligand-treated Treg into tumor-bearing mice elicited an enhanced anti-tumor immunity when compared with transfer of non-treated Treg. This observation highlights the fact that modulation of human Treg can be achieved by direct TLR8 ligation on Treg, thus has implications in immunotherapy of cancers, infections and autoimmunity (19).
Although TLR9 expression level in rat Teff and Treg was not very high compared with other TLRs, a direct stimulation by CpG enhanced dose-dependent proliferation of Teff and Treg in the absence of APCs, but without further increase of cytokine production in both subsets of CD4+ T cells. CpG completely abrogated the suppressiveness of Treg in a standard in vitro suppression assay, yet CpG didn’t alter the Foxp3 expression in Treg. Interestingly, pre-incubation of Teff, but not Treg, with CpG rendered Teff refractory to Treg suppression (21). Similarly, no change of Foxp3 expression in murine Treg was observed when treated with CpG. WT but not MyD88−/− Teff cells significantly produced more IL-2 in response to CpG. It was demonstrated that competent MyD88 signaling in either Teff or Treg was required for Teff resistance to Treg suppression in the presence of CpG (50). Whether IL-2 plays the same indispensable role in this process, as it does in the TLR2 on Treg scenario (24), remains enigmatic.
Little is known about TLR10 function. Human Treg expressed TLR10 ~200 fold higher than non-Treg cells and its protein was readily detected and could be further enhanced by TCR activation. A putative DNA binding site for Foxp3 was identified in a suspected cis-regulatory region proximal to the transcriptional start site of TLR10. An introduction of Fopx3 was sufficient to induced TLR10 expression in human non-Treg cells, which appeared to require cooperative complex formation of Foxp3:NF-AT (29).
Though less relevant to this review, it is worthwhile to mention that TLR signaling also plays an essential role for effector functions of CD4+CD25− Teff in vitro and in vivo (30, 32). Surface TLR2 is up-regulated in activated human Teff. Direct engagement of TLR2 on anti-CD3/IFN-α-activated Teff stimulated their proliferation and production of IFN-γ and IL-2 (24, 25). TLR2 ligand Pam3CSK4 has also been shown to induce IFN-γ production and proliferation and survival of pre-established mouse Th1 cells without TCR activation, which is dependent on MyD88-IRAK-4-MAPK pathway. In addition, Pam3CSK4 is capable of converting human quiescent naïve and memory CD4+ T cells into effector-like phenotype. Increased expression of a chemokine receptor CCR5 renders them more permissive to productive infection with X4 and R5 HIV-type 1 (51). Ligation with TLR3 (16), TLR4 (52), TLR5 (22, 23), TLR7/8 (22) or TLR9 (16, 17, 21, 30) all has direct roles on the proliferation/survival of and various cytokine production by Teff. For instance, LPS can induce human and mouse CD3+ T cells to adhere to fibronectin via TLR4-MyD88 pathway. An increase of suppressor of cytokine signaling 3 (SOC3) in T cells inhibited their CXCL12-mediated chemotaxis (52). Another example is a rescue of defective T cell functions in PKC-θ−/− CD4+ T cells by CpG (17). PKC-θ is primarily expressed in T cells and recruited to the immunological synapses upon TCR and anti-CD28 ligation (53, 54). In vitro, IL-2 production by PKC-θ−/−T cells is impaired, resulting in the defect of proliferation (55). In vivo, PKC-θ−/− appears to have differential roles in Th1, Th2 and Th17-mediated immune responses (56-59). It has been demonstrated that PKC-θ−/− mice are resistant to experimental autoimmune myocarditis (EAM), due to greatly reduced inflammatory infiltrates in the heart, Th17 production and failure to develop myosin-specific autoantibodies. Systemic injection of CpG renders PKC-θ−/− mice susceptible to EAM. In vitro, CpG restores PKC-θ−/− CD4+ T cell proliferative property and survival by up-regulation of Bcl-XL, and more so, the IL-17 production in conjunction with IL-6 and TGF-β (17).
Perspective and hypothesis: what is the missing link?
The field has progressed beyond the examination of TLR expression by Tregs. The biological relevance of direct TLR signaling in Treg, however, remains one of the key missing links. Moreover, it is equally perplexing that Treg function can be either enhanced or curtailed depending on which TLRs are engaged. For example, increased activities of bacteria-sensing cell surface TLRs, such as TLR2, TLR4 and TLR5, cause increased functions of Treg, whereas engaging nucleic acids-sensing intracellular TLRs, such as TLR7, TLR8 and TLR9, decrease Treg functions (Table I). The mechanism of the opposing effects of TLR2/4/5 on Treg from that of TLR7/8/9 is far from clear. We offered the following considerations.
First, in favor of host survival to acute massive inflammations induced by pathogens, timing in resolving such an inflammation could be more demanding in one type of infections than the other. In general, acute septic shock is primarily caused by a bacterial or fungal, not viral, infection. There is a much more heightened and immediate need for anti-inflammatory mechanisms to counterbalance bacterium/fungus-induced sepsis (Figure 1). Thus, in the setting of these infections, Treg function may be called upon to play more important anti-inflammatory roles. On the other hand, viral infection is primarily dealt with by adaptive immunity, which depends on optimal activation of Teff. Thus the removal of Treg hurdle appears to be more important for dealing with viral infections. There are obvious exceptions of pathogens that do not fit with this teleological argument.
Figure 1. A model on the direct roles of TLR stimulation on Treg function.

This model is based on the following conditions and predictions (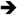 stimulatory,
stimulatory,  inhibitory): (i) functional outcome is dictated by the nature of PAMPs; (ii) acute sepsis is more often associated with bacterial and fungal but not viral infection; (iii) viral nucleic acids-sensing TLRs block Treg to amplify adaptive immunity, whereas bacterium/fungus-sensing TLRs enhance it to curtail acute sepsis.
inhibitory): (i) functional outcome is dictated by the nature of PAMPs; (ii) acute sepsis is more often associated with bacterial and fungal but not viral infection; (iii) viral nucleic acids-sensing TLRs block Treg to amplify adaptive immunity, whereas bacterium/fungus-sensing TLRs enhance it to curtail acute sepsis.
Second, differential regulation of Foxp3 in Treg by different TLR ligands could be another plausible explanation. Foxp3 is a transcription factor essential for the differentiation and function of Treg (60, 61). There are some evidence to suggest the interplay of TLR and Foxp3 in Treg. For example, flagellin up-regulates Foxp3 expression in Treg, which correlates with their heightened suppressive function (23). Similarly, Pam3CSK4 transiently down-regulates Foxp3 and activation of TLR2 on Tregs causes a temporary loss of their inhibitory property (24). Though the functionality of human TLR10 in Treg remains unclear, interplay between TLR10 and Foxp3 was suggested. TLR10 expression in human non-Treg cells was induced by transduction of Foxp3, which appeared to require Foxp3:NF-AT cooperative complex formation (29). Nonetheless, these surface TLRs demonstrated their potential to modulate Foxp3, whereas CpG ligation does not alter the Foxp3 level (21, 50).
Third, the relative expression level of each TLR in Treg is unclear due to different internal controls used in different studies. In general, surface TLRs (such as TLR1, 2, 4, 5, 6) were more readily detectable comparing with intracellular TLRs (3, 7, 8, 9), though variations in expression level exist among human, mouse and rat (20-22). It is plausible that the differential density/expression level of various TLRs, together with the nature of their ligands, may contribute to the differential modulating capacity of TLRs on Treg.
Fourth, both surface and intracellular TLRs showed a MyD88-dependent co-stimulatory receptor-like property as summarized above. It is possible that during the interaction process between Treg and Teff, surface TLRs on Treg provide an additional engagement force to Teff with unknown mechanism(s), therefore exerts more potent suppressive function, whereas intracellular TLRs do not have such an advantage. This point is entirely speculative.
Finally, there is a possibility that other TLR accessory molecules may exist which qualitatively and quantitatively modulate TLR signaling in Treg. One example is RP105-MD-1 complex for TLR4 signaling. RP105 is a TLR homolog without a functional intracellular signaling domain (62). It partners with MD-1 (a homolog to LPS co-receptor MD-2)(63), and is expressed on B cells, CD14+ monocytes and CD11c+ DCs, including plasmacytoid DCs (64). RP105 extracellular domain was able to specifically inhibit TLR4 signaling in HEK 293 cells, by its direct interference with the LPS binding to TLR4 signaling complex, in conjunction with MD-1. RP105-deficient mice showed much more severe endotoxinemia than WT mice (64). In addition, MD-1 blockade significantly lowered the threshold for LPS responsiveness to enhance Treg-mediated suppression in vitro and to increase numbers of CD4+CD25+Foxp3+ Treg in vivo, which consequently conferred a better protection against skin graft rejection (65). This piece of data again reinforces the concept that LPS can functionally enhance Treg activity in vitro and in vivo. Another category of the TLR accessory molecules is ER-resident chaperones, including Unc93B, PRAT4A and gp96. Mutation/deletion of each one of the genes results in functional inactivation of TLR3, 7 and 9 for UNC93B (so-called 3d mutation) (66), TLR1 and 4 for PRAT4A (67, 68), and universal defect in all TLRs in gp96 null cells (46, 69), respectively. It might be informative to closely examine the expression patterns of these accessory molecules in Treg and determine their functional relevance to the opposing effects of TLRs on Treg. Gp96 null mice are embryonically lethal. Lineage-specific conditional silencing of the TLR master chaperone gp96 shall offer a unique tool to define the roles of TLRs in T cell biology, namely, differentiation and functions of Th1, Th2, Th17 and Treg as well as CD8+ T cells. This line of investigation is ongoing in our laboratory.
Conclusive remarks
Most TLRs are expressed by T cells. It appears that Treg express more TLRs than Teff. The engagement of TLRs on Tregs can have both stimulatory and inhibitory consequences on Treg function, which may be related to the nature of pathogens, the expression level/density/subcellular localizations of TLRs, the roles of TLR accessory molecules (unc93b, gp96, PRAT4A, proteases etc.) and the downstream signaling cascades. Treg-specific ablation of each TLR, coupled with functional analysis of immune responses in the setting of infection and autoimmunity, shall be fruitful in uncovering the secrets of TLR biology in Tregs.
Acknowledgement
The cited work from our laboratory was supported in part by National Institutes of Health grant CA100191 and AI070603 (Z.L.). Z.L. is a clinical scholar of the Leukemia and Lymphoma Society, USA. J.D is sponsored by the postdoctoral fellowship from Schering-Plough Corp.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 3.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Willment JA, Brown GD. C-type lectin receptors in antifungal immunity. Trends Microbiol. 2008;16:27–32. doi: 10.1016/j.tim.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 6.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 8.Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 9.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 10.Mokuno Y, Matsuguchi T, Takano M, Nishimura H, Washizu J, Ogawa T, Takeuchi O, Akira S, Nimura Y, Yoshikai Y. Expression of toll-like receptor 2 on gamma delta T cells bearing invariant V gamma 6/V delta 1 induced by Escherichia coli infection in mice. J Immunol. 2000;165:931–940. doi: 10.4049/jimmunol.165.2.931. [DOI] [PubMed] [Google Scholar]
- 11.Rothenfusser S, Hornung V, Krug A, Towarowski A, Krieg AM, Endres S, Hartmann G. Distinct CpG oligonucleotide sequences activate human gamma delta T cells via interferon-alpha/-beta. Eur J Immunol. 2001;31:3525–3534. doi: 10.1002/1521-4141(200112)31:12<3525::aid-immu3525>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 13.Mansson A, Adner M, Cardell LO. Toll-like receptors in cellular subsets of human tonsil T cells: altered expression during recurrent tonsillitis. Respir Res. 2006;7:36. doi: 10.1186/1465-9921-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomita T, Kanai T, Fujii T, Nemoto Y, Okamoto R, Tsuchiya K, Totsuka T, Sakamoto N, Akira S, Watanabe M. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J Immunol. 2008;180:5291–5299. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]
- 16.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsland BJ, Nembrini C, Grun K, Reissmann R, Kurrer M, Leipner C, Kopf M. TLR ligands act directly upon T cells to restore proliferation in the absence of protein kinase C-theta signaling and promote autoimmune myocarditis. J Immunol. 2007;178:3466–3473. doi: 10.4049/jimmunol.178.6.3466. [DOI] [PubMed] [Google Scholar]
- 18.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 19.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 20.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiffoleau E, Heslan JM, Heslan M, Louvet C, Condamine T, Cuturi MC. TLR9 ligand enhances proliferation of rat CD4+ T cell and modulates suppressive activity mediated by CD4+ CD25+ T cell. Int Immunol. 2007;19:193–201. doi: 10.1093/intimm/dxl136. [DOI] [PubMed] [Google Scholar]
- 22.Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 23.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029–3034. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155–7163. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 27.Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 28.Rossi RJ, Muralimohan G, Maxwell JR, Vella AT. Staphylococcal enterotoxins condition cells of the innate immune system for Toll-like receptor 4 stimulation. Int Immunol. 2004;16:1751–1760. doi: 10.1093/intimm/dxh176. [DOI] [PubMed] [Google Scholar]
- 29.Bell MP, Svingen PA, Rahman MK, Xiong Y, Faubion WA., Jr. FOXP3 regulates TLR10 expression in human T regulatory cells. J Immunol. 2007;179:1893–1900. doi: 10.4049/jimmunol.179.3.1893. [DOI] [PubMed] [Google Scholar]
- 30.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174:2942–2950. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki N, Suzuki S, Millar DG, Unno M, Hara H, Calzascia T, Yamasaki S, Yokosuka T, Chen NJ, Elford AR, Suzuki J, Takeuchi A, Mirtsos C, Bouchard D, Ohashi PS, Yeh WC, Saito T. A critical role for the innate immune signaling molecule IRAK-4 in T cell activation. Science. 2006;311:1927–1932. doi: 10.1126/science.1124256. [DOI] [PubMed] [Google Scholar]
- 33.Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, Hartung T, Adema G, Kullberg BJ. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. 2004;172:3712–3718. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 34.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–31339. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 36.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 38.Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–20853. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 39.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 40.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 41.Warger T, Hilf N, Rechtsteiner G, Haselmayer P, Carrick DM, Jonuleit H, von Landenberg P, Rammensee HG, Nicchitta CV, Radsak MP, Schild H. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem. 2006;281:22545–22553. doi: 10.1074/jbc.M502900200. [DOI] [PubMed] [Google Scholar]
- 42.Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H, Hanau D. Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol. 2002;32:3708–3713. doi: 10.1002/1521-4141(200212)32:12<3708::AID-IMMU3708>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 43.Gao B, Tsan MF. Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem. 2003;278:22523–22529. doi: 10.1074/jbc.M303161200. [DOI] [PubMed] [Google Scholar]
- 44.Osterloh A, Kalinke U, Weiss S, Fleischer B, Breloer M. Synergistic and differential modulation of immune responses by Hsp60 and lipopolysaccharide. J Biol Chem. 2007;282:4669–4680. doi: 10.1074/jbc.M608666200. [DOI] [PubMed] [Google Scholar]
- 45.Liu B, Li Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood. 2008;112:1223–1230. doi: 10.1182/blood-2008-03-143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, Li Z. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol. 2006;177:6880–6888. doi: 10.4049/jimmunol.177.10.6880. [DOI] [PubMed] [Google Scholar]
- 48.Dai J, Liu B, Cua DJ, Li Z. Essential roles of IL-12 and dendritic cells but not IL-23 and macrophages in lupus-like diseases initiated by cell surface HSP gp96. Eur J Immunol. 2007;37:706–715. doi: 10.1002/eji.200636643. [DOI] [PubMed] [Google Scholar]
- 49.Dai J, Liu B, Ngoi SM, Sun S, Vella AT, Li Z. TLR4 hyperresponsiveness via cell surface expression of heat shock protein gp96 potentiates suppressive function of regulatory T cells. J Immunol. 2007;178:3219–3225. doi: 10.4049/jimmunol.178.5.3219. [DOI] [PubMed] [Google Scholar]
- 50.LaRosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunol Lett. 2007;108:183–188. doi: 10.1016/j.imlet.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thibault S, Tardif MR, Barat C, Tremblay MJ. TLR2 signaling renders quiescent naive and memory CD4+ T cells more susceptible to productive infection with X4 and R5 HIV-type 1. J Immunol. 2007;179:4357–4366. doi: 10.4049/jimmunol.179.7.4357. [DOI] [PubMed] [Google Scholar]
- 52.Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner-Fischer M, Lider O, Cohen IR. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol. 2007;179:41–44. doi: 10.4049/jimmunol.179.1.41. [DOI] [PubMed] [Google Scholar]
- 53.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 54.Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJ, Altman A. Antigen-induced translocation of PKC-theta to membrane rafts is required for T cell activation. Nat Immunol. 2001;2:556–563. doi: 10.1038/88765. [DOI] [PubMed] [Google Scholar]
- 55.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 56.Marsland BJ, Nembrini C, Schmitz N, Abel B, Krautwald S, Bachmann MF, Kopf M. Innate signals compensate for the absence of PKC-{theta} during in vivo CD8(+) T cell effector and memory responses. Proc Natl Acad Sci U S A. 2005;102:14374–14379. doi: 10.1073/pnas.0506250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsland BJ, Soos TJ, Spath G, Littman DR, Kopf M. Protein kinase C theta is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J Exp Med. 2004;200:181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Differential regulation of Th2 and Th1 lung inflammatory responses by protein kinase C theta. J Immunol. 2004;173:6440–6447. doi: 10.4049/jimmunol.173.10.6440. [DOI] [PubMed] [Google Scholar]
- 59.Tan SL, Zhao J, Bi C, Chen XC, Hepburn DL, Wang J, Sedgwick JD, Chintalacharuvu SR, Na S. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C theta-deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 60.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 61.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 62.Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J Exp Med. 1994;180:1217–1224. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagai Y, Shimazu R, Ogata H, Akashi S, Sudo K, Yamasaki H, Hayashi S, Iwakura Y, Kimoto M, Miyake K. Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood. 2002;99:1699–1705. doi: 10.1182/blood.v99.5.1699. [DOI] [PubMed] [Google Scholar]
- 64.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorczynski RM, Kai Y, Miyake K. MD1 expression regulates development of regulatory T cells. J Immunol. 2006;177:1078–1084. doi: 10.4049/jimmunol.177.2.1078. [DOI] [PubMed] [Google Scholar]
- 66.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 67.Takahashi K, Shibata T, Akashi-Takamura S, Kiyokawa T, Wakabayashi Y, Tanimura N, Kobayashi T, Matsumoto F, Fukui R, Kouro T, Nagai Y, Takatsu K, Saitoh S, Miyake K. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med. 2007;204:2963–2976. doi: 10.1084/jem.20071132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wakabayashi Y, Kobayashi M, Akashi-Takamura S, Tanimura N, Konno K, Takahashi K, Ishii T, Mizutani T, Iba H, Kouro T, Takaki S, Takatsu K, Oda Y, Ishihama Y, Saitoh S, Miyake K. A protein associated with toll-like receptor 4 (PRAT4A) regulates cell surface expression of TLR4. J Immunol. 2006;177:1772–1779. doi: 10.4049/jimmunol.177.3.1772. [DOI] [PubMed] [Google Scholar]
- 69.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 70.Liu G, Zhao Y. Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology. 2007;122:149–156. doi: 10.1111/j.1365-2567.2007.02651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]