

Abstract
Mammalian heat shock protein gp96 is an obligate chaperone for multiple integrins and TLRs, the mechanism of which is largely unknown. We have identified gp93 in Drosophila having high sequence homology to gp96. However, no functions were previously attributed to gp93. To determine whether gp93 and gp96 are functionally conserved, we have expressed gp93 in gp96-deficient mouse cells. Remarkably, the Drosophila gp93 is able to chaperone multiple murine gp96 clients including integrins α4, αL, and β2 and TLR2 and TLR9. This observation has led us to examine the structural basis of the chaperone function of gp96 by a close comparison between gp96 and gp93. We report that whereas gp96 undergoes intermolecular disulfide bond formation via Cys138, gp93 is unable to do so due to the absence of a cysteine near the same region. However, abrogation of disulfide bond formation by substituting C with A (C138A) in gp96 via site-directed mutagenesis did not compromise its chaperone function. Likewise, gp93 chaperone ability could not be improved by forcing intermolecular bond formation between gp93 N termini. We conclude that gp93 is the Drosophila ortholog of gp96 and that the chaperone function of the two molecules is conserved. Moreover, gp96 N-terminal disulfide bond formation is not critical for its function, underscoring the importance of N-terminal dimerization via non-disulfide bond-mediated interactions in client protein folding by gp96. Further study of gp96 from an evolutionary angle shall be informative to uncover the detailed mechanism of its chaperone function of client proteins in the secretory pathway.
The glycoprotein gp96 (1), known also as heat shock protein (HSP)5 90b1 (2), grp94 (3), endoplasmic reticulum (ER) p99 (4), endoplasmin (5), and HSPC4 (6), is the ER paralog of the cytosolic chaperone HSP90 (7). gp96 is thought to play general roles in both the unfolded protein response in the ER and the ER-associated degradation pathway to maintain protein homeostasis (8, 9). Up to 10% of the cytosolic proteins can be chaperoned by HSP90 (10, 11). By comparison, the number of confirmed client proteins of gp96 is limited. Although Ig is among the first implicated clients of gp96 (12), a recent study demonstrates that assembly and production of Abs are uncompromised in the absence of gp96 (13). Importantly, gp96 has been found to play essential functions in chaperoning multiple TLRs, including TLR1, TLR2, TLR4, TLR5, TLR7, and TLR9, and a variety of integrins such as α4, αL, and β2 (13–15). Similar to HSP90, gp96 can hydrolyze ATP (16–18), and it is thought to function as a homodimer (18). It has an N-terminal signal peptide, followed sequentially by an ATPase domain, a charged middle domain, a dimerization domain, and the C-terminal KDEL ER retention signal. Deletion of either the ATPase or the C-terminal dimerization domain results in total loss of chaperone function, whereas removal of the middle charged domain or C-terminal ER retention signal does not alter its chaperone function (14). Although the exact mode of the chaperone function of gp96 is unclear, it is thought that the binding of gp96 to client proteins leads to transformation of the low-energy-state open conformation of a gp96 to a closed conformation through N-terminal dimerization and final maturation of client protein folding (18).
The two major clients of gp96, integrins and TLRs, are evolutionarily conserved. Restricted to all species of metazoan (19), integrins are transmembrane proteins that link the extracellular matrix to the cytoskeleton and are important for various biological processes such as survival, proliferation, apoptosis, adhesion, chemotaxis, polarity, differentiation, and gene expression (20, 21). Integrins consist of α and β subunits which heterodimerize to form the receptor (22). Humans and mice have 18 α and 8 β subunits, which form 24 heterodimers and can be grouped as collagen receptors, laminin receptors, RGD receptors, and leukocyte receptors (19, 20, 23). They are capable of inside-out signaling as well as outside-in signaling (19, 20), and many are targeted by both viruses and bacteria as receptors (19).
Like integrins, TLRs are also type I transmembrane receptors. TLR1, TLR2, TLR4, TLR5, TLR6, TLR10, TLR11, and TLR12 are present at the cell surface; whereas TLR3, TLR7, TLR8, and TLR9 reside in the acidic endosomes. All TLRs are believed to homodimerize to function except TLR1 and TLR6 which need to heterodimerize with TLR2 (24). These receptors recognize bacterial and viral constituents, and the resulting signaling cascade activates the innate immune system. Exhibiting high similarity to the IL-1/IL-18 receptor family, TLRs have Toll/IL-1R domains in their cytoplasmic tails that are important for recruiting the downstream signaling adaptor molecules TIRAP, MyD88, TRAM, and TRIF (24). These pathways culminate in activation of the transcription factors NF-κB and IFN regulatory factor-3, and the resulting production of type 1 IFN and inflammatory cytokines such as IL-6. Although many of the same signaling molecules are involved, differential activation of TLRs can lead to different responses (24). Recent studies indicate that the ectodomain of TLR9 is further cleaved in the endolysosome to form the mature functional receptor (25, 26). TLR4 (27) was the first TLR found in mammals as a homolog of Toll in Drosophila, and it plays important roles in dorsal-ventral patterning and innate immunity in flies (28). Both Toll and TLRs have similar functions in the innate immune response, as well as similar structures with multiple leucine-rich repeats in the ectodomain (29). Whereas TLR4 requires MD-2 for signaling (30, 31), Toll requires Spatzle association for activation (28, 32).
The evolutional conservation of both TLRs and integrins argues for the need of conserved chaperone machinery for their folding. Indeed, gp96 is highly conserved (33). In this study, we have identified a previously unknown molecule, gp93 in Drosophila, with high sequence homology to gp96. To determine whether gp93 shares an evolutionarily conserved chaperone function with gp96, we have expressed gp93 in gp96 mutant murine cells. We found that Drosophila gp93 could efficiently rescue murine gp96null cells for the expression of a variety of natural clients of gp96 including integrins and TLRs. We further showed that unlike gp96, gp93 did not undergo disulfide bond-dependent N-terminal homodimerization. This distinction allows us to address for the first time the functional significance of intermolecular disulfide bond formation between gp96 monomers in client chaperoning. Our study demonstrates that gp96 and gp93 are orthologs of each other with conserved chaperone function, and suggests that the N-terminal homodimerization of gp96 through intermolecular interaction other than disulfide bond formation is important for the folding of TLRs and integrins.
Materials and Methods
Database search, sequence alignment, and conservation mapping
The National Center for Biotechnology Information (NCBI) protein database (http://www.ncbi.nlm.nih.gov/) was blasted and searched for gene products that share high sequence homology with mouse gp96 (NP_035761) and canine gp96 (NP_001003327). A protein with unknown function, gp93 (NP_651601), was identified from Drosophila melanogaster. All three sequences were aligned with ClusterW (34), and sequence conservation among them was mapped onto the molecular surface of the canine gp96 structure (PDB ID:2O1U) using the programs ConSurf (35) and PyMol (http://www/pymol.org/).
Constructs and site-directed mutagenesis
mRNA was isolated from Drosophila S2 cells (36) using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA with SuperScript II RNase H-reverse transcriptase (Invitrogen). gp93 cDNA was cloned by PCR (gp93s primer, 5′-accatgaagtactttttgctggt-3′; gp93as primer, 5′-ttta cagctcgtcgtgctgct-3′) into the pGEM-T Easy vector (Promega) and subcloned into the MigR1 retrovector which expresses GFP in a bicistronic fashion. gp93Flag was PCR amplified from wild-type (WT) gp93 in MigR1 using the following primers: gp93Flags, 5′-tagcggccgcgccaccatgaagtactttttgctggtg-3′; gp93Flagas, 5′-tagcggccgcttacaattcatccttcttgtcgtcatcgtctttgtagtcctgctgctcctcctcctc-3′. This construct has a KDEL instead of a HDEL ER retention signal to enable efficient ER targeting in a murine system, and the Flag epitope (DYKDDDDK) was placed immediately before the KDEL sequence (37). To generate the gp93A137C mutant, the following primers were designed with Stratagene's online primer design program: gp93A137Cs, 5′-ctgcacatccgcattaagtgcgacaaggagaacaagg-3′; gp93A137Cas 5′-ccttgttctccttgtcgcacttaatgcggatgtgcag-3′. Using gp93 as a template, the mutation was incorporated with the aid of the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) per manufacturer's protocol. It was later amplified by PCR with the same primers used to create gp93Flag. The C3A and C5A mutants of gp96 were also generated using the same mutagenesis kit. The mutant sequence necessary to create the desired cysteine to alanine substitution was incorporated into these primers. All cloned genes of interests were verified by sequencing.
Cells
Drosophila S2 cells were grown in Express Five SFM Medium (Life Technologies) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies) and l-glutamine. Cells were cultured at 27°C and shaken gently. gp96null E4.126 pre-B cells were a gift from Dr. B. Seed (Harvard University, Cambridge, MA), which were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, 10% FBS (Atlas Biologicals), and 0.055 mM 2-ME (Life Technologies). Phoenix Eco cells were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS and same concentration of penicillin and streptomycin as above. E4.126 and Phoenix Eco cells were cultured at 37°C with 5% CO2.
Virus production and transduction
Ecotropic retrovectors were transfected into HEK293-derived Phoenix Eco cells using Lipofectamine 2000 (Invitrogen). Two days later, virus-containing medium was collected and added to 2 × 105 E4.126 cells, along with hexadimethrine bromide (Sigma-Aldrich). Spin infection was achieved by centrifugation at 1900 × g for 1.5 h at 32°C to facilitate viral transduction.
Flow cytometry
Ab staining, flow cytometry instrumentation, and data analysis were performed essentially as described without significant modifications (13). In brief, single-cell suspensions were made, washed, and blocked with staining buffer containing serum. Cells were then incubated with primary Ab, followed by incubation with fluorochrome-labeled secondary Ab. Immediately before instrumentation, propidium iodide was added to stain and exclude dead cells. Cells were finally acquired with a FACSCalibur (BD Biosciences). For intracellular staining, cells were fixed and permeabilized with methanol and then stained with the appropriate Ab without the introduction of propidium iodide. Data analysis was done using FlowJo software (FlowJo).
SDS-PAGE, Western blot, and coimmunoprecipitation (co-IP)
For Western blot, cells lysates were prepared with radioimmunoprecipitation assay (RIPA) lysis buffer (0.01 M sodium phosphate (pH 7.2), 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS) and quantitated by Bradford protein assay (Bio-Rad). Fifty micrograms of total lysate were denatured by boiling for 5 min in the presence of 0.1 M DTT and SDS-loading buffer, followed by resolution on a 10% SDS-PAGE and transfer to a polyvinylidene difluoride membrane. After blocking, membranes were blotted with appropriate primary and HRP-conjugated secondary Abs and developed with chemiluminescent substrate (Pierce Chemical).
For coimmunoprecipitation, cells were treated with or without 0.1 mg/ml dithiobis-succinimidyl propionate (DSP; Thermo Scientific) for 30 min at room temperature before lysis with RIPA buffer. Lysate (1200 μg) was then precleared with protein G beads (GenScript), followed by incubating with Flag, hemagglutinin (HA), or isotype control Ab for 1 h at 4°C and subsequently with fresh protein G beads while rotating overnight at 4°C. Beads were vigorously washed with RIPA lysis buffer. Proteins were eluted by boiling the beads in the presence of 0.1 M DTT and SDS-loading buffer. Samples were then separated by 10% denaturing SDS-PAGE and immunoblotted using the Western protocol described here.
Results
Identification of gp93
Through a NCBI database search, a protein with unknown function, gp93 (NP_651601), was identified from D. melanogaster as sharing high sequence homology with gp96. A fly database search confirmed the identity of gp93, although no function had been attributed to this molecule previously. Sequence alignment of gp93 with both murine and canine gp96 revealed an overall 74% homology at the amino acid level. Like gp96, gp93 has a signal peptide at the N terminus for targeting to the secretory pathway (19% homology), followed by an ATPase domain (65% homology), a charged middle domain (48% homology), and the C-terminal dimerization domain (61% homology). The ATPase and the C-terminal dimerization domain of gp96 are essential for its chaperone function, whereas the middle charged domain is not critical (14). gp96 ends in KDEL, whereas gp93 terminates in HDEL; both are known canonical ER retention signals (38). Thus, the organization of these major functional domains is conserved between gp96 and gp93. Furthermore, the chaperone function of gp96 is dependent on several critical residues (Fig. 1A, highlighted yellow) which are involved in ATP binding and ATP hydrolysis (14, 18); these residues are Glu103, Asn149, and Arg448, all of which are completely conserved in gp93.
FIGURE 1.
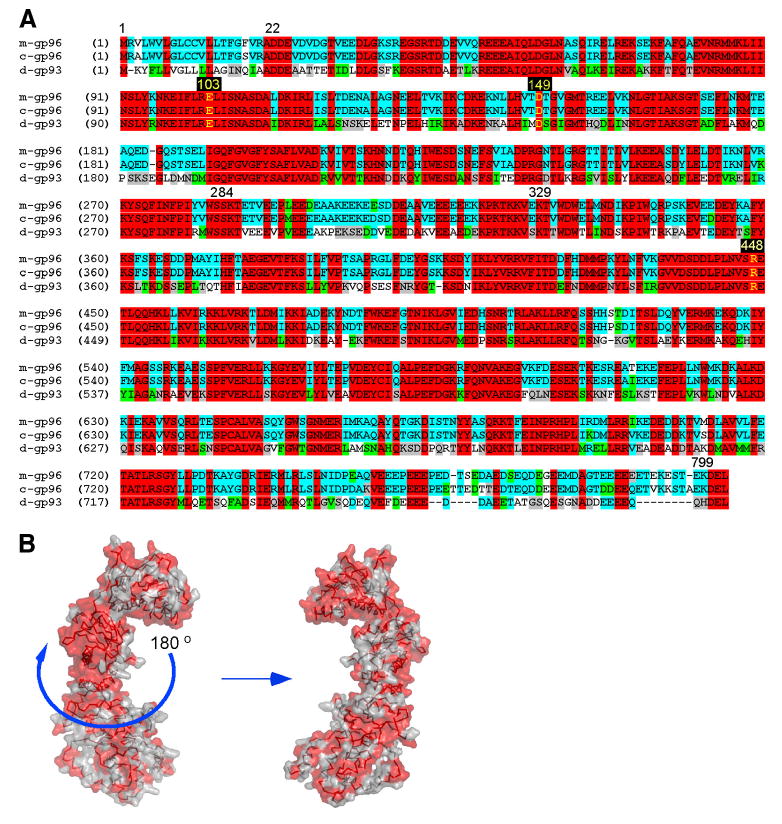
Sequence and structural conservation between Drosophila gp93 and the mammalian gp96. A, Sequence alignment of mouse gp96 (m-gp96), canine gp96 (c-gp96), and Drosophila gp93 (d-gp93) with amino acids corresponding to the ends of domains written numerically, and key residues involved in ATP binding and hydrolysis highlighted in yellow. Red, identical among all three; blue, conserved between two; green, similar class of amino acid; gray, weakly similar; white, nonconserved. B, Molecular surface representation of the canine gp96 monomer with N terminus pointing upward. Red solvent-accessible surfaces identify invariant residues between gp93 and gp96.
Given that the crystal structure of canine gp96 in complex with a nonhydrolyzable ATP, 5′-adenylyl-β,γ-imidodiphosphate has been recently determined (18), it is possible to map the sequence conservation among gp93 and the two mammalian gp96 proteins onto the structure. The nucleotide-bound gp96 dimer is open at the N terminus with tight dimerization at the C terminus, forming a twisted V-shaped conformation (18). To illustrate the structural conservation between gp96 and gp93, a monomer structure of gp96 is shown in Fig. 1B to reveal the residues in gp93 that are invariant throughout the structural domains of the canine gp96. The N-terminal ATP-binding pocket (pointing upwards) is conserved. Collectively, gp93 and gp96 share not only their primary sequences but also their domain organization and critical residues implicated in essential chaperone function. This finding prompted us to investigate the function of gp93 and to determine whether or not gp93 shares the evolutionarily conserved chaperone function of gp96.
Drosophila gp93 chaperones gp96 clients
We used a gp96 mutant cell line, E4.126 (14), for functional analysis of gp96 and gp93. It was shown previously that this gp96-deficient pre-B cell fails to express integrin α4, αL, β2, or TLR2 on the cell surface and that expression of these proteins is rescued by WT gp96 (13–15). We asked whether gp93 could functionally replace gp96 in this assay. To this end, we made a number of retroviral vectors for gp96 and gp93, which also express GFP in a bicistronic manner. To facilitate the detection of expression, a Flag-tagged version of both gp96 and gp93 was also generated by inserting the Flag epitope immediately upstream of the KDEL ER retention signal. All constructs were transduced into E4.126 cells. High-level transduction was accomplished with all constructs, indicated by GFP expression (Fig. 2A), intracellular staining, and immunoblot (IB; Fig. 2B).
FIGURE 2.
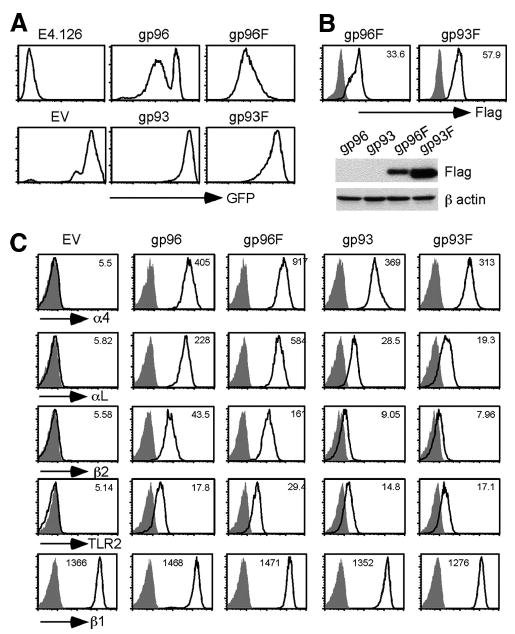
gp93 chaperones gp96 clients in mouse cells. A, GFP expression of gp96 mutant pre-B cells transduced with indicated retroviral vectors and empty vector (EV). B, Top, Intracellular stain of Flag (F; white histogram) and background (stained with isotype (iso) control Ab; gray histogram). Numbers denote MFI of Flag staining. Bottom, IB for Flag and β-actin (as a loading control). C, Restoration of cell surface expression of gp96 clients in gp96null cells by gp96 and gp93. Cells with similar intensity for GFP were gated on and analyzed for cell surface expression of gp96 clients (white histogram). Gray histograms, background stain with isotype control Ab. Numbers indicate MFI of client protein stain. The expression of a gp96 nonclient, β1 integrin, is also shown. Multiple experiments were performed with similar results.
As expected, empty vector-transduced E4.126 cells failed to express α4, αL, β2 integrins, or TLR2 on the cell surface. The surface expression of α4, αL, β2, and TLR2 was rescued by full-length gp96, consistent with the known function of gp96 in chaperoning these clients (13–15). Importantly, the cell surface expression of α4, αL, β2, and TLR2 were also rescued by both WT and Flag-tagged gp93 (Fig. 2C). The mean fluorescence intensity (MFI) of all gp96 clients tested in mutant cells was ∼5, which was increased significantly after gp93Flag expression to 313, 19.3, 7.96, and 17.1 for α4, αL, β2, and TLR2, respectively (Fig. 2C). The expression level of β1 integrin, a nonclient protein of gp96, was not increased from gp96 mutant cells (MFI 1366) by gp93Flag expression (MFI 1276). The fact that the C terminus Flag-tagged gp96 and gp93 retained chaperone function allowed us to compare the chaperone efficiency between the two. On the basis of both intracellular stain and IB using Flag-specific Ab, we found that gp93Flag was expressed considerably more than gp96Flag. Yet, the level of cell surface client proteins on gp96Flag-transduced cells was significantly higher than that on gp93Flag-expressing cells. We concluded thus that gp93 and gp96 shared conserved chaperone function for integrins and TLRs. The apparent quantitative inferiority of gp93 in folding gp96 clients created a useful experimental platform to understand the key structural features that determine the optimal function of gp96.
Similar to gp96, gp93 interacts with intracellular TLR9
We next determined whether gp93 is capable of chaperoning endosomal TLRs by investigating its interaction with TLR9. To this end, we further transduced gp96Flag and gp93Flag cells with TLR9 tagged at the C terminus with an HA epitope. As reported widely, C-terminal tagging of TLR9 does not alter its function (25, 26). Similar TLR9 expression levels in these two cells were verified by flow cytometry and IB (Fig. 3A). We then performed co-IP by immunoprecipitation for Flag followed by IB for HA. To stabilize the complex, we treated cells with DSP, a thiocleavable reversible cross-linker before cell lysis. We found indeed that both gp96 and gp93 were able to interact with TLR9 (Fig. 3, B and C). The requirement for DSP in the presence of strong detergent (Nonidet P-40, SDS, and deoxycholate) in this experiment was apparent for visualizing both gp96-TLR9 and gp93-TLR9 complexes. Thus, we concluded that gp93 is able to chaperone multiple-client proteins of gp96 that are destined for both cell surface and other compartments in the secretory pathway.
FIGURE 3.
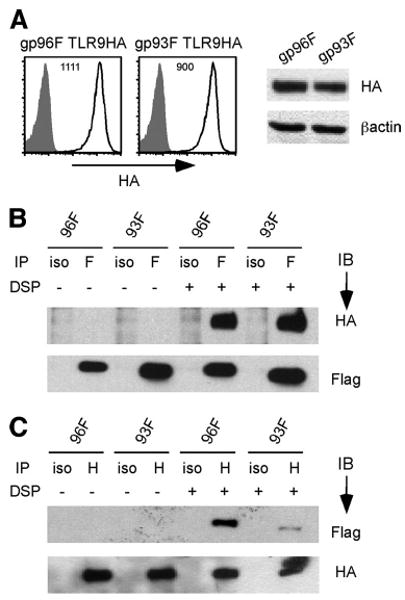
Similar to gp96, gp93 interacts with TLR9. A, Expression of TLR9-HA in gp96Flag and gp93Flag cells by flow cytometry (left, numbers denote MFI of HA staining) and by IB (right). Whole-cell lysates were subjected to SDS-PAGE followed by IB for HA and β-actin (as a loading control). B, Cells were subjected to DSP cross-linking or not, followed by co-IP with Flag (F) or isotype (iso)-matched control Ab. Immunoprecipitates (IP) were then resolved on SDS-PAGE and immunoblotted for HA or Flag. C, Experiment similar to that in B was performed, except co-IP was done first with HA (H) Ab, followed by IB for HA and Flag. Results are representative of two experiments.
gp96 forms homodimers via a single intermolecular disulfide bond
Although gp93 is able to complement gp96 in mutant murine cells to display cell surface clients of gp96 and to bind TLR9, it is not as efficient as gp96. In particular, the expression of surface αL and β2 integrins in gp96 mutant cells were not rescued to the same level by gp93 in comparison with gp96 (Fig. 2C). On further examination of the sequence difference between gp96 and gp93, we found that the cysteine at position 138 of gp96 is conserved across species in mammals but is not present in gp93, raising a question of potential importance of Cys138 of gp96 in regulating its chaperone function. There are five cysteines (Cys10, Cys11, Cys138, Cys576, and Cys645) within gp96, two of which (Cys10 and Cys11) are located in the 21-aa signal peptide. To determine whether gp96 indeed undergoes intermolecular disulfide bond formation, as a previous study suggested (39), we performed the following series of studies. We found that up to 50% of total gp96 from normal mouse organs migrated at ∼200 kDa on a nonreducing SDS-PAGE, which was confirmed by IB with multiple-gp96 Abs recognizing different regions of the gp96 molecule (Fig. 4, A and B), indicating that gp96 could indeed form an intermolecular disulfide bond. To further address this hypothesis and to determine which cysteine is engaged in disulfide bond formation, we performed a site-directed mutagenesis study. We individually mutated each cysteine to alanine. We found that the C138A mutation, but not C576A or C645A of gp96, resulted in the loss of the ∼200-kDa gp96 dimer on nonreducing SDS-PAGE (Fig. 4B), confirming unequivocally that Cys138 is involved in intermolecular disulfide bond formation between gp96 monomers.
FIGURE 4.
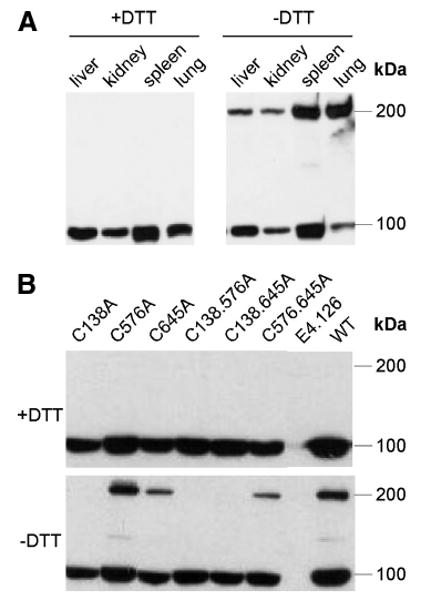
Cys138 is responsible for intermolecular disulfide bond formation of gp96. A, IB of gp96 from various mouse tissues in the presence or absence of DTT. B, Various gp96 mutants were transduced into gp96-deficient pre-B cells, followed by gp96 IB in the presence or absence of DTT. Two experiments were performed with similar results.
Roles of intermolecular disulfide bond formation on the chaperone function of gp96 and gp93
Cys138 in gp96 corresponds to Ala137 in gp93. Because we know that Cys138 is important for disulfide bond-dependent N-terminal dimerization of gp96, we hypothesized that gp93 would not dimerize due to lack of this cysteine. To probe whether gp93 function could be improved by disulfide bond formation, we created a mutant of gp93 using site-directed mutagenesis to change the alanine at position 137 in gp93 to a cysteine, gp93A137C. This was then cloned into the MigR1 retroviral vector and transduced into gp96 mutant cells. We confirmed high GFP and Flag-tagged protein expression by flow cytometry (Fig. 5A). More importantly, when cell lysate was blotted under nonreducing conditions, we saw that the gp93A137C mutant gained the ability to homodimerize (Fig. 5B). Clearly, no higher band at 200 kDa was seen when WT gp93 (Ala137) was resolved under nonreducing conditions.
FIGURE 5.
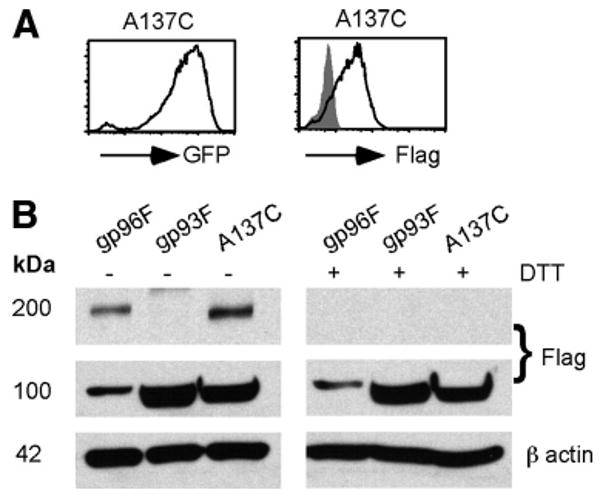
A137C mutant of gp93 but not natural gp93 dimerizes via a disulfide bond. A, Flow cytometric analysis of the expression of gp93A137C-retroviral transduced mutant pre-B cells by GFP and intracellular Flag staining. B, gp93A137C undergoes disulfide bond-dependent dimerization, whereas WT gp93 does not. gp96Flag, gp93Flag, or gp93A137C-Flag (A137C) were expressed in gp96-deficient pre-B cells, followed by Flag IB in the presence or absence of DTT. Two experiments were done with similar results.
We next performed functional analysis. To our surprise, we found that the C138A mutant of gp96 was able to fully restore cell surface expression of TLR2, αL, and α4 integrins to the same level as WT gp96 (Fig. 6A). Similarly, we found that the chaperone function of gp93A137C was not improved compared with WT gp93 (Fig. 6B). Our data indicate that disulfide bond-linked gp96 is not directly involved in folding client proteins, suggesting that N-terminal dimerization by non-disulfide bond-based interaction plays more important roles for the function of gp96 and gp93.
FIGURE 6.
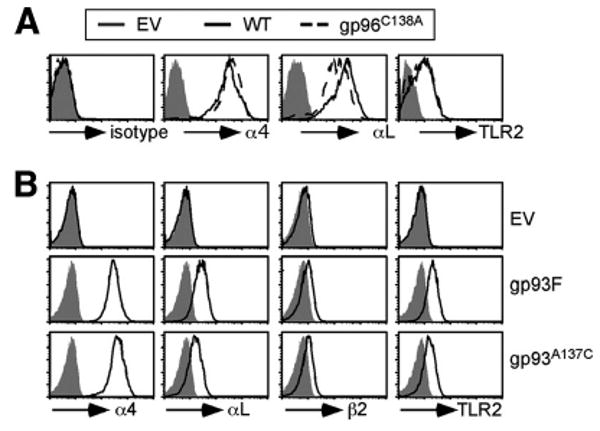
The chaperone function of gp96/gp93 is not dependent on intermolecular disulfide bond formation at the N terminus. A, Both WT gp96 and the gp96C138A mutant rescue the expression of the indicated gp96 clients. One representative result from multiple experiments is shown. B, Restoration of cell surface expression of gp96 clients by gp93 and gp93A137C. Cells with the same intensity for GFP were gated on and analyzed for cell surface expression of the indicated gp96 clients (white histogram). Gray histograms, background stain with isotype control Ab. At least two experiments were performed with similar results.
To further address the roles of inter- or intramolecular disulfide bond formation in gp96 function, we generated two additional gp96 mutants: gp96C5A in which all five cysteines (Cys10, Cys11, Cys138, Cys576, and Cys645) were changed to alanine and gp96C3A where only the cysteines in the mature protein were changed to alanine (C138A, C576A, and C645A). Both gp96 mutants were found to retain their ability to rescue cell surface expression of αL and β2 integrins in gp96-deficient cells in a dose-dependent manner, indicated by a direct correlation between the intracellular level of gp96 and the surface expression level of these clients (Fig. 7, A and B). Moreover, both C3A and C5A mutants were able to associate with TLR9 (Fig. 7C). We were not able to express C5A or C3A to the same level as WT gp96, suggesting that these cysteine residues of gp96 play more important roles in stabilizing the protein rather than qualitatively regulating the intrinsic chaperone function of gp96. We therefore conclude that the intermolecular N-terminal disulfide bond formation of gp96 is not essential for its ability to chaperone integrins or TLRs, although its roles in conferring protein stability and thus having an indirect impact on the chaperone function of gp96 could not be entirely ruled out.
FIGURE 7.

All Cys→Ala mutants of gp96 retain chaperone function. A, Both gp96-C5A and gp96-C3A rescue gp96-deficient cells for the cell surface expression of the indicated gp96 clients. Cell surface clients were stained followed by permeabilization and intracellular stain of gp96. Flow cytometric analyses were then performed. Numbers indicate percentage of cells in each quadrant. At least two experiments were done with similar findings. B, Cells in A were gated based on different expression levels of gp96 (high, medium, and low). Expression of cell surface clients on cells from each subpopulation was compared. At least two experiments were done with similar findings. C, Cells in A were cotransduced with TLR9 tagged at the C terminus with an HA epitope (TLR9HA), immunoprecipitated with HA Ab or with an isotype-matched Ab (iso), followed by IB for HA or gp96. Whole-cell lysates (WCL) were immunoblotted to indicate the expression level of gp96 and TLR9 tagged at the C terminus with HA epitope. Two experiments were performed with similar results.
Discussion
gp96 is the most abundant molecular chaperone in the ER lumen, and it is present ubiquitously in all nucleated cells. Although it is implicated in both the unfolded protein response and ER-associated degradation pathway to maintain global protein homeostasis in the secretory pathway, gp96null cells appear to lose a limited set of proteins, most notably integrins and TLRs (13–15). Because of the importance of integrins and TLRs in immune response, one might suggest that gp96 is a specialized immune chaperone (7). Detailed understanding of the mode of its action may thus facilitate the design of gp96 agonists or antagonists for the treatment of immune disorders. However, the basic mechanism of the regulation of the chaperone function of gp96 remains largely enigmatic. In this study, we have identified a previously uncharacterized Drosophila protein gp93 with ∼74% overall sequence homology to mammalian gp96. The two proteins share similar domain organization and possess critical residues for ATP binding and ATPase activity (Fig. 1). More importantly, we demonstrated that Drosophila gp93 is able to chaperone both integrins and TLRs in murine cells, in that 1) it was able to rescue the cell surface expression of αL, α4, and β2 integrins and TLR2 in the absence of gp96 (Fig. 2); and 2) like gp96, it has the capacity to directly interact with intracellular TLR9 (Fig. 3). Our data thus demonstrated that gp93 and gp96 have evolutionarily conserved chaperone function, which created an experimental platform for targeted structure-function analysis of gp96 based on the phylogenic sequence conservation of the two. We found that unlike gp96 (Fig. 4), gp93 was unable to form an intermolecular N-terminal disulfide bond (Fig. 5). Unexpectedly, our mutagenesis study of gp96 and gp93, coupled with functional analysis, revealed for the first time that N-terminal dimerization through intermolecular disulfide bond formation of gp96 is not necessary for its chaperone function (Figs. 6 and 7).
We examined the potential significance of N-terminal intermolecular disulfide bond formation on gp96 function, triggered primarily by the reasoning that the ER environment is largely reducing in nature and that the N-terminal dimerization of gp96 is necessary for completion of the folding cycle of client proteins. This point became even more important to address when we found that gp93 is weaker than gp96 in promoting client protein surface expression and that there is an absence of a disulfide bond-dependent dimer of gp93. We found that gp96 indeed dimerized at the N terminus through the formation of a disulfide bond at Cys138. A previous study clearly showed that the C-terminal 697–740 region of gp96 is sufficient for dimerization at the C terminus (40), which was confirmed by the recent crystal structure of near full-length gp96 (18). In other words, the N-terminal disulfide bound formation is unnecessary for gp96 to form a dimer. Does this mean that gp96 function is dependent on both noncovalent C-terminal and disulfide bond-mediated N-terminal dimerization? This conjecture, however, is not supported by our data; we found that neither loss of disulfide bond in gp96 nor gain of it in gp93 resulted in any significant alteration of their chaperone function. Therefore, contrary to our prediction, disulfide bond-dependent dimerization of gp96 is not necessary for the chaperone function of folding TLRs or integrins, at least in the steady state. It is unclear whether the disulfide bond mutant of gp96 is able to restore the function of other gp96 clients. It is plausible that C138 of gp96 is critical for the folding of some but not all gp96 clientele, particularly during oxidative stress conditions.
Our data should not be interpreted to argue against the prevailing notion that N-terminal dimerization of gp96 via other non-disulfide-dependent interaction is important for its function. In fact, the elegant work from the Gewirth group has revealed that the N-terminal open and the C-terminal closed twisted V-shaped dimer of gp96 could not possibly be enzymatically active, due to the unique arrangement of the N-M orientation of gp96, placing the catalytic residue Arg448 too far away from the γ phosphate of the ATP (18). The key question is how the N-terminal domain of the gp96 monomer is brought together. The high resolution of near full-length gp96 in complex with an ATP analog demonstrated convincingly that ATP binding itself is not the answer. Similarly, recent work from Agard's group unveiled that the HSP90 member undergoes conformational changes from the N-terminal open and the N-terminal closed state even in the absence of adenosine nucleotide (41). More functional and structural work is needed to understand the regulation and importance of N-terminal dimerization of HSP90 including gp96 in its function.
gp96 is known to chaperone both select integrins and TLRs. Because gp93 has conserved chaperone abilities, it is possible that gp93 is a natural chaperone for Toll and the various integrins expressed in Drosophila: αPS1, αPS2, αPS3, αPS4, αPS5, βPS, and βv (20). Further study is necessary to determine whether gp96 is able to complement gp93 function in Drosophila. More importantly, the quantitative difference between gp96 and gp93 in their chaperone function shall serve as a useful experimental platform to identify critical structural requirements for the optimal function of gp96. For example, domain swapping could be done between gp96 and gp93 to determine whether a weaker ATPase activity of gp93 was responsible for the decreased chaperone ability. More likely, the client-binding domain of gp96 might have changed significantly during evolution to accommodate the expansion and change of the client network in higher vertebrates. Detailed mapping of the potential client-binding domain of gp96 and gp93 might offer answers to these questions. Finally, several studies have indicated the presence of gp96 in the multimolecular chaperone complex in the ER including GRP78 and calreticulin to coordinate protein folding (42, 43). It is unclear whether gp93 is able to interact with these ER chaperones in the mammalian system to optimize its chaperone function.
gp96 is the ER paralog of the well-studied cytosolic HSP90. Known to chaperone a wide variety of clients, the chaperone function of HSP90 is further assisted by multiple cochaperones such as Hop, p23, Aha1, and CDC37 (44). Also a dimer, HSP90 is in a V-shaped conformation when bound to ADP to maintain an open N-terminal conformation and the C-terminal closed state, and then undergoes additional dimerization at the N terminus upon ATP binding (44–46). One of the long-standing puzzles of gp96 is that no single cochaperone has been discovered to specifically assist the function of gp96. Our experimental system, in conjunction with the evolutional conservation between gp96 and gp93, might facilitate the discovery of the elusive cochaperone of gp96. For instance, the gp93 cochaperones in Drosophila can be potentially discovered by expression cloning strategy based on the notion that such cochaperones should be able to enhance the chaperone function of gp93 in gp96-deficient murine cells.
In summary, our results demonstrate that Drosophila gp93 is an ortholog of mammalian gp96 both structurally and functionally. It is a bona fide chaperone in that it is able to compensate for the chaperone function of gp96 for both TLRs and integrins. Our structure-function studies reveal that the chaperone function of gp96/gp93 is not dependent on N-terminal intermolecular disulfide bond formation. Additional comparative studies between gp96 and gp93 shall be fruitful in uncovering their new client proteins as well as their essential molecular mechanisms in client folding. This effort might also contribute to the eventual generation of gp96-based pharmaceutics for the treatment of a variety of immune disorders.
Acknowledgments
We thank Dr. Bei Liu, Matthew Staron, Tao Wu, and Dr. Yi Li (all from Z.L.'s laboratory) for discussions and technical advice; the laboratory of Brenton Graveley for assistance in culturing Drosophila S2 cells; and Drs. Adam Adler, Pramod Srivastava, Linda Cauley, and Anthony Vella for helpful input.
Footnotes
This work was supported in part by National Institutes of Health Grants CA100191 and AI070603 (to Z.L.).
Abbreviations used in this paper: HSP, heat shock protein; ER, endoplasmic reticulum; WT, wild type; RIPA, radioimmunoprecipitation assay; DSP, dithiobis-succinimidyl propionate; HA, hemagglutinin; IB, immunoblot; MFI, mean fluorescence intensity; co-IP, coimmunoprecipitation.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci USA. 1986;83:3407–3411. doi: 10.1073/pnas.83.10.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–637. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Lee AS, Bell J, Ting J. Biochemical characterization of the 94- and 78-kilodalton glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 1984;259:4616–4621. [PubMed] [Google Scholar]
- 4.Mazzarella RA, Green M. ERp99, an abundant, conserved glycoprotein of the endoplasmic reticulum, is homologous to the 90-kDa heat shock protein (hsp90) and the 94-kDa glucose regulated protein (GRP94) J Biol Chem. 1987;262:8875–8883. [PubMed] [Google Scholar]
- 5.Koch G, Smith M, Macer D, Webster P, Mortara R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J Cell Sci. 1986;86:217–232. doi: 10.1242/jcs.86.1.217. [DOI] [PubMed] [Google Scholar]
- 6.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Li Z. Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol Cell. 2005;20:173–182. [PubMed] [Google Scholar]
- 8.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 9.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 11.McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 12.Melnick J, Dul JL, Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Li Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood. 2008;112:1223–1230. doi: 10.1182/blood-2008-03-143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for Toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Srivastava PK. Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein folding and antigen presentation. EMBO J. 1993;12:3143–3151. doi: 10.1002/j.1460-2075.1993.tb05983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. 2007;282:35612–35620. doi: 10.1074/jbc.M704647200. [DOI] [PubMed] [Google Scholar]
- 18.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 20.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–E90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 22.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 23.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 25.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 28.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 29.Gangloff M, Murali A, Xiong J, Arnot CJ, Weber AN, Sandercock AM, Robinson CV, Sarisky R, Holzenburg A, Kao C, Gay NJ. Structural insight into the mechanism of activation of the Toll receptor by the dimeric ligand Spätzle. J Biol Chem. 2008;283:14629–14635. doi: 10.1074/jbc.M800112200. [DOI] [PubMed] [Google Scholar]
- 30.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 31.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann JA. The immune response of Drosophila. Nature. 2003;426:33–38. doi: 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- 33.Robert J, Cohen N. Evolution of immune surveillance and tumor immunity: studies in Xenopus. Immunol Rev. 1998;166:231–243. doi: 10.1111/j.1600-065x.1998.tb01266.x. [DOI] [PubMed] [Google Scholar]
- 34.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J Embryol Exp Morphol. 1972;27:353–365. [PubMed] [Google Scholar]
- 37.Pöhner C, Hilbrig F, Jérôme V, Freitag R. Design and characterization of stimuli-responsive FLAG-tag analogues and the illumination-induced modulation of their interaction with antibody 4E11. Biotechnol Prog. 2006;22:1170–1178. doi: 10.1021/bp060011h. [DOI] [PubMed] [Google Scholar]
- 38.Pfeffer SR. Unsolved mysteries in membrane traffic. Annu Rev Biochem. 2007;76:629–645. doi: 10.1146/annurev.biochem.76.061705.130002. [DOI] [PubMed] [Google Scholar]
- 39.Qu D, Mazzarella RA, Green M. Analysis of the structure and synthesis of GRP94, an abundant stress protein of the endoplasmic reticulum. DNA Cell Biol. 1994;13:117–124. doi: 10.1089/dna.1994.13.117. [DOI] [PubMed] [Google Scholar]
- 40.Wearsch PA, Nicchitta CV. Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry. 1996;35:16760–16769. doi: 10.1021/bi962068q. [DOI] [PubMed] [Google Scholar]
- 41.Southworth DR, Agard DA. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meunier L, Usherwood YK, Chung KT, Hendershot LM. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandenbroeck K, Martens E, Alloza I. Multi-chaperone complexes regulate the folding of interferon-γ in the endoplasmic reticulum. Cytokine. 2006;33:264–273. doi: 10.1016/j.cyto.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 45.Harris SF, Shiau AK, Agard DA. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure. 2004;12:1087–1097. doi: 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Prodromou C, Piper PW, Pearl LH. Expression and crystallization of the yeast Hsp82 chaperone, and preliminary x-ray diffraction studies of the amino-terminal domain. Protein Struct Funct Genet. 1996;25:517–522. doi: 10.1002/prot.13. [DOI] [PubMed] [Google Scholar]