

Abstract
Lymphocyte activation requires Ca2+ influx through specialized Ca2+ channels in the plasma membrane. In T cells the predominant Ca2+ channel is the Ca2+ release activated Ca2+ (CRAC) channel encoded by the gene ORAI1. ORAI1 is activated by stromal interaction molecule (STIM) 1 that is localized in the ER where it senses the concentration of stored Ca2+. Following antigen binding to immunoreceptors such as the TCR, ER Ca2+ stores are depleted, STIM1 is activated and ORAI1-CRAC channels open resulting in what is referred to as store-operated Ca2+ entry (SOCE). Mutations in ORAI1 and STIM1 genes in human patients that lead to expression of non-functional ORAI1 or complete lack of ORAI1 or STIM1 protein are associated with a unique clinical phenotype that is characterized by immunodeficiency, muscular hypotonia and anhydrotic ectodermal dysplasia, as well as, in the case of STIM1 deficiency, autoimmunity and lymphoproliferative disease. The immunodeficiency in these patients is due to a severe defect in T cell activation but not in lymphocyte development. This review describes the immunological and non-immunological phenotypes of patients with defects in SOCE and CRAC channel function and discusses them in the context of similar immunodeficiency diseases and animal models of ORAI1 and STIM1 function.
Keywords: ORAI1, STIM1, CRAC, SOCE, store-operated calcium entry, Ca2+, T cells, lymphocytes, immunodeficiency, SCID, congenital myopathy, ectodermal dysplasia, amelogenesis imperfecta
1. INTRODUCTION
Mutations in about 80 different genes have been linked to defects in adaptive immune responses involving T and B cells [1; 2]. In the most dramatic form, severe combined immunodeficiency (SCID) is characterized by defective development or function of T and B cells, thus impairing the cellular and humoral arm of the adaptive immune response. Disease onset usually occurs within the first months of life with severe infections involving viral, bacterial and fungal pathogens. While the X-linked form of SCID due to mutations in the common γ chain (γc) of the IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 receptor accounts for about half the cases of SCID, mutations in more than a dozen other genes have been identified which are involved in cytokine signaling pathways (JAK3, IL-7Rα), purine nucleotide metabolism (ADA, PNP), VDJ recombination (RAG1, RAG2), DNA repair (DNA ligase IV, Artemis, Cernunnos), mitochondrial energy metabolism (AK2), thymic abnormalities (Coronin 1A, FOXN1), antigen presentation and signal transduction in lymphocytes [2]. Mutations in most signaling genes such as the CD3δ, εand ζ chain, ZAP-70, CD45 and IL-7Rα result in impaired T cell development (Figure 1)[3].
Figure 1. Molecules mutated in SCID and T cell activation defects.

Patients with mutations in ORAI1, STIM1, CD3γ and IκBα (yellow symbols) have impaired T cell activation and but normal numbers of lymphocytes. By contrast, SCID patients with mutations in other signaling molecules (red symbols) show severe defects in the development of T cells or T cell subsets [2]. Abbreviations: DAG, diacylglycerol; InsP3, inositol-1,4,5-trisphosphate; LAT, linker for activation of T cells; NFAT, nuclear factor of activated T cells; PLC, phospholipase; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; SLP76, SH2-domain-containing leukocyte protein of 76 kDa; STIM1, stromal interaction molecule 1; ZAP-70, ζ-chain-associated protein kinase of 70 kDa.
In addition, clinical phenotypes very similar to SCID have been described that result from severe defects in T cell activation in the presence of normal lymphycyte development. Several cases of patients with normal lymphocyte numbers but impaired T cell activation have been described in the literature [4; 5; 6; 7] but the genetic defect underlying most of these forms of Primary Immunodeficiencies (PIDs) has remained unresolved. In a few of these cases, the T cell activation defect has been attributed to impaired Ca2+ influx [8; 9; 10; 11]. Ca2+ influx is activated in response to TCR stimulation and results from the opening of specialized Ca2+ channels in the plasma membrane (Figure 2). The resulting increase in cytoplasmic Ca2+ concentration is required for T cell activation, proliferation and regulation of cytokine gene expression [12]. Ca2+ channels in T cells are activated following TCR engagement, activation of phospholipase C (PLC) γ1 and production of the second messanger inositol 1,4,5-triphosphate (InsP3) which binds to InsP3 receptors in the membrane of the ER and mediates release of Ca2+ from ER stores. PLCγ isoforms are also activated following engagement of other immunoreceptors such as the B cell receptor and Fc receptors on B cells, mast cells and monocytes, respectively. Emptying of Ca2+ stores results in a transient increase in the cytosolic Ca2+ concentration and is the trigger for activation of the Ca2+ release activated Ca2+ (CRAC) channel in the plasma membrane [13]. Ca2+ influx resulting from CRAC channel opening as previously noted is referred to as store-operated Ca2+ entry (SOCE) because it depends on the depletion of ER Ca2+ stores. The rise in intracellular [Ca2+] resulting from TCR engagement therefore is the sum of Ca2+ released from ER stores and Ca2+ influx from the extracellular space. Given the small volume of the ER in lymphocytes, the relative contribution of Ca2+ entering the T cell from the outside is typically much greater than that of Ca2+ released from stores. SOCE is therefore responsible for the sustained rise in cytosolic [Ca2+] required for activation of transcription factors such as NFAT, cytokine gene expression and induction of lymphocyte proliferation.
Figure 2. Store-operated Ca2+ entry (SOCE) in T cells.

T-cell receptor (TCR) engagement results in the activation of tyrosine kinases Lck and ZAP-70, assembly of the adaptor protein complex containing SLP76 and LAT and activation of PLCγ1. The latter hydrolyses PtdIns(4,5)P2 to InsP3 and DAG. InsP3 binds to and opens InsP3 receptors (InsP3Rs) in the ER, resulting in the release of Ca2+ from ER stores, reduction of [Ca2+]ER and transient increase in [Ca2+]i. The descrease in [Ca2+]ER is sensed by STIM1 resulting in binding of STIM1 to ORAI1 and opening of the CRAC channel. Ca2+ influx results in increased intracellular Ca2+ concentration [Ca2+]i from ~ 100 nM to ~1 μM. Sustained elevation of [Ca2+]i is required for activation of the phosphatase calcineurin, nuclear translocation of the transcription factor NFAT and cytokine gene expression. For abbreviations see Figure 1.
While the CRAC channel is the main gateway of Ca2+ influx in T cells [12], other Ca2+ channels such as voltage gated Ca2+ channels [14] and transient receptor potential (TRP) channels [15] have been proposed to be functional in T cells as well. Ion channels that do not conduct Ca2+ themselves are indirectly involved in the regulation of Ca2+ influx in lymphocytes. These include the nonselective, Na+ permeable cation channel TRPM4 and the potassium channels KCNN4 and KCNA3 which together regulate the plasma membrane potential and thereby the driving force for Ca2+ influx along an electrochemical gradient [16].
The importance of Ca2+ influx through CRAC channels for immunity against pathogens is highlighted by the existence of patients with severe combined immunodeficiency due to the lack of SOCE and ICRAC [10; 11; 17; 18; 19]. Affected children are susceptible to recurrent, potentially life-threatening infections early in life (< 1y) due to a severe T cell activation defect despite normal lymphocyte numbers. The causes of the combined immunodeficiency and the defect in Ca2+ influx in these patients have recently been identified. They are caused by mutations in genes responsible for CRAC channel function, ORAI1 as the pore forming subunit of the CRAC channel and stromal interaction molecule 1 (STIM1) as an essential CRAC channel activitating protein. Mutations in both genes are associated with a unique clinical phenotype that is characterized by immunodeficiency, anhydrotic ectodermal dysplasia and congenital myopathy.
2. ORAI1 DEFICIENCY
2.1. ORAI1: PORE FORMING SUBUNIT OF THE CRAC CHANNEL
The CRAC channel was identified almost 20 years ago based on its unique biophysical properties in patch clamp recordings while its molecular identity remained elusive. CRAC channel currents are highly selective for Ca2+ relative to other cations such as Na+ and are very small compared to many other cation channels [20; 21]. The gene encoding the CRAC channel was identified in 2006 in genome-wide RNAi screens for regulators of Ca2+ signaling and activation of the transcription factor NFAT [17; 22; 23] and by positional cloning for the gene mutation in two immunodeficient patients with defects in CRAC channel function [17]. Positional cloning of the CRAC channel gene was successful because of the functional identification of putative heterozygous carriers of the disease gene through in vitro tests for defects in Ca2+ influx (for details see Figure 3). RNAi and positional cloning screens identified the hypothetical genes olf-186F in drosophila and its human homolgue FLJ14466 on chromosome 12q24 as a candidate gene for the CRAC channel [17; 22; 23]. The human gene was termed ORAI1 or CRAC modulator (CRACM) 1 [22] and the drosophila gene dOrai, named after the Hours (Orai) in Greek mythology [24].
Figure 3. Phenotypic detection of putative heterozygous carriers.

Heterozygous carriers of an autosomal recessive mutation that impairs Ca2+ influx in immunodeficient patients were predicted based on in vitro analysis of SOCE in T cells from relatives of immunodeficient patients (see pedigree in Figure 4A). A, Ca2+ influx was measured in T cells stimulated with thapsigargin (TG) followed by readdition of lower than physiological extracellular Ca2+ (0.5 mM, black bar) to reveal a defect in Ca2+ influx. B, T cells from immunodeficient patients (purple trace in A and bar in B) lack SOCE, whereas T cells from relatives have normal (blue) or markedly reduced (green) Ca2+ influx, indicating that the latter, although healthy, may be heterozygous carriers of a recessive, disease-causing mutation. Not shown, The haplotypes of immunodeficient patients, predicted heterozygotes and predicted wild-type individuals from the same family were determined by genome-wide single nucleotide polymorphism (SNP) analysis and used to calculate multipoint parametric LOD scores in two independent analyses assuming an autosomal recessive and autosomal dominant mode of inheritance, respectively [17]. A 6.5 Mb interval on chromosome 12q24 containing the novel gene ORAI1 was identified to be linked to immunodeficiency disease with a LOD (log10 of the odds) score of 5.7. Identification of heterozygous carriers may be a useful tool for identification of gene defects in other rare diseases because it increases the number of haplotypes available for linkage analysis. DNA sequence analysis of ORAI1 revealed that all predicted carriers were indeed heterozygous for the ORAI1-R91W mutation. Figure modified from the version originally published in [17].
ORAI1 is a plasma membrane protein with four transmembrane domains (Figure 2). It is highly conserved throughout evolution but structurally unrelated to other ion channel proteins. Two paralogues, ORAI2 (or CRACM2) and ORAI3 (CRACM3), share a high degree of sequence similarity with ORAI1, especially in their transmembrane domains. ORAI1 serves as the pore forming subunit of the CRAC channel using a negatively charged glutamate residue in the first transmembrane domain, E106, as Ca2+ binding sites in the ion channel pore as shown by site-directed mutagenesis of glutamate and aspartate residues in ORAI1 [25; 26; 27] and cystein scanning mutagenesis of ORAI1 transmembrane domains for pore lining residues [28]. Functional CRAC channels likely consist of four ORAI1 subunits each of which contributes one or more negatively charged amino acid residues for the coordinated Ca2+ binding in the channel pore [29; 30; 31]. ORAI1 is expressed in many tissues and cell types in the human body [32; 33] consistent with the observation of SOCE and CRAC channel currents in many cell types (reviewed in [20]). The ORAI1 paralogues ORAI2 and ORAI3, too, are tetraspanning membrane proteins and form Ca2+ channels when ectopically expressed in vitro together with STIM1. The roles of endogenous ORAI2 and ORAI3 proteins in Ca2+ influx and CRAC channel function however have not yet been resolved.
2.2. ORAI1 MUTATIONS
Autosomal recessive mutations in the ORAI1 gene have been reported in six patients from three unrelated families. All patients lack store-operated Ca2+ entry in T cells and other cell types and suffer from severe immunodeficiency with recurrent and opportunistic infections. They have normal lymphocyte numbers and subsets but T cell activation is severely compromised with strongly impaired proliferative responses in vitro.
ORAI1-R91W missense mutation
Two patients were born to consanguineous parents of Turkish origin (Figure 4A). Polyclonal T cell lines established from the patients lack SOCE in reponse to TCR stimulation and thapsigargin, an inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), or ionomycin, both of which passively deplete ER Ca2+ stores and activate the CRAC channel [9]. A CRAC channel defect was established in these patients as ICRAC was undetectable ruling out indirect effects on SOCE as the cause of disease [9]. Both patients are homozygous for a missense mutation in ORAI1 that results in the substitution of a highly conserved arginine residue at the beginning of its first transmembrane domain with tryptophane (ORAI1-R91W) (Figure 5A)[17]. The R91W mutation disrupts CRAC calcium channel function but does not interfere with ORAI1 expression at the plasma membrane or ORAI1 interaction with STIM1 [17; 34]. It is likely that the mutation interferes with CRAC channel opening by curtailing the mobility of the first membrane domain in the lipid bilayer because substitution of R91 with hydrophobic but not charged or neutral amino acids abolishes channel function (SF unpublished)[35].
Figure 4. Pedigrees of patients lacking SOCE.

ORAI1-R91W patients were first reported in [17]. ORAI1-A88SfsX25 and ORAI1-A103E/L194P patients were first reported in [33](pedigrees reproduced with permission). STIM1-E128RfsX9 (E136X) patients were first reported in [19](pedigree reproduced with permission). Black symbols depict patients, strike-through symbols deceased individuals, dotted symbols heterozygous carriers. All heterozygous carriers are healthy.
Figure 5. Mutations in ORAI1 and STIM1 in immunodeficient patients lacking Ca2+ influx.

A, ORAI1 is a plasma membrane Ca2+ channel with four transmembrane domains and intracellular N- and C-termini (for details see text). Mutations: (i) An Arg→Trp (R91W) single amino acid substitution in ORAI1 at the beginning of the first transmembrane (TM1) domain results in expression of a nonfunctional protein [17]. (ii) A frameshift (fs) nonsense mutation in the first exon of ORAI1 (A88SfsX25) results in premature termination, nonsense mediated decay of ORAI1 mRNA and abolished ORAI1 protein expression [33]. (iii) Two independent single amino acid substitutions, A103E and L194P, in TM1 and TM3, respectively interfere with ORAI1 protein expression [33]. B, STIM1 is a single-pass transmembrane (TM) protein localized in the ER featuring an EF hand domain (EFh), sterile alpha motif (SAM), coiled-coil (CC), serin/proline (S/P) and lysine (K) rich domains (for details see text). Mutation: A frameshift nonsense mutation in exon 3 of STIM1 (E128RfsX9) results in premature termination, nonsense mediated mRNA decay of STIM1 and abolished STIM1 protein expression [19].
ORAI1 expression is abolished in patients from two families unrelated to the family described above. An ORAI1-A88EfsX25 frameshift nonsense mutation was identified in one of two patients born to consanguineous French parents (Figure 4B). Lymphocytes from one patient lacked CRAC channel currents and SOCE resulting in a severe T cell activation defect [11]. He is homozygous for an insertion mutation in exon 1 of ORAI1 which results in a frame shift and premature termination at position 112 (ORAI1-A88EfsX25) at the end of the first transmembrane domain (Figure 5A) [33]. No ORAI1 mRNA or ORAI1 protein were detectable in the patient’s cells indicating that the mutant transcript is degraded by nonsense mediated mRNA decay.
Two independent missense mutations (ORAI1--A103E/L194P) was identified in one of two patients born to unrelated parents from a third family (Figure 4C). The propositus of this family lacked SOCE in T cells, B cells, platelets and fibroblasts [10]. He is compound heterozygous for two missense mutations in exon 2 of ORAI1 resulting in single amino acid substitutions in the first (A103E) and third (L194P) transmembrane domain of ORAI1 (Figure 5A) [33]. ORAI1 protein was not detectable in the patient’s fibroblasts or in HEK293 cells ectopically expressing the ORAI1 mutants whereas ORAI1 mRNA levels were normal. Both mutations interfere with protein expression, presumably by destablizing the transmembrane domains in which they are located. The patient’s parents are heterozygous for either one of the two mutations, but the presence of one wild-type allele results in normal SOCE and prevents immunodeficiency.
2.3. CLINICAL PHENOTYPE
Immunodeficiency
Immunodeficiency in ORAI1 deficient patients is characterized by recurrent severe infections with viral, bacterial, mycobacterial and fungal pathogens causing pneumonia, meningitis, enteritis, gastrointestinal candidiasis and sepsis (Table 1)[10; 11; 18; 33; 36]. Antibiotic and intravenous immunoglobulin therapy are only partially effective in controlling infections in the ORAI1 deficient patients. One patient with ORAI1-R91W mutation suffered from recurrent infections since birth including BCGitis following vaccination, rotavirus enteritis, pneumonia and meningitis; he died from pneumonia and gastrointestinal sepsis at 11 months of age [18]. His younger brother was treated with recombinant IL-2 in the first month of his life because his T cells proliferated normally in vitro in the presence of IL-2. He received HSCT at 4 months of age and is free of severe infections at the current age of 16 years. Two brothers lacking ORAI1 expression due to a frameshift nonsense mutation (ORAI1-A88SfsX25) both suffered from recurrent infections since the first weeks of life including chronic diarrhea, mucocutaneous candidiasis, pneumonia and bacterial pyelonephritis. One patient died at 5 months of age from pneumonia, his younger brother succumbed to fever and progressive encephalopathy at 11 months of age. Lack of ORAI1 expression in a patient who is compound heterozygous for ORAI1-A103E and ORAI1-L194P mutations resulted in chronic diarrhea, pneumonia and CMV infections. He does not suffer from severe infections following HSCT at 4 months of age but developed an EBV-associated polymorphic B cell lymphoma. His older brother had suffered from protracted bloody diarrhea, chlamydia pneumonia and toxoplasma encephalitis from which he died of at 8 months of age.
Table 1.
Clinical phenotypes of patients with ORAI1 and STIM1 mutations
| ORAI1-R91W | ORAI1-A88SfsX25 | ORAI1-A103E/L194P | STIM1-E128RfsX9 | |
|---|---|---|---|---|
| Immunodeficiency | 2/2 | 2/2 | 2/2 | 3/3 |
| Viral, bacterial, fungal infections | ||||
| Autoimmunity | ||||
| AIHA | – | – | – | 3/3 |
| Thrombocytopenia | – | 1/2 | – | 3/3 |
| Neutropenia | – | 1/2 | – | – |
| Lymphoadenopathy/Hepatosplenomegaly | – | – | – | 3/3 |
| Myopathy | 2/2 | 2/2 | 2/2 | 3/3 |
| Ectodermal Dysplasia | ||||
| Amelogenesis imperfecta | 1/2* | –* | 1/2* | 3/3 |
| Anhydrosis | 2/2 | ? | 1/2 | – |
| Encephalopathy | – | 2/2 | – | – |
| Facial dysmorphy | – | 1/2 | – | – |
| Lethality in first year of life | 1/2 | 2/2 | 1/2 | – |
For details see text.
one or more patients died before dentition.
The clinical symptoms of immunodeficiency in all ORAI1 deficient patients resembled those observed in patients with severe combined immunodeficiency. In contrast to the majority of SCID patients, total lymphocyte counts and numbers of CD4+ T cells, CD8+ T cells and B cells were normal in patients lacking functional ORAI1. Numbers of Treg cells could not be evaluated after patients had been treated by HSCT. In contrast to normal lymphocyte numbers, T cell activation is severely compromised in all patients evidenced by impaired proliferative responses to TCR-dependent and independent stimuli in vitro and absent cutaneous delayed-type hypersensitivity reactions in vivo [10; 11; 18; 33; 36]. Interestingly, T cells from patients lacking ORAI1 expression proliferated normally in response to stimulation with PMA and ionomycin whereas T cells from patients expressing a non-functional form of ORAI1 (R91W) did not. The difference may be due to a potential compensatory effect of ORAI2 or ORAI3 that occurs in the absence of ORAI1, indicating that the R91W mutation may exert a moderate inhibitory effect on SOCE. This finding is consistent with partially impaired SOCE in T cells of heterozygous carriers of the R91W mutation in contrast to normal SOCE in T cells from individuals with monoallelic expression of wild-type ORAI1 [33]. It is important to note, however, that heterozygous carriers of the various mutations in ORAI1 (and STIM1) were healthy and did not show increased susceptibility to infections.
Defective T cell activation in the absence of SOCE manifests as an inability of ORAI1 deficient T cells to activate the transcription factor NFAT [10; 18; 36] and to produce cytokines such as IL-2, IL-4, IL10, IFNγ and TNFα [18; 36]. Despite the activation defect in vitro, T cells from ORAI1 deficient patients had an activated phenotype expressing HLA-DR+, CD45RO+ or CD29+ at their cell surface [33]. It is of note that autoimmune neutropenia and thrombocytopenia were observed in only one of six ORAI1 deficient patients [11; 33] which is in contrast to the higher prevalence of autoimmunity in patients lacking STIM1 as discussed further below.
Numbers of B cells and immunoglobulin levels were normal or moderately elevated in all ORAI1 deficient patients despite impaired SOCE observed in B cells from patients with ORAI1-R91W and ORAI1-A103E/L194P mutations [8; 10; 33]. A role for SOCE in B cell activation has been reported (reviewed in [37; 38; 39] and is consistent with ORAI1 expression in B cells [40]. Despite normal serum Ig levels, ORAI1 deficient patients failed to mount antigen specific antibody responses in response to vaccination with diphteria and pertussis or infection with candida which is a secondary effect caused by the defect in T cell activation. SOCE and ORAI1 expression have also been observed in NK cells [40; 41; 42]. While not directly tested, impaired SOCE in NK cells and attenuated NK function may contribute to the patients’ immunodeficiency.
In summary, the immunodeficiency in patients lacking expression of functional ORAI1 is dominated by a defect in T cell, and potentially also B and NK cell, activation while lymphocyte development is normal.
Nonimmunological symptoms
In addition to immunodeficiency, ORAI1 deficient patients are afflicted by non-immunological symptoms that are unique to patients with defects in SOCE and define a novel disease entity. Non-progressive muscular dysplasia and anhydrotic ectodermal dysplasia (EDA) are present from birth. In addition to providing important clues for differential diagnosis, both EDA and myopathy are clinically relevant.
EDA presented with an inability to sweat and recurrent fever episodes due to impaired thermoregulation, especially in the summer, in the majority of ORAI1 deficient patients [33]. In the absence of available skin biopsies, it remains unclear whether eccrine sweat glands do not develop or are non-functional in these patients. EDA is furthermore characterized by a defect in the calcification of the dental enamel matrix, termed hypocalcified amelogenesis imperfecta, resulting in the use-dependent loss of soft dental enamel and the painful exposure of the underlying dentin (Figure 5A). Due to its debilitating nature, this defect necessitates intensive restorative dental treatment. Other features often observed in ectodermal dysplasia such as hair or nail defects were not present in ORAI1 deficient patients.
Congenital myopathy was observed in all patients with ORAI1 mutations. Generalized muscular hypotonia resulted in poor head control, delayed ambulation and a positive Gower’s sign. Although not initially life-threatening, the hypotonia is clinically significant in the two ORAI1 deficient patients who survive after HSCT because it affects the respiratory muscles and impairs the patients’ ability to cough and clear mucus from their bronchial system causing superinfection, bronchiectasis and chronic pulmonary disease necessitating supplementary oxygen therapy [33]. A residual defect in B cell function in ORAI1 deficient patients may contribute to the development of bronchietasis given that HSCT in ORAI1 deficient patients resulted in mixed chimerism despite full conditioning prior to transplantation. Mixed chimerism is also observed in other PID patients that have received HSCT, for instance those with Wiskott Aldrich syndrome [93]. The histological correlate of the muscular hypotonia are atrophic fast twitch (type II) fibers found in one patient with ORAI1-R91W mutation. Other structural abnormalities found in other congenital myopathies such as cores and rod formation, ragged-red fibers or Oil-red-O positive lipid droplets were not observed. Although histology was available from only one patient, the consistent presence of muscular hypotonia in all ORAI1 deficient patients is consistent with the recently proposed role of SOCE in the development and/or function of skeletal muscle fibers [43; 44; 45; 46] and expression of ORAI1 in skeletal muscle [32; 33].
It is of note, that patients from one family who lacked ORAI1 expression because of a frameshift nonsense mutation (A88EfsX25) presented with progressive idiopathic encephalopathy characterized by developmental delay, seizures and a myelinization delay in magnetic resonance imaging at 5 months of age [11; 33]. One patient also showed facial dysmorphisms and a defect in posterior arch closing. Because encephalopathy occurred in patients from only one family with ORAI1 mutations and the neurological and mental development was normal in patients from the other two families, it is unlikely that CNS involvement is part of the clinical disease spectrum of ORAI1 deficiency. While SOCE has been described in neurons [47], ORAI1 expression in the CNS is very low compared to other tissues [33]. Alternative explanations for the encephalopathy observed in this family are infection with a neurotrophic pathogen, not uncommon in patients with T cell defects, or an additional mutation in a notably inbred family.
3. STIM1 DEFICIENCY
3.1. STIM1: ER Ca2+ SENSOR AND CRAC CHANNEL ACTIVATOR
STIM1 is a single-pass transmembrane protein localized predominantly in the ER. It has a dual role in sensing the Ca2+ concentration in the ER and activation of the CRAC channel by binding to ORAI1 (as well as other ion channels in the plasma membrane such as TRPC channels) [16; 48; 49]. STIM1 was initially speculated to function as a tumor suppressor implicated in the pathogenesis of rhabdomyosarcoma [50; 51] before being identified as an essential regulator of SOCE in RNAi screens [48; 49]. STIM1 binds Ca2+ in the ER through a pair of low-affinity EF hand calcium-binding domains in its N terminus adjacent to a sterile alpha motif (SAM) protein-protein interaction domain. Upon depletion of Ca2+ stores and reduction of [Ca2+]ER, Ca2+ dissociates from the EFh domain resulting in a conformational change in the EFh-SAM domain enabling STIM1 to multimerize and translocate to the plasma membrane where it binds to and activates ORAI1 CRAC channels [16]. A ~ 110 amino acid domain in the C terminus of STIM1 was identified as a minimal CRAC channel activation domain [52; 53; 54; 55] that interacts with ORAI1. STIM1 activation coincides with the formation of puncta that contain STIM1 and ORAI1 and which are the site of localized Ca2+ influx.
The protein sequence of STIM1 and its overall protein domain structure are conserved in its paralogue STIM2, which, like STIM1, is located in the ER and functions as a positive regulator of SOCE [56]. It activates Ca2+ influx upon smaller decreases in ER Ca2+ concentrations than STIM1 and was therefore suggested to regulate basal cytosolic Ca2+ concentrations [57]. In addition, STIM2 seems to be required for sustained SOCE as T cells from Stim2 deficient mice fail to maintain elevated intracellular Ca2+ concentrations and Ca2+ dependent nuclear translocation of the transcription factor NFAT at later time points after activation despite initially normal SOCE and CRAC channel function [58].
3.2. STIM1 MUTATION
Mutations in STIM1 have so far been reported in immunodeficient patients from only one consanguineous family. As in the ORAI1 deficient patients discussed above, lymphocyte numbers were normal in patients from this family but displayed a pronounced proliferation defect when stimulated in vitro [19]. Store-operated Ca2+ entry was undetectable in fibroblasts from the propositus following passive store-depletion. Two of the patients that could be analyzed were homozygous for a frameshift nonsense mutation in exon 3 of STIM1 resulting in premature termination of STIM1 protein at position 136 (E128RfsX9). The mutation results in nonsense mediated mRNA decay as STIM1 mRNA levels were greatly reduced or absent and a truncated, N-terminal STIM1 fragment was not detectable in the patient’s cells [19]. Ectopic expression of wild-type STIM1 in the patients’ cells restored SOCE, as did STIM2 overexpression confirming that STIM1 and STIM2 have overlapping functions as CRAC channel activators [57; 58]. Endogenous expression levels of STIM2 in the patient’s cells, however, are not sufficient to compensate for the lack of STIM1 in the patients.
3.3. CLINICAL PHENOTYPE
Immunodeficiency
Clinically, immunodeficiency in STIM1 deficient patients manifested through recurrent bacterial and viral infections in all three siblings. The propositus of the family suffered from multiple episodes of bacterial sepsis caused by Streptococcus pneumoniae and Escherichia coli, recurrent urinary tract infections, otitis media and pneumonia as well as Cytomegalovirus (CMV) and Varicella Zoster Virus (VZV) infections. Her younger sister, of whom DNA was not available to confirm the STIM1 mutation, suffered from prolonged diarrhea, EBV infection and enteroviral encephalitis. The third affected child in the family had one episode of sepsis while being treated with intravenous immunoglobulins since birth because of the known immunodeficiency in his older siblings. The propositus and her younger sister died at 9 years and 18 months of age from complications of HSCT and enteroviral encephalitis, respectively, whereas their younger brother survived after HSCT at 15 months of age and is free of severe infections. It is of note that the propositus survived for 9 years without HSCT which is in contrast to the apparent earlier lethality in ORAI1 deficient patients.
The immunodeficiency in the STIM1 deficient patients is due to severely impaired T cell activation. Proliferation in vitro in response to TCR crosslinking or recall antigens (VZV, Tetanus Toxoid) was barely detectable, that to mitogenic stimulation with phytohemagglutinin or phorbol 12-myristate 13-acetate plus ionomycin was markedly reduced. Serum Ig levels were normal or close to the normal range in two patients from the same family while IgG serum concentrations were significantly reduced in their sister who suffered from nephrotic syndrome.
Lymphocyte development appears to be unperturbed in patients lacking STIM1 expression. Total lymphocyte numbers were normal or slightly reduced. The relative distribution of T and B cells was normal, although percentages of CD8+ T cells were moderately decreased in two of three patients. The numbers of CD4+ CD45RA+ naive T cells and CD4+CD45RA+CD31+ recent thymic emigrants were decreased in the propositus [19], consistent with an activated or memory T cell phenotype in the presence of recurrent infections in the patient. Her T cell repertoire, however, was normal [19]. Normal lymphocyte counts were also observed in ORAI1 deficient patients. This is remarkable because a number of signaling molecules that are required for generating or transducing Ca2+ signals in T cells such as ZAP-70, Itk, LAT or calcineurin are associated with impaired T cell development when deleted in mice or mutated in human patients (Figure 1) [59; 60; 61; 62; 63]. In addition, Ca2+ signals have been observed in immature T cells and were implicated in positive selection of T cells in the thymus [64]. Normal CD4+ and CD8+ T cell numbers in ORAI1 and STIM1 deficient patients as well as Orai1−/− and Stim1−/− mice suggest, however, that SOCE may be dispensable for T cell development.
Autoimmunity and lymphoproliferation
In addition to immunodeficiency, all STIM1 deficient patients showed signs of lymphoproliferative and autoimmune disease Table 1) [19]. Lymphadenopathy and hepatosplenomegaly were present in two of the patients but not in their younger brother who had received HSCT at 15 months of age. Normal Fas-mediated apoptosis was observed in T cells from one of the patients indicating that lymphoproliferation is not due to impaired cell death. Autoimmune thrombocytopenia was present in all three, and autoimmune hemolytic anemia (AIHA) in two patients. Numbers of CD4+ CD25+ FoxP3+ Treg cells were reduced in the blood of one of the patients from whom material was available for analysis [19]. A similar reduction of Treg cells was found in mice with T cell specific deletion of both Stim1 and Stim2 (Figure 6C)[58]. While the reduced numbers of Treg cells provide a plausible explanation for the symptoms observed in STIM1 deficient patients, other factors such as impaired negative selection of autoreactive T cells in the thymus may contribute to the autoimmunity. It is of note that patients lacking STIM1 expression did not present with more severe symptoms typical of X-linked immune dysregulation, polyendocrinopathy, enteropathy (IPEX) syndrome which is caused by mutations in the FOXP3 gene [65]. This may be due to impaired effector T cell function in patients with STIM1 but not FOXP3 deficiency.
Figure 6. Non-immunological phenotypes in patients and mice lacking SOCE.
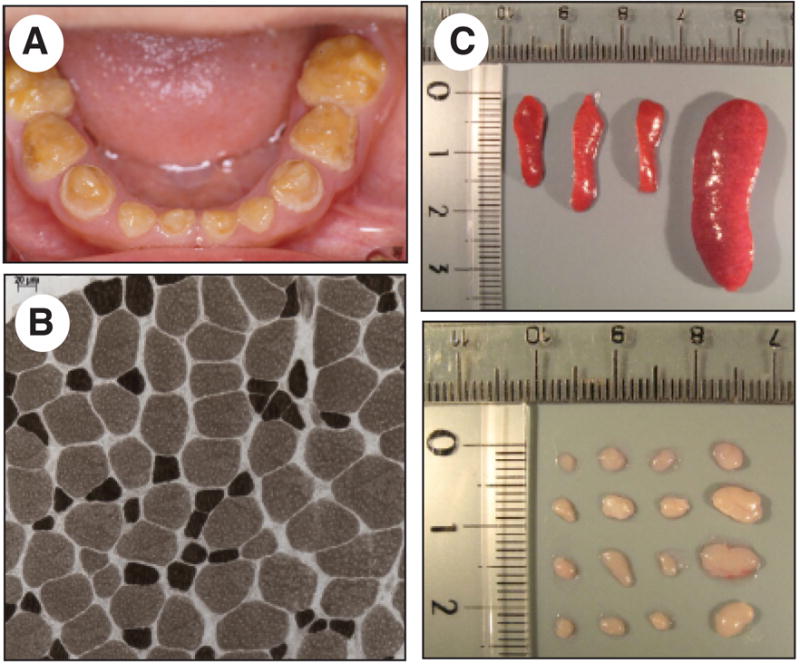
A, Amelogenesis imperfecta type III in a patient with ORAI1-R91W mutation. The dental enamel is hypocalcified resulting in use-dependent loss of the enamel layer. Shown are deciduous teeth at 6 years of age. B, Muscular dysplasia in a patient with ORAI1-R91W mutation. Atrophic type II muscle fibers in ATPase staining of a muscle biopsy at age 5 years (reproduced with permission from [33]. C, Splenomegaly and lymphadneopathy in Stim1f/f Stim2f/f Cd4-Cre mice [58]. Mice also show leukocytic infiltration of lung and liver due to reduced numbers and impaired function of Foxp3+ regulatory T cells. A similar lymphoproliferative phenotype was observed in patients with STIM1 mutation [19].
Nonimmunological symptoms
Patients lacking STIM1 expression – like ORAI1 deficient patients – suffer from ectodermal dysplasia and congenital myopathy [19]. Ectodermal dysplasia manifests with a severe defect in dental enamel formation. The anhydrosis observed in patients with ORAI1 mutations was not reported. Myopathy in STIM1 deficient patients is characterized by non-progressive global muscular hypotonia and partial iris hypoplasia. A muscle biopsy and electromyography failed to show abnormalities indicative of common neuropathies or myopathies. Given the similar myopathy in ORAI1 deficient patients, the structural abnormalities in skeletal muscle from Stim1−/− mice and the severely reduced resistance to fatigue in myoblasts from these mice, STIM1, ORAI1 and store-operated Ca2+ entry seem to play an important if somewhat unexpected role in skeletal muscle development and/or function.
4. DIFFERENTIAL DIAGNOSIS
T cell activation defects
Primary immunodeficiencies caused by a T cell activation defect but with normal lymphocyte counts as in ORAI1 and STIM1 deficiency present with similar if somewhat milder clinical features than the typical T−B+ or T−B− forms of SCID. ORAI1 and STIM1 deficient patients were initially diagnosed because of impaired T cell proliferation in vitro and impaired skin delayed type hypersensitivity responses to recall antigens in vivo. Similar T cell activation defects with attenuated proliferative responses and cytokine expression but normal lymphocyte numbers have been described but the underlying mutations have not been identified [4; 5; 7; 67; 68]. A clinical SCID-like phenotype resulting from impaired T cell activation in the presence of low to normal numbers of α β and γδ T cells was observed in patients with mutations in the CD3γ chain of the TCR complex (Figure 1)[66]. Moderately reduced numbers of all T cells or just CD8+ T cells and impaired T cell activation were also described in patients with mutations in STAT5b and a hypomorphic mutation in ZAP-70, respectively[72; 73; 74; 75]. While herpes virus and pneumocystis jiroveci infections, diarrhea and eczema in STAT5b deficient patients were associated with dwarfism and facial dysmorphy, a late onset (> 2 years of age) combined immunodeficiency with recurrent episodes of oral candidasis, upper respiratory tract infections, pneumonia and severe disseminated skin chicken-pox infection was observed in the patient with hypomorphic ZAP-70 mutation [75].
Immunodeficiency with lymphoproliferation and autoimmunity
Immunodeficiency in STIM1 deficient patients is associated with autoimmunity most likely due to reduced numbers of Treg cells. A similar but much more severe phenotype is observed in patients with Treg dysregulation due to mutations in FOXP3 and IL2RA (encoding for CD25) who suffer from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and autosomal recessive IPEX-like syndrome, respectively (reviewed in [69]). Disease in CD25 deficient patients is characterized by – in addtion to the IPEX phenotype – symptoms of T cell deficiency with chronic gastrointestinal, candida and CMV infections [70; 71], hepatosplenomegaly and lymphadenopathy. While total T cell numbers in the blood of these patients are moderately decreased or normal, T cell activation is impaired with reduced proliferative responses.
Immunodeficiency and EDA
The combination of immunodeficiency and EDA found in ORAI1 and STIM1 deficient individuals is also observed in patients with mutations in NEMO (or IKKγ) [76; 77; 78; 79] and IKBA [80; 81; 82; 83]. NEMO phosphorylates IκBα, resulting in its ubiquitination and degradation followed by activation of the transcription factor NFκB. T and B cell numbers are normal in NEMO deficient patients whereas mutation of IκBα is associated with a decrease in TCRγδ as well as memory CD8+ TCRαβ T cells. T cell proliferation in vitro in response to anti-CD3 or mitogen stimulation is impaired in some of the patients with IκBα and NEMO deficiency similar to ORAI1/STIM1 deficient patients. While a hyper-IgM phenotype is observed in some patients with IκBα [80; 81; 82; 83] and NEMO deficiency [76; 78; 79], it is absent in individuals lacking functional ORAI1 or STIM1. EDA in some NEMO [78] and IκBα [80] deficient individuals is characterized by conical teeth and hypodontia as well as sparse thin scalp hair [76; 77; 78; 80], in contrast to ORAI1 and STIM1 deficient patients who suffer from hypocalcified amelogenesis imperfecta. Importantly, congenital myopathy is observed only in patients lacking SOCE but in not those with impaired NFκB activation.
5. MOUSE MODELS OF ORAI1 AND STIM1 DEFICIENCY
Patients with mutations in ORAI1 or STIM1 fail to thrive and die of immunodeficiency in their first year of life. By contrast, ORAI1 and STIM1 deficient mice die perinatally even when housed under specific pathogen free conditions [58; 84; 85; 86; 87]. The lethality is more severe in completely inbred mouse strains and in Stim1 deficient mice was attributed to a defect in skeletal muscle function and development [88]. This is consistent with the myopathy found in patients lacking functional STIM1 and ORAI1, although their myopathy is relatively mild and resulted in respiratory insufficiency beginning only during adolescence of two ORAI1 deficient patients.
Similar to human patients lacking functional STIM1 or ORAI1, T cell activation is attenuated in mice lacking ORAI1 or STIM1. Their T cells have severely impaired SOCE, CRAC channel function and cytokine gene expression [58; 85; 89] whereas T cell proliferation in mice – unlike in human patients – in response to TCR crosslinking is normal. Total lymphocyte numbers in ORAI1 and STIM1 deficient mice are normal with no apparent defect in lymphocyte development in the T and B cell compartment suggesting that lymphocyte development occurs independently of SOCE – or at least Stim1 and Orai1 gene expression [58; 84; 85; 87; 89]. Conditional gene targeting of both Stim1 and Stim2 in murine T cells, however, interferes with the development and function of Treg but not conventional T cells [58](discussed in [40; 90]). Reduced numbers and impaired function of Treg cells in STIM1/STIM2 deficient mice results in lymphoproliferative disease that is characterized by lymphadenopathy, splenomegaly, leukocytic infiltration of liver and lung as well as dermatitis and blepharitis [58; 89]. This phenotype could largely be prevented by transfer of wild-type Treg cells to young STIM1/STIM2 deficient mice [58]. Additional factors such as impaired deletion of self-reactive T cells during negative selection in the thymus or impaired peripheral tolerance mechanisms may also contribute to lymphoproliferative disease because splenomegaly was observed in some Stim1−/− mouse strains despite normal numbers of Treg cells [89]. While autoimmunity in human STIM1 deficient patients is characterized by autoimmune thrombocytopenia and hemolytic anemia [19], mice lacking STIM1 were largely resistant to autoantibody mediated destruction of platelets and erythrocytes following injection of anti-platelet and anti-erythrocyte antibodies, respectively [91]. ORAI1 deficient mice, like human patients, do not consistently develop lymphoproliferative disease or autoimmunity and have normal numbers of Treg cells in contrast to mice and human patients lacking STIM1 expression [85; 87]. This difference is most likely due to residual SOCE in T cells from Orai1−/− mice, mediated presumably by ORAI2 or ORAI3, while Stim1−/− mice lack detectable SOCE.
6. CONCLUDING REMARKS
Patients with severe defects in T cell activation due to mutations in ORAI1 or STIM1 show an increased susceptibility to infections similar to that of patients with SCID although numbers of T cells and other lymphocytes are normal in these individuals. Impaired CRAC channel function results in a defect in T cell activation and function of other lymphoid cell types that require SOCE for full activation such as B cells, NK cells, mast cells, dendritic cells and other myeloid cells. CRAC channelopathy is therefore not strictly a T cell activation deficiency but rather a combined immunodeficiency affecting several different immune cell types.
The limited clinical phenotype of patients with CRAC channelopathy demonstrates the important role of ORAI1 and STIM1 mediated store-operated Ca2+ influx in cells of the immune system as well as skeletal muscle and ectodermal derived tissues. CRAC channel currents and SOCE were reported in many other cell types and organs outside the immune system, but the role of ORAI1 and STIM1 in these tissues may be more redundant or become apparent only under pathological conditions (discussed in [92]).
Finally, the positional cloning approach used to identify ORAI1 as the CRAC channel gene from a small number of patients [17] may be useful for the identification of mutations underlying other autosomal recessive diseases in which traditional linkage analysis is unlikely to yield significant LOD scores due to limiting numbers of available patients. This approach depends on (i) the reliable identification of putative heterozygous carriers of the disease gene through a quantitative and robust in vitro or in vivo test, and (ii) a sufficiently large pedigree with relatives available for functional and DNA haplotype analysis.
Acknowledgments
This work was supported by NIH grant AI066128 and a March of Dimes Foundations grant. Potential conflict of interest disclosure: The author is a scientific co-founder of CalciMedica, a biotechnology company that seeks to develop CRAC channel inhibitors.
Abbreviations in this paper
- CRAC
calcium release activated calcium (channel)
- EDA
anhydrotic ectodermal dysplasia
- SCID
severe combined immunodefiency
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- SOCE
store-operated calcium entry
- STIM1
stromal interaction molecule 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fischer A. Human Primary Immunodeficiency Diseases. Immunity. 2007;27:835–845. doi: 10.1016/j.immuni.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Hammartrom L, Nonoyama S, Ochs HD, Puck J, Roifman C, Seger R, Wedgwood J. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–655. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 4.Castigli E, Pahwa R, Good RA, Geha RS, Chatila TA. Molecular basis of a multiple lymphokine deficiency in a patient with severe combined immunodeficiency. Proc Natl Acad Sci U S A. 1993;90:4728–32. doi: 10.1073/pnas.90.10.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatila T, Castigli E, Pahwa R, Pahwa S, Chirmule N, Oyaizu N, Good RA, Geha RS. Primary combined immunodeficiency resulting from defective transcription of multiple T-cell lymphokine genes. Proc Natl Acad Sci U S A. 1990;87:10033–7. doi: 10.1073/pnas.87.24.10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg K, Parkman R. Severe combined immunodeficiency due to a specific defect in the production of interleukin-2. N Engl J Med. 1990;322:1718–23. doi: 10.1056/NEJM199006143222406. [DOI] [PubMed] [Google Scholar]
- 7.DiSanto JP, Keever CA, Small TN, Nicols GL, O’Reilly RJ, Flomenberg N. Absence of interleukin 2 production in a severe combined immunodeficiency disease syndrome with T cells. J Exp Med. 1990;171:1697–704. doi: 10.1084/jem.171.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–24. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 9.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–62. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, Belohradsky B, Choquet D, Fischer A. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–62. [PubMed] [Google Scholar]
- 11.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–35. [PubMed] [Google Scholar]
- 12.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 13.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–7. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 14.Matza D, Flavell RA. Roles of Ca(v) channels and AHNAK1 in T cells: the beauty and the beast. Immunol Rev. 2009;231:257–64. doi: 10.1111/j.1600-065X.2009.00805.x. [DOI] [PubMed] [Google Scholar]
- 15.Philipp S, Strauss B, Hirnet D, Wissenbach U, Mery L, Flockerzi V, Hoth M. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J Biol Chem. 2003;278:26629–38. doi: 10.1074/jbc.M304044200. [DOI] [PubMed] [Google Scholar]
- 16.Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 18.Feske S, Muller JM, Graf D, Kroczek RA, Drager R, Niemeyer C, Baeuerle PA, Peter HH, Schlesier M. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur J Immunol. 1996;26:2119–26. doi: 10.1002/eji.1830260924. [DOI] [PubMed] [Google Scholar]
- 19.Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–80. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 21.Prakriya M. The molecular physiology of CRAC channels. Immunol Rev. 2009;231:88–98. doi: 10.1111/j.1600-065X.2009.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart M. The Hours. Greek Mythology: From the Iliad to the Fall of the Last Tyrant. 2005 http://messagenet.com/myths/bios/hours.html.
- 25.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 26.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–9. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeromin AV, Zhang S, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–9. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNally B, Yamashita M, Engh A, Prakriya M. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci U S A. 2009;106:22516–22521. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–73. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–25. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penna A, Demuro A, Yeromin A, Zhang S, Safrina O, Parker I, Cahalan M. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–43. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 33.McCarl CA, Picard C, Khalil S, Kawasaki T, Röther J, Papolos A, Kutok J, Hivroz C, LeDeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy and ectodermal dysplasia. J Allergy and Clinical Immunology. 2009 doi: 10.1016/j.jaci.2009.10.007. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derler I, Fahrner M, Carugo O, Muik M, Bergsmann J, Schindl R, Frischauf I, Eshaghi S, Romanin C. Increased hydrophobicity at the N-terminus/membrane interface impairs gating of the SCID-related ORAI1 mutant. J Biol Chem. 2009;284:15903–15915. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feske S, Draeger R, Peter HH, Eichmann K, Rao A. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J Immunol. 2000;165:297–305. doi: 10.4049/jimmunol.165.1.297. [DOI] [PubMed] [Google Scholar]
- 37.King LB, Freedman BD. B-lymphocyte calcium influx. Immunol Rev. 2009;231:265–77. doi: 10.1111/j.1600-065X.2009.00822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scharenberg AM, Humphries LA, Rawlings DJ. Calcium signalling and cell-fate choice in B cells. Nat Rev Immunol. 2007;7:778–89. doi: 10.1038/nri2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engelke M, Engels N, Dittmann K, Stork B, Wienands J. Ca(2+) signaling in antigen receptor-activated B lymphocytes. Immunol Rev. 2007;218:235–46. doi: 10.1111/j.1600-065X.2007.00539.x. [DOI] [PubMed] [Google Scholar]
- 40.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caraux A, Kim N, Bell SE, Zompi S, Ranson T, Lesjean-Pottier S, Garcia-Ojeda ME, Turner M, Colucci F. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood. 2006;107:994–1002. doi: 10.1182/blood-2005-06-2428. [DOI] [PubMed] [Google Scholar]
- 42.Cassatella MA, Anegon I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J Exp Med. 1989;169:549–67. doi: 10.1084/jem.169.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darbellay B, Arnaudeau S, Konig S, Jousset H, Bader C, Demaurex N, Bernheim L. STIM1- and Orai1-dependent Store-operated Calcium Entry Regulates Human Myoblast Differentiation. Journal of Biological Chemistry. 2008;284:5370–5380. doi: 10.1074/jbc.M806726200. [DOI] [PubMed] [Google Scholar]
- 44.Kurebayashi N, Ogawa Y. Depletion of Ca2+in the sarcoplasmic reticulum stimulates Ca2+entry into mouse skeletal muscle fibres. J Physiol. 2001;533:185–99. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyfenko AD, Dirksen RT. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586:4815–24. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan Z, Yang D, Nagaraj RY, Nosek TA, Nishi M, Takeshima H, Cheng H, Ma J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat Cell Biol. 2002;4:379–83. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 47.Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal. 2009;2:ra67. doi: 10.1126/scisignal.2000522. [DOI] [PubMed] [Google Scholar]
- 48.Liou J, Kim M, Heo WD, Jones JT, Myers JW, Ferrell JE, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parker Molecular Cloning of a Novel Human Gene (D11S4896E) at Chromosomal Region 11p15.5. Genomics. 1996;37:4. doi: 10.1006/geno.1996.0553. [DOI] [PubMed] [Google Scholar]
- 51.Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57:4493–4497. [PubMed] [Google Scholar]
- 52.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muik M, Fahrner M, Derler I, Schindl R, Bergsmann J, Frischauf I, Groschner K, Romanin C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J Biol Chem. 2009;284:8421–6. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan J, Zeng W, Dorwart MR, Choi Y, Worley P, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–85. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oh-Hora M, Yamashita M, Hogan P, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, Koretzky GA. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science. 1998;281:416–419. doi: 10.1126/science.281.5375.416. [DOI] [PubMed] [Google Scholar]
- 60.Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity. 1995;3:757–769. doi: 10.1016/1074-7613(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 61.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 62.Pivniouk V, Tsitsikov EN, Swinton P, Rathbun G, Alt FW, Geha RS. Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell. 1998;94:229–238. doi: 10.1016/s0092-8674(00)81422-1. [DOI] [PubMed] [Google Scholar]
- 63.Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, Grinberg A, Tsay HC, Jacobs HM, Kessler CM, Long EO, Love PE, Samelson LE. Essential role of LAT in T cell development. Immunity. 1999;10:323–332. doi: 10.1016/s1074-7613(00)80032-1. [DOI] [PubMed] [Google Scholar]
- 64.Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol. 2005;6:143–51. doi: 10.1038/ni1161. [DOI] [PubMed] [Google Scholar]
- 65.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. 2007;38:112–21. doi: 10.1007/s12026-007-0022-2. [DOI] [PubMed] [Google Scholar]
- 66.Recio MJ, Moreno-Pelayo MA, Kilic SS, Guardo AC, Sanal O, Allende LM, Perez-Flores V, Mencia A, Modamio-Hoybjor S, Seoane E, Regueiro JR. Differential biological role of CD3 chains revealed by human immunodeficiencies. J Immunol. 2007;178:2556–64. doi: 10.4049/jimmunol.178.4.2556. [DOI] [PubMed] [Google Scholar]
- 67.Rijkers GT, Scharenberg JG, Van Dongen JJ, Neijens HJ, Zegers BJ. Abnormal signal transduction in a patient with severe combined immunodeficiency disease. Pediatr Res. 1991;29:306–9. doi: 10.1203/00006450-199103000-00017. [DOI] [PubMed] [Google Scholar]
- 68.Rodriguez-Gallego C, Arnaiz-Villena A, Corell A, Manzanares J, Timon M, Pacheco A, Regueiro JR. Primary T lymphocyte immunodeficiency associated with a selective impairment of CD2, CD3, CD43 (but not CD28)-mediated signal transduction. Clin Exp Immunol. 1994;97:386–91. doi: 10.1111/j.1365-2249.1994.tb06099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ochs HD, Oukka M, Torgerson TR. TH17 cells and regulatory T cells in primary immunodeficiency diseases. J Allergy Clin Immunol. 2009;123:977–83. doi: 10.1016/j.jaci.2009.03.030. quiz 984–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roifman CM. Human IL-2 receptor alpha chain deficiency. Pediatr Res. 2000;48:6–11. doi: 10.1203/00006450-200007000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482–7. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 72.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, Ornani A, Paz R, Rivarola MA, Zelazko M, Belgorosky A. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118:e1584–92. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 73.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–4. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 74.Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005;90:4260–6. doi: 10.1210/jc.2005-0515. [DOI] [PubMed] [Google Scholar]
- 75.Picard C, Dogniaux S, Chemin K, Maciorowski Z, Lim A, Mazerolles F, Rieux-Laucat F, Stolzenberg MC, Debre M, Magny JP, Le Deist F, Fischer A, Hivroz C. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur J Immunol. 2009;39:1966–76. doi: 10.1002/eji.200939385. [DOI] [PubMed] [Google Scholar]
- 76.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 77.Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–8. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 78.Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, Ferguson BM. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–62. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, Orange JS. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177. e16. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, Dupuis-Girod S, Bodemer C, Livadiotti S, Novelli F, Rossi P, Fischer A, Israel A, Munnich A, Le Deist F, Casanova JL. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–15. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, van de Vosse E, van Tol M, Bredius R, Ottenhoff TH, Weemaes C, van Dissel JT, Lankester A. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J Exp Med. 2004;200:559–68. doi: 10.1084/jem.20040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol. 2007;120:900–7. doi: 10.1016/j.jaci.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 83.Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, Quinones R, Gelfand EW, Jain A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29:861–8. doi: 10.1002/humu.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–8. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 85.Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–22. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, Renne T, Stoll G, Nieswandt B. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205:1583–91. doi: 10.1084/jem.20080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stiber J, Hawkins A, Zhang ZS, Wang SW, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, Williams RS, Eu JP, Rosenberg P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10:688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beyersdorf N, Braun A, Vogtle T, Varga-Szabo D, Galdos R, Kissler S, Kerkau T, Nieswandt B. STIM1-Independent T Cell Development and Effector Function In Vivo. The Journal of Immunology. 2009;182:3390–3397. doi: 10.4049/jimmunol.0802888. [DOI] [PubMed] [Google Scholar]
- 90.Oh-hora M. Calcium signaling in the development and function of T-lineage cells. Immunol Rev. 2009;231:210–24. doi: 10.1111/j.1600-065X.2009.00819.x. [DOI] [PubMed] [Google Scholar]
- 91.Braun A, Gessner JE, Varga-Szabo D, Syed SN, Konrad S, Stegner D, Vogtle T, Schmidt RE, Nieswandt B. STIM1 is essential for Fcγ receptor activation and autoimmune inflammation. Blood. 2008;113:1097–1104. doi: 10.1182/blood-2008-05-158477. [DOI] [PubMed] [Google Scholar]
- 92.Feske S. CRAC channelopathies. P flügers Archiv European Journal of Physiology. 2010 doi: 10.1007/s00424-009-0777-5. [Electronic publication ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E, Slatter MA, Le Deist F, Blanche S, Veys P, Fasth A, Bredius R, Sedlacek P, Wulffraat N, Ortega J, Heilmann C, O’Meara A, Wachowiak J, Kalwak K, Matthes-Martin S, Gungor T, Ikinciogullari A, Landais P, Cant AJ, Friedrich W, Fischer A. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111:439–45. doi: 10.1182/blood-2007-03-076679. [DOI] [PubMed] [Google Scholar]