

Abstract
Polyphosphazene-polyester blends are attractive materials for bone tissue engineering applications due to their controllable degradation pattern with non-toxic and neutral pH degradation products. In our ongoing quest for an ideal completely miscible polyphosphazene-polyester blend system, we report synthesis and characterization of a mixed-substituent biodegradable polyphosphazene poly[(glycine ethyl glycinato)1(phenyl phenoxy)1phosphazene] (PNGEG/PhPh) and its blends with a polyester. Two dipeptide-based blends namely 25:75 (Matrix1) and 50:50 (Matrix2) were produced at two different weight ratios of PNGEG/PhPh to poly(lactic acid-glycolic acid) (PLAGA). Blend miscibility was confirmed by differential scanning calorimetry, Fourier transform infrared spectroscopy, and scanning electron microscopy. Both blends resulted in higher tensile modulus and strength than the polyester. The blends showed a degradation rate in the order of Matrix2 < Matrix1 < PLAGA in phosphate buffered saline at 37°C over 12 weeks. Significantly higher pH values of degradation media were observed for blends compared to PLAGA confirming the neutralization of PLAGA acidic degradation by polyphosphazene hydrolysis products. The blend components PLAGA and polyphosphazene exhibited a similar degradation pattern as characterized by the molecular weight loss. Furthermore, blends demonstrated significantly higher osteoblast growth rates compared to PLAGA while maintaining osteoblast phenotype over a 21-day culture. Both blends demonstrated improved biocompatibility in a rat subcutaneous implantation model compared to PLAGA over 12 weeks.
Keywords: Polyphosphazenes, poly(lactic acid-glycolic acid), polymeric blends, biodegradation, biocompatibility, bone tissue engineering
Introduction
Fractures and loss of bone have significantly affected the quality of life for a large segment of the population. Due to the limitations associated with autografting and allografting, tissue engineering has emerged as an alternative strategy to regenerate or replace damaged or lost tissues [1]. In this approach, biodegradable polymers play a paramount role as transient substrates that can be replaced by the newly formed tissues in vivo. Biodegradable polymers such as poly(lactide), poly(glycolide), and their copolymers (PLAGA) are the most prevalent materials for tissue engineering applications [2,3]. However, the acidity from the bulk erosion of these materials has been reported to cause catastrophic failure of structure integrity, and adversely affect biocompatibility both in vitro and in vivo [4-8]. Inflammatory responses and foreign body reactions were reported for the patients after being treated with polyester implants [9,10]. Furthermore, these acidic degradation products potentially compromise the bioactivity of growth factors. One promising approach to circumvent these limitations is to blend PLAGA with other macromolecules that can buffer the acidic degradation products with a controlled degradation rate.
Biodegradable polyphosphazenes (PPHOS) have proved to be superior candidate materials to achieve this objective due to their unique buffering degradation products [11]. PPHOS are inorganic-organic hybrid polymers with a backbone of alternating phosphorus and nitrogen atoms and with each phosphorus atom bearing two organic side groups [12,13]. The synthetic flexibility provided by altering the side groups (more than 250 organic nucleophiles) enables access to a wide array of PPHOS with diverse properties. The type of side groups attached to the polyphosphazene backbone largely determines physical and chemical properties [14]. Thus the degradation rate and mechanical properties of the polymer can be altered by incorporating different side groups. For example, amino acid ester side groups sensitize the polymer backbone to hydrolysis while hydrophobic phenylphenoxy side groups protect the backbone from hydrolysis. In contrast to the polyester family, amino acid ethyl ester substituted PPHOS undergo hydrolysis generating non-toxic and buffering degradation products comprised mainly of phosphate, ammonia, and corresponding side groups [12,15]. Therefore, a co-substituted polymer with both amino acid ester groups and hydrophobic groups provides a high degree of tailorability in achieving required degradation pattern for a specific application. Further, studies have also demonstrated that bulkier side groups such as phenylphenoxy substituted PPHOS showed improved mechanical properties [16]. A wide spectrum of polymers developed on PPHOS platform have been reported to be osteocompatible substrates [17-19].
Our initial studies were primarily focused on the development of self-neutralizing systems where the buffering degradation products of amino acid ethyl ester substituted PPHOS neutralized the acidic hydrolysis products of polyester. However, most of these blend systems clearly lack the mechanical properties needed for bone tissue engineering applications [20-22]. Fabrication of completely miscible PPHOS-PLAGA blends with appropriate mechanical properties for bone tissue engineering applications still remains a significant challenge. For example, the mechanically sound phenylphenoxy side group hinders hydrogen bonding to produce miscible blends due to its steric bulk [23].
The objective of the present study was to develop and characterize mechanically strong miscible blends of PNGEG/PhPh and PLAGA. These dipeptide-based blends were characterized for mechanical behavior, degradation pattern, in vitro osteocompatibility, and in vivo biocompatibility. The present study systematically evaluated in vivo tissue responses to these blends over 12 weeks using a rat subcutaneous implantation model.
Materials and methods
Materials
Poly(lactic acid-glycolic acid), PLAGA (50:50 lactide to glycolide ratio, Mw=34 kDa) was purchased from Boehringer Ingelheim KG (Germany). Hexachlorocyclotriphosphazene (HCCTP) was procured from Nippon Fine Chemicals, Japan. HCCTP was purified by recrystallization from heptane and sublimation at 50°C. 4-Phenylphenol (Sigma) and glycyl-glycine ethyl ester hydrochloride (MP-Bio) were used as received. The polyphosphazene was synthesized with the use of standard Schlenk line techniques under an argon atmosphere. Tetrahydrofuran (THF) and chloroform were used as received from Fisher Scientific (Hampton, NH). The 31P NMR (145 MHz) and 1H NMR (360 MHz) spectra of the polymers were obtained using a Bruker 360 MHz NMR spectrometer in order to confirm the structure and side-group ratios. 31P NMR chemical shifts were reported in ppm relative to 85% phosphoric acid as an external reference. Molecular weights of the polymers were measured by gel permeation chromatography (GPC) using a Hewlett Packard LC 1100 series. Samples for GPC analysis were prepared in concentrations of approximately 1% (w/v) in THF and eluted at a rate of 1 mL/min through a size exclusion column (Phenomenex, Phenogel, USA) calibrated using a polystyrene standard (Polysciences, Inc., Warrington, PA).
Synthesis of polyphosphazenes
Poly(dichlorophosphazene) (20.00 g, 172.6 mmol) was dissolved in 2 L THF. 4-Phenylphenol (35.25 g, 207.1 mmol) was added to a suspension of sodium hydride (4.14 g, 172.6 mmol) in 1 L THF over a period of 30 min. The resulting solution was added dropwise to poly(dichlorophosphazene) solution over a period of 1 h. Glycyl-glycine ethyl ester hydrochloride (40.72 g, 207.1 mmol) was suspended in 1 L THF, and reacted with triethylamine (72.17 mL, 517.8 mmol). This suspension was refluxed for 24 h, then filtered and added to the polymer solution, which was then stirred at room temperature for 24 h, and refluxed for a further 48 h. The polymer was concentrated and purified by precipitation into methanol from THF three times and from THF into hexanes once. The solvent was removed under reduced pressure to yield a yellow solid. The yields were in the range of 70-80%. The polymers were dried under vacuum and stored under argon before use.
Polymer blend fabrication
Blends were prepared using a mutual solvent method [24]. The polymers with two weight ratios of PPHOS to PLAGA (25:75 and 50:50) with a total weight of 1 g, were dissolved in 14.5 mL of mutual solvent (chloroform) to obtain a homogeneous solution. Samples of the polymer solution were subsequently poured into petri dishes lined with Bytac paper and the solvent was allowed to evaporate slowly at 4°C for 48 h followed by freeze drying. Finally, circular disks of 10 mm diameter and 0.1 mm thickness were bored from the films. For in vitro and in vivo biodegradability and biocompatibility evaluation, a total weight of 5 g was used to fabricate the polymer matrices of 0.5 mm thickness.
Miscibility of polyphosphazene/PLAGA blends
Surface morphology of the blends
The surfaces of the blend films were sputter coated with gold using a Hummer V sputtering system (Technics Inc., Baltimore, MD) and examined under a JEOL 6700F scanning electron microscope (SEM) (JEOL, Boston, MA, USA).
Differential scanning calorimetry (DSC) measurements
The glass transition temperatures (Tg's) were determined by differential scanning calorimetry using TA instruments DSC Q1000 with Thermal Analysis software. Polymer samples were heated from -40°C to 100°C at a heating rate of 3°C/min under a nitrogen atmosphere. The Tg was determined from the half height point of the heat capacity change in the thermogram.
Fourier transform infrared (FTIR) analysis
The FTIR spectra of thin films of polymers and blends were recorded using a Brucker Vector 22 FTIR spectrophotometer at a resolution of 4 cm-1 and with an average of 50 scans. The blend films were cast on KBr plates and were sufficiently thin to be within an absorbance range where the Beer-Lambert law was obeyed.
Mechanical evaluation of polyphosphazene/PLAGA blends
Tensile tests were performed on the “dog bone shape” of 13.2 × 6.6 × 0.1 mm polymer strips cut from polymer films using an Instron mechanical testing machine (Instron model 5544, Canton, MA) with a crosshead speed of 2 mm/min. The samples were elongated to failure.
In vitro degradation
Blend disks with dimensions of 0.5 mm × 10 mm (T × D) and weighing ∼95 mg each were incubated in 10 mL of phosphate buffered saline (PBS) whose pH was adjusted to pH 7.4 prior to use. The vials were maintained at 37°C in a water bath/shaker for 12 weeks at 250 rpm. At specific time points (2, 4, 7, 10, and 12 weeks), the matrices were removed from the media and dried under vacuum for 2 weeks to constant weight. The results were reported as percentage mass loss versus time as calculated from the equation:
where wt is the dry weight of the matrix at predetermined time points, and w0 is the initial matrix dry weight. The molecular weight of the degraded matrices was also determined using GPC and reported as percentage molecular weight remaining: Percentage molecular weight remaining: where Mt is the molecular weight of polymer component at predetermined time points, and M0 is the initial molecular weight of polymer. Samples were visualized by SEM. Each week, the media were also collected and the pH values were recorded by a pH meter. The PBS was changed every week.
In vitro osteocompatibility evaluation of polyphosphazene/PLAGA blends
Cell isolation
Primary rat osteoblast (PRO) cells were isolated from calvarias of 2-day-old neonatal Sprague-Dawley rats according to a standard procedure [23]. Prior to cell seeding, all polymer films were sterilized with ultraviolet (UV) light for 15 min on each side after being treated with 70% ethanol for 30 min and distilled water twice for 15 min.
Cell seeding
The polymer films were pre-incubated in Ham's F-12 media (Gibco) for 1 h prior to cell seeding. PRO cells were seeded onto the polymer films at a density of 6 × 104 cells/cm2 after removing the pre-incubation media. The cells were cultured at 37°C in a 95% humidified air and 5% CO2 for various periods of time after adding 1 mL supplemented mineralized media (Ham's F-12 media supplemented with 12% FBS, 1% penicillin/streptomycin, 3 mM of β-glycerophosphate (Sigma) and 10 μg/ml of ascorbic acid (Fisher)) to each well. The media were changed every 2 days. The cultures were maintained for 21 days.
Live/Dead cell viability
Viability of PRO on polymer films was characterized with a live/dead cell viability kit (Molecular Probes, L-3224). In brief, calcein AM enters live cells and reacts with intracellular esterase to produce a bright green fluorescence, while ethidium homodimer-1 enters only dead cells with damaged membranes and produces a bright red fluorescence upon binding to nucleic acids. Polymer matrices were imaged at 2, 7, 14 and 21 days using a BioRad Radiance 2100 Multiphoton/Laser Scanning Confocal Microscope (LSCM) and a Nikon Eclipse E600 Fluorescent Microscope at different magnifications.
Cell morphology
At days 1, 3, 7, 14, and 21, the polymer films were gently washed with PBS to remove any unattached cells. Specimens were fixed at 4°C in 1% and 3% glutaraldehyde for 1 and 24 h, respectively. After being dehydrated with increasing concentration of ethanol (10, 30, 50, 70, 90, 95, and 100%), the films were allowed to air dry overnight at room temperature. SEM samples were sputter coated with Au/Pd and examined under a JEOL 6700F SEM.
Cell proliferation
The proliferation of PRO cells on the polymer films was analyzed using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium) (MTS, Promega) mitochondrial reduction [23]. Cells seeded onto tissue-culture polystyrene (TCPS) were used as a control. At days 1, 3, 7, 14, and 21, the films with cultured cells were washed with PBS, transferred into a new 48-well tissue-culture plate containing 500 μL culture medium and 100 μL MTS substrate and incubated for 2 h in a humidified atmosphere with 5% CO2 at 37°C. At the end of the incubation period, the reaction was stopped by adding 125 μL of 10% sodium dodecyl sulfate (SDS) solution. The resulting solution was diluted in a 4:1 ratio using distilled water and the absorbance was read at 490 nm using a Tecan SpectroFluo Plus reader (TECAN USA). Cell numbers were calculated based on a standard curve established with the incubation of different concentrations of PRO cells in 500 μL culture medium and 100 μL MTS for 2 h in a humidified atmosphere with 5% CO2 at 37°C.
Alkaline phosphatase activity
Alkaline phosphatase (ALP) activity of PRO cells on the polymer films was evaluated as an early marker of the retention of osteoblastic phenotype using an ALP substrate kit (Bio-Rad) [23]. In brief, at days 7, 14, and 21, the media were removed, the cells were washed with PBS, and then lysed with 1 mL of 1% Triton X-100 solution for 30 min. The cell lysates were collected and stored in a -70°C freezer. Three freeze-thaw cycles were performed before analysis. After the third thawing, 100 μL sample was added to 400 μL of P-NPP substrate and buffer solution mixture and incubated at 37°C for 30 min. At the end of the incubation time, the reaction was stopped by adding 500 μL of 0.4 N of sodium hydroxide. The optical density of the solution was determined by the absorbance at 405 nm using the TECAN. The intensity of the color formed is proportional to ALP activity. The results for ALP activity were further normalized by the amount of proteins on the scaffolds as determined in a companion protein assay. The protein concentration of cell lysates was determined with a BCA Protein Assay Reagent kit (Pierce) which is based on bicinchoninic acid (BCA) for the colorimetric detection and quantitation of total protein. Proteins of the cell suspension in an alkaline medium can reduce the cupric ions to cuprous ions which can form a chelation complex with BCA. The purple color of this complex is directly proportional to the protein concentration and the absorbance is read at 562 nm using TECAN.
In vivo biocompatibility
Circular blend matrices with dimensions of 0.5 mm × 10 mm (T × D) were sterilized by exposing to UV light for 30 min on each side and implanted subcutaneously in 45 male retired breeder Sprague-Dawley rats weighing ∼450 grams (Charles River Laboratories, Wilmington, MA) [19,25]. The animals were cared for according to the procedures approved by the Animal Care and Use Committee of the University of Virginia, following the guidelines established by the NIH. Two incisions (10 mm apart) of about 10 mm were made laterally on the dorsum using a No. 10 surgical blade. A subcutaneous pouch on opposite sides of the incision was created using blunt dissection technique and one disk implant was inserted into each pouch. Each rat was implanted with two polymer disks. The skin was closed using a sterile stapler. At specific time points (2, 4, 7, 10, and 12 weeks), the implants and the surrounding tissues were excised and were fixed in 10% formalin solution (Surgipath, USA) for 7 days. The samples were embedded in paraffin and sectioned using a microtome to about 4-5 μm thickness, and stained with hematoxylin and eosin (H&E) and Masson's trichrome stain (TRI). Samples were viewed using a light microscope and the presence of neutrophils, lymphocytes, macrophages, and giant cells were characterized as evidence of tissue response by an independent pathologist (W.A.K.) [19]. The thickness of the inflammatory zone (H&E) and collagen deposition (TRI) was expressed as the average value of at least six slides for each type of implant at each time point [25]. Only four readings were available for the thickness of collagen deposition for PLAGA at week 7. The degradation of the polymer in vivo was followed by measuring the mass, molecular weight of the polymer before implantation and those at different time points.
Statistical analysis
All the experiments were run in triplicate per sample. Quantitative data were reported as mean ± standard deviation (SD). Statistical analysis was performed using a one-way analysis of variance (one-way ANOVA). Comparison between means was determined using the Tukey post-hoc test with a minimum confidence level of p< 0.05 for statistical significance.
Results
Polymer characterization
The PNGEG/PhPh polymer was found to have a moderate molecular weight (Mw= 767 ± 37 kDa). The polymer had a relatively high glass transition temperature of 53°C, which is above the physiological temperature of 37°C. This is due to the steric bulk and the possibility of π − π stacking of the biphenyl units [23].
The structure and composition of the PNGEG/PhPh (Scheme I) were confirmed by 31P NMR and 1H NMR. 31P NMR peaks were obtained at 1.5 ppm, -3.5 ppm, -15.5 ppm; 1H NMR peaks were obtained at 1.30 ppm (3H), 3.54 ppm (2H), 4.12 ppm (4H), 6.79 ppm (2H), 7.31 ppm (5H), 7.48 ppm (2H).
Scheme I.
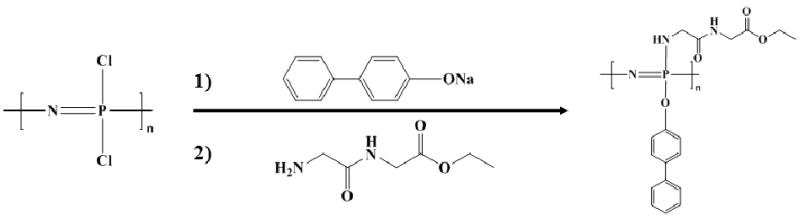
Synthesis of poly[(glycine ethyl glycinato)1(phenyl phenoxy)1phosphazene] (PNGEG/PhPh).
Miscibility of Polyphosphazene/PLAGA blends
Surface morphology of the blends
In contrast to colorless PLAGA, two different weight ratios of PNGEG/PhPh and PLAGA, namely 25:75 and 50:50, yielded Matrix1 (light yellow) and Matrix2 (yellow), respectively (Fig. 1a). SEM micrographs also illustrated that both blend matrices had smooth uniform surfaces with no evidence of phase separation.
Figure 1.
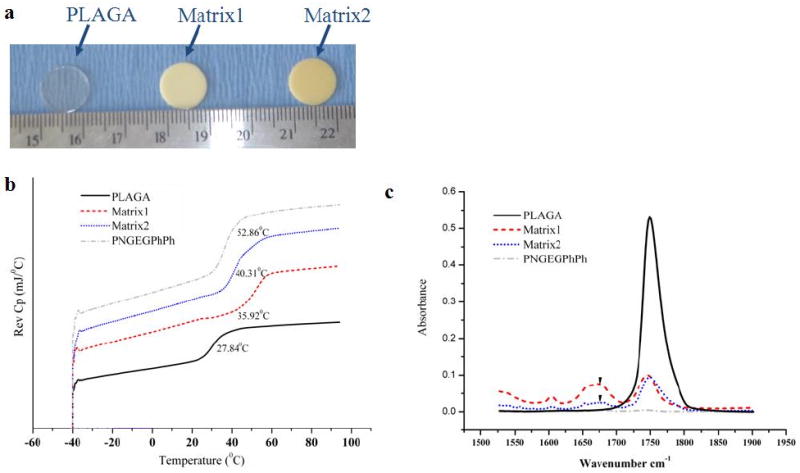
Miscibility of blend matrices. (a) Optical micrographs of blend matrices; (b) DSC thermograms of the parent polymers and blends where the blends show a single Tg intermediate between those of the parent polymers; (c) FTIR spectra presenting carbonyl absorption region for blends and parent polymers. The C=O stretching vibrations of PNGEG/PhPh and PLAGA occur at ∼1743 and ∼1749 cm-1, respectively. For both blends, a second band develops at ∼1677 cm-1 indicating hydrogen bonded carbonyl groups.
Differential scanning calorimetry (DSC) measurements
DSC thermograms for the parent polymers and blends are shown in Fig. 1b. Matrix1 and Matrix2 showed a single Tg of ∼35.9°C and ∼40.3°C, whereas PLAGA and PNGEG/PhPh showed Tg ∼27.8°C and ∼52.9°C, respectively. The single Tg value for both blend matrices was intermediate between those of the two parent polymers indicating blend miscibility.
Fourier transform infrared (FTIR) analysis
Fig. 1c shows the FTIR spectra of the polymers and the corresponding blends in the carbonyl stretching frequency region. The FTIR spectrum showed that the C=O stretching vibrations of PNGEG/PhPh and PLAGA occurred at ∼1743 and ∼1749 cm-1, respectively. Additionally for Matrix1 and Matrix2, a second band developed at ∼1677 cm-1, which indicated hydrogen bonded carbonyl groups.
Mechanical properties of polyphosphazene/PLAGA blends
Tensile testing is used as a means for assessing mechanical performance of the new scaffolding materials for bone tissue engineering. The average tensile Young's modulus and ultimate tensile strength of the polymer matrices determined by the stress-strain curves from tensile tests on thin strips of PLAGA and blends are shown in Fig. 2. Tensile Young's modulus values were found to be 290.4 ± 3.8 MPa, 466.4 ± 5.8 MPa, and 744.5 ± 58.9 MPa for PLAGA, Matrix1, and Matrix2, respectively. Ultimate tensile strengths were found to be 3.4 ± 0.1 MPa, 3.8 ± 0.1 MPa, and 6.1 ± 0.3 MPa for PLAGA, Matrix1, and Matrix2, respectively. It was revealed that the tensile modulus increased significantly (p<0.05) with the addition of polyphosphazene. A similar trend was also observed for ultimate tensile strength.
Figure 2.
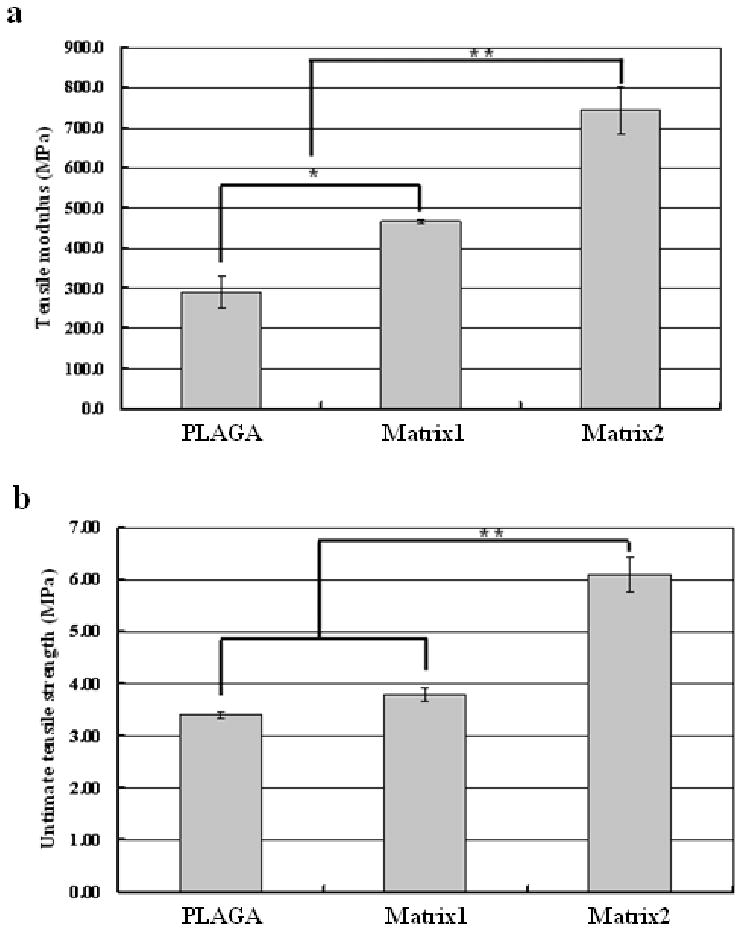
Effect of the polyphosphazene on the mechanical properties of the blends. (a) Tensile Young's modulus; (b) Tensile strength. (*) indicates significant increase compared to PLAGA and (**) indicates significant increase compared to both PLAGA and Matrix1. p<0.05. Tensile moduli of the blend matrices (Matrix1 and Matrix2) are in the range of trabecular bone (50-100 MPa) and cortical bone (17-20 GPa), whereas the tensile strengths are comparable to that of trabecular bone (5-10 MPa) but much lower than cortical bone (80-150 MPa). More interestingly, the mechanical properties (both tensile modulus and tensile strength) increase significantly (p<0.05) with the addition of polyphosphazene.
In vitro degradation
One of the major advantages of the blend system over PLAGA resides in its self-neutralizing ability with controllable degradation rate. The self-neutralizing ability of the blend system is assessed by recording the pH profiles of degradation in PBS. To mimic the physiological environment, the medium was adjusted to pH 7.4. In general, a significant increase in the pH of the degradation media was observed with blends compared to pristine PLAGA. For example, the pH of PBS decreased dramatically to ∼3.0 after 4 weeks for pristine PLAGA (Fig. 3a), which was due to the fast degradation of PLAGA to lactic acid and glycolic acid. It might also explain the PRO cell number decrease on PLAGA from in vitro cell studies. On the other hand, the pH was as high as ∼5.5 even after 4 weeks of degradation for Matrix2. The significant increase in the pH of the degradation media with Matrix2 was maintained until pristine PLAGA completely degraded by 7 weeks. This suggests that the acidity originated from the bulk degradation of PLAGA can be buffered by the phosphates and ammonia produced from the polyphosphazene backbone. This buffering effect can further reduce the acidic catalysis of hydrolysis of PLAGA.
Figure 3.
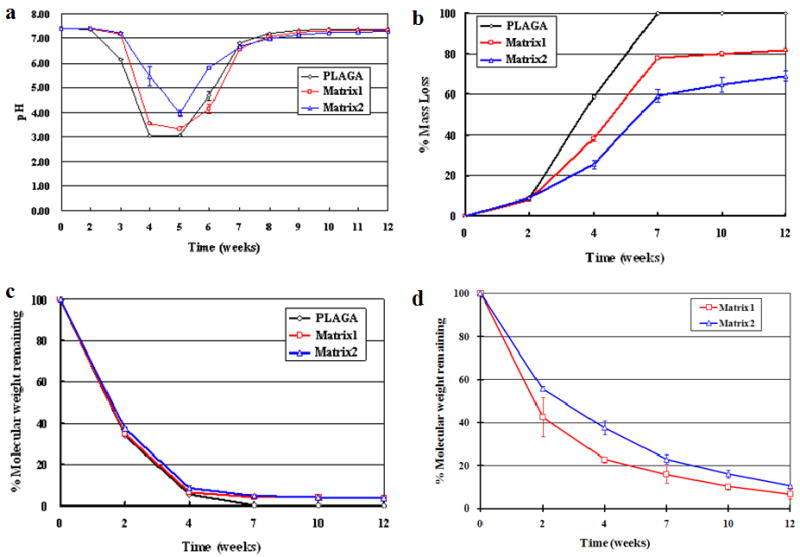
Degradation profiles of blends and pristine PLAGA in pH 7.4 PBS at 37°C over 12 weeks. (a) pH change; (b) Percentage of mass loss; (c) Percentage of PLAGA molecular weight remaining in the blends and pristine PLAGA over 12 weeks in PBS; (d) Percentage of polyphosphazene molecular weight remaining in the blends over 12 weeks in PBS. A significant increase in the pH of the degradation media was observed with blends compared to pristine PLAGA. The blend matrices showed slower degradation rate than pristine PLAGA. The changes of molecular weight for both the PLAGA and polyphosphazene components in the matrix further confirmed that the two components degraded in a similar pattern.
The blends degraded much slower than pristine PLAGA over 12 weeks (Fig. 3b). It was also observed that the increase of the polyphosphazene composition in the blend resulted in a slower degradation rate. Moreover, the difference in the mass loss between blends and pristine PLAGA increased with time and peaked at 7 weeks, by which the pristine PLAGA had completely degraded. It suggested that the polyphosphazene degradation products were able to neutralize the acidic products from PLAGA, and as a result, retarded the degradation of both PLAGA and polyphosphazene components in the blend. Following 7 weeks of continued degradation, the percentage of mass loss for Matrix1 and Matrix2 was 77.83 ± 0.23% and 59.18 ± 2.94%, respectively. After the rapid PLAGA hydrolysis phase, the degradation rate of the blends decreased dramatically and resulted in only ∼4% and 10% mass loss during the remaining 5 weeks of the degradation study, leading to a total mass loss of 81.72 ± 0.18% and 68.95 ± 2.39% for Matrix1 and Matrix2 after 12 weeks of degradation, respectively.
Fig. 3c shows that PLAGA component in Matrix1 and Matrix2 still retained ∼4% of original molecular weight even after 12 weeks of degradation, whereas all the PLAGA completely degraded in its pristine polymer after 7 weeks. After 12 weeks of degradation, the polyphosphazene component in Matrix1 and Matrix2 underwent 93% and 90% of loss in molecular weight, respectively, as evidenced from Fig. 3d. It was also observed that an increase in the PLAGA composition in the blend resulted in a faster degradation rate of the polyphosphazene. Comparison of molecular weight changes for both components in the matrix during degradation further confirmed that the two components degraded in a similar pattern. The acidic degradation products from PLAGA accelerated the degradation of both itself and the polyphosphazene. As a consequence, the degradation of the polyphosphazene neutralized the acidity and retarded the degradation of PLAGA.
In vitro osteocompatibility evaluation of polyphosphazene/PLAGA blends
To ascertain the osteocompatibility of the blends for bone tissue engineering, primary rat osteoblast (PRO) cells from neonatal rat calvaria were cultured on blends using PLAGA and tissue-culture polystyrene (TCPS) as controls. The blend matrices demonstrated robust cell growth with a well-spread morphology compared to PLAGA, as visualized by confocal micrographs (Fig. 4). Progressive cell growth was found on all the polymer matrices with increasing culture time, and the cells formed multilayers after 21 days of in vitro culture (Fig. 5a-c). However, during this period the PLAGA matrix could not maintain its structural integrity and cracks were found on the surface as indicated by the arrows in Fig. 5a. The blend matrices showed significantly higher osteoblast growth rates than PLAGA after 14 days (p<0.05) (Fig. 5d). By contrast, cell numbers decreased significantly on PLAGA matrices beyond 14 days due to the acidic degradation products. This is supported by our in vitro degradation data. Increase in the polyphosphazene content in the blend matrix improved cell growth. Furthermore, progressive growth on positive control TCPS indicated a healthy PRO culture. It was seen that the cell numbers on TCPS were significantly higher than on all the polymeric matrices after 7 days. Fig. 6 shows the ALP activity of cells on different matrices normalized to the total protein content.
Figure 4.
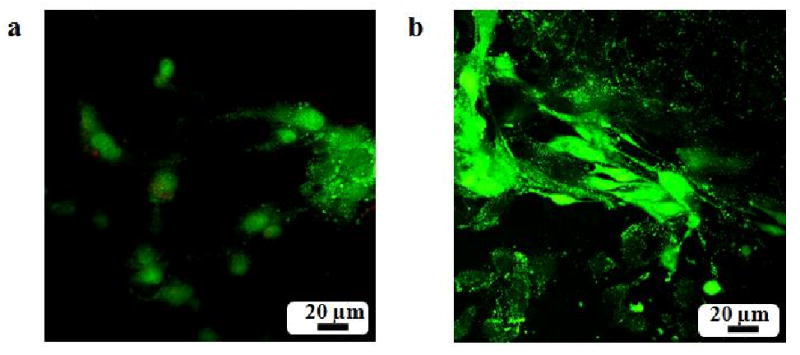
Fluorescence images of PRO cells on polymer matrices with the Live/Dead cell stain after 48 hours of cell seeding. (a) PLAGA; (b) Matrix1. The osteoblast cells exhibit robust growth with a well-spread morphology on the blend surface compared to PLAGA.
Figure 5.
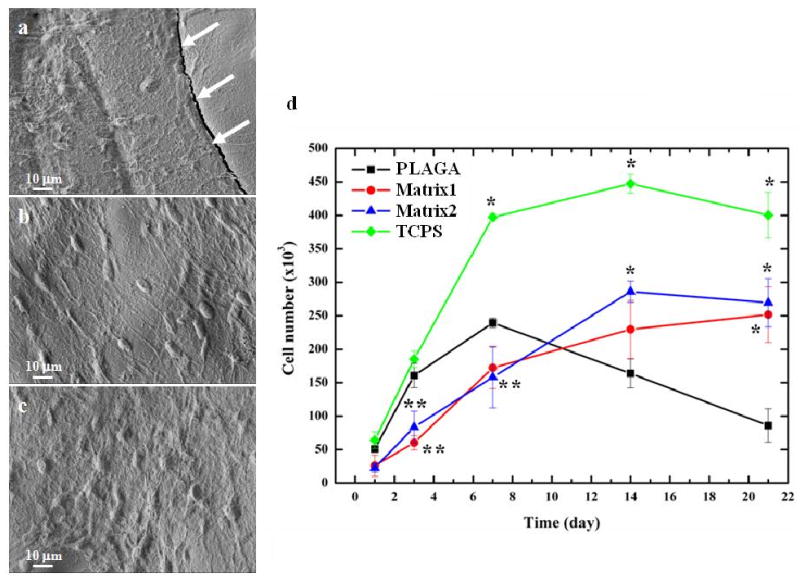
In vitro osteocompatibility of polymer matrices. (a-c): SEM micrographs of PRO cultured on polymer matrices after 21 days where (a) PLAGA; (b) Matrix1; (c) Matrix2; (d) Cell proliferation by MTS assay. (*) indicates significant increase compared to PLAGA and (**) indicates significant decrease compared to PLAGA. p< 0.05.
Figure 6.
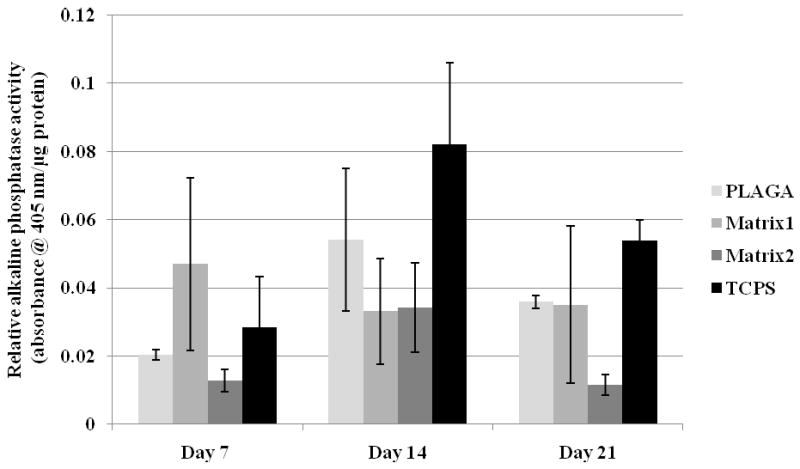
ALP activity for osteoblastic phenotype expression over 21 days. ALP activity of the cells on blends is comparable to PLAGA throughout 21 days of cell culture.
Throughout the culture period, osteoblasts maintained their phenotype and expressed similar levels of ALP on the blend matrices and PLAGA. In addition, no significant differences in ALP activity were observed for the osteoblasts grown on TCPS and on the polymeric matrices. These experiments suggest that the blend matrices enhanced cell growth while maintaining osteoblast phenotype.
In vivo biocompatibility
Subcutaneous implantation in Sprague-Dawley rats was used to evaluate the in vivo biocompatibility of the two blends with PLAGA as the control. No behavioral changes or visible signs of physical impairment were observed during the 12-week post-implantation period. All the polymer implants showed the presence of fibrous encapsulation. Gross observation of the implant sites at each time point showed that blend implants were surrounded by a relatively thinner fibrous capsule than PLAGA. The tissue responses for both blend implants were minimal, which was characterized by the presence of few neutrophils, erythorocytes, and lymphocytes. PLAGA implants were found to be completely absorbed by 7 weeks.
Matrix tissue compatibility was characterized by the extent of inflammation and fibrous capsule formation. As shown in Fig. 7, both blend matrices were found to possess better histological responses with thinner fibrous capsules than PLAGA. The inflammatory responses diminished with time for both blends. After the first two weeks, the thickness of inflammatory zones of Matrix1 and Matrix2 were 14% and 22% thinner than that of PLAGA sites (Fig. 8a-c). At week 4, the thickness of the inflammatory zones in both blend implantation sites were further reduced to 30% and 54% thinner than that of PLAGA sites (Fig. 7 and 8d-f). At week 7, the thickness of the inflammatory zones of both blend implantation sites became 87% and 91% thinner than that of PLAGA sites. Noticeably, the chronic inflammatory response of PLAGA implant increased dramatically at week 7 presumably because of the bulk degradation to lactic acid and glycolic acid (Fig. 7). Thereafter, the thickness of chronic inflammatory response for blend matrices continued to decrease. For example, the thickness for Matrix2 reduced to ∼22 μm after 12 weeks of implantation. As seen in Fig. 9, a minimal grade response was observed for the blend matrices after 12 weeks of implantation. No granulation or scar tissue formation was found at the implantation sites. Furthermore, fibrous capsules surrounding blend implants were also found to be thinner than those of PLAGA (Fig. 7 and 10). The thickness of fibrous capsules surround PLAGA was found to be ∼167 μm after first two weeks and slightly reduced to ∼90 μm at week 4. At week 7, it increased to ∼213 μm. Such reported values for fibrous capsule thickness were in line with the literature reports for PLAGA [25]. The thickness of the collagen layer for Matrix1 and Matrix2 was very thin and remained ∼40 μm for the first 7 weeks. It further reduced to ∼27 μm for Matrix2 after 12 weeks of implantation. In vivo degradation characteristics of the blends were also examined by measuring the change in molecular weight. It was also found that the molecular weight for both PLAGA and polyphosphazene components in the blend decreased in a similar trend throughout the post-implantation period, which is in line with our in vitro findings.
Figure 7.
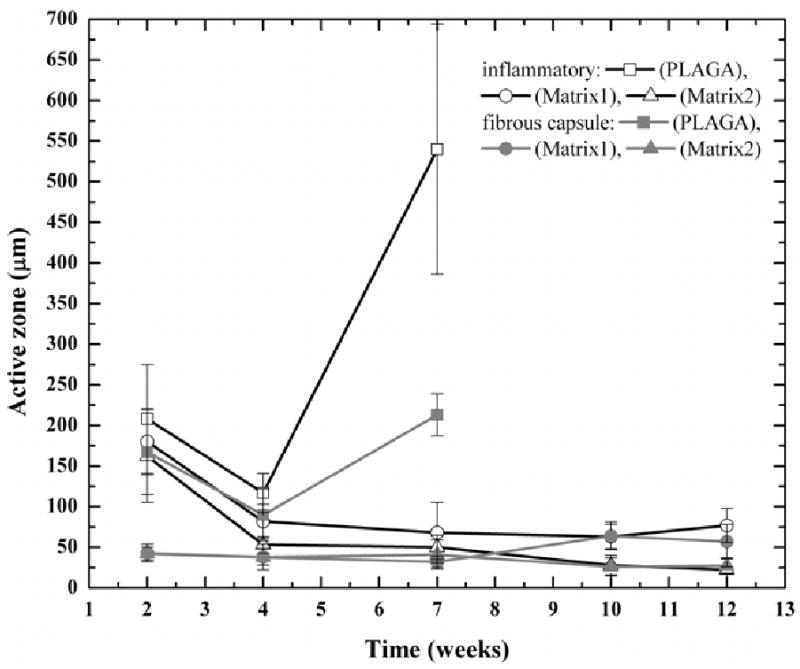
Change of thickness of the tissue responses with time for PLAGA and blends. The blend matrices show less inflammatory responses than PLAGA and the inflammatory responses diminish with time for the blend matrices. Furthermore, Matrix2 shows less inflammatory response than Matrix1. A similar trend is observed for the thickness of fibrous capsules.
Figure 8.
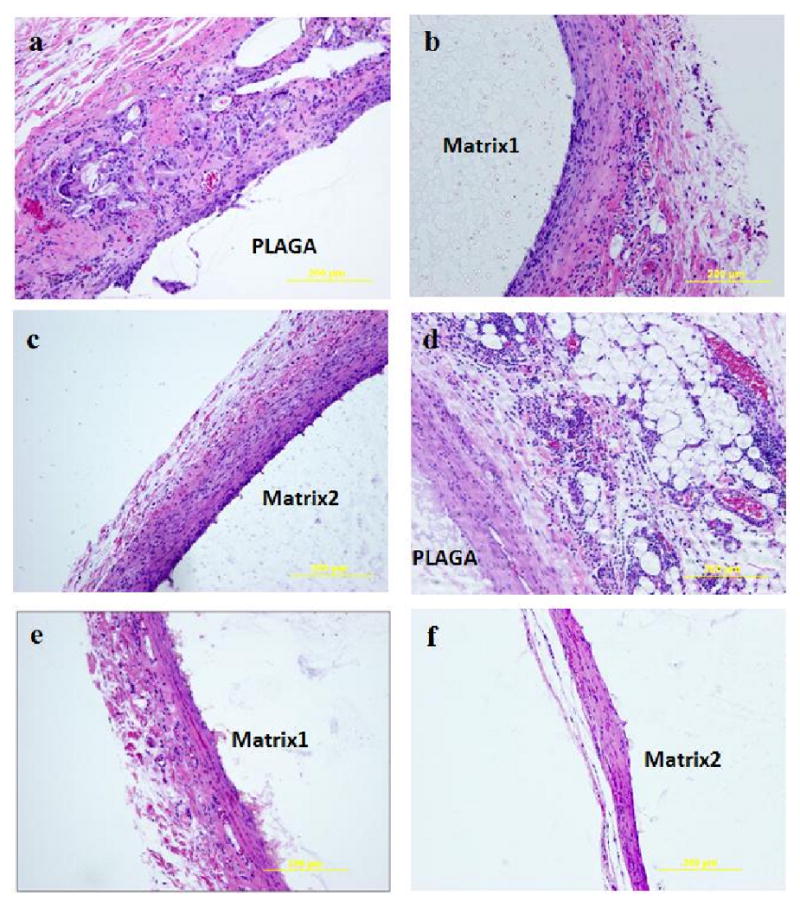
Representative photomicrographs of H&E stained section illustrating the chronic inflammatory response to PLAGA and blend matrices during the post-implantation period. After 2 weeks, (a) PLAGA, (b) Matrix1, (c) Matrix2; After 4 weeks, (d) PLAGA, (e) Matrix1, (f) Matrix2. Both blend matrices elicited less inflammatory responses than PLAGA.
Figure 9.
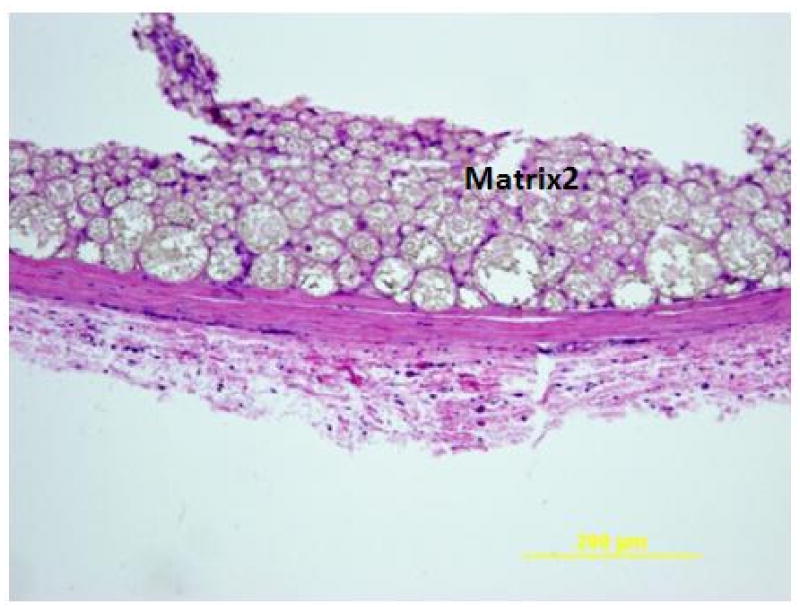
Representative photomicrographs of H&E stained section illustrating the minimal inflammatory response to Matrix2 after 12 weeks of implantation. In addition, Matrix2 was extensively infiltrated with connective tissue.
Figure 10.
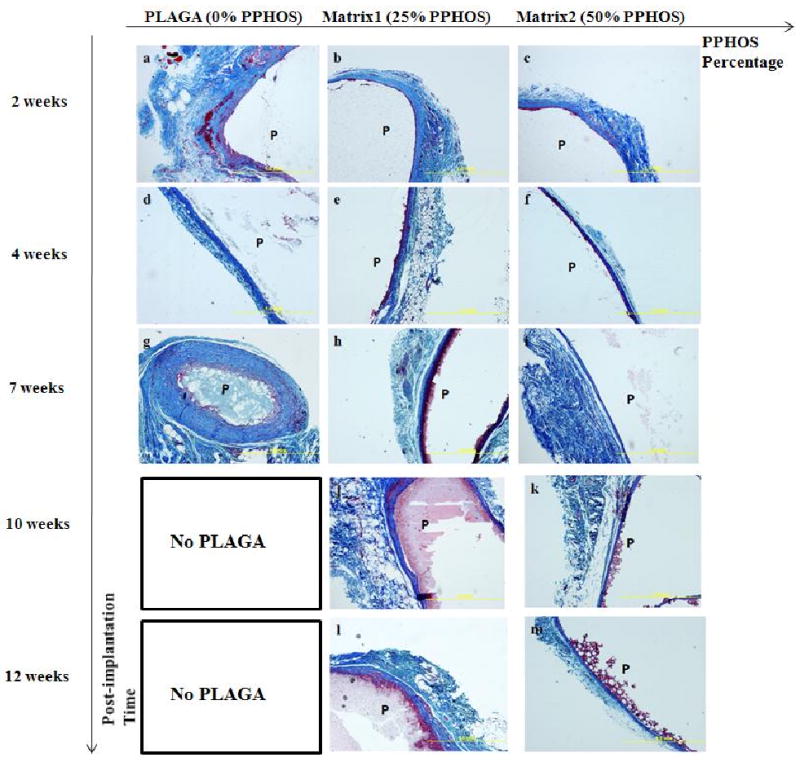
Photomicrographs of histological sections via TRI staining illustrating changes of fibrous capsule surrounding polymer matrices as a function of post-implantation time and blend composition. (a-c): Comparisons of lumen wall characteristics at implantation sites after 2 weeks where (a) PLAGA; (b) Matrix1; (c) Matrix2; (d-f): Comparisons of lumen wall characteristics at implantation sites after 4 weeks where (d) PLAGA; (e) Matrix1; (f) Matrix2; (g-i): Comparisons of lumen wall characteristics at implantation sites after 7 weeks where (g) PLAGA; (h) Matrix1; (i) Matrix2; (j-k): Comparisons of lumen wall characteristics at implantation sites after 10 weeks where (j) Matrix1; (k) Matrix2; (l-m): Comparisons of lumen wall characteristics at implantation sites after 12 weeks where (l) Matrix1; (m) Matrix2. PLAGA had completely degraded after 7 weeks of implantation. P, polymer. Fibrous capsules surrounding blend implants were found to be thinner than those of PLAGA.
Discussion
Currently there is an increasing need for alternative biodegradable polymers with the rapid growth of musculoskeletal regeneration applications in tissue engineering. Polymer blending is a practical strategy to design new materials that synergistically combine the beneficial features of polymers due to the ease of blend preparation and efficient control of material properties via compositional change [11]. Our previous studies have demonstrated that biodegradable PPHOS-PLAGA blends are attractive materials for bone tissue engineering applications due to their controllable degradation pattern with non-toxic and neutral pH degradation products [23]. However, a significant challenge exists in introducing sufficient hydrogen bonding network to ensure blend miscibility while at the same time maintaining appropriate mechanical strength. Thus, a mixed-substituent polyphosphazene with both glycylglycine dipeptide and phenylphenoxy groups was designed for the present study. The glycylglycine dipeptide has been shown to provide two proton donors per monomer for hydrogen bonding with PLAGA while the phenylphenoxy is beneficial for maintaining the mechanical stability of the polymer [16,22]. In addition, a mixed-substituent polymer with both hydrolysis sensitive groups and hydrophobic groups provides efficient tunability to achieve a suitable degradation rate. Specifically the combination of the two side groups in a 50:50 ratio results in a complete degradation time in the range of 12-24 weeks for bone tissue regeneration. Furthermore, the glycylglycine dipeptide is biocompatible, with degradation into glycine units, one of the common amino acids in the body [26,27]. Two different compositions of PNGEG/PhPh and PLAGA blends, namely 25:75 (Matrix1) and 50:50 (Matrix2), were fabricated to determine the effect of polyphosphazene composition on the physical, chemical, and biological properties of the materials. However, we were unable to use higher polyphosphazene compositions due to the difficulty of handling the materials as a result of the high glass transition temperatures.
As demonstrated in our earlier studies, miscibility is an important factor in creating a uniform binary material system with superior mechanical performance as well as predictable degradability [11]. SEM, DSC, and FTIR techniques confirm the formation of miscible blends through the hydrogen bonding between the amide and amine protons in the dipeptide units of PNGEG/PhPh and the carbonyl groups in PLAGA. Blending PLAGA with PNGEG/PhPh resulted in polymer matrices with higher Tg's (36°C for Matrix1 and 40°C for Matrix2) than PLAGA (28°C). For a miscible blend system, the Tg of the blend can be predicted by the Fox equation: Tg= Tg1Tg2/(w1 Tg2+ w2Tg1) where wi and Tgi are the weight fraction and the Tg of polymer i (1 and 2 designate PNGEG/PhPh and PLAGA, respectively) [23]. The glass transition temperatures estimated using the Fox equation for Matrix1 and Matrix2 are 32°C and 37°C, respectively, which are very close to the experimental values from DSC thermograms. Both blends exhibited higher tensile moduli and tensile strength than PLAGA. In addition to the increased mechanical properties, the contact angles increased significantly with the addition of the polyphosphazene (data not shown). These observations are consistent with earlier literature reports where both phenylphenoxy side group and hydrogen bonding interactions have been shown to improve mechanical properties of polymeric materials [16,28,29]. The tensile Young's moduli of the blends (Matrix1 and Matrix2) were in the range of trabecular bone (50-100 MPa) and cortical bone (17-20 GPa), whereas the tensile strength were comparable to that of trabecular bone (5-10 MPa) but much lower than cortical bone (80-150 MPa) [30]. Further studies are currently underway to characterize the compressive strengths of the blends.
In vitro degradation profiles in PBS revealed a slow degradation rate with near-neutral pH degradation products for both blend matrices compared to PLAGA. This suggests that the phosphates and ammonia produced from the hydrolyzed polyphosphazene backbone were able to neutralize the acidic degradation of PLAGA and further reduce the auto-catalyzed PLAGA hydrolysis. The multiphase degradation profiles of blend matrices could be important in new bone regeneration, which involves sequential healing processes in the range of 12 to 24 weeks [31]. Furthermore, comparison of the degradation rate of the polymer matrices suggested that the order of degradation rate is PLAGA > Matrix1 > Matrix2. The resulting low pH environment from the degradation of the polyester materials is detrimental to implant functions in anatomical defect sites, where there is less fluid in tissues such as articular cartilage. The blend degradation rate and the resulting pH environment can be well tuned for better regeneration by simply adjusting the blend composition. In addition, blending polyesters with polyphosphazenes can also potentially circumvent the dramatic losses in structural integrity caused by the bulk degradation of polyesters.
The osteocompatibility of the blends was investigated using in vitro PRO cell culture. PRO cells showed a normal morphological sequence of adhesion and proliferation on the blend matrices as reported on the surfaces of other osteocompatible polymers (Fig. 4). A multilayer of osteoblast cells was found on the blend surfaces after 21 days of cell culture as evident from Fig. 5b and c. This observation was further supported by viability and proliferation results of osteoblast cells on polymer matrices as presented in Fig. 5d. The cell numbers on the blends increased with culture time. In addition, Matrix1 and Matrix2 showed significantly higher cell numbers than PLAGA after 14 days. The significant decrease in cell numbers on PLAGA started at day 14 presumably due to the fast degradation and the acidic degradation products. In addition, a trend of better cell growth was observed with the increase of polyphosphazene in the blend. Furthermore, ALP activity of the cells on blends was comparable to PLAGA throughout the cell culture suggesting the maintenance of osteoblast phenotypic expression. As shown in Fig. 6, cells expressed higher ALP activity on Matrix1 compared to PLAGA at day 7, which indicates increased osteoblast differentiation. During the 21-day culture period, the gradually elevated phenotypic expressions of osteoblast cells on PLAGA, Matrix2, and TCPS suggested the progression of cell maturity, whereas osteoblasts matured much earlier on Matrix1 and were proliferating throughout the later time points. Such findings are in line with sequential proliferation and differentiation phases of osteoblasts in vitro [23,24].
One of the most important requirements for the potential use of any new biodegradable materials for tissue engineering applications is tissue compatibility. Matrix1 and Matrix2 were evaluated for tissue biocompatibility for 12 weeks. PLAGA matrices were used as the control due to its recognized biocompatibility. Both blend matrices were well tolerated throughout the duration of the 12-week of subcutaneous implantation period. No acute inflammation, tissue necrosis or abscess formation was observed around either polymer matrices. While complete degradation of PLAGA by 7 weeks resulted in a thick fibrous capsule around the implant site, both blend matrices showed diminished minimal tissue inflammation with thin fibrous capsules during the 12-week implantation. A minimal inflammatory response for both blend matrices indicates an apparent absence of local toxicity, which could be due to the non-toxicity and near-neutral pH of the degradation products. Furthermore, in comparing the biocompatibility of Matrix1 and Matrix2, it was observed that Matrix2 elicited a less intense inflammatory reaction with thinner fibrous capsules. The thickness of the fibrous capsule surrounding the blend matrices was much smaller than the fibrous capsule around PLAGA and other reported polyesters (over a hundred microns) [25,32]. Reduction in fibrous capsule thickness is highly advantageous to improve mass transfer between the matrices and surrounding tissues. In contrast to most biocompatible polymers that are steadily encapsulated in a fibrous capsule, both of the blend matrices were extensively infiltrated with collagen tissue as shown in Fig. 9. This dynamic matrix-tissue interface could be attributed to the unique degradation characteristics of the blend matrix, where the fast degradation of PLAGA facilitated cell infiltration and tissue in-growth while the degradation of polyphosphazene maintained the structural integrality of the matrix. It is known that inflammation and fibrous tissue encapsulation represent a process of normal host defense mechanisms triggered by the presence of implanted materials [33]. For a polymeric material, the inflammatory reaction depends on factors such as degradation characteristics, physico-chemical and mechanical properties of the materials [34]. Many biodegradable polymers including polyesters and polyanhydrides have been reported to produce inflammatory responses [25,35]. Here both of the blend matrices displayed improved subcutaneous tissue compatibility compared to PLAGA due to the controlled degradation with near-neutral pH degradation products of the blends.
The results presented in this study have illustrated great tunability in physical, chemical, and biological properties of dipeptide-based blend matrices. The synthetic flexibility of polyphosphazenes has allowed us to design specific side group chemistry that enables strong hydrogen bonding interactions as well as mechanical integrity. The degradation rates of blends were in the optimal range of 12-24 weeks for bone regeneration purposes. The excellent in vitro and in vivo biocompatibility demonstrated the great potential of the blend matrices for tissue engineering applications. Furthermore, an appropriate combination of mechanical stability and degradation pattern coupled with excellent biocompatibility can be achieved by simply adjusting the blend composition. For example, increasing the ratio of polyphosphazene in the blend results in a slower degradation rate, which is required for long-term healing. Thus, the blends can be adopted for developing three-dimensional scaffolds for specific orthopaedic applications based on the composition.
Conclusions
In this study, a mixed-substituent polyphosphazene (PNGEG/PhPh) with both glycylglycine dipeptide and phenylphenoxy group has been synthesized and characterized. Two blends namely 25:75 (Matrix1) and 50:50 (Matrix2) were produced at two weight ratios of PNGEG/PhPh to PLAGA. Both of the blends were confirmed to be completely miscible. It was shown that the physico-chemical properties, mechanical behavior, and degradation pattern of the blend can be modulated by altering the blend composition. The degradation products of the polyphosphazene neutralized the acidic degradation of PLAGA and resulted in significantly higher degradation media pH values. The blend components, PLAGA and polyphosphazene, exhibited a similar degradation pattern as characterized by molecular weight loss. Both of the blends enhanced PRO cell growth while maintaining osteoblast phenotype compared to the polyester. Furthermore, both blends showed improved biocompatibility in a rat subcutaneous implantation model compared to PLAGA over 12 weeks. Therefore, biodegradable dipeptide-based polyphosphazene-PLAGA blends are very promising candidate materials for developing mechanically competent scaffolds for bone tissue engineering. Further studies are underway to construct three-dimensional scaffolds using these blend materials for in vivo bone regeneration.
Acknowledgments
This work was supported by NIH RO1 EB004051 and NSF EFRI-0736002. Dr. Laurencin was the recipient of Presidential Faculty Fellow Award from the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laurencin CT, Ambrosio AMA, Borden MD, Cooper JA. Tissue engineering: orthopedic applications. Annu Rev Biomed Eng. 1999;1:19–46. doi: 10.1146/annurev.bioeng.1.1.19. [DOI] [PubMed] [Google Scholar]
- 2.Jabbarzadeh E, Starnes T, Khan YM, et al. Induction of angiogenesis in tissue-engineered scaffolds designed for bone repair: a combined gene therapy-cell transplantation approach. Proc Natl Acad Sci U S A. 2008;105:11099–11104. doi: 10.1073/pnas.0800069105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper JA, Jr, Sahota JS, Gorum IIWJ, Carter J, Doty SB, Laurencin CT. Biomimetic tissue-engineered anterior cruciate ligament replacement. Proc Natl Acad Sci U S A. 2007;104:3049–3054. doi: 10.1073/pnas.0608837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MS, Daniels AU, Andriano KP, Heller J. Six bioabsorbable polymers: in vitro acute toxicity of accumulated degradation products. J Appl Biomater. 1994;5:151–157. doi: 10.1002/jab.770050208. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal CM, Athanasiou KA. Technique to control pH in vicinity of biodegrading PLA-PGA implants. J Biomed Mater Res. 1997;38:105–114. doi: 10.1002/(sici)1097-4636(199722)38:2<105::aid-jbm4>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 7.Bostman OM. Osteoarthritis of the ankle after foreign-body reaction to absorbable pins and screws: a three- to nine-year follow-up study. J Bone Joint Surg Br. 1998;80-B:333–338. doi: 10.1302/0301-620x.80b2.8302. [DOI] [PubMed] [Google Scholar]
- 8.Sung HJ, Meredith C, Johnson C, Galis ZS. The effect of scaffold degradation rate on three-dimensional cell growth and angiogenesis. Biomaterials. 2004;25:5735–5742. doi: 10.1016/j.biomaterials.2004.01.066. [DOI] [PubMed] [Google Scholar]
- 9.Bostman O, Pihlajamaki H. Clinical biocompatibility of biodegradable orthopaedic implants for internal fixation: a review. Biomaterials. 2000;21:2615–2621. doi: 10.1016/s0142-9612(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 10.Landes CA, Ballon A, Roth C. Maxillary and mandibular osteosyntheses with PLGA and P(L/DL)LA implants: a 5-year inpatient biocompatibility and degradation experience. Plast Reconstr Surg. 2006;117:2347–2360. doi: 10.1097/01.prs.0000218787.49887.73. [DOI] [PubMed] [Google Scholar]
- 11.Deng M, Nair LS, Krogman NR, Allcock HR, Laurencin CT. Biodegradable polyphosphazene blends for biomedical applications. In: Andrianov A, editor. Polyphosphazenes for biomedical applications. Hoboken, NJ: Wiley-Interscience; 2009. pp. 139–154. [Google Scholar]
- 12.Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester)phosphazenes]: synthesis, crystallinity, and hydrolytic sensitivity in solution and the solid state. Macromolecules. 1994;27:1071–1075. [Google Scholar]
- 13.Lakshmi S, Katti DS, Laurencin CT. Biodegradable polyphosphazenes for drug delivery applications. Adv Drug Deliv Rev. 2003;55:467–482. doi: 10.1016/s0169-409x(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 14.Allcock HR. Chemistry and applications of polyphosphazenes. Hoboken, NJ: Wiley Interscience; 2003. [Google Scholar]
- 15.Kumbar S, Bhattacharyya S, Nukavarapu S, Khan Y, Nair L, Laurencin C. In vitro and in vivo characterization of biodegradable poly(organophosphazenes) for biomedical applications. J Inorg Organomet Polym Mater. 2006;16:365–385. [Google Scholar]
- 16.Singh A, Krogman NR, Sethuraman S, et al. Effect of side group chemistry on the properties of biodegradable L-alanine cosubstituted polyphosphazenes. Biomacromolecules. 2006;7:914–918. doi: 10.1021/bm050752r. [DOI] [PubMed] [Google Scholar]
- 17.Laurencin CT, Norman ME, Elgendy HM, et al. Use of polyphosphazenes for skeletal tissue regeneration. J Biomed Mater Res. 1993;27:963–973. doi: 10.1002/jbm.820270716. [DOI] [PubMed] [Google Scholar]
- 18.Laurencin CT, El-Amin SF, Ibim SE, et al. A highly porous 3-dimensional polyphosphazene polymer matrix for skeletal tissue regeneration. J Biomed Mater Res. 1996;30:133–138. doi: 10.1002/(SICI)1097-4636(199602)30:2<133::AID-JBM1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 19.Sethuraman S, Nair LS, El-Amin S, et al. In vivo biodegradability and biocompatibility evaluation of novel alanine ester based polyphosphazenes in a rat model. J Biomed Mater Res A. 2006;77:679–687. doi: 10.1002/jbm.a.30620. [DOI] [PubMed] [Google Scholar]
- 20.Ibim SEM, Ambrosio AMA, Kwon MS, El-Amin SF, Allcock HR, Laurencin CT. Novel polyphosphazene/poly(lactide-co-glycolide) blends: miscibility and degradation studies. Biomaterials. 1997;18:1565–1569. doi: 10.1016/s0142-9612(97)80009-9. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosio AMA, Allcock HR, Katti DS, Laurencin CT. Degradable polyphosphazene/poly(alpha-hydroxyester) blends: degradation studies. Biomaterials. 2002;23:1667–1672. doi: 10.1016/s0142-9612(01)00293-9. [DOI] [PubMed] [Google Scholar]
- 22.Krogman NR, Singh A, Nair LS, Laurencin CT, Allcock HR. Miscibility of bioerodible polyphosphazene/poly(lactide-co-glycolide) blends. Biomacromolecules. 2007;8:1306–1312. doi: 10.1021/bm061064q. [DOI] [PubMed] [Google Scholar]
- 23.Deng M, Nair LS, Nukavarapu SP, et al. Miscibility and in vitro osteocompatibility of biodegradable blends of poly[(ethyl alanato) (p-phenyl phenoxy) phosphazene] and poly(lactic acid-glycolic acid) Biomaterials. 2008;29:337–349. doi: 10.1016/j.biomaterials.2007.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng M, Nair LS, Nukavarapu SP, et al. Biomimetic, bioactive etheric polyphosphazene-poly(lactide-co-glycolide) blends for bone tissue engineering. J Biomed Mater Res A. 2010;92A:114–125. doi: 10.1002/jbm.a.32334. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002;20:602–606. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 26.Adibi SA. Glycyl-dipeptides: new substrates for protein nutrition. J Lab Clin Med. 1989;113:665–673. [PubMed] [Google Scholar]
- 27.El-Amin SF, Kwon MS, Starnes T, Allcock HR, Laurencin CT. The biocompatibility of biodegradable glycine containing polyphosphazenes: a comparative study in bone. J Inorg Organomet Polym Mater. 2006;16:387–396. [Google Scholar]
- 28.Sivalingam G, Karthik R, Madras G. Blends of poly(epsilon-caprolactone) and poly(vinyl acetate): mechanical properties and thermal degradation. Polym Degrad Stab. 2004;84:345–351. [Google Scholar]
- 29.Gajria AM, Dav V, Gross RA, McCarthy SP. Miscibility and biodegradability of blends of poly(lactic acid) and poly(vinyl acetate) Polymer. 1996;37:437–444. [Google Scholar]
- 30.Anseth KS, Shastri VR, Langer R. Photopolymerizable degradable polyanhydrides with osteocompatibility. Nat Biotechnol. 1999;17:156–159. doi: 10.1038/6152. [DOI] [PubMed] [Google Scholar]
- 31.Kofron MD, Griswold A, Kumbar SG, Martin K, Wen XJ, Laurencin CT. The implications of polymer selection in regenerative medicine: a comparison of amorphous and semi-crystalline polymer for tissue regeneration. Adv Funct Mater. 2009;19:1351–1359. [Google Scholar]
- 32.Mainil-Varlet P, Gogolewski S, Nieuwenhuis P. Long-term soft tissue reaction to various polylactides and their in vivo degradation. J Mater Sci Mater Med. 1996;7:713–721. [Google Scholar]
- 33.Menei P, Daniel V, Montero-Menei C, Brouillard M, Pouplard-Barthelaix A, Benoit JP. Biodegradation and brain tissue reaction to poly(D,L-lactide-co-glycolide) microspheres. Biomaterials. 1993;14:470–478. doi: 10.1016/0142-9612(93)90151-q. [DOI] [PubMed] [Google Scholar]
- 34.Homsy CA. Biocompatibility in selection of materials for implantation. J Biomed Mater Res. 1970;4:341–356. doi: 10.1002/jbm.820040306. [DOI] [PubMed] [Google Scholar]
- 35.Ibim SM, Uhrich KE, Bronson R, El-Amin SF, Langer RS, Laurencin CT. Poly(anhydride-co-imides): in vivo biocompatibility in a rat model. Biomaterials. 1998;19:941–951. doi: 10.1016/s0142-9612(98)00019-2. [DOI] [PubMed] [Google Scholar]