

Abstract
Bone fillers have emerged as an alternative to the invasive surgery often required to repair skeletal defects. Achieving controlled release from these materials is desired for accelerating healing. Here, oppositely-charged Poly (d,l-lactic-co-glycolic acid) (PLGA) nanoparticles were used to create a cohesive colloidal gel as an injectable drug-loaded filler to promote healing in bone defects. The colloid self-assembled through electrostatic forces resulting in a stable 3-D network that may be extruded or molded to the desired shape. The colloidal gel demonstrated shear-thinning behavior due to the disruption of interparticle interactions as the applied shear force was increased. Once the external force was removed, the cohesive property of the colloidal gel was recovered. Similar reversibility and shear-thinning behavior were also observed in colloidal gels loaded with dexamethasone. Near zero-order dexamethasone release was observed over two months when the drug was encapsulated in PLGA nanoparticles and simply blending the drug with the colloidal gel showed similar kinetics for one month. Surgical placement was facilitated by the pseudoplastic material properties and in vivo observations demonstrated that the PLGA colloidal gels stimulated osteoconductive bone formation in rat cranial bone defects.
Keywords: PLGA, colloidal gel, drug delivery, bone defect
1.Introduction
Injectable biomaterials that form three-dimensional (3-D) structures in situ are being pursued as tissue engineering scaffolds [1], biosensors [2] and drug delivery systems [3]. Colloidal gels composed of oppositely-charged nanoparticles at high concentration can exhibit pseudoplastic behavior facilitating the fabrication of shape-specific macroscale materials with 3-D architectures [4, 5]. Emerging applications in materials science have also aimed to leverage the unique properties of colloidal gels [6-12]. For example, freeform printing of colloidal gels may be utilized to produce 3-D, microperiodic networks exhibiting precise structure [13-16]. The cohesive strength of these materials depends upon interparticle interactions such as electrostatic forces, van der Waals attraction, steric hindrance, etc. [17] which may be leveraged to achieve unique bulk material properties for many different applications [18-23]. The application of moldable colloidal gels towards generating tissues has also been proposed [24, 25], but has not yet to be applied with the integration of biodegradable and non-cytotoxic nanoparticles composed of materials commonly used as tissue scaffolds.
The injectability of 3-D scaffolds is desired as a means to fill tissue defects of irregular size and shape. Injectable scaffolds are particularly desired in order to avoid the invasive surgery typically required for tissue implantation [26]. From a clinical perspective, the use of injectable scaffolds is an attractive alternative to surgery as it reduces the risk of infection, scar formation, patient discomfort and the cost of treatment [27]. Recently, many scaffolds that stiffen or solidify in vivo have been applied as injectable scaffolds [28]. Often injectable scaffolds are polymerized or chemically crosslinked to stiffen the material. Chemically crosslinked scaffolds normally form via an in situ reaction induced by the presence of water, heat, light or other stimuli. During solidification, However, toxic chemical agents are sometimes employed which may adversely affect the scaffolds, destabilize encapsulated biomolecules, or pose toxicity concerns. On the contrary, colloidal gels that stiffen through interactions such as electrostatic forces, van der Waals attraction and steric hindrance may overcome some of these limitations.
Poly (D,L-lactic-co-glycolic acid) (PLGA) is a biodegradable polymer, which has been widely used in pharmaceutical products and in tissue engineering scaffolds [29-32]. Previously, oppositely-charged PLGA nanoparticles were used to create a cohesive colloidal gel. The colloid self-assembled through electrostatic forces resulting in a stable, porous 3-D network that was easily molded to the desired shape [33]. The colloidal gel demonstrated shear-thinning behavior due to the disruption of interparticle interactions as the applied shear force was increased. Once the external force was removed, the strong cohesive property of the colloidal gel was recovered. This material also demonstrated negligible toxicity to human umbilical cord matrix stem cells (hUCMSCs). The biodegradability, biocompatibility and reversibility of this unique material make it a potential injectable scaffold for tissue engineering.
In this paper, PLGA colloidal gels were studied as an injectable controlled release system to deliver dexamethasone (DEX). DEX is a glucocorticoid which is usually used as an anti-inflammatory and immunosuppressive agent in bone tissue engineering [34-36]. In addition, previous reports suggest that glucocorticoids such as DEX may facilitate osteogenesis [37, 38]. Here, DEX was used to investigate the drug release properties of PLGA colloidal gels. The rheological properties of the drug-loaded colloidal gel were also evaluated and its application in rat cranial bone defects was assessed.
2.Materials and methods
2.1. Materials
All materials were purchased from Sigma-Aldrich unless otherwise stated. PLGA (75:25) (inherent viscosity: 0.47 dL/g in chloroform at 30 °C) was purchased from Absorbable Polymers. DEX was obtained from Alfa Aesar Co. Poly (ethylene-co-maleic acid) (PEMA) was purchased from Polysciences Inc. Surfactant PEMA [39] and polyvinylamine (PVAm) [40] were synthesized through reported protocols.
2.2. Preparation of blank PLGA nanoparticles
The oppositely-charged blank PLGA nanoparticles were prepared by a solvent diffusion method. 100 mg of PLGA was dissolved in 10 mL acetone and then the solution was added into 0.2 % PVAm or PEMA (150 mL) surfactant solution through a syringe pump (20 mL/h) under stirring at 200 rpm overnight to evaporate acetone. Nanoparticles were collected by centrifugation (Beckman Co., Avanti 30) (16,000 rpm, 20 min). The nanoparticles were centrifuged and resuspended using deionized water three times to remove excess surfactant. A fine powder of nanoparticles was obtained by lyophilization for ~2 days.
2.3. Preparation of drug loaded PLGA nanoparticles
PLGA naoparticles loaded with DEX were prepared by a single oil-in-water (O/W) emulsion/solvent evaporation method [41]. In order to maximize the encapsulation efficiency (EE) of the DEX in the nanoparticles, different conditions were tested. 90 mg of PLGA was dissolved in 9 ml dichloromethane and then 10 mg DEX was dissolved in 1 ml acetone. The DEX in acetone was added to the PLGA in dichloromethane to form the oil phase. Then 10 ml of the oil phase was added dropwise to a 0.2 % PVAm or PEMA (30 mL) surfactant solution through a syringe pump (1 mL/min). The oil-in-water (O/W) emulsion was formed using a high speed homogenizer (Biosepc Products, Inc.) at 16,000 rpm in an ice bath to prevent overheating. Then the emulsion was added to a 160 ml 0.2 % PVAm or PEMA surfactant solution under stirring at 200 rpm overnight to evaporate the organic phase. DEX-loaded nanoparticles were collected by centrifugation (16,000 rpm, 20 min). The nanoparticles were centrifuged and resuspended using deionized water three times to remove excess surfactant and free drug. A fine powder of drug loaded nanoparticles was obtained by lyophilization for ~2 days.
2.4. Preparation of colloidal gels
Lyophilized nanoparticles (PLGA-PVAm or PLGA-PEMA, blank or drug loaded) were dispersed in deionized water at 20 % wt/vol. These dispersions were mixed in different ratios to obtain the different PLGA-PEMA: PLGA-PVAm ratios studied. Homogeneous colloid mixtures were prepared in a bath sonicator for 3 minutes and stored at 4 °C for 2 h before use. Several gels with different mass ratios of PLGA-PEMA nanoparticles to PLGA-PVAm nanoparticles were designated as AB73, AB55 and AB37 (A: PLGA-PEMA nanoparticles; B: PLGA-PVAm nanoparticles; the mass ratio of PLGA-PEMA nanoparticles to PLGA-PVAm nanoparticles is 70:30, 50:50 and 30:70, respectively).
2.5. Characterization of nanoparticles and colloidal gels
The sizes and zeta potentials of the different PLGA nanoparticles were determined using a ZetaPALS dynamic light scattering system (Brookhaven, ZetaPALS). All samples were analyzed in triplicate. Scanning electron microscopy (SEM) was performed using a Jeol JSM-6380 field emission scanning electron microscope at an accelerating voltage of 10 kV.
2.6. Rheological experiments
Rheological experiments were performed by a controlled stress rheometer (AR2000, TA Instrument Ltd.). 2° cone steel plates (20 mm diameter) were used and the 500 μm gap was filled with colloidal gel. A solvent trap was used to prevent evaporation of water. The viscoelastic properties of the sample were determined at 20°C by forward-and-backward stress sweep experiments. The viscosity (η) was monitored while the stress was increased and then decreased (frequency = 1 Hz) in triplicate with 10 minutes between cycles. The gel recoverability was assessed using the defined time break between cycles. All samples were analyzed in triplicate.
2.7. In vitro drug release tests
The encapsulation efficiency of DEX in the drug loaded nanoparticles was determined by dissolving 10 mg of drug-loaded nanoparticles powder in 2 ml trifluoroethanol (TFE). Samples were rotated for at least 24 h at 10 rpm to ensure complete dissolution in TFE. Blank nanoparticles were treated identically. The concentration of DEX in the resulting solution was determined by measuring the absorbance at 242 nm in a spectrophotometer (Agilent Technologies, 89090A) and then subtracting the absorbance values for the blank nanoparticles. All samples were analyzed in triplicate.
DEX release profiles were determined by suspending 800 mg of drug-loaded nanoparticles in 50 mL of PBS (pH 7.4). The samples were incubated at 37 °C while shaking at 50 rpm in an incubator/shaker (New Brunswick Scientific C24). At selected time points, the supernatants were removed and replaced with fresh buffer. The concentration of DEX in the supernatant was determined using the UV detection method described above. The absorption of supernatant collected from blank PLGA nanoparticles was negligible at 242 nm throughout the release study. The amount of drug in each sample was summed with the amount at each previous time point to obtain the cumulative drug release amount and the total was divided by the amount of drug in the nanoparticles (encapsulation efficiency times mass of drug-loaded nanoparticles) to calculate the cumulative drug release percentage. Each release experiment was performed in triplicate.
2.8. Animals and surgical implantation of colloidal gels
The use of animals and the surgical procedures used in this study were approved by the Animal Care and Use Committee at the University of Kansas Medical Center, Kansas City, KS. Male Sprague-Dawley rats, 7 to 8 weeks old, were used in this study. All surgical procedures were performed under general anesthesia and sterile conditions. A longitudinal skin incision was made centered over the mid-sagittal suture of the skull. Following careful dissection and removal of the periosteum, an 8-mm diameter full thickness defect was produced in the parietal bone using a dental burr. The wound was carefully rinsed with normal saline to remove any bone debris in the defect, which was an important step in this procedure for evaluating the formation of new bone within the cranial bone defect as previously described [42, 43]. The cranial defects were filled with injectable PLGA colloidal gel, PLGA colloidal gel with DEX, or left untreated. The wound was closed with a 5-0 nylon suture. The day of surgery was designated as day “0”. Animals were sacrificed at 4 weeks after surgery and operated calvaria were harvested for histological and histochemical analyses.
2.9. Histology and histochemistry
To evaluate the cellular and matrix responses to the implants, implanted materials were retrieved with the surrounding host bone and residual periosteum. Tissue samples were fixed in 2% paraformaldehyde (Boston BioProducts), pH 7.4, decalcified in 25% formic acid (Sigma), embedded in paraffin, sectioned at 5 μm, and stained with either hematoxylin and eosin (H&E) or safranin-O that identifies cartilage cells and cartilage matrix. At least three animals per treatment group were examined histologically and histochemically.
3.Results and discussions
3.1. Characterization of PLGA nanoparticles and colloidal gels
A schematic representation of the DEX-loaded colloidal gels provides an overview of the concept (Figure 1). Drug-free PLGA nanoparticles were prepared by a solvent diffusion method. PLGA dissolved in acetone was titrated into a water phase containing PVAm or PEMA. The surface charge of particles resulted from the precipitation of PLGA nanoparticles coated with the respective polyelectrolyte. The particle sizes of drug-free nanoparticles were 181 ± 15 nm (PLGA-PEMA) and 144 ± 12 nm (PLGA-PVAm). The zeta potentials of drug-free nanoparticles were -20.1 ± 1.0 mV (PLGA-PEMA) and +32.2 ± 1.3 mV (PLGA-PVAm) [33]. The DEX-loaded PLGA nanoparticles were prepared by a single oil-in-water (O/W) emulsion/solvent evaporation method. PLGA and DEX dissolved in a mixture of dichloromethane and acetone was emulsified into a water phase containing PVAm or PEMA. The sizes of drug-loaded nanoparticles were 241 ± 26 nm (PLGA-PEMA) and 182 ± 21 nm (PLGA-PVAm). The zeta potentials of drug-loaded nanoparticles were -28.5 ± 2.1 mV (PLGA-PEMA) and +34.6 ± 2.5 mV (PLGA-PVAm).
Figure 1.

Schematic representation of the properties of DEX-loaded colloidal gels.
Zeta potential and particle size are two critical factors influencing the cohesive properties of colloidal gel systems [33]. Small nanoparticles (~100-200 nm) were selected to provide the desired cohesive strength of colloidal gels. Larger particles yield fewer particle-particle contacts, thus reducing the gel strength. The large zeta potential of the DEX-loaded nanoparticles also facilitated tight particle packing as a result of strong electrostatistic interparticle attraction. These features led to the formation of stable, drug-loaded colloidal gels. DEX-loaded nanoparticles were linked together into micrometer-scale, ring-like structures resulting in a bulk porous structure with microchannels. The formation of these structures is hypothesized to result from an equilibrium of nanoparticle attraction (tight agglomerates) and repulsion (pores) (Figure 2). Microchannels were ~1 μm in the dry state; however, these are large in the hydrated state [33]. The entire drug-loaded colloidal gel exhibited a loosely organized, micro-porous structure which was consistent for different gel compositions and drug loading percentages. Scanning electron micrographs (SEM) of dried colloidal gels showed little difference in the structure of the gels containing different mass ratios of drug-loaded nanoparticles (Figure 2A and 2B), despite the different zeta potential and particle size of the DEX-loaded PLGA-PEMA and PLGA-PVAm nanoparticles. DEX loading percentage also did not affect the dry structure of the drug-loaded colloidal gels (Figure 2C). The appearance of drug-loaded colloidal gels was similar to the unloaded colloidal gels [33]; molded scaffolds still exhibited stable structure and shape retention when handled (Figure 2D).
Figure 2.

Scanning electron micrographs of AB55 colloidal gel with 10% DEX loading (A), AB73 colloidal gel with 10% DEX loading (B), AB55 colloidal gel with 20% DEX loading (C) and shaped tissue scaffold made from AB55 colloidal gel with 10% DEX loading (D) (scale bar = 1μm).
3.2. Rheological properties of PLGA colloidal gels
The strength of colloidal gels depends upon interparticle interactions such as electrostatic forces and van der Waals attraction [17]. Here, the nanoparticle colloid presumably self-assembled through long-range electrostatic interactions resulting in a stable 3-D network as was reported previously [33]. When an external force was applied to disrupt the interparticle interactions, the colloidal gel demonstrated shear-thinning behavior. Once the external force was removed, the strong cohesive property of the colloidal gel was recovered. This reversibility makes the gel an excellent material for molding, extrusion, or injection of tissue scaffolds. The cohesive strength and the reversibility of colloidal gels depended on the mass ratios of oppositely-charged nanoparticles and the concentration of the nanoparticles in the gels [33].
The dependence of lag time on the recovery of colloidal gel viscosity after accelerating/decelerating shear force cycles was determined for drug-loaded colloidal gels (Figure 3). The colloidal gels did not rapidly recover during consecutive acceleration/deceleration cycles of shear force. When no recovery time was allowed, the viscosity of the colloidal gel only recovered about 30% of the original value. On the contrary, if more time (30 minutes) was provided for the recovery process, the cohesive property of the colloidal gel was restored to around 65% of the original value. The results suggested good recoverability for sheared colloidal gels.
Figure 3.
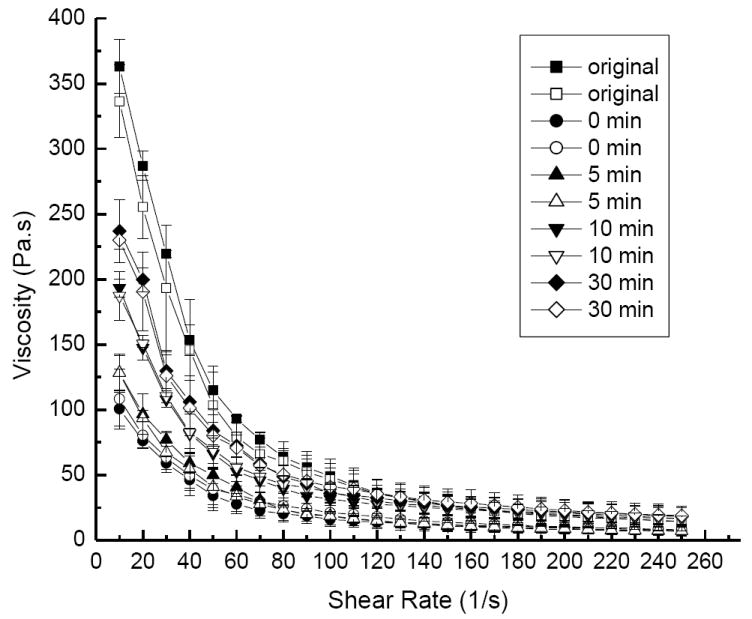
Viscosity profiles demonstrate the shear-thinning behavior of AB55 colloidal gel with 10% DEX loading for accelerating (solid symbols) and decelerating (open symbols) shear force using different lag times between cycles.
The percentage of drug-loading did not appreciably affect the rheological behavior of colloidal gels (Figure 4). Meanwhile, colloidal gels with different compositions showed different viscosity profiles as expected. Equal mass ratios (50:50) of nanoparticles yielded a higher viscosity gel (AB55) than an unequal ratio (70:30) of nanoparticles (AB73). The result confirmed the importance of equilibrating charge as a means to increase cohesion.
Figure 4.
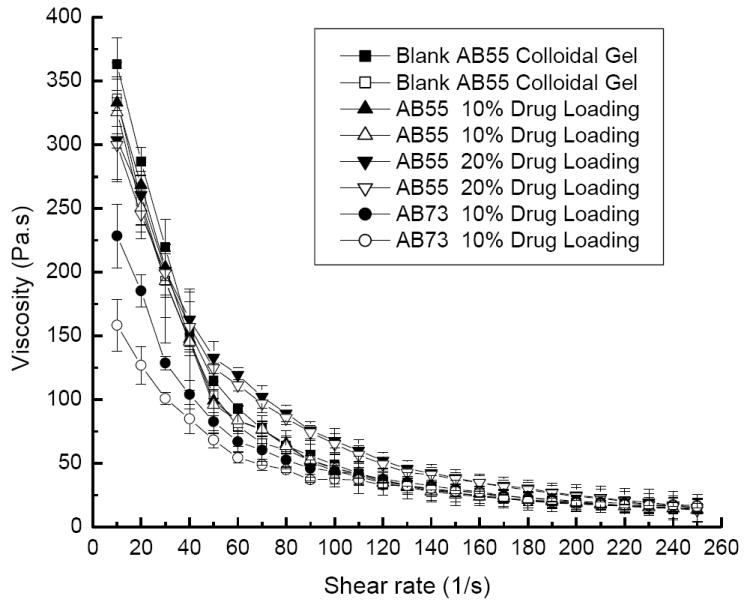
Viscosity profiles demonstrate the shear-thinning behavior of AB55 and AB73 colloidal gel with different DEX loading for accelerating (solid symbols) and decelerating (open symbols) shear force.
3.3. Drug release tests
The encapsulation efficiency of DEX in the nanoparticles depended upon the drug loading (Figure 5). Because of the slight solubility of DEX in water, some was lost during the fabrication process. The PLGA-PEMA nanoparticles seemed to encapsulate DEX slightly more efficiently than PLGA-PVAm nanoparticles; however, the differences were not statistically significant.
Figure 5.
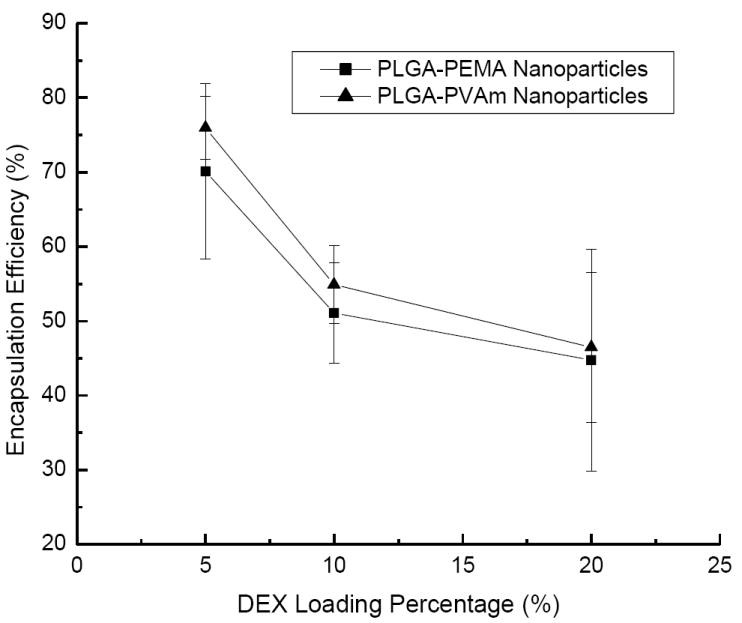
Encapsulation efficiency of DEX in PLGA-PEMA and PLGA-PVAm nanoparticles with different loading percentage.
DEX released from the colloidal gels with different drug loading for more than 60 days when the drug was encapsulated in the PLGA nanoparticles (Figure 6). More drug was released from the colloidal gel with the highest drug loading (20%) and followed a near zero order release profile. However, only about half of the drug was released over 60 days due to the larger amount of drug present at this high loading. On the other hand, almost all drug was released from 5% drug-loading colloidal gels within 60 days.
Figure 6.
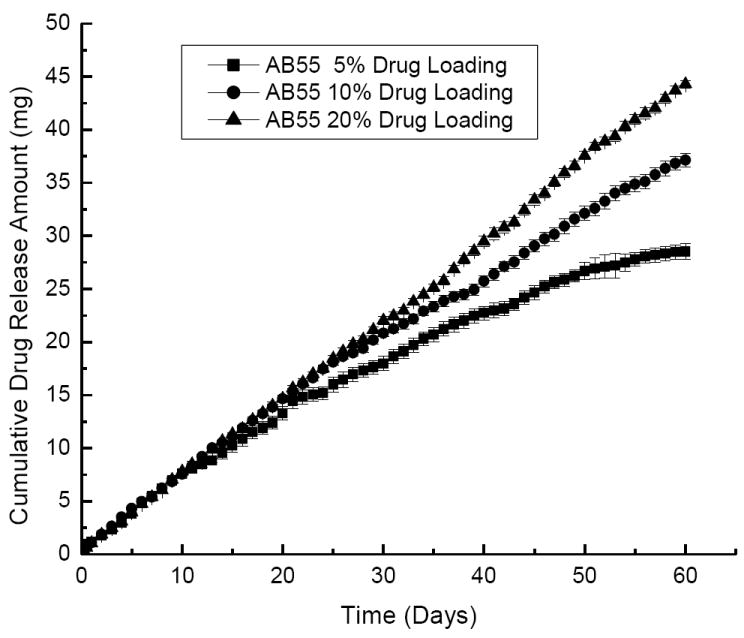
Drug release profiles of AB55 colloidal gels with different DEX loading. Drug was loaded into both positively charged and negatively charged PLGA nanoparticles.
Suspensions of purely cationic or anionic drug-loaded nanoparticles released DEX very quickly (Figure 7). The entire release process only lasted about 15-25 days. Colloidal gels composed of mixtures of the exact same drug-loaded nanoparticles released the drug for more than 2 months. Again, the kinetics were near linear regardless of the mass ratio of charged, drug-loaded nanopaticles in the colloidal gels. For comparison, when the drug was directly mixed with the colloidal gels, the material still performed as a controlled release drug delivery system for about 35 days (Figure 8). Slow diffusion of the drug through the porous microstructure of colloidal gels and, perhaps, adsorption of the drug onto the nanoparticles, led to the sustained release profile of DEX directly mixed with the colloidal gel. Furthermore, the time required for drug release from suspensions of purely charged drug-loaded nanoparticles plus the time required for drug release from colloidal gels mixed with drug was nearly equal to the duration of drug release from colloidal gels composed of DEX-loaded nanoparticles. This result was supportive of a two-stage drug release process from this material.
Figure 7.
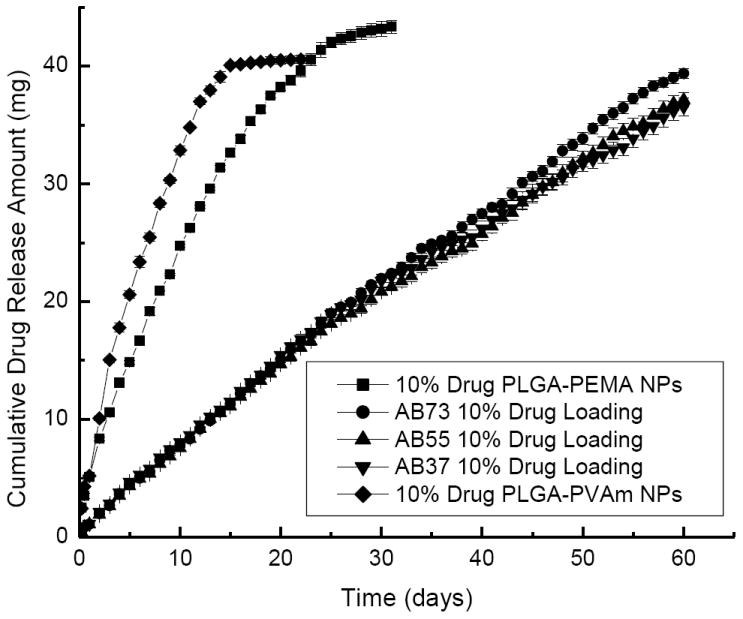
Drug release profiles of colloidal gels mixed at different ratios with 10% DEX loading. Drug was loaded into both positively charged and negatively charged PLGA nanoparticles (NPs).
Figure 8.
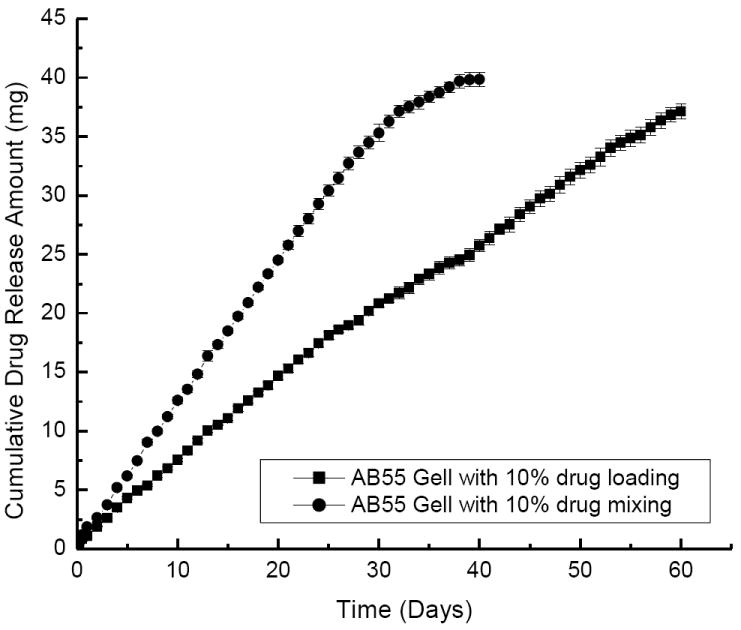
Drug release profiles of AB55 colloidal gels with 10% DEX directly mixed into the bulk material (round) compared to the drug encapsulated in both nanoparticle types (square).
In previous reports, porous drug delivery systems composed of nanoparticles with microchannels showed drug release with near zero-order kinetics [44, 45]. Presumably, the drug released from the colloidal gels via a two-stage process. Here, DEX was first released from the nanoparticles into the micropores of the colloidal gel. In this process, the drug release rate was faster for colloidal gels containing nanoparticles with higher drug loading, presumably due to the larger concentration of DEX within nanoparticles driving diffusion. Then, the released drug diffused through the microchannels and was ultimately transferred to the media. It appears that, the second process may have been the rate limiting step of this two step release mechanism. The entire release process exhibited near zero-order release and colloidal gels with higher drug loading released more drug for a longer duration, but with similar kinetics.
3.4. Histological and histochemical analyses
Rat cranial defects were used as an in vivo model to test the efficacy of PLGA colloidal gels. Previous reports suggested that sustained delivery of low doses of DEX may enhance osteogenesis; therefore, PLGA colloidal gels containing 5% DEX were selected for in vivo studies [37, 38]. Cranial defects were chosen as a model since the current colloidal gel materials would not be amenable to load-bearing bone regeneration. These materials are desired for cranial defects, however, since they may conform to irregular defect shapes and the viscosity is appropriate for placement and subsequent stiffening in situ. Colloidal gels are also malleable, which may facilitate the ingrowth of new tissue.
Untreated rat cranial defects were filled with a thin layer of fibrous tissue resulting in a collapse of the defect area. In contrast, the defects treated with either PLGA colloidal gel or colloidal gel with DEX were filled with residual implant materials surrounded by either new bone or fibrous tissue, which effectively prevented the defects from collapsing (Figure 9, Top row). The formation of new bone, which was observed in the areas adjacent to the host bone, was substantially more abundant in the defects treated with PLGA colloidal gel (with or without DEX) compared to the untreated control defects, indicating that PLGA colloidal gel can stimulate osteoconductive bone formation (Figure 9, Middle and Bottom rows). The central regions of the defects treated with PLGA colloidal gel were filled with residual implant materials and fibrous tissue without new bone, suggesting that complete bone healing was not achieved in 8-mm critical-sized cranial defects even in the presence of the colloidal gel implants. A combination of osteoconductive biomaterials and osteoinductive factor(s)/osteogenic cells may be required to achieve complete bone regeneration of critical-sized rat cranial bone defects. No cartilage formation was observed in the PLGA colloidal gel-treated or untreated defects. These results suggest that PLGA colloidal gels are non-cytotoxic in vivo and highly osteoconductive for the repair of rat cranial bone defects.
Figure 9.
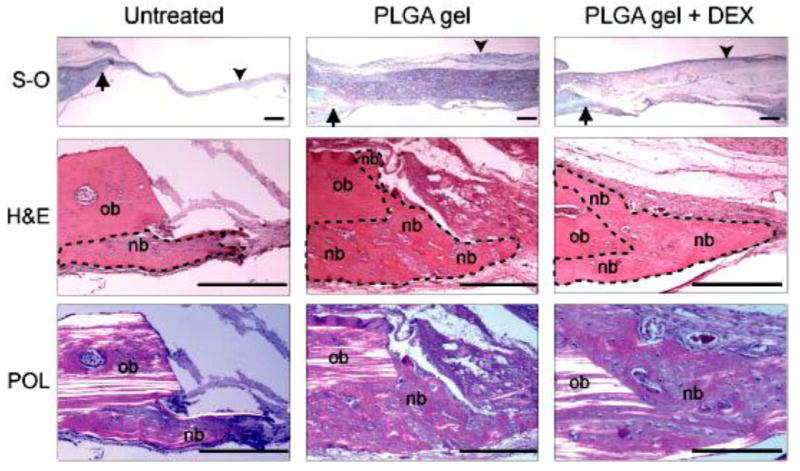
Photomicrographs of tissue sections prepared from untreated, PLGA colloidal gel (PLGA gel)-treated, or PLGA colloidal gel with encapsulated DEX (PLGA gel + DEX)-treated rat cranial bone defects. Top row: Low magnification of tissue sections including the bone defect margins (arrows) and the mid-portion of the defects (arrow heads). S-O, safranin-O and fast-green staining. Middle row: Higher magnification of tissue sections showing both the original host bone (ob) and the new bone (nb) formed in the areas adjacent to the host bone. New bone is outlined by a dotted line. H&E, hematoxylin and eosin staining. Bottom row: Polarizing photomicrographs (POL) of the cranial defect margins demonstrate that collagen fibers of the new bone (nb) display non- or low-polarizing orientation compared to that of the highly polarized parallel lamellae of the original host bone (ob) in each treatment group. H&E staining. Scale bar = 100 μm for all photos in this figure.
4.Conclusions
Cohesive colloidal gels made by mixing oppositely-charged PLGA nanoparticles were investigated as potential bone defect fillers. The strength of the colloidal gels resulted from electrostatic interparticle interactions. The pseudoplastic, shear-thinning behavior of the colloidal gels was desired for applications as an injectable scaffold for tissue repair. The PLGA colloidal gel also exhibited recovery of viscosity after shear shinning suggesting that the material may stiffen after placement in vivo. Drug release tests revealed that DEX was slowly released at a constant rate for more than two months. In vivo results demonstrated that PLGA colloidal gels were osteoconductive fillers capable of controlled release for the repair of rat cranial bone defects.
Acknowledgments
We gratefully acknowledge Prof. Stevin Gehrke for the use of equipment and the Microscopy Laboratory for assistance with imaging. We are grateful for support from funding agencies such as the American Heart Association, the Cystic Fibrosis Foundation, the Institute for Advancing Medical Innovation, the DOD and the NIH (1R03 AR054035-01A1, P20 RR016443 and P20 RR015563 (CB) and 1R21DE017673-01A1 (MSD). This study was also supported by the Paul R. Harrington, M.D. and Mary Alice Harrington Distinguished Professorship endowment (JW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Griffith LG, Naughton G. Tissue engineering--current challenges and expanding opportunities. Science. 2002;295:1009–1014. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- 2.Asher SA, Alexeev VL, Goponenko AV, Sharma AC, Lednev IK, Wilcox CS, et al. Photonic crystal carbohydrate sensors: low ionic strength sugar sensing. J Am Chem Soc. 2003;125:3322–3329. doi: 10.1021/ja021037h. [DOI] [PubMed] [Google Scholar]
- 3.Li YY, Barlow S, Sailor MJ, Tan KH, Zehner RW, Marder SR. Polymer replicas of photonic porous silicon for sensing and drug delivery applications. Science. 2003;299:2045–2047. doi: 10.1126/science.1081298. [DOI] [PubMed] [Google Scholar]
- 4.Johnson SA, Oliver PJ, Mallouk TE. Ordered mesoporous polymers of tunable pore size from colloidal silica templates. Science. 1999;283:963–965. doi: 10.1126/science.283.5404.963. [DOI] [PubMed] [Google Scholar]
- 5.Holtz JH, Asher SA. Polymerized colloidal crystal hydrogel films as intelligent chemical sensing materials. Nature. 1997;389:829–832. doi: 10.1038/39834. [DOI] [PubMed] [Google Scholar]
- 6.Blaaderen AV, Ruel R, Wiltzius P. Template-directed colloidal crystallization. Nature. 1997;385:321–324. [Google Scholar]
- 7.Braun PV, Wiltzius P. Electrochemically grown photonic crystals. Nature. 1999;402:603–604. [Google Scholar]
- 8.Joannopoulos JD, Villeneuve PR, Fan S. Photonic crystals: putting a new twist on light. Nature. 1997;386:143–149. [Google Scholar]
- 9.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27:4356–4373. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 10.Xia XH, Hu ZB, Marquez M. Physically bonded nanoparticle networks: a novel drug delivery system. J Control Release. 2005;103:21–30. doi: 10.1016/j.jconrel.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Van Tomme SR, van Steenbergen MJ, De Smedt SC, van Nostrum CF, Hennink WE. Self-gelling hydrogels based on oppositely charged dextran microspheres. Biomaterials. 2005;26:2129–2135. doi: 10.1016/j.biomaterials.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 12.Van Tomme SR, van Nostrum CF, de Smedt SC, Hennink WE. Degradation behavior of dextran hydrogels composed of positively and negatively charged microspheres. Biomaterials. 2006;27:4141–4148. doi: 10.1016/j.biomaterials.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Gratson GM, Xu M, Lewis JA. Microperiodic structures: direct writing of three-dimensional webs. Nature. 2004;428:386. doi: 10.1038/428386a. [DOI] [PubMed] [Google Scholar]
- 14.Smay JE, Graston GM, Shepherd RF, Cesarano JI, Lewis JA. Directed colloidal assembly of 3D periodic sStructures. Adv Mater. 2002;14:1279–1283. [Google Scholar]
- 15.Wu H, Odom TW, Chiu DT, Whitesides GM. Fabrication of complex three-dimensional microchannel systems in PDMS. J Am Chem Soc. 2003;125:554–559. doi: 10.1021/ja021045y. [DOI] [PubMed] [Google Scholar]
- 16.Liu JW, Luijten E. Stabilization of colloidal suspensions by means of highly charged nanoparticles. Phys Rev Lett. 2004;93:247802–247805. doi: 10.1103/PhysRevLett.93.247802. [DOI] [PubMed] [Google Scholar]
- 17.Tohver V, Chan A, Sakurada O, Lewis JA. Phase behavior and 3D structure of strongly attractive microsphere–nanoparticle mixtures. Langmuir. 2001;17:8414–8421. doi: 10.1021/la051998k. [DOI] [PubMed] [Google Scholar]
- 18.Therriault D, White SR, Lewis JA. Chaotic mixing in three-dimensional microvascular networks fabricated by direct-write assembly. Nat Mater. 2003;2:265–271. doi: 10.1038/nmat863. [DOI] [PubMed] [Google Scholar]
- 19.Chrisey DB. Materials processing: the power of direct writing. Science. 2000;289:879–881. doi: 10.1126/science.289.5481.879. [DOI] [PubMed] [Google Scholar]
- 20.Klajn R, Bishop KJM, Fialkowski M, Paszewski M, Campbell CJ, Gray TP, et al. Plastic and moldable metals by self-assembly of sticky nanoparticle aggregates. Science. 2007;316:261–264. doi: 10.1126/science.1139131. [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Cuddihy MJ, Cater GM, Kotov NA. Engineering liver tissue spheroids with inverted colloidal crystal scaffolds. Biomaterials. 2009;30:4687–4694. doi: 10.1016/j.biomaterials.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Hu ZB, Lu XH, Gao J. Hydrogel opals. Adv Mater. 2001;13:1708–1712. [Google Scholar]
- 23.Katz E, Willner I. Integrated nanoparticle-biomolecule hybrid systems: synthesis, properties, and applications. Angew Chem Int Ed. 2004;43:6042–6108. doi: 10.1002/anie.200400651. [DOI] [PubMed] [Google Scholar]
- 24.Xie B, Parkhill RL, Warren WL, Smay JE. Direct writing of three-dimensional polymer scaffolds using colloidal gels. Adv Funct Mater. 2006;16:1685–1693. [Google Scholar]
- 25.Dellinger JG, Cesarano JI, Jamison RD. Robotic deposition of model hydroxyapatite scaffolds with multiple architectures and multiscale porosity for bone tissue engineering. J Biomed Mater Res A. 2007;82A:383–394. doi: 10.1002/jbm.a.31072. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Cuddihy MJ, Kotov NA. Three-dimensional cell culture matrices: state of the art. Tissue Eng Part B. 2008;14:61–86. doi: 10.1089/teb.2007.0150. [DOI] [PubMed] [Google Scholar]
- 27.Hou QP, De Bank PA, Shakesheff KM. Injectable scaffolds for tissue regeneration. J Mater Chem. 2004;14:1915–1923. [Google Scholar]
- 28.Cushing MC, Anseth KS. Hydrogel Cell Cultures. Science. 2007;316:1133–1134. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 29.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 30.Kim DH, Martin DC. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials. 2006;27:3031–3037. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 31.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 32.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Wang L, Detamore MS, Berkland C. Biodegradable colloidal gels as moldable tissue engineering scaffolds. Adv Mater. 2008;20:236–239. [Google Scholar]
- 34.Wu X, Yu G, Parks H, Hebert T, Goh BC, Dietrich MA, et al. Circadian mechanisms in murine and human bone marrow mesenchymal stem cells following dexamethasone exposure. Bone. 2008;42:861–870. doi: 10.1016/j.bone.2007.12.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oshina H, Sotome S, Yoshii T, Torigoe I, Sugata Y, Maehara H, et al. Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone. 2007;41:575–583. doi: 10.1016/j.bone.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 36.Kubota S, Moritani NH, Kawaki H, Mimura H, Minato M, Takigawa M. Transcriptional induction of connective tissue growth factor/hypertrophic chondrocyte-specific 24 gene by dexamethasone in human chondrocytic cells. Bone. 2003;33:694–702. doi: 10.1016/s8756-3282(03)00227-8. [DOI] [PubMed] [Google Scholar]
- 37.Nuttelman CR, Tripodi MC, Anseth KS. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. J Biomed Mater Res A. 2005;76A:183–195. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JE, Gersbach CA, Wojtowicz AM, García AJ. Glucocorticoid-induced osteogenesis is negatively regulated by Runx2/Cbfa1 serine phosphorylation. J Cell Sci. 2006;19:581–591. doi: 10.1242/jcs.02758. [DOI] [PubMed] [Google Scholar]
- 39.Shi L, Berkland C. pH-triggered dispersion of nanoparticle clusters. Adv Mater. 2006;18:2315–2319. [Google Scholar]
- 40.Keegan ME, Falcone JL, Leung TC, Saltzman WM. Biodegradable microspheres with enhanced capacity for covalently bound surface ligands. Macromol. 2004;37:9779–9784. [Google Scholar]
- 41.Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, Shebuski RJ, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release. 1997;43:197–212. [Google Scholar]
- 42.Wang J, Zhou HY, Salih E, Xu L, Wunderlich L, Gu X, et al. Site-specific in vivo calcification and osteogenesis stimulated by bone sialoprotein. Calcif Tissue Int. 2006;79:179–189. doi: 10.1007/s00223-006-0018-2. [DOI] [PubMed] [Google Scholar]
- 43.Xu L, Anderson AL, Lu Q, Wang J. Role of fibrillar structure of collagenous carrier in bone sialoprotein-mediated matrix mineralization and osteoblast differentiation. Biomaterials. 2007;28:750–761. doi: 10.1016/j.biomaterials.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 44.Gareth AH. Nanostructure-mediated drug delivery. Nanomedicine. 2005;1:22–30. doi: 10.1016/j.nano.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Tao SL, Desai TA. Microfabricated drug delivery systems: from particles to pores. Adv Drug Deliv Rev. 2003;55:315–328. doi: 10.1016/s0169-409x(02)00227-2. [DOI] [PubMed] [Google Scholar]