

Abstract
Study Objectives
To evaluate whether patients with human immunodeficiency virus (HIV) infection who were receiving protease inhibitor therapy had altered bile acid concentrations compared with noninfected control subjects, and whether bile acid concentrations could predict the onset of hepatotoxicity caused by protease inhibitors.
Design
Retrospective sample analysis from a prospectively conducted clinical trial.
Setting
Academic research center.
Patients
Eleven adults with advanced HIV disease who were taking protease inhibitor–based antiretroviral therapy, one of whom had developed protease inhibitor–induced hepatotoxicity.
Measurements and Main Results
Plasma concentrations of cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), lithocholic acid (LCA), ursodeoxycholic acid (UDCA), and taurocholic acid (TC) were analyzed by using a novel high-performance liquid chromatography with tandem mass spectrometry detection method. Comparisons of the relative contribution of each bile acid to the total bile acid pool were made with previously published values and with bile acid concentrations contained in two pooled plasma samples from healthy, non–HIV-infected volunteers analyzed in our laboratory. Each pooled plasma sample used for this analysis contained contributions from three non–HIV-infected volunteers. The LCA and TC concentrations in HIV-infected patients were 3–4-fold higher than those previously reported for non–HIV-infected subjects; concentrations of other bile acids were similar to those of previous reports. The relative contribution of CDCA to the total bile acid pool was 9% in HIV-infected patients compared with 30–50% in noninfected subjects. Total and individual bile acid concentrations in the HIV-infected patient who developed hepatotoxicity were similar to the bile acid concentrations from the other study patients who did not develop hepatotoxicity.
Conclusion
These data suggest that bile acid concentrations may be altered by HIV infection and/or protease inhibitor therapy. However, further investigations should be performed to assess whether antiretroviral-associated hepatotoxicity can be predicted by alterations in individual bile acid concentrations.
Keywords: bile acids, human immunodeficiency virus, HIV, protease inhibitor, hepatotoxicity, cholic acid
Liver dysfunction and disease can result in elevations in plasma bile acid concentrations, as well as perturbations in the relative contribution of each bile acid to the total bile acid pool.1-8 Total bile acid concentrations and the cholic acid:chenodeoxycholic acid ratio are elevated in primary sclerosing cholangitis3-6 and cholestasis.5 Hepatotoxicity implicated from occupational and drug exposure has also been associated with elevations in total bile acid concentrations.9-13
Recently, we published the results of a clinical investigation conducted in 11 human immunodeficiency virus (HIV)–infected patients on the pharmacokinetics, safety, and efficacy of an antiretroviral regimen that included three protease inhibitors: amprenavir, ritonavir, and saquinavir.14 During that study, drug-induced hepatotoxicity was diagnosed in one of the 11 patients. In this current study, we used blood samples obtained from that study to determine whether the average plasma concentrations of individual bile acids of HIV-infected patients differ compared with a population of non–HIV-infected individuals, whether protease inhibitor therapy influences bile acid concentrations during a dosing interval, and whether elevations in total or individual plasma bile acid concentrations might be an early signal of drug-induced hepatotoxicity.
Methods
Our previously published study was designed to evaluate the safety, efficacy, and pharmacokinetic changes occurring with triple protease inhibitor–based therapy that included saquinavir, amprenavir, and ritonavir.14 Real-time pharmacokinetic analyses of drug exposure and subsequent dosage adjustments of protease inhibitor therapy were performed in that study. The study was approved by the University of North Carolina at Chapel Hill institutional review board. Informed consent was obtained from each participant, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Highly active antiretroviral therapy–experienced HIV-infected patients who were failing their current regimen were enrolled.14 Patients were prescribed a new regimen of nucleoside reverse transcriptase inhibitors (based on HIV genotyping results) and were randomly assigned to receive either a combination protease inhibitor regimen of saquinavir 1000 mg–ritonavir 100 mg twice/day or amprenavir 600 mg–ritonavir 100 mg twice/day. After taking this regimen for 7–10 days, a full pharmacokinetic profile was obtained for each patient (PK1). The third protease inhibitor (either amprenavir or saquinavir) was added to the regimen, and pharmacokinetic analysis was repeated after 14 days of receiving triple–protease inhibitor therapy (PK2). Based on the results of PK2, the dosages of each protease inhibitor were adjusted in order to achieve amprenavir and saquinavir exposures similar to those seen in patients receiving amprenavir-ritonavir or saquinavir-ritonavir alone. Additional pharmacokinetic analyses (PK3, PK4) were performed over the next 6 months, after any necessary dosage adjustments were made to achieve the desired protease inhibitor concentrations. The PK3 analysis occurred at a median of 15.0 weeks (range 10.6–23 wks) and PK4 occurred at a median of 23.9 weeks (range 16.6–27.7 wks).
In the current study, we analyzed blood samples obtained from the patients who had participated in this previously published study14 for quantification of their bile acid concentrations.
Quantification of Bile Acids
A novel liquid chromatography with tandem mass spectrometry detection method was developed to simultaneously quantify six predominant bile acids—cholic acid (CA), chenodeoxycholic acid CDCA, deoxycholic acid (DCA), lithocholic acid (LCA), ursodeoxycholic acid (UDCA), and taurocholic acid (TC)—in plasma collected from the blood samples. All chemicals and solvents were of analytic grade and were used with no further purification. Methanol was purchased from JT Baker (Phillipsburg, NJ) and ammonium acetate salt was purchased from Fisher Scientific (Fair Lawn, NJ). Bile acid standards (CA, CDCA, DCA, LCA, TC, UDCA) and formic acid were purchased from Sigma Chemical (St. Louis, MO). The N-1 naphthylph-thalamic acid ([NPA] Sigma Chemical) was used as an internal standard.
Preparation of Plasma Samples
Whole blood was collected in ethylenediamine-tetraacetic acid Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ) and centrifuged at 2600 rpm at 4°C within 30 minutes of collection. Plasma was stored at −70°C until analysis. Plasma samples were subjected to a solid-phase extraction method. Briefly, extraction cartridges (1.0 ml, 100-mg Bond Elute-C18; Varian, Harbor City, CA) were conditioned with 1.0 ml of methanol and equilibrated with ammonium acetate (pH 7.0) 25 mM. Subsequently, 0.4 ml of internal standard (NPA 0.25 μg/ml in ammonium acetate [pH 7.0] 25 mM) and 0.2 ml of plasma were added to the cartridges and passed through with minimal suction (1–3 mm Hg). The cartridges were washed with 1.0 ml of ammonium acetate (pH 7.0) 25 mM, and the cartridge bed was suctioned dry for 1 minute. The bile acids were eluted with 0.8 ml of 100% methanol. The eluent was evaporated to dryness under a nitrogen stream at 40°C and reconstituted in 100 μl of a 50:50 mixture of methanol:water. The resultant solutions were mixed by vortex and centrifuged at 12,000 rpm for 5 minutes. The supernatants were transferred to 200-μl high-performance liquid chromatography microvials (Agilent Technologies, Santa Clara, CA), and 3 μl was injected onto the column.
Bile Acid Analysis
Bile acids were analyzed with a triple quadrupole mass spectrometer API 4000 (Applied Biosystems–Sciex Instruments, Foster City, CA) in the negative-ion mode. Quantitative data were analyzed in the multiple reaction monitoring mode by using optimal parameters for each bile acid. Analysis was performed using two acquisition periods to increase the signal:noise ratio for LCA, the only bile acid within acquisition period 2.
Chromatography was achieved by using a reverse-phase C18 Pinnacle II column (100 × 4.6-mm internal diameter, 3-μm particle size; Restek, Bellefonte, PA) with a flow rate of 1 ml/minute. Mobile phase A consisted of 100% NANOpure water and mobile phase B consisted of 90% methanol–0.05% formic acid. The chromatographic separation of analytes was performed with gradient elution of mobile phase B (75% from 2–10 min, 95% from 10–17 min, 75% from 17–19 min).
Calibration standard curves ranged from 1.0–1000 ng/ml and were linear throughout this range except for CDCA, which was linear from 1.0–500 ng/ml. Intra- and interday coefficients of variation across the range of concentrations were less than 15% for all analytes. The lower limit of quantification was 10 ng/ml for CA, UDCA, and DCA; 2.5 ng/ml for LCA; and 1.0 ng/ml for TC and CDCA.
Data Analysis
Comparisons of the relative contribution of each bile acid to the total bile acid pool were made with previously published values15, 16 and with bile acid concentrations contained in two pooled plasma samples from healthy, non–HIV-infected volunteers analyzed in our laboratory. Each pooled plasma sample used for this analysis contained contributions from three non–HIV-infected volunteers.
Interpatient variability was calculated among patients for individual bile acids by using PK1, PK2, and PK3 visits. Intrapatient variability was calculated based on individual mean bile acid concentrations at PK2 and PK3 for each patient on whom data were available (8 of 11 patients). Area under the concentration-time curve (AUC in μmol•hr/L) during a 12-hour dosing interval was calculated by using the linear-log trapezoidal rule (WinNonlin version 4.1; Pharsight Corp., Mountain View, CA). Data are presented as mean and SD unless otherwise indicated.
Statistical Analysis
Statistical analysis was performed by using a mixed model with compound symmetry covariance structure with the SAS PROC MIXED software, version 9 (SAS Institute Inc., Cary, NC). Logarithmic transformation was performed on the bile acid concentration data in order to meet the assumptions used in the analysis.
Results
In the previously published study, nine male and two female patients with advanced HIV disease were evaluated.14 Seven patients were African-American and four were Caucasian. Median age was 44 years (range 26–64 yrs), and mean ± SD HIV-1 RNA and CD4+ T-cell counts were 5.16 ± 5.33 log copies/ml and 195 ± 128 cells/mm3, respectively, at the time of enrollment. Patients were highly active antiretroviral therapy experienced, with previous exposure to a median of 3.5 (range 1–6) protease inhibitors, 1 (0–2) nonnucleoside reverse transcriptase inhibitors, and 4.5 (2–5) nucleoside reverse transcriptase inhibitors. The median concentrations at 12 hours for amprenavir, saquinavir, and ritonavir at PK2 were 1.81 μg/ml (range 0.98–4.78 μg/ml), 0.15 μg/ml (0.05–0.48 μg/ml), and 0.15 μg/ml (0.04–0.63 μg/ml), respectively.
Mean bile acid concentrations during a 12-hour dosing interval at PK1 (dual–protease inhibitor therapy) and PK2 (triple–protease inhibitor therapy) are shown in Figure 1. There was a trend toward differences in LCA and TC concentrations in these HIV-infected patients compared with noninfected subjects.16 Mean LCA concentrations for HIV-infected patients (15.8–36.3 ng/ml; Figure 1) were greater at all time points than average fasting concentrations of LCA for non–HIV-infected subjects (1.1–7.5 ng/ml).16 Plasma concentrations for TC were generally greater in the HIV-infected patients during the 12-hour dosing interval (105.2–429.7 ng/ml), although some concentrations fell within the published range for non–HIV-infected subjects (0–128.5 ng/ml).16 The CA, CDCA, UDCA, and DCA concentrations were similar between HIV-infected patients and noninfected control subjects.
Figure 1.

Concentration versus time data for the six bile acids: cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), lithocholic acid (LCA), taurocholic acid (TC), and ursodeoxycholic acid (UDCA). Data are mean ± SD from the first pharmacokinetic analysis (PK1) after dual–protease inhibitor therapy (open circles) and from PK2 after triple–protease inhibitor therapy (closed triangles). Dotted lines (PK1) and dashed lines (PK2) represent data from the patient who developed hepatotoxicity. Horizontal solid lines represent the normal range of the bile acid concentration in healthy control subjects.
Mean intrapatient variability in AUC for these individual bile acids were 25.9% (range 1.4–61.9%) for CA, 34.3% (1.1–98.9%) for CDCA, 31.8% (0.2–100.6%) for DCA, 50.3% (10.6–141.4%) for LCA, 42.6% (8.8–82.4%) for UDCA, and 44.3% (3.9–101.7%) for TC. Interpatient variability within each of the three PK visits ranged from 53–84% for CA, 49–92% for CDCA, 79–94% for DCA, 108–157% for LCA, 48-60% for UDCA and 109–215% for TC. The interpatient variability for CA, DCA, and UDCA was less than what has been previously reported in noninfected persons (103–115%, 120–125%, and 79%, respectively),15, 16 whereas the interpatient variability for CDCA and TC was similar to that of historical reports (62–84% and 110%, respectively).15, 16 Only LCA interpatient variability was greater in our study patients (108–157%) than what has been previously reported for noninfected persons (50%).16
The relative contribution of individual bile acids compared with the total bile acid pool is listed in Table 1. The relative contribution of bile acids was calculated by using the average AUC values for each bile acid during the three PK visits. The CDCA contribution, a primary bile acid which normally constitutes 30–50% of the total plasma bile acid pool,15, 16 represented only 9% of the major bile acids in our HIV-infected patients. The contribution of TC was 3–4-fold higher in these HIV-infected patients than that of previous reports of non–HIV-infected subjects.16
Table 1.
Comparison of the Relative Contribution of Individual Plasma Bile Acids to the Total Bile Acid Pool
| Bile Acid | HIV-Infected Patients |
Noninfected Pooled Plasmaa |
Historical Controls15, 16 |
|---|---|---|---|
| CDCA | 9 | 37 | 31–50 |
| DCA | 34 | 18 | 20–37 |
| UDCA | 17 | 7 | 18 |
| CA | 5 | 22 | 13–17 |
| TC | 28 | 3 | 11 |
| LCA | 6 | 4 | 2 |
Data are percentages.
HIV = human immunodeficiency virus; CDCA = chenodeoxycholic acid; DCA = deoxycholic acid; UDCA = ursodeoxycholic acid; CA = cholic acid; TC = taurocholic acid; LCA = lithocholic acid.
Mean result of two pooled plasma samples (consisting of contributions from three non–HIV-infected individuals each) evaluated in our assay.
Bile acid concentrations were similar with dual (PK1) and triple (PK2) protease inhibitor therapy (Figures 1 and 2) for all patients. In the one patient who developed hepatotoxicity, elevations in aspartate (AST) and alanine aminotransferase (ALT) levels occurred between day 27 (PK2) and day 103 (PK3) of therapy (Figure 3). However, neither individual (Figure 1) nor total (Figure 2) bile acid concentrations increased in this patient, even after AST and ALT concentrations exceeded 3-fold the upper limit of normal (noted on day 103 of therapy; Figure 3). Similarly, there was no clear relationship between the CA:CDCA ratio and the development of AST and ALT elevations in our investigation. This ratio increased slightly in the patient who developed hepatotoxicity (PK1 0.39, PK2 0.40, PK3 0.48), but was within the range of ratios for all patients (Figure 4). No association between bile acid concentrations and such as CD4+ count, concurrent disease states, concomitant drugs, and cholesterol concentrations was found (data not shown).
Figure 2.
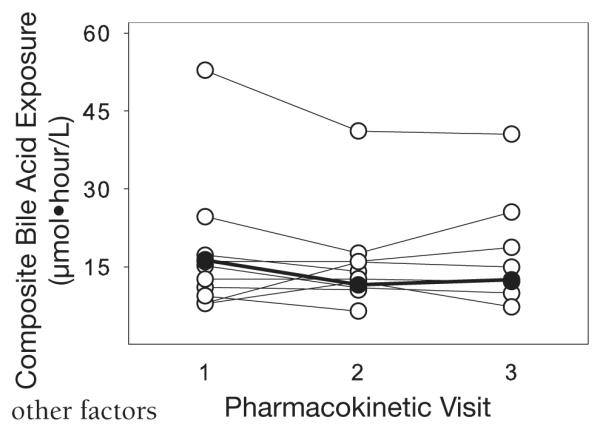
Composite total bile acid exposure in all patients at pharmacokinetic visits 2 and 3. Closed circles represent the patient who developed hepatotoxicity between PK2 and PK3. Composite exposure was calculated as the combination of the 12-hour area under the concentration-time curve for the six major bile acids (cholic acid, chenodeoxycholic acid, deoxycholic acid, lithocholic acid, taurocholic acid, and ursodeoxycholic acid).
Figure 3.
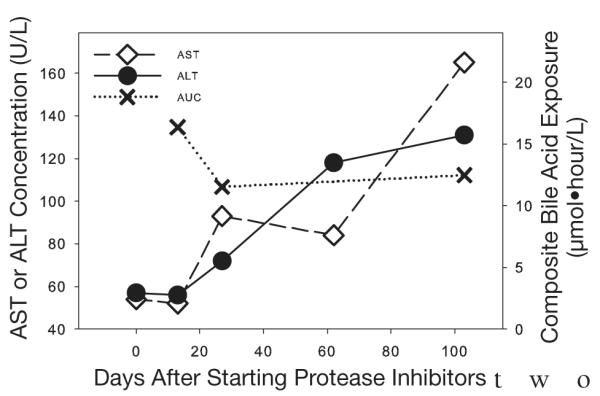
Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) concentrations and composite bile acid exposure (area under the concentration-time curve [AUC]) for the one patient who developed drug-induced hepatotoxicity. The composite bile acid exposure was calculated as the combination of the 12-hour AUC values for the six major bile acids (cholic acid, chenodeoxycholic acid, deoxycholic acid, lithocholic acid, taurocholic acid, and ursodeoxycholic acid).
Figure 4.
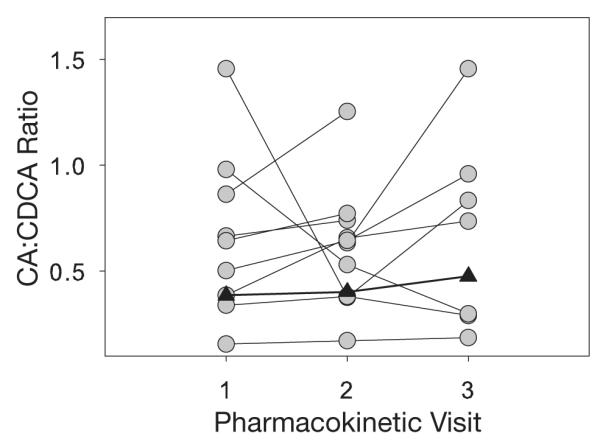
Cholic acid (CA):chenodeoxycholic acid (CDCA) area under the concentration-time curve ratios for individual patients at pharmacokinetic (PK) visits 1–3. Closed triangles represent the patient who developed hepatotoxicity between PK2 and PK3.
Discussion
This investigation was performed to simultaneously measure six plasma bile acid concentrations in patients taking combination protease inhibitors. Several analyses were conducted to describe individual bile acid concentrations in these patients. Intra- and interindividual variability in individual bile acid concentrations were quantified. We also evaluated whether protease inhibitors altered individual plasma bile acid concentrations, whether there were differences in plasma bile acid concentrations in HIV-infected patients relative to noninfected controls, and whether elevations in bile acid concentrations occur before elevations in serum transaminase levels.
Interpatient variability among our HIV-infected patients was greater for LCA, similar for CDCA and TC, and smaller for CA, UDCA, and DCA than those in historical non–HIV-infected controls. To our knowledge, this is the first study to use multiple sampling of individual bile acid concentrations to calculate intraindividual variability. To determine this information, we used data from PK2 and PK3. Two confounders may make these results larger than the nominal value: the protease inhibitor dosage adjustment between PK2 and PK3, and the sampling of only time points.
In addition, this is the first investigation, to our knowledge, describing individual plasma bile acid concentrations in HIV-infected patients. Results of this study suggest that plasma bile acid composition is altered in individuals with advanced HIV infection who are taking protease inhibitor–based therapy, with the concentrations of LCA and TC being greater than what has previously been reported for non–HIV-infected individuals.15-17 There are several possible explanations for these observations. Bile acids are formed from the catalysis of cholesterol and serve as a major excretory pathway for cholesterol. It has been reported previously that protease inhibitor therapy (and HIV infection itself) can result in deleterious elevations in plasma lipid concentrations.18, 19 Therefore, it is possible that an increase in cholesterol concentrations also could increase bile acid concentrations. Analyses were performed to explore whether or not cholesterol concentrations influenced the concentrations of bile acids in the plasma. The mean ± SD fasting cholesterol concentration in these patients was elevated (205 ± 102 mg/dl), but no association between cholesterol and bile acid concentrations was noted.
This investigation examined serum concentrations of the five major unconjugated bile acids (CA, CDCA, DCA, LCA, UDCA) as well as one conjugated bile acid (TC). If sulfation, glucuronidation, glycine, or taurine conjugation processes are altered, the relative contribution of conjugated and unconjugated bile acids can change. We found no published data on the influence of HIV infection on expression or activity of the enzymes involved in the conjugation of bile acids, although it is known that HIV can affect drug metabolizing enzyme activity.
Select antiretroviral agents can affect glucuronidation pathways. Ritonavir induces bilirubin uridine 5′-diphospho– glucuronosyl-transferase (UGT), whereas indinavir and atazanavir inhibit the activity of this enzyme.20-23 Inhibition of UGT by indinavir and atazanavir can lead to asymptomatic elevations in unconjugated bilirubin and is not generally considered to be true hepatotoxicity. Treatment with ritonavir-containing regimens increases the risk for the development of hepatotoxicity.23, 24
In our investigation, the plasma concentrations of the taurine conjugate of CA (i.e., TC) in HIV-infected patients were greater than in noninfected subjects despite normal CA concentrations. This suggests a higher taurine conjugation of cholate in HIV-infected patients. These findings also could be due to the inhibition of hepatic TC uptake by antiretroviral agents. The patient who developed hepatotoxicity exhibited elevated TC concentrations at the early time points of the dosing interval compared with the mean TC concentrations for all study patients. It is plausible that TC uptake into the hepatocyte through the predominant bile acid uptake transporter, sodium-dependent taurocholate cotransporting polypeptide, was inhibited to a greater extent in this patient, resulting in elevated plasma concentrations and predisposing this individual to the development of hepatotoxicity.
The hepatotoxic drugs bosentan and troglitazone inhibit hepatic bile acid transport.12, 25, 26 Patients who developed hepatotoxicity while taking bosentan exhibited increased total plasma bile acid concentrations.12 In rats, the administration of the hepatotoxic drug nefazodone resulted in the inhibition of hepatic bile acid transport as well as a transient increase in serum bile acid concentrations.27 In addition, in vitro data from our laboratory have demonstrated that selected protease inhibitors inhibit bile acid transport in the liver.28 Since one patient in this clinical investigation developed hepatotoxicity, it was of interest to measure individual bile acid concentrations to determine whether elevations might be used as an early indicator of hepatotoxicity. The LCA is considered a toxic bile acid in several species.29 Elevated concentrations of LCA can result in the downregulation of UGT, which could slow the elimination of LCA and may lead to toxicity.30 In addition, HIV infection or protease inhibitor therapy may result in an alteration in the detoxification pathways of LCA, which could result in increased toxicities in HIV-infected patients. Of interest, in this investigation, LCA concentrations for the patient who developed hepatotoxicity were noticeably lower than the mean LCA concentrations for all the patients in this study. It is conceivable that this patient may have exhibited elevated hepatic LCA concentrations but was unable to excrete LCA into the plasma, therefore predisposing this patient to hepatotoxicity.
It is plausible that individual protease inhibitors would have variable effects on bile acid concentrations. In our investigations, we have demonstrated that ritonavir and saquinavir inhibited bile acid transport in vitro and that ritonavir is a more potent inhibitor than saquinavir.28 However, the relative effects of saquinavir and amprenavir (the drugs used in this investigation) on bile acid transport have not yet been investigated.
To our knowledge, this is the first investigation to describe the intra- and interindividual variability in the concentrations of six major bile acids in a small population of HIV-infected patients, and the first to investigate the utility of bile acid concentrations as indicators of drug-induced liver toxicity associated with protease inhibitor therapy. The two major limitations of this study are its small sample size and its retrospective design. The study was not originally designed to have statistical power to detect differences in bile acid concentrations in a hepatotoxic patient(s) versus those who did not experience hepatotoxicity. Therefore, using inferential statistics to interpret the information presented here has inherent limitations. However, it is rare to find robust serial pharmacokinetic sampling in a study that contains antiretroviral-induced hepatotoxicity, and the small sample size does not negate the potential importance of the trends found in this study.
Conclusion
Although no changes in bile acid concentrations were noted in the patient who developed hepatotoxicity, distinctive alterations in individual bile acid concentrations and in the relative contribution of bile acids to the bile acid pool were observed in HIV-infected patients with advanced disease compared with non–HIV-infected control subjects. These findings may prove to be significant for antiretroviral-induced hepatotoxicity in larger, prospectively designed studies.
Acknowledgments
Funded by University of North Carolina Center for AIDS Research, National Institutes of Health grants (P30 AI50410, K23 AI54980, RR00046, and RO1 GM41935), and American Foundation for Pharmaceutical Education predoctoral fellowship.
References
- 1.Bremmelgaard A, Sjovall J. Hydroxylation of cholic, chenodeoxycholic, and deoxycholic acids in patients with intrahepatic cholestasis. J Lipid Res. 1980;21:1072–81. [PubMed] [Google Scholar]
- 2.Bloomer JR, Allen RM, Klatskin G. Serum bile acids in primary biliary cirrhosis. Arch Intern Med. 1976;136:57–61. [PubMed] [Google Scholar]
- 3.Magyar I, Loi HG, Feher T. Plasma bile acid levels and liver disease. Acta Med Acad Sci Hung. 1981;38:109–15. [PubMed] [Google Scholar]
- 4.Azer SA, Coverdale SA, Byth K, Farrell GC, Stacey NH. Sequential changes in serum levels of individual bile acids in patients with chronic cholestatic liver disease. J Gastroenterol Hepatol. 1996;11:208–15. doi: 10.1111/j.1440-1746.1996.tb00064.x. [DOI] [PubMed] [Google Scholar]
- 5.Okuda H, Obata H, Nakanishi T, Hisamitsu T, Matsubara K, Watanabe H. Quantification of individual serum bile acids in patients with liver diseases using high-performance liquid chromatography. Hepatogastroenterology. 1984;31:168–71. [PubMed] [Google Scholar]
- 6.Morita T, Matsuyama Y, Fujimoto T, Higuchi M, Tsujii T, Matsuoka Y. Clinical significance of serum bile acid measurement in liver diseases. Gastroenterol Jpn. 1978;13:491–502. doi: 10.1007/BF02774915. [DOI] [PubMed] [Google Scholar]
- 7.Laatikainen T. Postprandial serum bile acids in cholestasis of pregnancy. Ann Clin Res. 1978;10:307–12. [PubMed] [Google Scholar]
- 8.Laatikainen T, Ikonen E. Serum bile acids in cholestasis of pregnancy. Obstet Gynecol. 1977;50:313–18. [PubMed] [Google Scholar]
- 9.Galeazzi R, Lorenzini I, Orlandi F. Rifampicin-induced elevation of serum bile acids in man. Dig Dis Sci. 1980;25:108–12. doi: 10.1007/BF01308307. [DOI] [PubMed] [Google Scholar]
- 10.Neghab M, Qu S, Bai CL, Caples J, Stacey NH. Raised concentration of serum bile acids following occupational exposure to halogenated solvents, 1,1,2-trichloro-1,2,2-trifluoroethane and trichloroethylene. Int Arch Occup Environ Health. 1997;70:187–94. doi: 10.1007/s004200050205. [DOI] [PubMed] [Google Scholar]
- 11.Shima T, Tada H, Morimoto M, et al. Serum total bile acid level as a sensitive indicator of hepatic histological improvement in chronic hepatitis C patients responding to interferon treatment. J Gastroenterol Hepatol. 2000;15:294–9. doi: 10.1046/j.1440-1746.2000.02126.x. [DOI] [PubMed] [Google Scholar]
- 12.Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–31. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- 13.Tripodi V, Nunez M, Carducci C, Mamianetti A, Carreno C Agost. Total serum bile acids in renal transplanted patients receiving cyclosporine A. Clin Nephrol. 2002;58:350–5. doi: 10.5414/cnp58350. [DOI] [PubMed] [Google Scholar]
- 14.Corbett AH, Eron JJ, Fiscus SA, Rezk NL, Kashuba AD. The pharmacokinetics, safety, and initial virologic response of a triple–protease inhibitor salvage regimen containing amprenavir, saquinavir, and ritonavir. J Acquir Immune Defic Syndr. 2004;36:921–8. doi: 10.1097/00126334-200408010-00005. [DOI] [PubMed] [Google Scholar]
- 15.Ahlberg J, Angelin B, Bjorkhem I, Einarsson K. Individual bile acids in portal venous and systemic blood serum of fasting man. Gastroenterology. 1977;73:1377–82. [PubMed] [Google Scholar]
- 16.Tagliacozzi D, Mozzi AF, Casetta B, et al. Quantitative analysis of bile acids in human plasma by liquid chromatography-electrospray tandem mass spectrometry: a simple and rapid one-step method. Clin Chem Lab Med. 2003;41:1633–41. doi: 10.1515/CCLM.2003.247. [DOI] [PubMed] [Google Scholar]
- 17.Costarelli V, Sanders TA. Plasma deoxycholic acid concentration is elevated in postmenopausal women with newly diagnosed breast cancer. Eur J Clin Nutr. 2002;56:925–7. doi: 10.1038/sj.ejcn.1601396. [DOI] [PubMed] [Google Scholar]
- 18.Hui DY. Effects of HIV protease inhibitor therapy on lipid metabolism. Prog Lipid Res. 2003;42:81–92. doi: 10.1016/s0163-7827(02)00046-2. [DOI] [PubMed] [Google Scholar]
- 19.El-Sadr WM, Mullin CM, Carr A, et al. Effects of HIV disease on lipid, glucose and insulin levels: results from a large antiretroviral-naive cohort. HIV Med. 2005;6:114–21. doi: 10.1111/j.1468-1293.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 20.Zucker SD, Qin X, Rouster SD, et al. Mechanism of indinavir-induced hyperbilirubinemia. Proc Natl Acad Sci U S A. 2001;98:12671–6. doi: 10.1073/pnas.231140698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abbott Laboratories . Norvir (ritonavir) product Information. Abbott Park, IL: 2005. [Google Scholar]
- 22.Bristol-Myers Squibb . Reyataz (atazanavir) product Information. Princeton, NJ: 2006. [Google Scholar]
- 23.Sulkowski MS. Drug-induced liver injury associated with antiretroviral therapy that includes HIV-1 protease inhibitors. Clin Infect Dis. 2004;38(suppl 2):S90–7. doi: 10.1086/381444. [DOI] [PubMed] [Google Scholar]
- 24.Sulkowski MS, Thomas DL, Chaisson RE, Moore RD. Hepatotoxicity associated with antiretroviral therapy in adults infected with human immunodeficiency virus and the role of hepatitis C or B virus infection. JAMA. 2000;283:74–80. doi: 10.1001/jama.283.1.74. [DOI] [PubMed] [Google Scholar]
- 25.Funk C, Ponelle C, Scheuermann G, Pantze M. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol. 2001;59:627–35. [PubMed] [Google Scholar]
- 26.Kemp DC, Zamek-Gliszczynski MJ, Brouwer KL. Xenobiotics inhibit hepatic uptake and biliary excretion of taurocholate in rat hepatocytes. Toxicol Sci. 2005;83:207–14. doi: 10.1093/toxsci/kfi020. [DOI] [PubMed] [Google Scholar]
- 27.Kostrubsky SE, Strom SC, Kalgutkar AS, et al. Inhibition of hepatobiliary transport as a predictive method for clinical hepatotoxicity of nefazodone. Toxicol Sci. 2006;90:451–9. doi: 10.1093/toxsci/kfj095. [DOI] [PubMed] [Google Scholar]
- 28.McRae MP, Lowe CM, Tian X, et al. Ritonavir, saquinavir, and efavirenz, but not nevirapine, inhibit bile acid transport in human and rat hepatocytes. J Pharmacol Exp Ther. 2006;318:1068–75. doi: 10.1124/jpet.106.102657. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–58. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y, Heydel JM, Li X, Bratton S, Lindblom T, Radominska-Pandya A. Lithocholic acid decreases expression of UGT2B7 in Caco-2 cells: a potential role for a negative farnesoid X receptor response element. Drug Metab Dispos. 2005;33:937–46. doi: 10.1124/dmd.104.003061. [DOI] [PMC free article] [PubMed] [Google Scholar]