

Abstract
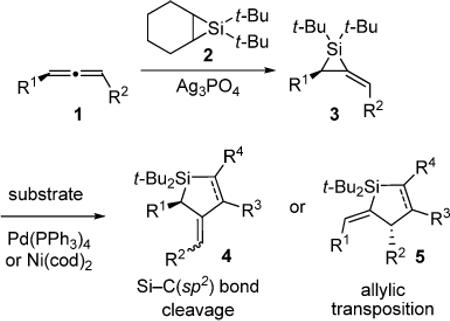
Palladium and nickel catalysts promoted highly selective carbon–carbon bond insertion reactions with di-tert-butyl-alkylidenesilacyclopropanes. Pd(PPh3)4 was demonstrated to be the optimal catalyst, allowing for a variety of carbon–carbon π-bond insertion reactions. Depending on the nature of the carbon–carbon π bond, the insertion reaction proceeded with either direct insertion into the carbon(sp2)–silicon bond or with allylic transposition. Ring-substituted alkylidenesilacyclopropanes required a nickel catalyst to afford insertion products. Using Ni(cod)2 as the carbon–carbon bond insertion catalyst, new double alkyne insertion products and alkene isomerization products were observed.
Introduction
Strained-ring silanes undergo a variety of ring-expanding reactions. For example, metal-catalyzed insertions with carbon–carbon π bonds have been reported for various three-1-12 and four-membered ring silacyclic compounds.13-17 These reactions, however, suffer from low selectivity or limited substrate scope.
In this Article, we report selective and general metal-catalyzed carbon–carbon π-bond insertion reactions with alkylidenesilacyclopropanes (3) derived from allenes (1) (eq 1).18 We also report the synthesis of a silacyclopropane derived from an allene ether and its high reactivity that allows subsequent two-atom ring-expansion reactions to occur with high regio- and diastereoselectivity. Electronic effects are examined and mechanistic considerations are discussed.
 |
(1) |
Results and Discussion
Attempted Synthesis of an Unsubstituted Alkylidenesilacyclopropane
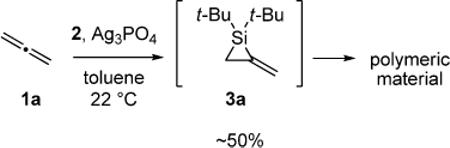
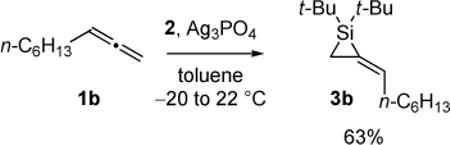
An unactivated alkylidenesilacyclopropane was desired for preliminary experiments. Unsubstituted alkylidenesilacyclopropane 3a was formed by silylene transfer to allene (1a), but it could not be isolated (eq 2).19 Because monosubstituted alkylidenesilacyclopropane 3b can be isolated (eq 3),18 it was employed in the initial carbon–carbon bond insertion reactions.
 |
(2) |
 |
(3) |
Pd(0)-Catalyzed Insertion Reaction with a Monosubstituted Alkylidenesilacyclopropane
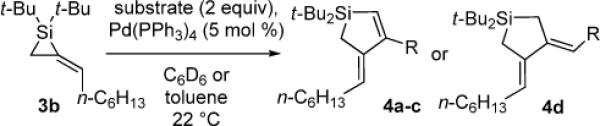
To determine the potential of metal-catalyzed carbon–carbon π-bond insertion reactions, alkylidenesilacyclopropane 3b was tested with various substrates in the presence of Pd(PPh3)4. For all reactions, two equivalents of substrate were required for the reaction to proceed to completion. Insertions of terminal alkynes into substrate 3b afforded Si–C(sp2) bond cleavage products selectively, as did the insertion of an allene (Table 1). Instead of direct insertion, allylic transposition products were formed in moderate yields in the presence of both internal alkynes and terminal alkenes (Table 2). Many of the resultant silacyclopentene and -pentane products exhibited moderate instability, so they were difficult to purify. All reactions went to completion at ambient temperature, except for the reaction of substrate 3b with 1-octene (entry 4), which required heating at 70 °C. Attempts to react alkyl-substituted allenes, internal alkenes, and sterically hindered internal alkynes with alkylidenesilacyclopropane 3b led to decomposition.
Table 1.
Pd(0)-Catalyzed Carbon–Carbon Bond Insertion with 3b to Yield Si–C(sp2) Bond Cleavage Products
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3). Isolated yields are shown in parentheses.
Unstable towards isolation attempts.
Table 2.
Pd(0)-Catalyzed Carbon–Carbon Bond Insertion with 3b to Yield Allylic Transposition Products
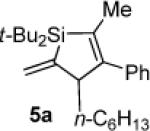
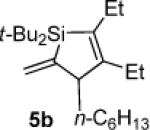

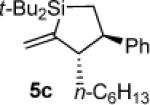
The stereo- and regiochemistry of the products were assigned by nOe analyses. Details are provided as Supporting Information.
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3). Isolated yields are shown in parentheses.
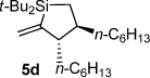
Formed as a minor product in a 29:71 mixture with silacyclopentane 6.
Required heating at 70 °C to go to completion, and was formed as a 56:44 mixture with dimeric material derived from 3b.
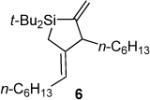
In addition to the observed allylic transposition products, side products were observed for some of the reactions shown in Table 2. Silacyclopentane 6 was observed as the major product in entry 1. The formation of this product results from the formal incorporation of a second equivalent of alkylidenesilacyclopropane 3b in conjunction with di-tert-butylsilylene extrusion.8,9 Dimeric material20,21 derived from alkylidenesilacyclopropane 3b appeared when heating the reaction mixture in entry 4.5,7,16,22,23 No reactions (including decomposition and byproduct formation) were observed in the absence of catalyst.
Pd(0)-Catalyzed Insertion Reaction with an Electronically Activated Alkylidenesilacyclopropane
Alkylidenesilacyclopropane 3c was synthesized to investigate possible electronic effects in the Pd(0)-catalyzed reactions. No silylene transfer was observed α- to the methoxy group, which is surprising considering the electrophilic nature of the silver silylenoid generated in situ.24,25 Control experiments demonstrated that silylene transfer does not occur with enol ether 9 (Scheme 1), although it proceeded readily in the presence of acetylenic ethers.26,27
Scheme 1.

Silacyclopropanation of Allene Ether 1c and a Control Experiment with Enol Ether 8
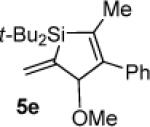
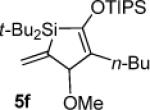
When subjected to the Pd(0)-catalyzed carbon–carbon π-bond insertion reaction, alkylidenesilacyclopropane 3c was shown to be more reactive than alkyl-substituted analogue 3b while maintaining high regio- and diastereoselectivity. Terminal alkynes and allenes afforded Si–C(sp2) bond cleavage products (Table 3), but internal alkynes and terminal alkenes gave allylic transposition products (Table 4). A sterically hindered internal alkyne (Table 4, entry 2) and an alkyl-substituted allene (Table 3, entry 4) were also viable substrates in this reaction. Insertion with 1-octene proceeded to completion at ambient temperature (Table 4, entry 4). Analogously to silacyclopropane 3b, internal alkenes were unreactive, and a dimeric compound derived from 3c was observed as a byproduct in the reaction with 1-octene (Table 4, entry 4).20,21 Although alkylidenesilacyclopropane 3c exhibited greater reactivity and substrate scope, most of the insertion products were found to be too unstable to isolate and purify.
Table 3.
Pd(0)-Catalyzed Carbon–Carbon Bond Insertion with 3c to Yield Si–C(sp2) Bond Cleavage Products
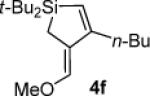
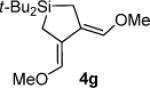
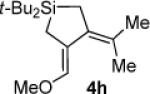
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3). Isolated yields are shown in parentheses.
Hydrolysis gave (α,β-unsaturated aldehyde 4h′ when subjected to chromatography.
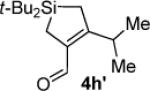
Table 4.
Pd(0)-Catalyzed Carbon–Carbon Bond Insertion with 3c to Yield Allylic Transposition Products
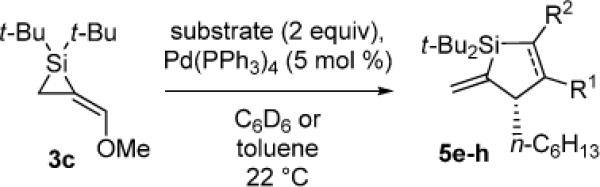

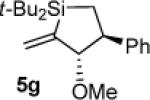
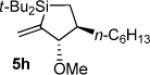
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3). Isolated yields are shown in parentheses.
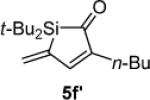
Hydrolysis and elimination gave α,β,γ,δ-unsaturated acyl silane 5f′ upon attempted chromatography. Difficulties with purification prevented accurate determination of the isolated yield.
Formed as a 68:32 mixture with dimeric material derived from 3c.
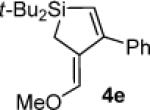
In an effort to circumvent the isolation of the air-sensitive alkylidenesilacyclopropane, a one-flask, two-step silacyclopropanation/π-bond insertion reaction (eq 4) was attempted with alkylidenesilacyclopropane 3c in a manner similar to carbonyl insertion reactions.18 Residual silver salts inhibited the Pd(0)-catalyzed reaction, providing product 4e in low yield. As a result, all further experiments were conducted using isolated samples of alkylidenesilacyclopropanes.
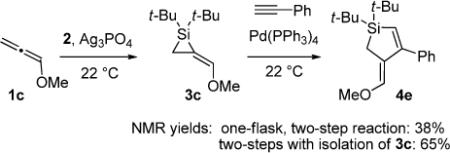 |
(4) |
Pd(0)-Catalyzed Insertion Reaction with Disubstituted Alkylidenesilacyclopropanes
To extend these reactions to alkylidenesilacyclopropanes derived from chiral allenes, alkylidenesilacyclopropane 3d was also subjected to the Pd(0)-catalyzed insertion reaction (Scheme 2). Only the reaction with phenylacetylene led to Si–C(sp2) bond cleavage product 4i as a mixture with silole byproduct 10. Attempted insertion of 1-hexyne gave silole isomers 11a and 11b exclusively. Silole product formation has been reported for metal-catalyzed reactions of silacyclopropenes and -propanes, and regeneration of the starting alkyne or alkene was observed.1,5,6,10-12 In contrast, no allene was observed after silole formation from alkylidenesilacyclopropane 3d. All other substrates either gave no reaction or resulted in decomposition of the starting materials.
Scheme 2.

Pd(0)-Catalyzed Carbon–Carbon Bond Insertion with 3d
Electronic Effects in the Pd(0)-Catalyzed Insertion Reactions
To investigate electronic effects, two electronically different alkynes were employed in the carbon–carbon bond insertion reaction with alkylidenesilacyclopropane 3d. An electron-deficient terminal alkyne required a longer reaction time compared to an electron-rich alkyne (Scheme 3). Although a difference in reactivity was observed, competitive silole formation was still observed.
Scheme 3.

Electronic Effects in the Pd(0)-Catalyzed Carbon–Carbon Bond Insertion Reaction with 3d
A control experiment demonstrated that steric effects were responsible for the low reactivity of alkylidenesilacyclopropane 3d. This substrate has both an increase in branching at its allylic carbon as well as additional substitution of its three-membered ring compared to alkylidenesilacyclopropane 3b. The increased substitution on the ring, not the allylic branching, was found to impede insertion reactions. The insertion reaction was repeated with alkylidenesilacyclopropane 3e (eq 5), and the results were comparable with those obtained for alkylidenesilacyclopropane 3d. Because the stereogenic center in the three-membered ring, which is necessary for developing an asymmetric variant of this reaction, also inhibited the reactivity of alkylidenesilacyclopropane 3d in the Pd(0)-catalyzed reaction, alternative reaction conditions were investigated.
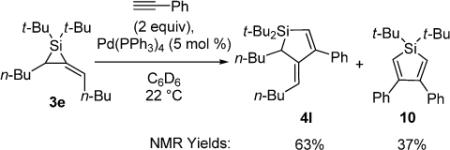 |
(5) |
Optimization and Catalyst Screen
Initial attempts to increase the rate of the insertion reactions and avoid byproduct formation by heating the reaction mixture resulted in increased dimerization of the starting alkylidenesilacyclopropanes (eq 6).20,21 Control experiments with both alkylidenesilacyclopropanes 3b and 3c showed rapid dimerization upon heating in the absence of substrate (eq 7), and suggested that formation of dimeric material is a thermodynamically favored process. An attempt to dimerize disubstituted alkylidenesilacyclopropane 3d resulted in decomposition of starting material.
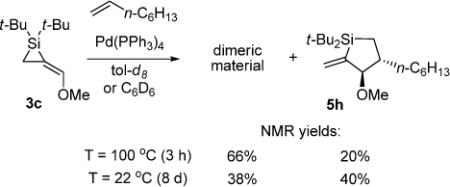 |
(6) |
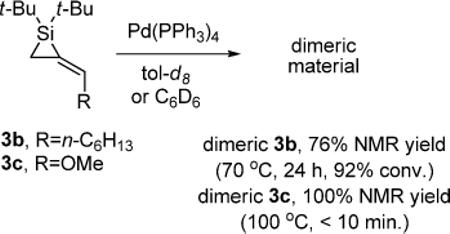 |
(7) |
Because heating the reactions led to an increase in both byproduct formation and decomposition, alternative catalysts were investigated. To find a more reactive catalytic system that remained competent in the carbon–carbon π-bond insertion reaction, we screened a variety of ligands with metal catalysts at various temperatures.28 Most of the catalyst studies were directed towards the reaction of 1-phenyl-1-propyne with alkylidenesilacyclopropane 3d in an effort to develop an asymmetric version of the metal-catalyzed carbon–carbon bond insertion reaction. Palladium catalysts did not afford products in this reaction, and all ligands examined were demonstrated to inhibit reactivity. Only Ni(cod)2 gave an insertion product, although in moderate yield (eq 8).
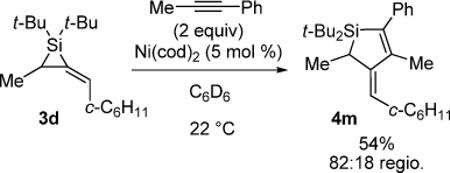 |
(8) |
Ni(0)-Catalyzed Insertion Reaction with a Disubstituted Alkylidenesilacyclopropane
The scope of the Ni(0)-catalyzed carbon–carbon π-bond insertion reaction was investigated with alkylidenesilacyclopropane 3d. Reactivity was observed with phenylacetylene, 1-phenyl-1-propyne, and 3-hexyne (Scheme 4). All other substrates (e.g., electron-rich terminal alkynes, alkenes, and allenes) led to decomposition of starting material. The increased reactivity exhibited by the Ni(0) catalyst allowed for double insertion29,30 of phenylacetylene to give a mixture of silacycloheptadienes 13a and 13b (Scheme 4a). If only one equivalent of phenylacetylene were used, decomposition of starting materials was observed. Alkyl-substituted internal alkynes led to Si–C(sp2) bond cleavage products with isomerization of alkene geometry (Scheme 4b).
Scheme 4.

Ni(0)-Catalyzed Carbon–Carbon Bond Insertion with 3d
aFormed as an 82:18 mixture of regioisomers with 4m′ (R1 = Me, R2 = Ph). bAlkene migration occurred during purification to give silole 4n′ in 52% yield. Silacyclopentene 4n could be obtained in 10% yield if purified in the presence of NEt3.
 For the latter two reactions, alkene isomerization from 3d to 3d′ was observed prior to insertion of the corresponding alkyne (eq 9).31 Alkene isomerization did not occur in the absence of alkyne. The general applicability of alkylidenesilacyclopropane 3d in the carbon–carbon π-bond insertion reaction remains limited.
For the latter two reactions, alkene isomerization from 3d to 3d′ was observed prior to insertion of the corresponding alkyne (eq 9).31 Alkene isomerization did not occur in the absence of alkyne. The general applicability of alkylidenesilacyclopropane 3d in the carbon–carbon π-bond insertion reaction remains limited.
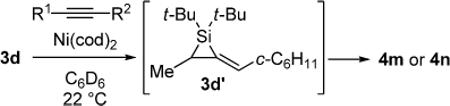 |
(9) |
Ni(0)-Catalyzed Insertion Reaction with a Monosubstituted Alkylidenesilacyclopropane
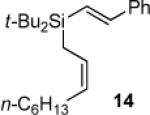
For comparison, monosubstituted alkylidenesilacyclopropane 3b was also examined in the Ni(0)-catalyzed reaction (Table 5). As a result, the carbon–carbon bond insertion reaction gave Si–C(sp2) bond cleavage product 4a with phenylacetylene (entry 1) and allylic transposition product 5b with 3-hexyne (entry 2). These products were analogous to those obtained in the Pd(0)-catalyzed reaction. The insertion of styrene led to acyclic product 14.1-3,8,32 Unlike the reaction with alkylidenesilacyclopropane 3d, neither double insertion nor alkene isomerization products were observed in any of the reactions. In contrast to the Pd(0)-catalyzed reaction, no byproducts were observed.
Table 5.
Ni(0)-Catalyzed Carbon–Carbon Bond Insertion with 3b
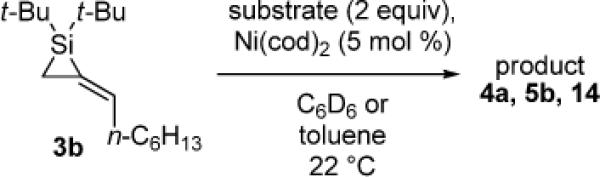

As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3). Isolated yields are shown in parentheses.
In the 1H NMR spectra of the reaction mixture, diagnostic peaks corresponding to 14 overlapped with peaks from residual styrene, making accurate determination of the NMR yield difficult.
Reaction Mechanism
Although different products were observed depending on either the substitution pattern of the alkylidenesilacyclopropane or the π-bond substrate, all of the carbon–carbon bond insertion reactions share similar mechanistic characteristics. Regiochemistry can best be explained by insertion to a metal–silicon bond,33 and each reaction involves oxidative addition and reductive elimination processes.
To explain the substrate-dependent product differentiation that was observed in the Pd(0)-catalyzed reaction, catalytic cycles are proposed (Schemes 5 and 6). Reversible oxidative addition of alkylidenesilacyclopropane 3 provides palladasilacyclobutane 15.2,3,5,10,12 For terminal alkynes and allenes, migratory insertion into the palladium–silicon bond3,8,10,12,14,17,34-36 affords intermediate 16. Reductive elimination then provides product (Z)-4. Internal alkynes and terminal alkenes are slow to undergo migratory insertion37-40 with intermediate 15, so alkylidenesilacyclopropane 3 instead proceeds through a less favorable and irreversible simultaneous allylic transposition and oxidative addition to afford palladasilacyclobutane 17. Coordination of an alkyne followed by migratory insertion would afford palladasilacyclohexene 18. Reductive elimination from intermediate 18 leads to product 5. Previously, allylic transposition had not been observed for a carbon–carbon bond insertion reaction with an alkylidenesilacyclopropane.8,9
Scheme 5.

Proposed Catalytic Cycle for the Pd(0)-Catalyzed Carbon–Carbon π-Bond Insertion Reaction with Terminal Alkynes
Scheme 6.

Proposed Catalytic Cycle for the Pd(0)-Catalyzed Carbon–Carbon π-Bond Insertion Reaction with Internal Alkynes
For disubstituted alkylidenesilacyclopropane 3d, the Ni(0)-catalyzed reaction displayed an increase in reactivity41 that resulted in both alkene isomerization and double alkyne insertion. A possible catalytic cycle to explain the former process is depicted in Scheme 7. In the presence of internal alkynes, alkylidenesilacyclopropane 3d undergoes alkene isomerization31 to generate 3d′. Oxidative addition and migratory insertion afford nickelasilacyclohexene intermediate 21,42,43 which undergoes reductive elimination to yield Si–C(sp2) bond cleavage product (E)-4. For double insertion, a catalytic cycle is proposed (Scheme 8) comparable to that depicted for the Pd(0)-catalyzed reaction in Scheme 5. Oxidative addition by Ni(0) into alkylidenesilacyclopropane 3d gives nickelasilacyclobutane intermediate 22,6,7,44,45 which undergoes migratory insertion into the nickel–silicon bond.42,43 A second insertion into the less sterically hindered nickel–carbon(sp2) bond46 affords intermediate 24, which reductively eliminates to yield product 13a. It is assumed that Ni(0) facilitates the migratory insertion that, with Pd(0) catalysis, would have been inhibited in the transformations leading from intermediates 20 to 21 and from 22 to 23.41
Scheme 7.

Proposed Catalytic Cycle for the Ni(0)-Catalyzed Carbon–Carbon Bond Insertion with 3d and Internal Alkynes
Scheme 8.

Proposed Catalytic Cycle for the Ni(0)-Catalyzed Carbon–Carbon Insertion Reaction with 3d and Phenylacetylene
The subtle substrate-ligand effects observed in the Ni(0)-catalyzed reaction suggested that a more electron-rich ligand (either π-bond substrate or alkylidenesilacyclopropane) would lead to an increase in reactivity. For the Ni(0)-catalyzed reaction, we propose that hyperconjugation of σC-Me into π*C=C makes alkylidenesilacyclopropane 3d a slightly better ligand for Ni(0) and allows for unusual reactivity not observed for alkylidenesilacyclopropane 3b.
Conclusion
The reported insertion reactions with alkylidenesilacyclopropanes 3b, 3c, and 3d constitute general and selective examples of metal-catalyzed carbon–carbon π-bond insertion reactions into cyclic organosilicon compounds. New reactivity was observed, and catalytic cycles were proposed to explain these observations.
Experimental Section
General Procedures
General experimental details are provided as Supporting Information.
Palladium-Catalyzed Synthesis of Carbon–Carbon Bond Insertion Products
Method A (isolation)
To a solution of alkyne, alkene, or allene (2.0 mmol) and silacyclopropane 3 (1.0 mmol) in toluene (5 mL) was added Pd(PPh3)4 (0.060 g, 0.05 mmol). When the reaction had proceeded to completion as determined by 1H NMR spectroscopic analysis, the reaction mixture was filtered through SiO2 and concentrated in vacuo. The resulting oil was purified by flash chromatography (hexanes, 1:99 EtOAc/hexanes, or 0.5:1:98.5 NEt3/EtOAc/hexanes) to afford carbon–carbon bond insertion products. Unless otherwise indicated, all spectral data was obtained using Method A.
Method B (NMR observation)
Alkyne, alkene, or allene (0.254 mmol) was added to a solution of silacyclopropane 3 (0.550 mL, 0.127 mmol, 0.230 M solution of 3 and 0.0465 M solution of PhSiMe3 in C6D6), followed by the addition of Pd(PPh3)4 (0.007 g, 0.006 mmol). The progress of the reaction was monitored and yields were determined by 1H NMR spectroscopic analysis (compared to the PhSiMe3 internal standard) using a single scan.
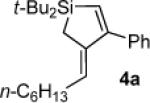
Silacyclopentene 4a
1H NMR (500 MHz, CDCl3) δ 7.35–7.25 (m, 5H), 5.96 (s, 1H), 5.41 (t, J = 7.2, 1H), 2.17 (q, J = 7.1, 2H), 1.57 (s, 2H), 1.36–1.27 (m, 8H), 1.04 (s, 18H), 0.90 (t, J = 7.0, 3H); 13C NMR (125 MHz, CDCl3) δ 165.8, 143.7, 141.9, 129.9, 129.2, 128.5, 127.8, 127.0, 31.8, 30.0, 29.28, 29.26, 28.7, 22.7, 19.6, 14.2, 10.2; IR (neat) 3057, 2931, 2858, 1601, 1577, 1470 cm−1; HRMS (APCI) m / z calcd for C25H41Si (M + H)+ 369.2978, found 369.2975.
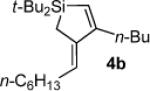
Silacyclopentene 4b
1H NMR (500 MHz, C6D6) δ 5.89 (s, 1H), 5.78 (tt, J = 7.2, 2.3, 1H), 2.41 (appar t, J = 7.7, 2H), 2.28 (q, J = 7.3, 2H), 1.58 (quint, J = 7.6, 2H), 1.57 (s, 2H), 1.47 (sext, J = 7.5, 2H), 1.34–1.26 (m, 8H), 1.08 (s, 18H), 0.91 (t, J = 7.4, 3H), 0.90 (t, J = 7.0, 3H); 13C NMR (125 MHz, C6D6) δ 165.0, 143.3, 125.4, 124.6, 32.4, 31.1, 29.8, 29.6, 29.3, 29.2, 28.5, 28.5, 22.83, 22.77, 19.1, 14.0, 13.9, 10.1; IR (neat) 2957, 2856, 1543, 1469, 1363, 1141 cm−1; HRMS (GCMS) m / z calcd for C23H45Si (M + H)+ 349.3290, found 349.3290.
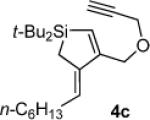
Silacyclopentene 4c
1H NMR (500 MHz, C6D6) δ 6.38 (s, 1H), 5.68 (t, J = 7.2, 1H), 4.36 (s, 2H), 3.97 (d, J = 2.4, 2H), 2.23 (q, J = 7.3, 2H), 2.00 (t, J = 2.4, 1H), 1.57 (s, 2H), 1.37–1.26 (m, 8H), 1.02 (s, 18H), 0.91 (t, J = 6.8, 3H); 13C NMR (125 MHz, C6D6) δ 159.6, 141.6, 128.5, 125.2, 80.0, 74.3, 69.3, 57.1, 31.9, 29.7, 29.5, 29.3, 29.1, 28.4, 22.8, 19.1, 14.0, 10.2; IR (neat) 2957, 2856, 2362, 1553, 1469, 1119 cm−1; HRMS (GCMS) m / z calcd for C23H41OSi (M + H)+ 361.2927, found 361.2925.
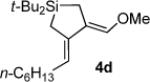
Silacyclopentane 4d: 47
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.29 (s, 1H), 5.63 (t, J = 7.2, 1H), 3.26 (s, 3H), 2.22 (q, J = 7.1, 2H), 1.77 (s, 2H), 1.57 (s, 2H), 1.01 (s, 18H).
Silacyclopentene 4e
1H NMR (400 MHz, C6D6) δ 7.38 (d, J = 7.2, 2H), 7.21–7.16 (m, 3H), 6.21 (s, 1H), 5.83 (s, 1H), 3.06 (s, 3H), 1.93 (s, 2H), 1.09 (s, 18H); 13C NMR (125 MHz, C6D6) δ 163.8, 147.7, 141.8, 128.3, 127.2, 126.4, 124.3, 59.0, 31.6, 28.5, 19.4, 8.2; HRMS (ESI) m / z calcd for C20H31OSi (M + H)+ 315.2144, found 315.2141.
Silacyclopentene 4f
1H NMR (400 MHz, C6D6) δ 6.30 (s, 1H), 5.74 (s, 1H), 3.26 (s, 3H), 2.29 (t, J = 7.5, 2H), 1.82 (s, 2H), 1.59 (quint, J = 7.6, 2H), 1.35 (sext, J = 7.5, 2H), 1.07 (s, 18H), 0.92 (t, J = 7.4, 3H); 13C NMR (125 MHz, C6D6) δ 162.4, 143.9, 123.7, 122.3, 59.0, 32.1, 30.9, 28.5, 22.8, 19.2, 14.0, 7.9.
Silacyclopentane 4g
1H NMR (500 MHz, C6D6) δ 6.11 (t, J = 2.0, 2H), 3.29 (s, 6H), 1.79 (d, J = 2.1, 4H), 1.01 (s, 18H); 13C NMR (125 MHz, C6D6) δ 138.0, 120.0, 58.8, 28.1, 19.0, 9.7.
Silacyclopentane 4h: 47
1H NMR (400 MHz, C6D6, distinctive peaks) δ 5.90 (t, J = 2.0, 1H), 3.26 (s, 3H), 1.95 (s, 3H), 1.82, (s, 3H), 1.75 (d, J = 2.0, 2H), 1.01 (s, 18H).
Silacyclopentene 4h′: 48
mp 50–54 °C; 1H NMR (500 MHz, C6D6) δ 10.21 (s, 1H), 3.30–3.26 (m, 1H), 1.77 (s, 2H), 1.39 (s, 2H), 0.87 (s, 18H), 0.81 (d, J = 6.8, 6H); 13C NMR (125 MHz, C6D6) δ 188.1, 168.9, 138.2, 28.2, 27.8, 20.9, 18.4, 13.5, 11.3; IR (thin film) 2964, 2931, 2858, 1666, 1595, 1468 cm−1; HRMS (ESI) m / z calcd for C16H31OSi (M + H)+ 267.2144, found 267.2149.
Silacyclopentene 4i
1H NMR (500 MHz, C6D6) δ 7.41 (dd, J = 7.6, 1.4, 2H), 7.21 (t, J = 7.3, 2H), 7.15 (td, J = 7.5, 1.7, 1H), 6.02 (s, 1H), 5.38 (d, J = 10.2, 1H), 2.46–2.39 (m, 2H), 1.78–1.54 (m, 8H), 1.42 (d, J = 7.8, 3H), 1.33–1.25 (m, 2H), 1.18 (s, 9H), 1.09 (s, 9H); 13C NMR (125 MHz, C6D6) δ 165.4, 149.1, 141.7, 133.9, 128.7, 128.1, 128.0, 127.3, 38.4, 33.6, 32.6, 29.6, 28.8, 25.99,49 25.95, 21.8, 21.6, 20.2, 18.5; IR (neat) cm−1 3076, 3021, 2929, 1538, 1470, 1363 cm−1; HRMS (GCMS) m / z calcd for C26H41Si (M + H)+ 381.2978, found 381.2982.
Silacyclopentene 4j: 50
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.00 (s, 1H), 5.41 (d, J = 10.3, 1H), 3.37 (s, 3H), 1.41 (d, J = 7.8, 3H); HRMS (GCMS) m / z calcd for C27H43OSi (M + H)+ 411.3083, found 411.3093.
Silacyclopentene 4k: 50
1H NMR (400 MHz, C6D6, distinctive peaks) δ 5.93 (s, 1H), 5.15 (d, J = 10.3, 1H), 1.36 (d, J = 7.8, 3H); HRMS (GCMS) m / z calcd for C27H40F3Si (M + H)+ 449.2851, found 449.2837.
Silacyclopentene 4l: 47
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.04 (s, 1H), 5.51 (t, J = 7.1, 1H), 2.34 (t, J = 7.3, 1H).
Silacyclopentene 5a
1H NMR (400 MHz, C6D6) δ 7.21 (t, J = 7.7, 2H), 7.11 (t, J = 7.5, 1H), 7.07 (d, J = 6.8, 2H), 5.90 (t, J = 2.2, 1H), 5.73 (t, J = 2.5, 1H), 3.55–3.51 (m, 1H), 1.83 (d, J = 2.2, 3H), 1.75–1.66 (m, 1H), 1.53–1.44 (m, 1H), 1.39–1.27 (m, 2H), 1.26–1.03 (m, 6H), 1.21 (s, 9H), 1.14 (s, 9H), 0.82 (t, J = 7.0, 3H); 13C NMR (125 MHz, C6D6, distinctive peaks) δ 129.5, 129.1, 129.0, 128.3, 128.2, 126.9, 126.4, 123.1, 47.9, 36.0; IR (neat) 3057, 2929, 2856, 1581, 1468, 1389 cm−1; HRMS (GCMS) m / z calcd for C26H43Si (M + H)+ 383.3134, found 383.3145.
Silacyclopentene 5b
1H NMR (500 MHz, C6D6) δ 5.85 (t, J = 2.1, 1H), 5.68 (t, J = 2.4, 1H), 3.16 (br s, 1H), 2.48 (dq, J = 13.7, 7.5, 1H), 2.40–2.16 (m, 5H), 2.10, (quint, J = 7.2, 1H), 1.91 (dq, J = 13.7, 7.5, 1H), 1.84–1.75 (m, 1H), 1.65–1.56 (m, 1H), 1.53–1.34 (m, 4H), 1.19 (s, 9H), 1.10 (s, 9H), 1.08 (appar t, J = 5.7, 3H), 0.96 (t, J = 7.5, 3H), 0.89 (t, J = 7.0, 3H); 13C NMR (125 MHz, C6D6) δ 160.9, 151.9, 135.1, 122.4, 50.9, 32.1, 30.4, 29.4, 29.1, 28.8, 26.5, 25.4, 22.7, 21.4, 20.3, 20.0, 15.2, 14.0, 12.7; IR (neat) 2960, 2857, 1580, 1469, 1387, 820 cm−1; HRMS (GCMS) m / z calcd for C23H45Si (M + H)+ 349.3290, found 349.3296.
Silacyclopentane 5c
1H NMR (400 MHz, C6D6) δ 7.24–7.22 (m, 4H), 7.14–7.10 (m, 1H), 5.80 (t, J = 2.5, 1H), 5.65 (t, J = 2.7, 1H), 2.76 (td, J = 12.6, 6.4, 1H), 2.53 (dtd, J = 12.5, 4.7, 1.6, 1H), 1.68–1.60 (m, 1H), 1.53–1.44 (m, 1H), 1.39–1.30 (m, 2H), 1.26–1.16 (m, 8H), 1.11 (s, 9H), 1.09 (s, 9H), 0.83 (t, J = 6.9, 3H); 13C NMR (125 MHz, C6D6) δ 153.3, 147.8, 128.5, 127.1, 126.0, 121.2, 53.4, 49.2, 31.7, 30.0, 28.7, 28.5, 27.9, 24.7, 22.7, 19.6, 18.9, 18.7, 14.0; IR (neat) 3026, 2929, 2856, 1601, 1470, 1363 cm−1; HRMS (GCMS) m / z calcd for C25H43Si (M + H)+ 371.3134, found 371.3135. Anal. Calcd for C25H42Si: C, 81.00; H, 11.42. Found: C, 80.87; H, 11.49.
Silacyclopentane 5d
1H NMR (500 MHz, C6D6) δ 5.78 (d, J = 2.2, 1H), 5.61 (d, J = 1.0, 1H), 2.33–2.27 (m, 1H), 2.00–1.92 (m, 2H), 1.84 (appar t, J = 11.8, 2H), 1.78–1.71 (m, 2H), 1.63–1.58 (m, 4H), 1.45–1.33 (m, 13H), 1.10 (s, 9H), 1.07 (s, 9H), 0.93–0.89 (m, 6H); 13C NMR (125 MHz, C6D6) δ 154.3, 120.3, 52.9, 41.3, 36.4, 32.1, 31.9, 30.4, 29.8, 28.7, 28.5, 27.5, 27.3, 24.2, 22.85, 22.82, 19.5, 18.7, 14.1, 14.0, 13.4; IR (neat) 3041, 2927, 1468, 1387, 1363 cm−1; HRMS (GCMS) m / z calcd for C25H51Si (M + H)+ 379.3760, found 379.3764.
Silacyclopentene 5e
1H NMR (400 MHz, C6D6) δ 7.30–7.21 (m, 5H), 6.10 (dd, J = 2.9, 1.8, 1H), 5.78 (t, J = 2.5, 1H), 5.01 (q, J = 1.9, 1H), 3.02 (s, 3H), 1.84 (d, J = 1.9, 3H), 1.17 (s, 9H), 1.08 (s, 9H); 13C NMR (125 MHz, C6D6) δ 156.5, 146.7, 139.4, 137.7, 128.7, 128.7, 126.7, 126.2, 87.6, 52.7, 29.0, 28.6, 20.2, 19.8, 17.5; HRMS (ESI) m / z calcd for C21H32OSiNa (M + Na)+ 351.2120, found 351.2118.
Silacyclopentene 5f: 47
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.10 (s, 1H), 5.74 (s, 1H), 4.87 (s, 1H), 3.11 (s, 3H), 2.05 (t, J = 6.2, 2H).
Silacyclopentene 5f′
1H NMR (500 MHz, C6D6, distinctive peaks) δ 7.23 (s, 1H), 5.68 (d, J = 2.6, 1H), 5.42 (d, J = 2.3, 1H), 2.20 (t, J = 7.4, 2H), 1.58 (m, 2H), 1.07 (s, 18H), 0.83 (t, J = 7.0, 3H); 13C NMR (125 MHz, C6D6, distinctive peaks) δ 172.9, 157.2, 154.2, 144.3, 125.3, 35.6, 27.8, 24.9, 18.7, 13.7; HRMS (ESI) m / z calcd for C17H30SiONa (M + Na)+ 301.1964, found 301.1962.
Silacyclopentane 5g
1H NMR (500 MHz, C6D6) δ 7.33 (d, J = 7.1, 2H), 7.24 (t, J = 7.6, 2H), 7.14 (t, J = 7.3, 1H), 6.16 (t, J = 2.9, 1H), 5.65 (t, J = 3.0, 1H), 4.04 (dt, J = 11.4, 2.7, 1H), 3.06 (s, 3H), 2.95 (ddd, J = 12.7, 11.5, 7.0, 1H), 1.050 (s, 9H), 1.045 (s, 9H), 0.89 (appar t, J = 6.8, 1H), 0.83 (dd, J = 15.2, 13.0, 1H); 13C NMR (125 MHz, C6D6) δ 149.5, 146.2, 128.3, 127.4, 126.2, 122.4, 91.2, 57.9, 50.0, 28.34, 28.33, 19.3, 18.7, 14.1; IR (neat) 3059, 3028, 2931, 2858, 1470, 1105 cm−1; HRMS (ESI) m / z calcd for C20H32OSiNa (M + Na)+ 339.2120, found 339.2126.
Silacyclopentane 5h
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.01 (t, J = 2.8, 1H), 5.57 (t, J = 3.2, 1H), 3.49 (dt, J = 11.2, 2.7, 1H), 3.34 (s, 3H), 1.03 (s, 9H), 1.01 (s, 9H); 13C NMR (125 MHz, C6D6, distinctive peaks) δ 151.3, 121.2, 91.2, 56.9, 42.9, 35.9, 32.1, 27.4, 14.0; HRMS (APCI) m / z calcd for C14H28OSi (M + H)+ 325.2927, found 325.2932.
Silacyclopentane 6
1H NMR (500 MHz, C6D6) δ 5.78 (t, J = 1.5, 1H), 5.53 (t, J = 2.2, 1H), 5.26 (td, J = 7.2, 1.8, 1H), 2.97–2.95 (m, 1H), 2.44 (q, J = 6.8, 1H), 2.29 (q, J = 7.6, 1H), 2.25–2.12 (m, 3H), 1.81 (d, J = 15.6, 1H), 1.65–1.56 (m, 4H), 1.52–1.44 (m, 8H), 1.41–1.36 (m, 4H), 1.10 (s, 9H), 1.09 (s, 9H), 0.93–0.91 (m, 6H); 13C NMR (125 MHz, C6D6) δ 154.7, 142.6, 122.88, 122.86, 58.3, 36.6, 32.1, 31.9, 30.0, 29.5, 29.3, 29.0, 28.9, 28.5, 28.1, 27.5, 22.8, 20.0, 18.9, 14.03, 14.02, 11.3; IR (neat) 3037, 2927, 2856, 1468, 1389, 1363 cm−1; HRMS (GCMS) m / z calcd for C26H51Si (M + H)+ 391.3760, found 391.3756.
Silole 10
mp 103–104 °C; 1H NMR (400 MHz, C6D6) δ 7.13–7.11 (m, 4H), 7.00–6.97 (m, 6H), 6.16 (s, 2H), 1.18 (s, 18H); 13C NMR (125 MHz, C6D6) δ 162.1, 141.3, 130.7, 128.01, 128.00, 126.8, 28.8, 19.3; IR (thin film) 3061, 2958, 1597, 1470, 1387, 1190 cm−1; HRMS (GCMS) m / z calcd for C24H31Si (M + H)+ 347.2195, found 347.2193. Anal. Calcd for C24H30Si: C, 83.17; H, 8.72. Found: C, 83.09; H, 8.89.
Silole 11a
1H NMR (500 MHz, C6D6) δ 5.74 (s, 2H), 2.25 (t, J = 7.6, 4H), 1.51 (quint, J = 7.3, 4H), 1.34 (sext, J = 7.3, 4H), 1.18 (s, 18H), 0.91 (t, J = 7.4, 6H); 13C NMR (125 MHz, C6D6) δ 162.7, 122.3, 32.8, 32.4, 29.0, 26.5, 22.6, 14.0; IR (neat) 2956, 2856, 1543, 1512, 1468, 1363 cm−1; HRMS (GCMS) m / z calcd for C20H39Si (M + H)+ 307.2821, found 307.2824.
Silole 11b
1H NMR (500 MHz, C6D6) δ 5.88 (s, 1H), 5.74 (s, 1H), 2.38 (t, J = 7.6, 2H), 2.21 (t, J = 7.3, 2H), 1.60–1.55 (m, 4H), 1.46 (sext, J = 7.4, 4H), 1.07 (s, 18H), 0.96 (t, J = 7.3, 3H), 0.91 (t, J = 7.4, 3H); 13C NMR (125 MHz, C6D6) δ 165.0, 143.5, 125.4, 124.3, 32.4, 31.8, 28.5, 26.2, 22.82, 22.78, 19.1, 18.9, 13.94, 13.92.
Silole 12a: 50
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.13 (s, 2H), 3.25 (s, 6H); HRMS (GCMS) m / z calcd for C26H35O2Si (M + H)+ 407.2406, found 407.2391.
Silole 12b: 50
1H NMR (400 MHz, C6D6, distinctive peaks) δ 6.09 (s, 2H); HRMS (GCMS) m / z calcd for C26H29F6Si (M + H)+ 483.1943, found 483.1923.
Nickel-Catalyzed Synthesis of Carbon–Carbon Bond Insertion Products
Method A (isolation)
To a solution of alkyne, alkene, or allene (2.0 mmol) and silacyclopropane 3 (1.0 mmol) in toluene (5 mL) was added Ni(cod)2 (0.014 g, 0.05 mmol). When the reaction had proceeded to completion as determined by 1H NMR spectroscopic analysis, the reaction mixture was filtered through SiO2 and concentrated in vacuo. The resulting oil was purified by flash chromatography (hexanes) to afford carbon–carbon bond insertion products. Unless otherwise indicated, all of the spectral data was obtained using Method A.
Method B (NMR observation)
Alkyne, alkene, or allene (0.254 mmol) was added to a solution of silacyclopropane 3 (0.550 mL, 0.127 mmol, 0.230 M solution of 3 and 0.0465 M solution of PhSiMe3 in C6D6), followed by the addition of Ni(cod)2 (0.002 g, 0.006 mmol). The progress of the reaction was monitored and yields were determined by 1H NMR spectroscopic analysis (compared to the PhSiMe3 internal standard) using a single scan.
Silacyclopentene 4m
1H NMR (500 MHz, C6D6) δ 7.23–7.19 (m, 4H), 7.08–7.03 (m, 1H), 5.10 (dd, J = 9.7, 2.1, 1H), 2.74 (qt, J = 10.2, 3.4, 1H), 2.19 (qd, J = 7.1, 2.1, 1H), 2.07 (s, 3H), 1.90–1.41 (m, 10H), 1.43 (d, J = 7.2, 3H), 1.11 (s, 9H), 1.09 (s, 9H); 13C NMR (125 MHz, C6D6) δ 155.2, 146.0, 145.1, 144.0, 128.7, 128.4, 128.2, 125.3, 37.5, 34.5, 34.2, 29.5, 29.3, 27.0, 26.2, 26.2, 26.1, 23.0, 20.5, 18.9, 14.4; IR (neat) 3020, 2925, 1597, 1541, 1468, 1365 cm−1; HRMS (GCMS) m / z calcd for C27H43Si (M + H)+ 395.3134, found 395.3129.
Silacyclopentene 4m′
1H NMR (400 MHz, C6D6, distinctive peaks) δ 4.97 (d, J = 8.9, 1H).
Silacyclopentene 4n
1H NMR (500 MHz, C6D6, distinctive peaks) δ 4.90 (dd, J = 9.4, 2.1, 1H), 1.36 (d, J = 7.1, 3H), 1.10 (s, 9H), 1.09 (s, 9H); 13C NMR (125 MHz, C6D6) δ 160.9, 144.3, 142.4, 124.4, 37.4, 34.3, 34.2, 29.3, 29.0, 28.2, 23.1, 22.71, 22.68, 22.4, 20.0, 14.0, 13.8, 13.7; IR (neat) 2927, 2854, 1470, 1448, 1363, 820 cm−1; HRMS (GCMS) m / z calcd for C22H45Si (M + H)+ 361.3290, found 361.3285.
Silacyclopentene 4n′
1H NMR (500 MHz, C6D6) δ 2.35 (q, J = 7.3, 2H), 2.34 (q, J = 7.5, 2H), 2.23 (d, J = 7.0, 2H), 1.96 (s, 3H), 1.79–1.37 (m, 11H), 1.16 (s, 18H), 1.14 (t, J = 7.6, 3H), 1.01 (t, J = 7.5, 3H); 13C NMR (125 MHz, C6D6) δ 157.9, 153.4, 134.7, 131.1, 39.1, 35.3, 33.7, 29.0, 26.6, 26.5, 22.6, 21.1, 19.3, 16.6, 15.3, 14.4; IR (neat) 2927, 2854, 1468, 1448, 1363, 820cm−1; HRMS (GCMS) m / z calcd for C24H45Si (M + H)+ 361.3290, found 361.3287.
Silacycloheptadiene 13a
mp 64–67 °C; 1H NMR (500 MHz, C6D6) δ 7.41–7.37 (m, 4H), 7.03 (s, 1H), 7.00 (appar t, J = 7.5, 4H), 6.90–6.85 (m, 2H), 6.58 (s, 1H), 5.21 (d, J = 9.9, 1H), 3.18 (q, J = 7.7, 1H), 2.42 (appar q, J = 10.8, 1H), 1.84–1.53 (m, 10H), 1.48 (d, J = 7.8, 3H), 1.21 (s, 9H), 1.15 (s, 9H); 13C NMR (125 MHz, C6D6) δ 155.7, 144.3, 144.1, 140.6, 140.1, 136.0, 131.6, 129.0, 128.13, 128.06, 126.92,49 126.89, 126.8, 37.5, 34.4, 33.1, 30.6, 30.0, 28.6, 26.2, 26.0, 25.9, 23.0, 20.1, 17.1; IR (thin film) 2926, 2854, 1599, 1566, 1470, 1444 cm−1; HRMS (GCMS) m / z calcd for C34H47Si (M + H)+ 483.3447, found 483.3439.
Silacycloheptadiene 13b
mp 68–70 °C; 1H NMR (500 MHz, C6D6) δ 7.74 (d, J = 7.4, 2H), 7.47 (d, J = 7.0, 2H), 7.27–7.11 (m, 6H), 6.65 (s, 1H), 6.45 (s, 1H), 5.11 (d, J = 9.8, 1H), 3.33 (q, J = 8.1, 1H), 2.56–2.48 (m, 1H), 1.73–1.57 (m, 10H), 1.22 (d, J = 8.2, 3H), 1.18 (s, 9H), 1.13 (s, 9H); 13C NMR (125 MHz, C6D6) δ 154.6, 147.1, 144.4, 141.2, 138.6, 131.7, 129.3, 128.3, 128.3, 128.2, 128.2, 127.4, 126.5, 126.2, 37.8, 32.0, 30.6, 28.7, 26.2, 26.0, 25.8, 22.4, 19.9, 17.3; IR (thin film) 3078, 2926, 1597, 1562, 1491, 1470 cm−1; HRMS (GCMS) m / z calcd for C34H47Si (M + H)+ 483.3447, found 483.3445. Anal. Calcd for C34H46Si: C, 84.58; H, 9.60. Found: C, 84.36; H, 9.75.
Diene 14
1H NMR (400 MHz, C6D6) δ 7.37 (d, J = 7.9, 2H), 7.17 (d, J = 19.2, 1H), 7.13–7.06 (m, 3H), 6.60 (d, J = 19.3, 1H), 5.81 (appar q, J = 9.0, 1H), 5.43 (dt, J = 10.9, 7.7, 1H), 2.22 (q, J = 6.9, 2H), 1.93 (d, J = 9.0, 2H), 1.47–1.40 (m, 2H), 1.38–1.26 (m, 6H), 1.13 (s, 18H), 0.89 (t, J = 6.7, 3H); 13C NMR (125 MHz, C6D6) δ 146.3, 138.6, 128.5, 128.1, 128.0, 126.48, 126.45, 123.7, 31.9, 29.7, 29.2, 28.8, 27.4, 22.7, 19.9, 14.0, 10.9; IR (neat) 3059, 2958, 1603, 1575, 1495, 1464 cm−1; HRMS (GCMS) m / z calcd for C25H43Si (M + H)+ 371.3134, found 371.3130. Anal. Calcd for C25H42Si: C, 81.00; H, 11.42. Found: C, 80.80; H, 11.61.
Supplementary Material
Acknowledgment
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM-54909). K.M.B. thanks the Department of Education (GAANN) for a predoctoral fellowship. K.A.W. thanks Amgen and Lilly for awards to support research. We thank Dr. P. Dennison (UCI) for assistance with NMR spectroscopy, Dr. J. W. Ziller and M. K. Takase (UCI) for X-ray crystallography, and Dr. J. Greaves and S. Sorooshian (UCI) for mass spectrometry.
Footnotes
Supporting Information Available. X-ray data for 13a, additional experimental procedures, spectroscopic and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Seyferth D, Duncan DP, Vick SC. J. Organomet. Chem. 1977;125:C5–C10. [Google Scholar]
- 2.Seyferth D, Vick SC, Shannon ML, Lim TFO, Duncan DP. J. Organomet. Chem. 1977;135:C37–C44. [Google Scholar]
- 3.Seyferth D, Shannon ML, Vick SC, Lim TFO. Organometallics. 1985;4:57–62. [Google Scholar]
- 4.Sakurai H, Imai T. Chem. Lett. 1975:891–894. [Google Scholar]
- 5.Sakurai H, Kamiyama Y, Nakadaira Y. J. Am. Chem. Soc. 1977;99:3879–3880. [Google Scholar]
- 6.Ishikawa M, Sugisawa H, Harata O, Kumada M. J. Organomet. Chem. 1981;217:43–50. [Google Scholar]
- 7.Ishikawa M, Matsuzawa S, Higuchi T, Kamitori S, Hirotsu K. Organometallics. 1985;4:2040–2046. [Google Scholar]
- 8.Saso H, Ando W. Chem. Lett. 1988:1567–1570. [Google Scholar]
- 9.Saso H, Ando W, Ueno K. Tetrahedron. 1989;45:1929–1940. [Google Scholar]
- 10.Palmer WS, Woerpel KA. Organometallics. 1997;16:1097–1099. [Google Scholar]
- 11.Palmer WS, Woerpel KA. Organometallics. 1997;16:4824–4827. [Google Scholar]
- 12.Palmer WS, Woerpel KA. Organometallics. 2001;20:3691–3697. [Google Scholar]
- 13.Terunuma D, Shibuya N, Nohira H. Bull. Chem. Soc. Jpn. 1982;55:2287–2288. [Google Scholar]
- 14.Takeyama Y, Nozaki K, Matsumoto K, Oshima K, Utimoto K. Bull. Chem. Soc. Jpn. 1991;64:1461–1466. [Google Scholar]
- 15.Kuniyasu H, Kurosawa H. Chem. Eur. J. 2002;8:2660–2665. [Google Scholar]
- 16.Agenet N, Mirebeau J-L, Petit M, Thouvenot R, Gandon V, Malacria M, Aubert C. Organometallics. 2007;26:819–830. [Google Scholar]
- 17.Liu J, Sun X, Miyazaki M, Liu L, Wang C, Xi Z. J. Org. Chem. 2007;72:3137–3140. doi: 10.1021/jo062583m. [DOI] [PubMed] [Google Scholar]
- 18.Buchner KM, Clark TB, Loy JMN, Nguyen TX, Woerpel KA. Org. Lett. 2009;11:2173–2175. doi: 10.1021/ol900456v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silacyclopropane 3a was observed by 1H NMR spectroscopic analysis of the crude reaction mixture. The yield was calculated relative to an internal standard (PhSiMe3).
- 20.The dimeric material could not be isolated due to its instability and its structure could not be determined in the reaction mixture. The exact mass of the dimer was determined by HRMS (GCMS). Additional details are provided as Supporting Information.
- 21.Atwell WH. Organometallics. 2009;28:3573–3586. [Google Scholar]
- 22.Ishikawa M, Sugisawa H, Kumada M, Higuchi T, Matsui K, Hirotsu K. Organometallics. 1982;1:1473–1477. [Google Scholar]
- 23.Ishikawa M, Fuchikami T, Kumada M. J. Chem. Soc., Chem. Commun. 1977:352. [Google Scholar]
- 24.Driver TG, Woerpel KA. J. Am. Chem. Soc. 2003;125:10659–10663. doi: 10.1021/ja0301370. [DOI] [PubMed] [Google Scholar]
- 25.Driver TG, Woerpel KA. J. Am. Chem. Soc. 2004;126:9993–10002. doi: 10.1021/ja0306563. [DOI] [PubMed] [Google Scholar]
- 26.Clark TB, Woerpel KA. Organometallics. 2005;24:6212–6219. [Google Scholar]
- 27.Clark TB, Woerpel KA. Org. Lett. 2006;8:4109–4112. doi: 10.1021/ol061652g. [DOI] [PubMed] [Google Scholar]
- 28.Metals included in the screen were Pd(0), Pd(II), Ni(0), Ni(II), Rh(I), and Pt(0). Also included were a variety of bimetallic systems with gold catalysts. Ligands included a series of mono-, di-, and tridentate ligands containing some combination of either phosphorous, nitrogen, or oxygen chelation. Pd(PPh3)4 and Ni(cod)2 proved to be the best catalysts in our catalytic system by consistently giving products in modest to high yields. PdCl2(PPh3)2, Pt(PEt3)4, and [RhCl(CO)2]2 were each observed to give products in low yields (<10%). All other catalysts gave no reaction or decomposition of starting materials.
- 29.Nickel-catalyzed incorporation of two equivalents of a terminal alkyne has been reported for an acyclic example. See reference 30.
- 30.Ananikov VP, Orlov NV, Kabeshov MA, Beletskaya IP, Starikova ZA. Organometallics. 2008;27:4056–4061. [Google Scholar]
- 31.Nakao Y, Oda S, Yada A, Hiyama T. Tetrahedron. 2006;62:7567–7576. [Google Scholar]
- 32.Seyferth D, Duncan DP, Shannon ML, Goldman EW. Organometallics. 1984;3:574–578. [Google Scholar]
- 33.Braunstein P, Knorr M. J. Organomet. Chem. 1995;500:21–38. [Google Scholar]
- 34.Osakada K, Tanabe M. Bull. Chem. Soc. Jpn. 2005;78:1887–1898. [Google Scholar]
- 35.Tanabe M, Osakada K. Chem. Eur. J. 2004;10:416–424. doi: 10.1002/chem.200305344. [DOI] [PubMed] [Google Scholar]
- 36.Horn KA. Chem. Rev. 1995;95:1317–1350. [Google Scholar]
- 37.Allen A, Jr., Lin W. Organometallics. 1999;18:2922–2925. [Google Scholar]
- 38.Canovese L, Visentin F, Chessa G, Uguagliati P, Bandioli G. Organometallics. 2000;19:1461–1463. [Google Scholar]
- 39.Onozawa S.-y., Tanaka M. Organometallics. 2001;20:2956–2958. [Google Scholar]
- 40.Reddy KR, Surekha K, Lee G-H, Peng S-M, Liu S-T. Organometallics. 2001;20:5557–5563. [Google Scholar]
- 41.Strömberg S, Zetterberg K, Siegbahn PEM. J. Chem. Soc., Dalton Trans. 1997:4147–4152. [Google Scholar]
- 42.Hirano K, Yorimitsu H, Oshima K. Chem. Commun. 2008:3234–3241. doi: 10.1039/b803172j. [DOI] [PubMed] [Google Scholar]
- 43.Hirano K, Yorimitsu H, Oshima K. J. Am. Chem. Soc. 2007;129:6094–6095. doi: 10.1021/ja070938t. [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa M, Ohshita J, Ito Y, Iyoda Y. J. Am. Chem. Soc. 1986;108:7417–7419. [Google Scholar]
- 45.Ohshita J, Isomura Y, Ishikawa M. Organometallics. 1989;8:2050–2054. [Google Scholar]
- 46.The latter migratory insertion suffers from low regioselectivity, which ultimately leads to products 13a and 13b in a 72:28 mixture. The catalytic cycle is shown for the major regioisomer.
- 47.Spectrum for material obtained from Method B.
- 48.Obtained upon attempted chromatography of the desired product.
- 49.Corresponds to two chemically unequivalent carbons.
- 50.Spectra for material obtained from Method B.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.