

Abstract
Normal pregnancy is associated with significant vascular remodeling in the uterine and systemic circulation in order to meet the metabolic demands of the mother and developing fetus. The pregnancy-associated vascular changes are largely due to alterations in the amount/activity of vascular mediators released from the endothelium, vascular smooth muscle and extracellular matrix. The endothelium releases vasodilator substances such as nitric oxide, prostacyclin and hyperpolarizing factor as well as vasoconstrictor factors such as endothelin, angiotensin II and thromboxane A2. Vascular smooth muscle contraction is mediated by intracellular free Ca2+ concentration ([Ca2+]i), and [Ca2+]i sensitization pathways such as protein kinase C, Rho-kinase and mitogen-activated protein kinase. Extracellular matrix and vascular remodeling are regulated by matrix metalloproteases. Hypertension in pregnancy and preeclampsia are major complications and life threatening conditions to both the mother and fetus, precipitated by various genetic, dietary and environmental factors. The initiating mechanism of preeclampsia and hypertension in pregnancy is unclear; however, most studies have implicated inadequate invasion of cytotrophoblasts into the uterine artery, leading to reduction in the uteroplacental perfusion pressure and placental ischemia/hypoxia. This placental hypoxic state is thought to induce the release of several circulating bioactive factors such as growth factor inhibitors, anti-angiogenic proteins, inflammatory cytokines, reactive oxygen species, hypoxia-inducible factors, and vascular receptor antibodies. Increases in the plasma levels and vascular content of these factors during pregnancy could cause an imbalance in the vascular mediators released from the endothelium, smooth muscle and extracellular matrix, and lead to severe vasoconstriction and hypertension. This review will discuss the interactions between the various circulating bioactive factors and the vascular mediators released during hypertension in pregnancy, and provide an insight into the current and future approaches in the management of preeclampsia.
Keywords: cytokines, endothelium, nitric oxide, vascular smooth muscle, calcium, preeclampsia
INTRODUCTION
Normal pregnancy (Norm-Preg) is associated with increased heart rate, plasma volume and cardiac output, as well as decreased vascular resistance and late gestational decrease in blood pressure (BP). These beneficial hemodynamic changes ensure critical blood and nutrient supply to the growing fetus. In 5–10% of pregnancies women develop hypertension in pregnancy (HTN-Preg) [1]. HTN-Preg may take one of four forms: chronic HTN that predates pregnancy, preeclampsia (PE)-eclampsia, chronic HTN with superimposed PE, and nonproteinuric gestational HTN [2]. The World Health Organization Antenatal Care Trial has shown that 9.1% of pregnant women have either PE or gestational HTN [3]. PE is characterized by a high BP 140/90 mmHg or more in two separate readings taken at least 4 hours apart, 300 mg of protein in a 24-hour urine sample, and pitting edema. Also, if untreated, PE can lead to eclampsia, and life-threatening maternal neurovascular complications including convulsions and severe HTN [4]. Although HTN-Preg represents a major cause of maternal and fetal morbidity/mortality, the exact mechanisms of this disorder have not been clearly identified. Analysis of samples of plasma, body fluids, and postpartum placenta from PE women has been useful in determining some of the underlying biochemical changes. Because of the difficulty to perform mechanistic studies in pregnant women, animal models of HTN-Preg with characteristics similar to those observed in human PE have been developed. Studies in animal models of HTN-Preg have demonstrated significant changes in the renal control mechanisms of BP, and highlighted potential glomerular injury and alteration in kidney functions as possible causes of the increased BP [5]. However, HTN is a multifactorial disorder that involves additional alterations in the neurohumoral and vascular control mechanisms of BP as well as changes in lipid metabolism and the blood coagulation system. Also, several genetic, dietary and environmental risk factors are thought to play a role in the development of HTN-Preg. These maternal risk factors may lead to poor placentation, placental ischemia/hypoxia, and increased release of biologically active circulating factors that eventually cause vascular dysfunction and HTN-Preg (Fig. 1). Understanding the mechanistic interactions between circulating bioactive factors and vascular mediators in non-pregnant conditions characterized by hypoxia could also be useful, although such interactions may differ from those occurring during pregnancy due to the influence of sex hormones. The purpose of this review is to discuss the interactions between the various biologically active circulating factors and vascular mediators, released during PE as compared to those released in other conditions characterized by hypoxia, in order to provide insight into the sequence of events that lead to PE and other forms of HTN-Preg. Throughout the review we will discuss the changes in Norm-Preg followed by those in PE, evidence in human followed by data from experimental animals, and data from large vessels followed by small vessels and isolated vascular cells.
Fig. 1.
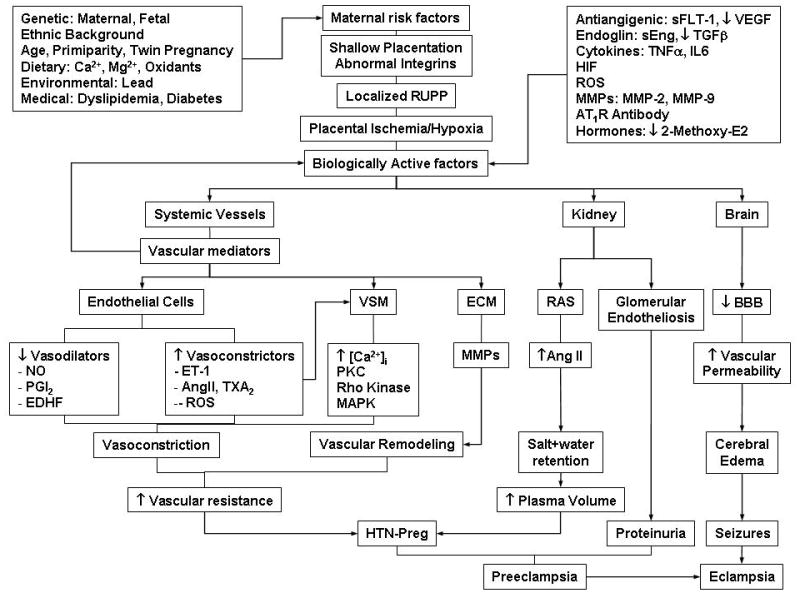
Cellular Mechanisms of HTN-Preg. Maternal genetic, dietary and environmental factors lead to shallow placentation and RUPP during late pregnancy and trigger the release of biologically active factors, which affect the systemic blood vessels and lead to generalized vasoconstriction, increased vascular resistance and HTN-Preg. The vascular mediators could in turn affect the biologically active factors and further promote their biological effects. The bioactive factors could also injure the kidney leading to increased plasma volume and severe HTN, as well as glomerular endotheliosis and proteinuria, leading to clinically manifested PE. Increases in cerebral vascular permeability and edema lead to seizures and life-threatening eclampsia.
Genetic Risk Factors of PE
Maternal Genes
Women offspring of a pregnancy complicated by PE may have a higher risk of developing PE in their own pregnancies. The increased risk may reflect the inheritance of genes that enhance maternal susceptibility to PE [6]. Genome-wide scans and linkage studies have implicated more than one gene locus, and identified loci on chromosomes 1, 2p13, 2q, 3p, 4q, 9, 10q, 11q23–24, 12q, 15q, 18 and 22q, suggesting that PE could be a multigenic disorder [7]. Also, genes encoding novel molecules such as sialic acid binding Ig-like lectin 6 (Siglec-6), a potential leptin receptor, and pappalysin-2 (PAPP-A2), a protease that cleaves insulin-like growth factor (IGF) binding proteins, are overexpressed in PE [8]. Additionally, genes encoding different components of the renin-angiotensin system (RAS) have been studied, and variations in the angiotensinogen, angiotensin type 1 receptor (AT1R) and endothelial nitric oxide synthase (NOS) genes have been associated with HTN-Preg. Also, mutation in factor V Leiden gene, which results in resistance to activated protein C, has been found in 10% of Caucasian women with HTN-Preg compared with only 4% of Caucasian Norm-Preg women [9].
Fetal Genes
Another important factor in the development of HTN-Preg could be the fetal genes. Mouse models have been used to study the fetal genetic factors associated with HTN-Preg. The cyclin-dependent kinase inhibitor cdkn1c (p57Kip2) is a major regulator of embryonic growth, and mutant embryos demonstrate altered placental architecture and histological changes indicative of reduced utero-placental blood flow. Also, pregnant mice expressing wild-type levels of p57Kip2 develop a PE-like condition when carrying p57kip2-deficient pups [7], suggesting that fetal and paternal genes play a role in the development of HTN-Preg.
Ethnic Background
Ethnic background may also be a factor in the development of HTN-Preg. A study of pregnant women in New York found that African-American women had the highest rates of HTN-Preg, while Asian women had the lowest incidence rates [10]. Also, African-American and Hispanic women had much higher rates of PE compared to Caucasian and Asian women.
Dietary Risk Factors
Dyslipidemia and Hyperglycemia
Diet can also be a risk factor for the development of HTN-Preg. There has been a progressive increase in the prevalence of obesity and metabolic syndrome among the United States population. Metabolic syndrome is characterized by glucose intolerance, dyslipidemia, insulin resistance, diabetes, obesity, and HTN. Patients with metabolic syndrome also have vascular endothelial dysfunction, and their blood vessels could be more sensitive to circulating factors released during HTN-Preg. PE is also characterized by changes in lipid metabolism, and such changes may play a role in the endothelial cell (EC) damage characteristic of PE. Recent epidemiological studies have shown elevated maternal plasma concentration of triglycerides in women who develop PE. Women with PE also have higher serum levels of free fatty acids and higher concentrations of cholesterol, phospholipids, and lipid peroxides in the placenta [11]. Furthermore, increased intake of energy, sucrose, or polyunsaturated fatty acids is associated with increased risk of PE [1]. Increased sugar consumption may also lead to hyperglycemia, which in turn could inhibit endothelium-dependent vasorelaxation. Thus both dyslipidemia and hyperglycemia have been associated with EC dysfunction, an important feature of PE. Also, increased intake of polyunsaturated fat, which is easily oxidized, may enhance the oxidative environment that exists in PE.
Calcium, Vitamin D, Magnesium, Potassium
Dietary intake of micro- and macronutrients may influence the risk of HTN-Preg. Increased calcium (Ca2+) intake has been associated with lower incidence of HTN-Preg; however, the beneficial effect of Ca2+ supplementation may depend on the mother’s age and socioeconomic status (12). Experimental studies on pregnant ewes showed that restriction of Ca2+ intake is associated with a decrease in uterine blood flow, increased BP and elevated urine protein; all symptoms similar to those of PE [13]. Other studies suggested that increasing Ca2+ intake during pregnancy may not lower the risk of PE [14]. Randomized controlled clinical trials by the World Health Organization on Ca2+ supplementation during pregnancy showed that Ca2+ may not decrease the risk of PE but may help in lowering the severity of the condition [15]. Vitamin D is a major regulator of Ca2+ absorption and metabolism. Vitamin D deficiency in early pregnancy is a strong, independent risk factor for PE. Studies have shown that women with PE have lower levels of Vitamin D compared to Norm-Preg women [16]. Also, experimental studies examining the influence of vitamin D on the renin-angiotensin system in mice have shown that renin expression and plasma AngII production are increased in vitamin D-deficient mice, and are associated with HTN [17]. Magnesium (Mg2+) is an essential dietary mineral needed in a relatively large amount. Mg2+ is a cofactor of many enzymes that regulate temperature and protein synthesis, and maintain electrical potential in nerves and muscle membrane [18]. Observational studies based on medical records reported that Mg2+ supplementation during pregnancy was associated with a reduced risk of fetal growth retardation and PE. Maternal Mg2+ intake is also positively associated with birth weight. Also, diets high in potassium and fiber are associated with a reduced risk of HTN [19]. Thus, the maternal intake of recommended amounts of Ca2+, vitamin D, Mg2+, potassium and fiber may lead to a reduced risk of HTN-Preg.
Oxidative Stress
Oxidant-antioxidant imbalance may also play a pathogenic role in PE. Vitamin E is an important antioxidant, and its level and relation with circulating levels of lipid peroxides in women with HTN-Preg have been studied. As with other antioxidants, studies have shown lower levels of vitamin E in serum of women with HTN-Preg or PE compared to Norm-Preg women. Also, studies have shown decreased level of Vitamin C in plasma taken from women with PE, suggesting that supplemental intake of Vitamin C and E may decrease the incidence of PE in women at high risk [20]. On the other hand, the role of vitamin A in HTN-Preg and PE is unclear. Some clinical studies found markedly lower serum vitamin A levels in PE than Norm-Preg women in the third trimester. However, no clinical trials have assessed the effect of vitamin A supplementation on HTN-Preg or PE. Furthermore, the potential teratogenic effects of vitamin A should be considered.
Other Environmental Risk Factors of HTN-Preg
Lead
Environmental exposure to toxic compounds may cause undesirable health effects to the mother and fetus. Exposure to lead and high lead accumulation in the bones are associated with the development of HTN. In experimental animals, chronic exposure to low levels of lead affects the electrical and mechanical properties of the heart and alters vascular smooth muscle (VSM) function. Studies have also shown a dose-dependent relationship between lead exposure and HTN-Preg, and demonstrated a 4% increase in the risk of HTN per 0.05 mg/m3 increase in the seasonal average lead level [21]. Additionally, some lead is absorbed into the bones, where it is stored and accumulated with age. Thus women who have had past lead exposure or chronic low level exposure may have a higher risk of HTN-Preg, even after a long period of no exposure [21,22].
Medical Factors
PE and HTN-Preg are also associated with medical risk factors such as diabetes, renal or cardiac disease, PE in a previous pregnancy, urinary tract infection, extreme maternal age (<16 or >40 years old), and twin pregnancy. Risk factors associated with HTN-Preg also include a previous neonatal macrosomia, history of reproductive tract surgery, and antepartum hemorrhage. Primiparity and a history of chronic respiratory conditions may also be risk factors for PE [3].
Abnormal Placentation as an Initiating Event of HTN-Preg
During the early stages of Norm-Preg, the cytotrophoblasts invade the uterine spiral arteries, progressively replacing ECs, medial elastic tissue, VSM, and neural tissue. Consequently, the uterine spiral arteries undergo marked transformation from small muscular arterioles to low resistance large capacitance vessels. By the end of the second trimester, the spiral arteries become dilated tubes lined by cytotrophoblasts, which ensures sufficient blood supply and nutrition to the growing fetus. During PE, abnormal expression of the cell adhesion molecules integrins as well as apoptosis of cytotrophoblasts lead to limited invasion of the uterine spiral arteries to only the superficial layers of the decidua [23,24]. The shallow cytotrophoblast invasion and the inadequate vascular remodeling of the uterine spiral arteries do not meet the fetal blood flow and nutrition demands, resulting in fetal intrauterine growth retardation (IUGR) [25,26]. Placental ischemia also initiates a cascade of events leading to marked changes in the maternal circulation. The ischemic placenta may induce the release of bioactive circulating factors that cause the harmful changes in the maternal circulation and EC function observed in PE. It is also likely that the ischemic placenta may not be capable of releasing potentially beneficial biological factors that are necessary for the normal hemodynamic effects observed in Norm-Preg.
Animal models of HTN-Preg have been useful in understanding the role of placental ischemia as a possible initiating event of the elevated BP in PE. Although experimental induction of uteroplacental ischemia during pregnancy has shown variable effects in different species, it is still considered one of the most promising animal models of HTN-Preg. Reduction of uteroplacental perfusion pressure (RUPP) in late pregnant sheep, dogs, rabbits and rats results in a hypertensive state that resembles PE [27].
Biologically Active Factors Involved in HTN-Preg
Placental hypoxia is thought to induce the release of various bioactive circulating factors. Elevated plasma and vascular tissue levels of these bioactive factors during pregnancy could cause EC and VSM dysfunction, severe vasoconstriction and PE in human (Table 1) and HTN-Preg in rats (Table 2). Studies have searched for a unique factor as a single cause of the HTN-Preg, but this concept has been challenged, and numerous factors have been suggested [27]. Understanding the effects of these biologically active factors on vascular-derived mediators is crucial in determining the specific compounds involved in the development of HTN-Preg.
Table 1.
Plasma and Vascular Levels of Bioactive Factors and Vascular Mediators during Normal Pregnancy (Norm-Preg) and Preeclampsia (PE) in Humans.
| Site of Action | Effect | Norm-Preg | PE | Reference | |
|---|---|---|---|---|---|
| Bioactive Factor | |||||
| VEGF | Endothelium | Vasorelaxation | 184 pg/mL | 396 pg/mL | [28] |
| sFlt-1 | Endothelium | Anti VEGF | 339.4 pg/mL | 7247pg/mL | [29] |
| Endoglin | Endothelium | Anti TGF-β | 9.8 ng/mL | 46.4 ng/mL | [30] |
| Cytokines TNF-α IL-6 |
Endothelium Endothelium |
EC Dysfunction EC Dysfunction |
3.31 pg/ml 4.9±1.1 pg/mL |
4.68 pg/mL 16.5±2.1 pg/mL |
[31] [32] |
| HIF | DNA | Gene Transcription | ↑ | ↑↑ | [33] |
| ROS | Endothelium | EC Dysfunction | -- | ↑ | [34] |
| MMP-2 | ECM, Endothelium | Remodeling | 779±194 ng/mL | 783±322.3 ng/mL | [35] |
| MMP-9 | ECM | Remodeling | 121±190.5 ng/mL | 262.2±398.6 ng/mL | [35] |
| 2-Methoxy-E2 | Estrogen receptor | Vasodilator | ↑ | ↓ | [36] |
| Vascular Mediator | |||||
| NO | VSM | Vasorelaxation | 13.0±4.3 μmol/L | 18.1±6.2 μmol/L | [37] |
| PGI2 | VSM | Vasorelaxation | ↑ | ↓ | [38] |
| EDHF | VSM | Vasorelaxation | ↑ | ↓ | [39] |
| ET-1 | VSM Endothelium | Vasoconstriction Vasorelaxation | 12±1.0 pmol/L | 22.6±2.0 pmol/L | [40] |
| AngII | VSM | Vasoconstriction | 30.2 fmol/mL | 53.3 fmol/mL | [41] |
↑↑, Greatly increased;
↑, Increased;
↓, Decreased;
--, No change, relative to nonpregnant.
Table 2.
Plasma and Vascular Levels of Bioactive Factors and Vascular Mediators during Norm-Preg and HTN-Preg in rats.
| Site of Action | Effect | Norm-Preg | HTN-Preg | Reference | |
|---|---|---|---|---|---|
| Bioactive Factor | |||||
| VEGF | Endothelium | Vasorelaxation | 830±33 pg/mL | 594±34 pg/mL | [42] |
| sFlt-1 | Endothelium | Anti VEGF | 82±26 pg/mL | 660±270 pg/mL | [43] |
| Endoglin | Endothelium | Anti TGF-β | 8 ng/mL | 17 ng/mL | [44] |
| Cytokines TNF-α IL-6 |
Endothelium Endothelium |
EC Dysfunction EC Dysfunction |
8±1 pg/mL 36.6±7 pg/mL |
48±13 pg/mL 104.5±28.6 pg/mL |
[45] [46] |
| HIF | DNA | Gene Transcription | ↑ | ↑↑ | [27] |
| ROS | Endothelium | EC Dysfunction | -- | ↑ | [27] |
| Vascular Mediator | |||||
| NO (Urinary Nitrite/Nitrate) | VSM | Vasorelaxation | 46.4±5.3 μmol/24 hr | 49.8±6.4 μmol/24 hr | [47] |
| PGI2 | VSM | Vasorelaxation | ↑ | ↓ | [48] |
| ET-1 | VSM, Endothelium | Vasoconstriction, Vasorelaxation | 1.7 pmol/L | 2.2 pmol/L | [49] |
| AngII | VSM | Vasoconstriction | ↑ | ↑ | [50] |
| VSM [Ca2+]i - Basal - PHE (10−5M) - AngII (10−7M) |
Myofilaments Myofilaments Myofilaments |
Basal Tone Contraction Contraction |
63 ± 5 nM 149 ±8 nM 149 ±8 nM |
109 ± 8 nM 234 ±1 nM 225 +/− 9 nM |
[51] [51] [52] |
↑↑, Greatly increased;
↑, Increased;
↓, Decreased;
--, No change, relative to nonpregnant.
The circulating biologically active factors in PE have not been fully characterized. Placental trophoblasts may be a potential source of the factors that are released into the maternal circulation as result of placental ischemia, and consequently cause the increased vascular permeability observed in PE. Fragments of syncytiotrophoblast microvillous membranes have been detected in blood of PE women and may cause EC dysfunction as a part of a more generalized intravascular inflammatory response of the maternal immune system to pregnancy [53]. Also, a complex purified from syncytiotrophoblast microvillous membranes inhibits the proliferation of cultured human EC [54]. Studies on human umbilical vein ECs co-cultured with trophoblast cells isolated from normal and PE placentas have suggested that endothelial cell-to-cell junctions are sensitive targets for trophoblast-derived factors, leading to marked increase in ECs permeability, and may translate into increased vascular permeability in vivo [55]. Other studies do not support the trophoblast deportation theory and have shown that intraluminal perfusion of isolated myometrial arteries of Norm-Preg women with syncytiotrophoblast microvillous membranes at concentrations up to 100 times those reported in PE does not affect bradykinin-induced dilation or cause significant damage to the endothelium [56]. The preliminary characterization of the putative biologically active factor(s) in PE plasma suggested a high-molecular-weight protein/glycoprotein, and possible contribution of a hydrophobic, lipophilic factor [57]. Plasma VEGF, antiangiogenic factors, cytokines, oxidative stress, hypoxia-inducible factors (HIFs), matrix metalloproteases (MMPs), and numerous modulators of endogenous hormones and vascular receptors have been implicated as possible biologically active factors in PE (Fig. 2).
Fig. 2.
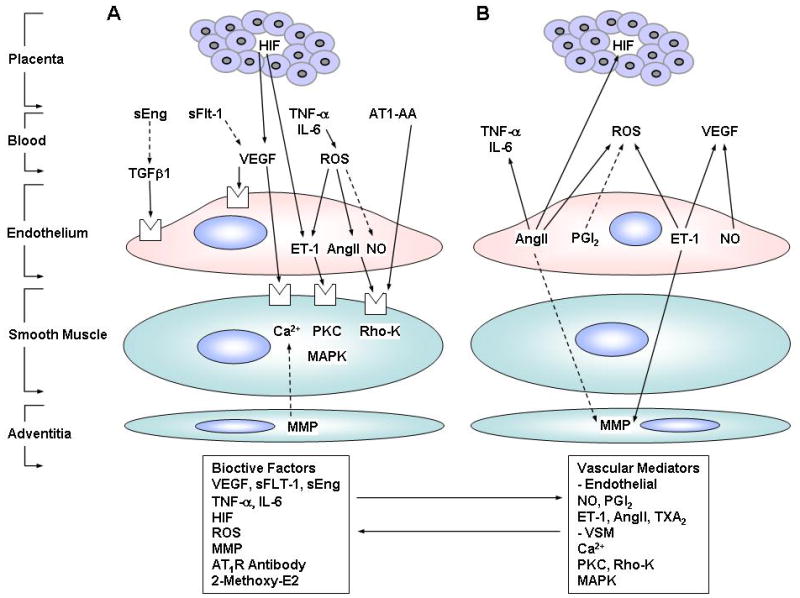
Interactions between biologically active factors and vascular mediators during HTN-Preg. Increases in biologically active factors result in changes in vascular mediators and increases in other biologically active factors (A). Increases in vascular mediators causes feedback changes in biologically active factors (B). Solid arrows indicate stimulation. Interrupted arrows indicate inhibition.
Vascular Endothelial Growth Factor
VEGF is a glycoprotein that acts as a mitogen and mediator of EC growth and tissue neovascularization. VEGF expression is induced by hypoxia in various cell types, including ECs and VSM cells. The main mechanism of regulation of VEGF by hypoxia occurs at the level of VEGF mRNA stability [58]. Some studies suggest that serum VEGF immunoreactivity is suppressed during Norm-Preg, and may further decrease during PE [59]. Other studies have shown that elevated serum VEGF levels may be involved in the EC activation/dysfunction that occurs in PE [60]. Maternal plasma VEGF increases before the clinical onset of PE and is further elevated during the vasoconstriction phase of the disease. It is possible that the hyperdynamic circulation in the latent phase of PE may induce vascular shear stress, which in turn increases circulating VEGF. Because VEGF normally acts as a vasodilator, its increase may represent an unsuccessful vascular rescue response during PE [61]. Studies on PE women have shown that treatment with VEGF impairs bradykinin-mediated vascular relaxation, but enhances basal tone and permeability, supporting a role for VEGF in the EC dysfunction in PE [62]. The vasodilator effects of VEGF may be due to stimulation of the NO-cGMP vascular relaxation pathway. Studies have shown that VEGF increases [Ca2+]i, which promotes calmodulin binding to eNOS and stimulates NO production. Also, VEGF-induced NO production has been associated with upregulation of eNOS activity in ECs [63].
sFlt-1
Soluble fms-like tyrosine kinase-1 (sFlt-1) is a VEGF and placental growth factor (PLGF) antagonist protein that binds VEGF and prevents its interaction with its endogenous receptors. Compared with Norm-Preg women, circulating levels of sFlt-1 are increased in PE women [42,64]. The excess production of sFlt-1 in PE may be secondary to placental ischemia/hypoxia [43]. Increased placental expression of sFlt-1 appears to play a central role in the pathogenesis of PE. sFlt-1 administered to pregnant rats induces albuminurea, HTN, and renal pathologic changes in the form of glomerular endotheliosis [30]. In vitro, removal of sFlt-1 from supernatant of PE tissue culture restores EC function and angiogenesis to normal levels [29]. Interestingly, two karyotypic anomalies, trisomy 13 and hydatiform mole, are associated with a risk for PE. Redman and coworkers suggested an association between trisomy 13 and PE [65], and predicted the involvement of fetal factors and proteins encoded by genes located on chromosome 13. In effect, the sFlt-1 gene is localized on chromosome 13 [65,66].
Endoglin
Endoglin is a transmembrane receptor for transforming growth factor-β (TGF-β) that is expressed on ECs. A novel placenta-derived soluble endoglin (sEng) is an anti-angiogenic protein that appears to be an important factor in PE [67]. sEng inhibits TGF-β1 signaling in ECs and blocks TGF-β1 mediated activation of eNOS and vasodilation, suggesting that dysregulated TGF-β signaling may be involved in the pathogenesis of PE [68]. sEng is elevated in sera of PE women 2 to 3 months before the onset of clinical signs of PE, correlates with the severity of the disease, and falls after delivery. An increased level of sEng accompanied by an increased ratio of sFlt-1:PLGF is predictive of developing PE [69]. In pregnant rats, sEng increases vascular permeability and induces HTN [68]. Also, in pregnant rats, sEng potentiates the vascular effects of sFlt-1 to induce a severe PE-like state, including the development of HELLP syndrome and restriction of fetal growth [69].
Cytokines
Various cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-8, are involved in cell proliferation and hypoxia-mediated cell activation. Hypoxia has been shown to induce marked increases in IL-6 and IL-8 synthesis in VSM cells [46]. RUPP during pregnancy and the ensuing placental ischemia are also thought to increase the release of cytokines such as TNF-α and IL-6 into the maternal circulation. The increased plasma cytokines could then lead to maternal EC dysfunction, generalized vascular changes and HTN [70]. This is consistent with the observation that the plasma concentrations of TNF-α and IL-6 are elevated in women with PE [70]. IL-10 may be involved in the maintenance of pregnancy by inducing corpus luteum maturation and progesterone production, and the level of IL-10 is decreased in PE women [71].
Cytokines have been shown to affect vascular mediators as well as other biologically active factors (Fig. 2A). TNF-α induces structuralas well as functional alterations in ECs. TNF-α also enhances the formation of a number of EC substances such as endothelin-1 (ET-1), reduces acetylcholine-induced vasodilatation, and destabilizes the mRNA of endothelial eNOS [45]. Also, both TNF-α and IL-6 induce VEGF expression in ECs [72].
Infusion of cytokines has been used to develop animal models of HTN-Preg. Experimental studies have shown that chronic infusion of TNF-α or IL-6 in late pregnant rats to increase their plasma 2–3 fold results in marked elevation of renal vascular resistance and BP [73–75]. Also, the vascular contraction is greater in TNF-α and IL-6 infused pregnant rats compared with Norm-Preg rats [74,75]. Furthermore, endothelium-dependent vascular relaxation is reduced in TNF-α and IL-6 infused compared with Norm-Preg rats possibly due to inhibition of the endothelium-dependent NO-cGMP pathway [74,75].
Reactive Oxygen Species (ROS)
An increase in oxidative stress is another factor that could provide the link between localized RUPP and the generalized EC dysfunction in PE [76]. Hypoxia is known to cause increases in the production of ROS, including superoxide anion (O2•−) and hydrogen peroxide in ECs and VSM cells [77]. Free radicals and placental lipid peroxides are increased in PE. Also, a deficiency in the plasma concentration of the antioxidant ascorbate is typical of PE [78]. It has been shown that brachial artery flow-mediated and endothelium-dependent vasodilation is reduced in women who previously developed PE compared with controls. Administration of ascorbic acid increases flow-mediated vasodilation in women who previously developed PE but not in controls, suggesting that the EC dysfunction in women with previous PE is possibly due to increased oxidative stress and ROS [79]. However, whether antioxidants are beneficial in preventing the impaired EC function associated with PE needs to be further examined [80].
Neutrophils are another possible source of EC damage through their ability to produce ROS. Stimulated neutrophils from women with PE produce more O2•− compared to neutrophils from Norm-Preg women. Neutrophils also produce NO, which can react with O2•− and protect cells from O2•−-induced damage in Norm-Preg. However, in PE O2•− production may dominate neutrophil and EC NO production, resulting in EC injury [34]. Furthermore, the plasma concentration of lipid peroxidation markers is increased in women with PE. This supports the hypothesis that poor uteroplacental perfusion results in an increase in placental free radical synthesis and maternal oxidative stress [81].
Oxidant species, including O2•− and peroxynitrite, are potent stimulators of matrix metalloproteases MMP-1, -2, and -9 in VSM [82]. Also, upregulation of ROS during hypoxia is associated with increased degradation of HIF-1α [83]. ROS also cause increases in VSM [Ca2+]i via activation of voltage-gated Ca2+ channels, leading to an influx of extracellular Ca2+. ROS can also increase [Ca2+]i by inhibiting Ca2+-ATPase activity and promoting inositol triphosphate (IP3)-induced Ca2+ release from intracellular stores. The increases in [Ca2+]i induce VSM contraction, and activate Ca2+-dependent protein kinase C (PKC) to further promote vascular contraction. ROS can also promote vasoconstriction by activating Rho-A/Rho-kinase signaling mechanisms [84].
Hypoxia Inducible Factors
Induction of HIF-1 by hypoxia requires synthesis of both the α and β-subunits, which are quickly degraded after reoxygenation. HIF-1 binds to the enhancer region of genes that encode glycolytic enzymes as well as VEGF [85]. Because placental hypoxia likely plays a role in PE, the role of HIFs in the human placenta has been investigated. The expression of HIF-1α and HIF-2α, but not HIF-1β, is selectively increased in PE placenta [33]. The molecular mechanisms underlying the changes in HIF expression and the genes affected during PE are unclear. However, a HIF-1α binding site exists in the promoter region for the ET-1 gene and acts, in combination with other transcription factors, to regulate ET-1 expression in response to hypoxia [86]. Upregulation of HIF-1 also enhances the expression of VEGF [83].
Matrix Metalloproteases
MMPs are zinc-dependent endopeptidases that are capable of degrading extracellular matrix proteins and to process a number of bioactive molecules. MMPs may play a role in the vascular dysfunction associated with HTN-preg. MMP-2 is the main collagenolytic enzyme in umbilical cord artery (UCA) wall. MMP-2 also cleaves big endothelin-1 into ET-11–32, which is a potent vasodilator. MMP-2 also inhibits relaxin and thrombin. The levels of MMP-2 in PE have been controversial; some studies suggest an increase [87], while other studies suggest that a reduced level of MMP-2 in UCA reduces the breakdown of collagen in the arterial wall. The accumulation of collagen and with a reduction of elastin in the UCA may decrease the blood flow in the fetus of women with PE [88]. Hypoxia has been shown to increase MMP-9 mRNA and enzymatic activity 3- to 4-fold in ECs [85]. Also, the higher net MMP-9 activity in HTN-Preg compared with Norm-Preg suggests that MMP-9 may play a role in the pathophysiology of PE. Recent studies have suggested that MMP-2 and MMP-9 inhibit increases in vascular [Ca2+]i, through reduction of Ca2+ entry from extracellular spaces, but not from the intracellular stores [89]. Interestingly, the relationship between MMP and VEGF is bi-directional. Studies have shown that VEGF upregulates MMP expression; while membrane type-1 MMP promotes VEGF expression [90].
2-Methoxy-estradiol (2-methoxy-E2)
2-Methoxy-E2, a natural metabolite of E2, is elevated during the third trimester of Norm-Preg and has recently been linked to PE. Studies have shown that pregnant mice deficient in catechol-O-methyltransferase (COMT) show a PE-like phenotype resulting from an absence of 2-methoxy-E2. 2-Methoxy-E2 ameliorated all PE-like features without toxicity in the COMT deficient pregnant mice and suppressed placental hypoxia, HIF-1α expression and sFLT-1 elevation. The plasma levels of 2-methoxy-E2 are markedly lower in women with severe PE. These findings suggested that measurement of 2-methoxy-E2 in plasma and urine may be used as a diagnostic marker for PE. Also, 2-methoxy-E2 may serve as a therapeutic measure to prevent or treat PE [36].
Vascular Mediators in HTN-Preg
Blood vessels maintain their tone through a balance between vasodilator and vasoconstrictor mediators. The release of biologically active factors in response to placental hypoxia/ischemia could result in EC and VSM dysfunction and influence the release of various vascular mediators (Fig. 1). ECs regulate vascular tone through the release of vasodilators including NO, prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor (EDHF) as well as vasoconstrictor mediators such as ET-1 and AngII in human (Table 1) and in rats (Table 2).
Endothelial Mediators
Nitric Oxide
NO is a powerful vasodilator and relaxant of VSM. NO is produced from the transformation of L-arginine to L-citrulline by neuronal nNOS, inducible iNOS, and endothelial eNOS. It has been reported that low oxygen tension in vivo results in a substantial decrease in eNOS mRNA expression and eNOS protein and NO production [91]. The vascular changes during Norm-Preg have been attributed, in part, to the increased NO synthesis by various maternal cells including vascular ECs [92]. NOS expression and activity are increased in human uterine artery during Norm-Preg [93]. Also, the plasma concentration and urinary excretion of cGMP, a second messenger of NO and cellular mediator of VSM relaxation, are increased in Norm-Preg. Interestingly, cGMP production is markedly increased during the first trimester when the maternal circulation is rapidly vasodilating, while the whole body NO production is not proportionately elevated, suggesting additional sources of cGMP [94].
Measurements of total NO production during HTN-Preg have not produced consistent findings. Some studies have shown that the concentration of nitrite/nitrate, stable byproducts of NO, is reduced in the sera of women with PE [95], while other studies have shown increased levels [96]. The difference in the nitrite/nitrate levels during PE could be related to dietary nitrate intake. However, in a study of women with PE, during which dietary nitrate/nitrite intake was carefully controlled, unequivocal support for reduced NO production was not demonstrated [94]. The apparent dissociation between whole body NO production and the decreased vascular relaxation during HTN-Preg suggest that tissue-specific differences in NOS expression and NO bioavailability may occur during pregnancy. For instance, NOS expression may not be different in the placentas of Norm-Preg and PE women [97]. However, in late pregnant rats the amount of renal eNOS is decreased by 39% while iNOS and nNOS increase by 31% and 25%, respectively [98].
Although the total nitrate/nitrite production may be unchanged in the plasma of PE women, the availability of NO to produce vascular relaxation may be reduced. Ascorbate is essential for the decomposition of S-nitrosothiols and the release of NO. Ascorbate deficiency is typical of PE plasma and might result in decreased decomposition rates of S-nitrosothiols [20]. This is supported by reports that PE plasma contains higher concentrations of total S-nitrosothiols and S-nitrosoalbumin than plasma taken during Norm-Preg, possibly due to insufficient NO release from these major NO reservoirs in PE [78].
While measurements of nitrate/nitrite have not produced consistent findings in PE, the increase in NO production and the reduction of vascular resistance during Norm-Preg suggests that a decrease in NO production during pregnancy should significantly affect the vascular resistance and BP. Studies have suggested endothelial dysfunction in PE [99]. Also, NOS blockade with Nω-nitro-L-arginine methyl ester (L-NAME) during mid to late gestation in rats has been shown to produce pathological changes similar to those observed in women with PE including renal vasoconstriction, proteinuria, thrombocytopenia and IUGR [100,101]. Also, BP is markedly increased in pregnant rats treated with L-NAME compared with virgin rats treated with equal doses of L-NAME [101].
Prostacyclin
PGI2 is an anti-platelet aggregator and a vasodilator with significant beneficial effects in the maternal circulation. PGI2 is produced from the metabolism of arachidonic acid by the cyclooxygenase (COX)-1 and COX-2. Hypoxia has been shown to down-regulate COX-1 and PGI2 production [102]. Endothelium-derived PGI2 may contribute to the hemodynamic and vascular changes during pregnancy. The plasma and urinary levels of 6-keto-prostaglandin F1α (PGF1α), a hydration product of PGI2, are increased during Norm-Preg [103]. Alterations in PGI2 production have been reported in PE [103]. The plasma and urinary concentrations of PGF1α are lower in women with severe PE than in Norm-Preg women, suggesting that overall PGI2 synthesis is diminished in PE. Endothelial PGI2 production may also be decreased in PE [104]. Interestingly, in vascular strips of RUPP rats, acetylcholine-induced relaxation is not completely inhibited by L-NAME or methylene blue, suggesting changes in other endothelium-derived vasodilator substances such as PGI2 in animal models of HTN-preg [38].
Endothelium-Derived Hyperpolarizing Factor
While the exact nature of EDHF is unclear, it is a prominent relaxing factor particularly in the small resistance vessels. Studies have shown that EDHF is the predominant relaxing factor in female mice, while NO and PGI2 are the dominant vasodilators in male mouse vessels [105]. This suggests that an increase in EDHF production/activity may play a role in the vascular adaptation during Norm-Preg. Studies on the uterine vascular beds of pregnant rats have suggested that EDHF release is activated by a delayed rectifier type of voltage-sensitive K+ channel [106], and cause vasodilation by affecting myoendothelial gap junctions alone or in combination with H2O2 or cytochrome P-450 epoxygenase, a metabolite of arachidonic acid. Although EDHF does not appear to be reduced by chronic hypoxia [107,108], its release may be impaired during PE [109,110].
While NO, PGI2, and EDHF act as vasodilators, the interactions between these molecules has not been clearly defined. PGI2 analogue has been shown to upregulate eNOS and NO production in ECs [111]. Studies have also shown that PGI2 may activate the inward rectifier potassium channels through the release of EDHF [112].
Endothelin-1
ET-1 is produced by ECs, and acts on VSM cells to cause marked vasoconstriction. Hypoxia is one of the most potent stimuli of ET-1 production. Hypoxia induces the synthesis and secretion of ET-1 from ECs by a mechanism that is antagonized by NO and carbon monoxide [86]. ET-1 production also appears to be increased in women with PE [113], perhaps via activation of AT1R [40]. ET-1 secretion is 4- to 8-fold higher in umbilical cord ECs of PE compared to Norm-Preg women [114]. Also, the concentration of immunoreactive ET-1 is elevated in plasma of PE women and rapidly returns to a normal value within 48 h of delivery, suggesting that ET-1 may contribute to the vasoconstriction associated with PE [115]. Typically, plasma ET-1 levels are highest during the later stage of the disease, suggesting that ET-1 may not be involved in the initiation of PE, but rather in the progression into the malignant hypertensive phase of the disease [40].
ET-1 interacts with ETAR and ETB2R in VSM, initiating a cascade of biochemical events that leads to vascular contraction. Also, chronic infusion of ET-1 results in increased plasma concentrations of AngII [116]. ET-1 also activates ETB1R in ECs and causes the release of vasodilator substances such as NO, PGI2, and EDHF that promote vascular relaxation [117]. ET-1, via activation of ETBR, may also mediate the reduced myogenic reactivity and vasodilation of renal arteries as well as hyperfiltration during pregnancy in rats [118,119]. Under normoxic conditions, ET-1 stimulates Ca2+ release from the intracellular stores, PKC-dependent inhibition of voltage-gated K+ channels, and subsequent membrane depolarization and Ca2+ influx through voltage-dependent Ca2+ channels, leading to increased [Ca2+]i and VSM contraction. In contrast, in hypoxic conditions, up to 80% of ET-1 induced vascular contraction occurs independently of [Ca2+]i and PKC [120].
Angiotensin II
AngII is an important circulating hormone for BP regulation and electrolyte homeostasis. Also, up to 40% of AngII is produced locally by non-angiotensin converting enzyme (ACE) pathways in blood vessels [121]. Hypoxia has been shown to cause an increase in plasma levels of AngII in rabbits, although this increase is temporary [122]. Also, local levels of ACE, as well as AT1R and angiotensin type 2 receptor (AT2R) are increased during chronic hypoxia [122]. Plasma Ang-(1–7) concentration and most other components of the rennin-angiotensin system, except serum ACE, are reduced in PE compared with Norm-Preg subjects. Only plasma AngII remains elevated in PE compared to nonpregnant women. In PE the decreased plasma Ang-(1–7) in the presence of elevated AngII may contribute to the development of HTN [123]. AngII may cause vascular remodeling by activating signal transduction cascades that promote vasoconstriction, vascular growth, and inflammation. AngII may also activate the Rho/Rho-kinase system, a prominent regulator of VSM contraction, the cytoskeleton and vascular remodeling [124]. Studies have shown increased sensitivity and vascular responses to AngII in PE. Other studies have suggested that the decreased response to AngII in Norm-Preg is caused by increased inactivation of AngII by angiotensinase in serum and placenta. Angiotensinase activity increases with advancing gestation in Norm-Preg sera, but the enzyme activity is lower in sera of women with severe PE leading to decreased degradation and increased sensitivity to AngII [125]. A fraction of circulating IgG antibodies activate the AT1R and are considered a factor in PE. The plasma level of AT1R autoantibodies (AT-1AA) rises at the time the symptoms of PE develop and subsides within 6 weeks after delivery. AT-1AA activity may promote Ca2+ signaling and VSM contraction. Also, the AT-1AA induced mobilization of intracellular Ca2+ initiates a signaling cascade that leads to activation of NF-κB and activator protein-1 and increased tissue factor expression which in turn cause vasoconstriction and lead to HTN (126).
TNF-α stimulates AngII production in the female reproductive tract [127] and IL-6 upregulates the expression of AT1R in VSM cells [128]. Studies have also suggested an interaction between AngII and ET-1, such that AngII induces the production of ET-1 [129]. While AngII is often thought of as a vasoconstrictor mainly by activating AT1R in VSM, it also stimulates AT2R in ECs and thereby promotes the release of various endothelium-derived relaxing factors. AngII causes the release of NO through activation of eNOS [121]. In vascular tissues AngII also stimulates synthesis of prostanoid compounds, including PGI2, which act in opposition to AngII-induced vasoconstriction [130]. It has been shown that AngII infusion in mice does not cause any changes in EDHF-mediated vascular relaxation [131]. However, administration of an ACE inhibitor or an AT1R antagonist results in improved EDHF-mediated vascular relaxation in rats [132,133].
Thromboxane A2
TXA2 is a potent vasoconstrictor and platelet aggregator released from the platelets, and was thought to play a major role in the pathogenesis of PE. TXB2 metabolites, markers of TXA2 synthesis, were found to be higher in PE compared to Norm-Preg women [134]. Platelet TXA2 synthesis may be particularly increased in severe PE, suggesting that TXA2 might be used to determine the severity of PE. Experimental studies suggested that enhanced production of TXA2 may not play a major role in mediating the HTN and renal vasoconstriction produced by chronic RUPP in pregnant rats [135]. Some clinical studies suggested the use of a low dose of aspirin as an approach to prevent PE [136]; however, a randomized controlled clinical trial on women at high risk for PE showed no benefit of low dose aspirin when used as a preventive measure of the disease [137].
Vascular Smooth Muscle Mediators
VSM Ca2+
[Ca2+]i is a major determinant of vascular tone. During VSM activation, increased [Ca2+]i is initiated by IP3-induced Ca2+ release and ryanodine-sensitive Ca2+-induced Ca2+ release mechanisms. Vasoconstrictor agonists also stimulate Ca2+ entry through ligand-gated and voltage-gated Ca2+ channels. Relaxing factors released from ECs act on VSM and inhibit phospholipase C, open K+ channels or stimulate [Ca2+]i extrusion, and thereby decrease [Ca2+]i. EC dysfunction is associated with decreased release of relaxing factors, and decreased VSM Ca2+ extrusion mechanisms. EC dysfunction also causes an increase in VSM contracting factors, which stimulate Ca2+ mobilization from the intracellular stores and Ca2+ entry from the extracellular space. VSM contraction in response to hypoxia is also mediated by increased [Ca2+]i [138]. It is generally thought that vasoconstrictors may play a role in PE. However, studies on women myometrial and subcutaneous resistance vessels have shown that microvessel reactivity to high KCl depolarizing solution, phenylephrine (PHE] and AngII were not increased in PE compared with Norm-Preg women, and suggested that this is an unlikely mechanism of the increased peripheral vascular resistance in PE [139]. Studies on arterial VSM cells of rats have shown that basal and agonist-stimulated [Ca2+]i are reduced in Norm-Preg compared with virgin rats, but significantly elevated in pregnant rats treated with L-NAME (51). [Ca2+]i may also play a role in the changes in VSM contraction in RUPP rats. In renal arterial VSM, enhanced mechanisms of Ca2+ entry, rather than Ca2+ release from the intracellular stores, may be a contributing factor to the increased VSM contraction and [Ca2+]i in response to AngII and KCl in RUPP rats as compared to Norm-Preg rats (52).
VSM Protein Kinase C (PKC)
PKC plays a role in VSM contraction. Activation of PKC by phorbol esters causes sustained contraction of VSM with no detectable change in [Ca2+]i, suggesting that PKC increases the Ca2+ sensitivity of the contractile proteins. Studies on uterine artery from pregnant sheep and the aorta of late pregnant rats have demonstrated that the decreased vascular contraction during Norm-Preg is associated with a decrease in vascular PKC activity [140,141]. Studies have also shown that the expression and subcellular redistribution of the Ca2+-dependent α-PKC and the Ca2+-independent δ- and ζ-PKC are reduced in aortic VSM isolated from late pregnant compared with nonpregnant rats [141,142].
Increased VSM PKC expression/activity has been identified in HTN (143,144). Also, the Ca2+ sensitivity of VSM contractile elements is increased in women with PE, suggesting that VSM PKC activity may be involved in the vascular changes observed in PE (145). PKC may also play a role in the changes in AngII and AT1R-mediated signaling associated with PE. Studies on cultured neonatal rat cardiomyocytes have shown that IgG in plasma from PE women enhances AT1R-mediated chronotropic response, whereas IgG from control subjects has no effect. Treatment of cardiomyocytes with the PKC inhibitor calphostin C prevented the stimulatory effect of IgG from PE rats. Also, examination of VSM cells with confocal microscopy have shown colocalization of purified IgG from PE women and AT1R antibody. These studies concluded that PE women develop stimulatory AT-1AA, a process that appears to be mediated via PKC [146]. We have recently shown that PHE-induced contraction is increased in aortic segments from L-NAME treated compared to non-treated pregnant rats [101]. Also, the expression, subcellular redistribution and activity of α- and δ-PKC isoforms are enhanced in L-NAME treated compared with Norm-Preg rats, suggesting a role of α- and δ-PKC in the increased vasoconstriction and vascular resistance observed in HTN-Preg [141,142].
VSM Rho/Rho-Kinase
Rho is a family of small GTP-binding proteins that are involved in many cellular functions including cell proliferation, migration, cytoskeletal reorganization and contraction. Rho-kinase is activated by GTP binding and is inactivated by hydrolyzing GTP to GDP, and this process is influenced by various stimuli including growth factors and vasoactive substances [147]. The importance of Rho-kinase in PE is supported by a study comparing subcutaneous resistance arteries from PE, Norm-Preg and nonpregnant women, and showing that Ca2+ sensitivity of the contractile elements is increased in women with PE [145]. Also, stimulation of AT1R induces upregulation of RhoA/Rho-kinase activity in hypertensive rats, and an increase in AngII activity during PE may activate Rho-kinase and promote vasoconstriction [148]. Other studies have shown that the expression of Rho-kinase mRNA may be downregulated in umbilical arteries of PE women [149].
VSM MAP kinase (MAPK)
MAPKs are serine/thrionine-specific protein kinases that respond to extracellular stimuli and regulate various cellular activities such as gene expression, mitosis, differentiation, and apoptosis [150]. MAPK activities have been identified in differentiated VSM, suggesting a role in VSM contraction [151]. Tyrosine phosphorylation of MAPK targets it to the contractile myofilaments, where it phosphorylates the actin-binding protein caldesmon [152,153]. The phosphorylation of caldesmon increases the actin-myosin crossbridge cycling and enhances VSM contraction [151]. MAPK promotes vascular remodeling in many pathological conditions [154] and may be involved in PE. A decrease in VSM PKC expression during Norm-Preg and potential increase in HTN-Preg likely change the MAPK-caldesmon phosphorylation pathway and thereby VSM contraction.
Effects of Vascular Mediators on Bioactive Factors
The relationship between biologically active factors and vascular mediators is complex. In many cases the increases in vascular mediators may cause stimulation or inhibition of bioactive factors (Fig. 2B). For example, AngII, through stimulation of AT1R, stimulates the expression of TNF-α, IL-6, and IL-1β mRNA and increases the plasma levels of IL-6 and IL-1β [155]. Also, AngII stimulates ROS production through an ET-1 mediated pathway [116]. AngII stimulates VEGF production through HIF-1α activation in VSM cells [156], and may also inhibit MMP-1 activity [157]. ET-1 is also an important factor in the regulation of vascular MMPs. Increases in plasma and EC ET-1 are associated with increases in expression and activity of vascular MMPs [158]. Also, ET-1, via activation of ETAR, stimulates the synthesis and release of VEGF in VSM cells, and both ET-1 and VEGF play a synergistic role in neovascularization [159]. ET-1 also stimulates NADPH oxidase, the main vascular source of O2•− [129], while PGI2 attenuates ROS production by reducing NADPH oxidase activity [83].
Management of HTN-Preg
Currently, the management of mild and moderate HTN-Preg consists of bed rest and anti-hypertensive drugs. Methyldopa, hydralazine, β-blockers and nifedipine are the preferred antihypertensives because they are safe and have few side effects on the mother and fetus [160]. The antihypertensive drugs mainly decrease the risk of developing acute HTN, but may not have a clear effect on fetal mortality, premature birth, or fetal growth restriction. If PE worsens into eclampsia, maintenance of airway patency and prevention of fluid aspiration should be the first measure, then anticonvulsants should be given, with magnesium sulfate infusion being the drug of choice.
The current management of HTN-Preg is mainly symptomatic, and the most effective way to reverse the manifestations is induction of prompt delivery. Future approaches for management of HTN-Preg should focus on preventing the disease rather than treating the symptoms. Some of the potential approaches include methods to minimize placental hypoxia. Other possible approaches include cytokine antagonists, antioxidants, and blockers of the release or the effects of vasoconstrictors such as ET-1. Control of dietary and environmental factors could also reduce the incidence of PE in susceptible women. Advanced gene analysis and manipulation may also provide an alternative gene therapy approach and a more specific measure to prevent PE.
In summary, genetic predisposition, dietary and environmental factors affect the normal development of the placenta and may lead to placental ischemia/hypoxia, which in turn causes an increase in the release of biologically active factors such as growth factors, cytokines, ROS, HIF-1 and antibodies to vasoconstrictor receptors. These bioactive factors in turn decrease the vasodilator mediators and increase the vasoconstrictive substances leading to severe vasoconstriction, increased systemic vascular resistance and HTN-Preg. Understanding the cellular mechanisms of PE should provide more specific and efficient measures for treatment and prevention of this disorder.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998 and HL-70659), and The Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD-60702).
List of Abbreviations
- AngII
angiotensin II
- BP
blood pressure
- [Ca2+]i
intracellular free Ca2+ concentration
- cGMP
cyclic guanosine monophosphate
- COX
cyclooxygenase
- EC
endothelial cell
- ECM
extracellular matrix
- EDHF
endothelium-derived hyperpolarizing factor
- Eng
endoglin
- ET-1
endothelin-1
- HIF
hypoxia-inducible transcription factor
- HTN-Preg
hypertension in pregnancy
- IL
interleukin
- L-NAME
Nω-nitro-L-arginine methyl ester
- MMP
matrix metalloproteinase
- 2-methoxy-E2
2-methoxy-estradiol
- MAPK
mitogen-activated protein kinase
- MLC
myosin light chain
- NO
nitric oxide
- NOS
NO synthase
- Norm-Preg
normal pregnant
- O2−•
superoxide anion
- PE
preeclampsia
- PGI2
prostacyclin
- PHE
phenylephrine
- PKC
protein kinase C
- RAS
renin-angiotensin system
- ROS
reactive oxygen species
- RUPP
reduced uterine perfusion pressure
- sFlt-1
soluble fms-like tyrosine kinase-1
- TNF-α
tumor necrosis factor-α
- TXA2
thromboxane A2
- VEGF
vascular endothelial growth factor
- VSM
vascular smooth muscle
References
- 1.Clausen T, Slott M, Solvoll K, Drevon CA, Vollset SE, Henriksen T. High intake of energy, sucrose, and polyunsaturated fatty acids is associated with increased risk of preeclampsia. Am J Obstet Gynecol. 2001;185:451–458. doi: 10.1067/mob.2001.116687. [DOI] [PubMed] [Google Scholar]
- 2.Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46(6):1243–1249. doi: 10.1161/01.HYP.0000188408.49896.c5. [DOI] [PubMed] [Google Scholar]
- 3.Villar J, Carroli G, Wojdyla D, Abalos E, Giordano D, Ba’aqeel H, Farnot U, Bergsjø P, Bakketeig L, Lumbiganon P, Campodónico L, Al-Mazrou Y, Lindheimer M, Kramer M World Health Organization Antenatal Care Trial Research Group. Preeclampsia, gestational hypertension and intrauterine growth restriction, related or independent conditions? Am J Obstet Gynecol. 2006;194(4):921–931. doi: 10.1016/j.ajog.2005.10.813. [DOI] [PubMed] [Google Scholar]
- 4.Stennett AK, Khalil RA. Neurovascular mechanisms of hypertension in pregnancy. Curr Neurovasc Res. 2006;3(2):131–148. doi: 10.2174/156720206776875885. [DOI] [PubMed] [Google Scholar]
- 5.Sholook MM, Gilbert JS, Sedeek MH, Huang M, Hester RL, Granger JP. Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am J Physiol Heart Circ Physiol. 2007;293(4):H2080–H2084. doi: 10.1152/ajpheart.00667.2007. [DOI] [PubMed] [Google Scholar]
- 6.Skjaerven R, Vatten LR, Wilcox AJ, Ronning T, Irgens LM, Lie RT. Recurrence of pre-eclampsia across generations: Exploring fetal and maternal genetic components in a population based cohort. Obstetrical & Gynecological Survey. 2006;61(3):162–163. doi: 10.1136/bmj.38555.462685.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross JC. The genetics of pre-eclampsia: a feto-placental or maternal problem? Clin Genet. 2003;64:96–103. doi: 10.1034/j.1399-0004.2003.00127.x. [DOI] [PubMed] [Google Scholar]
- 8.Winn VD, Gormley M, Paquet AC, Kjaer-Sorensen K, Kramer A, Rumer KK, Haimov-Kochman R, Yeh RF, Overgaard MT, Varki A, Oxvig C, Fisher SJ. Severe preeclampsia-related changes in gene expression at the maternal-fetal interface include sialic acid-binding immunoglobulin-like lectin-6 and pappalysin-2. Endocrinology. 2009;150(1):452–62. doi: 10.1210/en.2008-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobashi G. Genetic and environmental factors associated with the development of hypertension in pregnancy. J Epidemiol. 2006;16:1–8. doi: 10.2188/jea.16.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg TJ, Garbers S, Lipkind H, Chiasson MA. Maternal obesity and diabetes as risk factors for adverse pregnancy outcomes: differences among 4 racial/ethnic groups. Am J Public Health. 2005;95(9):1545–1551. doi: 10.2105/AJPH.2005.065680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray J, Diamond P, Singh G, Bell C. Brief overview of maternal triglycerides as a risk factor for pre-eclampsia. BJOG. 2006;113:379–386. doi: 10.1111/j.1471-0528.2006.00889.x. [DOI] [PubMed] [Google Scholar]
- 12.Thomas M, Weisman SM. Calcium supplementation during pregnancy and lactation: effects on the mother and the fetus. Am J Obstet Gynecol. 2006;194(4):937–945. doi: 10.1016/j.ajog.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 13.Prada JA, Tsang RC, Clark KE. Hypocalcemia and pregnancy-induced hypertension produced by low-calcium diet. Hypertension. 1994;23(6):695–702. doi: 10.1161/01.hyp.23.6.695. [DOI] [PubMed] [Google Scholar]
- 14.Oken E, Ning Y, Rifas-Shiman SL, Rich-Edwards JW, Olsen SF, Gillman MW. Diet during pregnancy and risk of preeclampsia or gestational hypertension. Ann Epidemiol. 2007;17(9):663–668. doi: 10.1016/j.annepidem.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villar J, Abdel-Aleem H, Merialdi M, Mathai M, Ali MM, Zavaleta N, Purwar M, Hofmeyr J, Nguyen TN, Campodónico L, Landoulsi S, Carroli G, Lindheimer M World Health Organization Calcium Supplementation for the Prevention of Preeclampsia Trial Group. World Health Organization randomized trial of calcium supplementation among low calcium intake pregnant women. Am J Obstet Gynecol. 2006;194(3):639–649. doi: 10.1016/j.ajog.2006.01.068. [DOI] [PubMed] [Google Scholar]
- 16.Bodnar LM, Catov JM, Simhan HN, Holick MF, Powers RW, Roberts JM. Maternal vitamin D deficiency increases the risk of preeclampsia. J Clin Endocrinol Metab. 2007;92(9):3517–3522. doi: 10.1210/jc.2007-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts JM, Balk JL, Bodnar LM, Belizan JM, Bergel E, Martinez A. Nutrition as a preventive strategy against adverse pregnancy outcomes. J Nutr. 2003;133:1684S–1692S. doi: 10.1093/jn/133.5.1684S. [DOI] [PubMed] [Google Scholar]
- 19.Frederick IO, Williams MA, Dashow E, Kestin M, Zhang C, Leisenring WM. Dietary fiber, potassium, magnesium and calcium in relation to the risk of preeclampsia. J Reprod Med. 2005;50:332–344. [PubMed] [Google Scholar]
- 20.Holmes VA, McCance DR. Could antioxidant supplementation prevent pre-eclampsia? Proc Nutr Soc. 2005;64:491–501. doi: 10.1079/pns2005469. [DOI] [PubMed] [Google Scholar]
- 21.Chen XK, Yang Q, Smith G, Krewski D, Walker M, Wen SW. Environmental lead level and pregnancy-induced hypertension. Environ Res. 2006;100:424–430. doi: 10.1016/j.envres.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Vigeh M, Yokoyama K, Mazaheri M, Beheshti S, Ghazizadeh S, Sakai T, Morita Y, Kitamura F, Araki S. Relationship between increased blood lead and pregnancy hypertension in women without occupational lead exposure in Tehran. Iran Arch Environ Health. 2004;59(2):70–75. doi: 10.3200/AEOH.59.2.70-75. [DOI] [PubMed] [Google Scholar]
- 23.Fisher SJ. The placental problem: linking abnormal cytotrophoblast differentiation to the maternal symptoms of preeclampsia. Reprod Biol Endocrinol. 2004;2:53. doi: 10.1186/1477-7827-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMaster MT, Zhou Y, Fisher SJ. Abnormal placentation and the syndrome of preeclampsia. Semin Nephrol. 2004;24(6):540–547. doi: 10.1016/s0270-9295(04)00124-x. [DOI] [PubMed] [Google Scholar]
- 25.Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R1–R10. doi: 10.1152/ajpregu.00417.2005. [DOI] [PubMed] [Google Scholar]
- 26.Ojeda NB, Grigore D, Alexander BT. Intrauterine growth restriction: fetal programming of hypertension and kidney disease. Adv Chronic Kidney Dis. 2008;15(2):101–106. doi: 10.1053/j.ackd.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;294(2):H541–H550. doi: 10.1152/ajpheart.01113.2007. [DOI] [PubMed] [Google Scholar]
- 28.Shaarawy M, Al-Sokkary F, Sheba M, Wahba O, Kandil HO, Abdel-Mohsen I. Angiogenin and vascular endothelial growth factor in pregnancies complicated by preeclampsia. Int J Gynaecol Obstet. 2005;88:112–117. doi: 10.1016/j.ijgo.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Stepan H, Geide A, Faber R. Soluble fms-like tyrosine kinase 1. N Engl J Med. 2004;351(21):2241–2242. doi: 10.1056/NEJM200411183512123. [DOI] [PubMed] [Google Scholar]
- 30.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, Karumanchi SA CPEP Study Group. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355(10):992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi M, Ueda Y, Yamaguchi T, Sohma R, Shibazaki M, Ohkura T, Inaba N. Tumor Necrosis Factor-α in the placenta is not elevated in the pre-eclamptic patients despite its elevation in peripheral blood. Am J Reprod Immunol. 2005;53:113–119. doi: 10.1111/j.1600-0897.2005.00253.x. [DOI] [PubMed] [Google Scholar]
- 32.Casart YC, Tarrazzi K, Camejo MI. Serum levels of interleukin-6, interleukin-1beta and human chorionic gonadotropin in pre-eclamptic and normal pregnancy. Gynecol Endocrinol. 2007;23(5):300–303. doi: 10.1080/09513590701327638. [DOI] [PubMed] [Google Scholar]
- 33.Rajakumar A, Whitelock KA, Weissfeld LA, Daftary AR, Markovic N, Conrad KP. Selective overexpression of the hypoxia-inducible transcription factor, HIF-2alpha, in placentas from women with preeclampsia. Biol Reprod. 2001;64:499–506. doi: 10.1093/biolreprod/64.2.499. [DOI] [PubMed] [Google Scholar]
- 34.Tsukimori K, Fukushima K, Tsushima A, Nakano H. Generation of reactive oxygen species by neutrophils and endothelial cell injury in normal and preeclamptic pregnancies. Hypertension. 2005;46:696–700. doi: 10.1161/01.HYP.0000184197.11226.71. [DOI] [PubMed] [Google Scholar]
- 35.Narumiya H, Zhang Y, Fernandez-Patron C, Guilbert LJ, Davidge ST. Matrix metalloproteinase-2 is elevated in the plasma of women with preeclampsia. Hypertens Pregnancy. 2001;20(2):185–194. doi: 10.1081/PRG-100106968. [DOI] [PubMed] [Google Scholar]
- 36.Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, Strauss JF, Kalluri R. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature. 2008;453(7198):1117–1121. doi: 10.1038/nature06951. [DOI] [PubMed] [Google Scholar]
- 37.Ranta V, Viinikka L, Halmesmäki E, Ylikorkala O. Nitric oxide production with preeclampsia. Obstet Gynecol. 1999;93(3):442–445. doi: 10.1016/s0029-7844(98)00465-7. [DOI] [PubMed] [Google Scholar]
- 38.Crews JK, Herrington JN, Granger JP, Khalil RA. Decreased endothelium-dependent vascular relaxation during reduction of uterine perfusion pressure in pregnant rat. Hypertension. 2000;35:367–372. doi: 10.1161/01.hyp.35.1.367. [DOI] [PubMed] [Google Scholar]
- 39.Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. Differential mechanisms of endothelium-dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre-eclampsia. Clin Sci (Lond) 2002;103:67–73. doi: 10.1042/cs1030067. [DOI] [PubMed] [Google Scholar]
- 40.Clark BA, Halvorson L, Sachs B, Epstein FH. Plasma endothelin levels in preeclampsia: elevation and correlation with uric acid levels and renal impairment. Am J Obstet Gynecol. 1992;166(3):962–968. doi: 10.1016/0002-9378(92)91372-h. [DOI] [PubMed] [Google Scholar]
- 41.Brosnihan KB, Neves LA, Anton L, Joyner J, Valdes G, Merrill DC. Enhanced expression of Ang-(1–7) during pregnancy. Braz J Med Biol Res. 2004;37:1255–1262. doi: 10.1590/s0100-879x2004000800017. [DOI] [PubMed] [Google Scholar]
- 42.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension. 2007;50(6):1142–1147. doi: 10.1161/HYPERTENSIONAHA.107.096594. [DOI] [PubMed] [Google Scholar]
- 44.Purwosunu Y, Sekizawa A, Farina A, Wibowo N, Koide K, Okazaki S, Nakamura M, Okai T. Evaluation of physiological alterations of the placenta through analysis of cell-free messenger ribonucleic acid concentrations of angiogenic factors. Am J Obstet Gynecol. 2008;198(1):124.e1–7. doi: 10.1016/j.ajog.2007.06.079. [DOI] [PubMed] [Google Scholar]
- 45.LaMarca BB, Bennett WA, Alexander BT, Cockrell K, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-alpha. Hypertension. 2005;46(4):1022–1025. doi: 10.1161/01.HYP.0000175476.26719.36. [DOI] [PubMed] [Google Scholar]
- 46.LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep. 2007;9(6):480–485. doi: 10.1007/s11906-007-0088-1. [DOI] [PubMed] [Google Scholar]
- 47.Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension. 2001;37(4):1191–1195. doi: 10.1161/01.hyp.37.4.1191. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Walsh S, Guo J, Zhang J. The imbalance between thromboxane and prostacyclin in preeclampsia. Curr Hypertens. 1991;165:1695–1700. doi: 10.1016/0002-9378(91)90017-l. [DOI] [PubMed] [Google Scholar]
- 49.LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension. 2005;46(1):82–86. doi: 10.1161/01.HYP.0000169152.59854.36. [DOI] [PubMed] [Google Scholar]
- 50.Lamarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor-alpha in pregnant rats. Hypertension. 2008;52(6):1168–1172. doi: 10.1161/HYPERTENSIONAHA.108.120576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy JG, Fleming JB, Cockrell KL, Granger JP, Khalil RA. [Ca2+]i signaling in renal arterial smooth muscle cells of pregnant rat is enhanced during inhibition of NOS. Am J Physiol Regul Integr Comp Physiol. 2001;280(1):R87–R99. doi: 10.1152/ajpregu.2001.280.1.R87. [DOI] [PubMed] [Google Scholar]
- 52.Murphy JG, Herrington JN, Granger JP, Khalil RA. Enhanced [Ca2+]i in renal arterial smooth muscle cells of pregnant rats with reduced uterine perfusion pressure. Am J Physiol Heart Circ Physiol. 2003;284(1):H393–H403. doi: 10.1152/ajpheart.00247.2002. [DOI] [PubMed] [Google Scholar]
- 53.Sargent IL, Germain SJ, Sacks GP, Kumar S, Redman CW. Trophoblast deportation and the maternal inflammatory response in pre-eclampsia. J Reprod Immunol. 2003;59(2):153–160. doi: 10.1016/s0165-0378(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 54.Kertesz Z, Hurst G, Ward M, Willis AC, Caro H, Linton EA, Sargent IL, Redman CW. Purification and characterization of a complex from placental syncytiotrophoblast microvillous membranes which inhibits the proliferation of human umbilical vein endothelial cells. Placenta. 1999;20(1):71–79. doi: 10.1053/plac.1998.0351. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Lewis DF, Gu Y, Zhang Y, Alexander JS, Granger DN. Placental trophoblast-derived factors diminish endothelial barrier function. J Clin Endocrinol Metab. 2004;89(5):2421–2428. doi: 10.1210/jc.2003-031707. [DOI] [PubMed] [Google Scholar]
- 56.Van Wijk MJ, Boer K, Nisell H, Smarason AK, Van Bavel E, Kublickiene KR. Endothelial function in myometrial resistance arteries of normal pregnant women perfused with syncytiotrophoblast microvillous membranes. Br J Obstet Gynecol. 2001;108:967–972. doi: 10.1111/j.1471-0528.2001.00221.x. [DOI] [PubMed] [Google Scholar]
- 57.Hayman R, Warren A, Johnson I, Baker P. The preliminary characterization of a vasoactive circulating factor(s) in preeclampsia. Am J Obstet Gynecol. 2001;184:1196–1203. doi: 10.1067/mob.2001.113130. [DOI] [PubMed] [Google Scholar]
- 58.Brownbill P, Mills TA, Soydemir DF, Sibley CP. Vasoactivity to and endogenous release of vascular endothelial growth factor in the in vitro perfused human placental lobule from pregnancies complicated by preeclampsia. Placenta. 2008;29(11):950–955. doi: 10.1016/j.placenta.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 59.Munaut C, Lorquet S, Pequeux C, Blacher S, Berndt S, Frankenne F, Foidart JM. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum Reprod. 2008;23(6):1407–1415. doi: 10.1093/humrep/den114. [DOI] [PubMed] [Google Scholar]
- 60.Akercan F, Cirpan T, Terek MC, Ozcakir HT, Giray G, Sagol S, Karadadas N. The immunohistochemical evaluation of VEGF in placenta biopsies of pregnancies complicated by preeclampsia. Arch Gynecol Obstet. 2008;277(2):109–114. doi: 10.1007/s00404-007-0430-5. [DOI] [PubMed] [Google Scholar]
- 61.Bosio PM, Wheeler T, Anthony F, Conroy R, O’herlihy C, McKenna P. Maternal plasma vascular endothelial growth factor concentrations in normal and hypertensive pregnancies and their relationship to peripheral vascular resistance. Am J Obstet Gynecol. 2001;184:146–152. doi: 10.1067/mob.2001.108342. [DOI] [PubMed] [Google Scholar]
- 62.VanWijk MJ, Kublickiene K, Boer K, VanBavel E. Vascular function in preeclampsia. Cardiovasc Res. 2000;47(1):38–48. doi: 10.1016/s0008-6363(00)00087-0. [DOI] [PubMed] [Google Scholar]
- 63.Serraf A, Aznag H, Baudet B, Détruit H, Séccatore F, Mazmanian MG, Planché C. Pulmonary vascular endothelial growth factor and nitric oxide interaction during total cardiopulmonary bypass in neonatal pigs. J Thorac Cardiovasc Surg. 2003;125:1050–1057. doi: 10.1067/mtc.2003.402. [DOI] [PubMed] [Google Scholar]
- 64.Woolcock J, Hennessy A, Xu B, Thornton C, Tooher J, Makris A, Ogle R. Soluble Flt-1 as a diagnostic marker of pre-eclampsia. Aust N Z J Obstet Gynaecol. 2008;48(1):64–70. doi: 10.1111/j.1479-828X.2007.00804.x. [DOI] [PubMed] [Google Scholar]
- 65.Boyd PA, Lindenbaum RH, Redman C. Pre-eclampsia and trisomy 13: a possible association. Lancet. 1987;2:425–427. doi: 10.1016/s0140-6736(87)90960-3. [DOI] [PubMed] [Google Scholar]
- 66.Tuohy JF, James DK. Pre-eclampsia and trisomy 13. Br J Obstet Gynaecol. 1992;99:891–894. doi: 10.1111/j.1471-0528.1992.tb14436.x. [DOI] [PubMed] [Google Scholar]
- 67.Hirashima C, Ohkuchi A, Matsubara S, Suzuki H, Takahashi K, Usui R, Suzuki M. Alteration of serum soluble endoglin levels after the onset of preeclampsia is more pronounced in women with early-onset. Hypertens Res. 2008;31(8):1541–1548. doi: 10.1291/hypres.31.1541. [DOI] [PubMed] [Google Scholar]
- 68.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;2:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 69.ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11(1):79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 70.Conrad KP, Miles TM, Benyo DF. Circulating levels of immunoreactive cytokines in women with preeclampsia. Am J Reprod Immunol. 1998;40:102–111. doi: 10.1111/j.1600-0897.1998.tb00398.x. [DOI] [PubMed] [Google Scholar]
- 71.Hashii K, Fujiwara H, Yoshioka S, Kataoka N, Yamada S, Hirano T, Mori T, Fujii S, Maeda M. Peripheral blood mononuclear cells stimulate progesterone production by luteal cells derived from pregnant and non-pregnant women: possible involvement of interleukin-4 and interleukin-10 in corpus luteum function and differentiation. Hum Reprod. 1998;13(1O):2738–2744. doi: 10.1093/humrep/13.10.2738. [DOI] [PubMed] [Google Scholar]
- 72.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension. 2006;48(4):711–716. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 73.Alexander BT, Cockrell KL, Massey MB, Bennett WA, Granger JP. Tumor necrosis factor-α-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens. 2002;15:12–19. doi: 10.1016/s0895-7061(01)02255-5. [DOI] [PubMed] [Google Scholar]
- 74.Davis JR, Giardina JB, Green GM, Alexander BT, Granger JP, Khalil RA. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-α-induced hypertension in pregnant rats. Am J Physiol Reg Int Comp Physiol. 2002;282:R390–R399. doi: 10.1152/ajpregu.00270.2001. [DOI] [PubMed] [Google Scholar]
- 75.Orshal JM, Khalil RA. Reduced endothelial NO-cGMP-mediated vascular relaxation and hypertension in IL-6-infused pregnant rats. Hypertension. 2004;43:434–444. doi: 10.1161/01.HYP.0000113044.46326.98. [DOI] [PubMed] [Google Scholar]
- 76.Sedeek M, Gilbert JS, LaMarca BB, Sholook M, Chandler DL, Wang Y, Granger JP. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am J Hypertens. 2008;21(10):1152–1156. doi: 10.1038/ajh.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sato H, Sato M, Kanai H, Uchiyama T, Iso T, Ohyama Y, Sakamoto H, Tamura J, Nagai R, Kurabayashi M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc Res. 2005;67(4):714–722. doi: 10.1016/j.cardiores.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 78.Tyurin VA, Liu SX, Tyurina YY, Sussman NB, Hubel CA, Roberts JM, Taylor RN, Kagan VE. Elevated levels of S-nitrosoalbumin in preeclampsia plasma. Circ Res. 2001;88(11):1210–1215. doi: 10.1161/hh1101.092179. [DOI] [PubMed] [Google Scholar]
- 79.Chambers JC, Fusi L, Malik IS, Haskard DO, De Swiet M, Kooner JS. Association of maternal endothelial dysfunction with preeclampsia. JAMA. 2001;285:1607–1612. doi: 10.1001/jama.285.12.1607. [DOI] [PubMed] [Google Scholar]
- 80.Roberts JM, Speer P. Antioxidant therapy to prevent preeclampsia. Semin Nephrol. 2004;24:557–64. doi: 10.1016/s0270-9295(04)00126-3. [DOI] [PubMed] [Google Scholar]
- 81.Parra M, Rodrigo R, Barja P, Bosco C, Fernández V, Muñoz H, Soto-Chacón E. Screening test for preeclampsia through assessment of uteroplacental blood flow and biochemical markers of oxidative stress and endothelial dysfunction. Am J Obstet Gynecol. 2005;193(4):1486–1491. doi: 10.1016/j.ajog.2005.02.109. [DOI] [PubMed] [Google Scholar]
- 82.Egi K, Conrad NE, Kwan J, Schulze C, Schulz R, Wildhirt SM. Inhibition of inducible nitric oxide synthase and superoxide production reduces matrix metalloproteinase-9 activity and restores coronary vasomotor function in rat cardiac allografts. Eur J Cardiothorac Surg. 2004;26:262–269. doi: 10.1016/j.ejcts.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 83.Chang TC, Huang CJ, Tam K, Chen SF, Tan KT, Tsai MS, Lin TN, Shyue SK. Stabilization of hypoxia-inducible factor-1{alpha} by prostacyclin under prolonged hypoxia via reducing reactive oxygen species level in endothelial cells. J Biol Chem. 2005;280(44):36567–36574. doi: 10.1074/jbc.M504280200. [DOI] [PubMed] [Google Scholar]
- 84.Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol. 2004;287:H1495–H1500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- 85.Semenza GL, Agani F, Iyer N, Kotch L, Laughner E, Leung S, Yu A. Regulation of cardiovascular development and physiology by hypoxia-inducible factor 1. Ann N Y Acad Sci. 1999;874:262–8. doi: 10.1111/j.1749-6632.1999.tb09241.x. [DOI] [PubMed] [Google Scholar]
- 86.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276(16):12645–53. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 87.Kelly BA, Bond BC, Poston L. Aortic adaptation to pregnancy: elevated expression of matrix metalloproteinases-2 and -3 in rat gestation. Mol Hum Reprod. 2004;10:331–337. doi: 10.1093/humrep/gah045. [DOI] [PubMed] [Google Scholar]
- 88.Galewska Z, Bañkowski E, Romanowicz L, Jaworski S. Pre-eclampsia (EPH-gestosis)-induced decrease of MMP-s content in the umbilical cord artery. Clin Chim Acta. 2003;335(1–2):109–115. doi: 10.1016/s0009-8981(03)00296-1. [DOI] [PubMed] [Google Scholar]
- 89.Chew DKW, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–1010. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Olszewski B, Rosebury W, Wang D, Robertson A, Keiser JA. Impaired angiogenesis in SHR is associated with decreased KDR and MT1-MMP expression. Biochem Biophys Res Commun. 2004;315:363–368. doi: 10.1016/j.bbrc.2004.01.059. [DOI] [PubMed] [Google Scholar]
- 91.Faller DV. Endothelial cell responses to hypoxic stress. Clin Exp Pharmacol Physiol. 1999;26:74–84. doi: 10.1046/j.1440-1681.1999.02992.x. [DOI] [PubMed] [Google Scholar]
- 92.Anumba DO, Robson SC, Boys RJ, Ford GA. Nitric oxide activity in the peripheral vasculature during normotensive and preeclamptic pregnancy. Am J Physiol. 1999;277:H848–H854. doi: 10.1152/ajpheart.1999.277.2.H848. [DOI] [PubMed] [Google Scholar]
- 93.Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ Res. 2000;87:406–411. doi: 10.1161/01.res.87.5.406. [DOI] [PubMed] [Google Scholar]
- 94.Conrad KP, Kerchner LJ, Mosher MD. Plasma and 24-h NO(x) and cGMP during normal pregnancy and preeclampsia in women on a reduced NO(x) diet. Am J Physiol. 1999;277:F48–F57. doi: 10.1152/ajprenal.1999.277.1.F48. [DOI] [PubMed] [Google Scholar]
- 95.Mutlu-Turkoglu U, Aykac-Toker G, Ibrahimoglu L, Ademoglu E, Uysal M. Plasma nitric oxide metabolites and lipid peroxide levels in preeclamptic pregnant women before and after delivery. Gynecol Obstet Invest. 1999;48:247–250. doi: 10.1159/000010192. [DOI] [PubMed] [Google Scholar]
- 96.Schiessl B, Strasburger C, Bidlingmaier M, Mylonas I, Jeschke U, Kainer F, Friese K. Plasma- and urine concentrations of nitrite/nitrate and cyclic guanosinemonophosphate in intrauterine growth restricted and preeclamptic pregnancies. Arch Gynecol Obstet. 2006;274(3):150–154. doi: 10.1007/s00404-006-0149-8. [DOI] [PubMed] [Google Scholar]
- 97.Conrad KP, Davis AK. Nitric oxide synthase activity in placentae from women with pre-eclampsia. Placenta. 1995;16:691–699. doi: 10.1016/0143-4004(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 98.Alexander BT, Miller MT, Kassab S, Novak J, Reckelhoff JF, Kruckeberg WC, Granger JP. Differential expression of renal nitric oxide synthase isoforms during pregnancy in rats. Hypertension. 1999;33(1 Pt 2):435–439. doi: 10.1161/01.hyp.33.1.435. [DOI] [PubMed] [Google Scholar]
- 99.Roberts JM. Endothelial dysfunction in preeclampsia. Semin Reprod Endocrinol. 1998;16:5–15. doi: 10.1055/s-2007-1016248. [DOI] [PubMed] [Google Scholar]
- 100.Molnar M, Suto T, Toth T, Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol. 1994;170:1458–1466. doi: 10.1016/s0002-9378(94)70179-2. [DOI] [PubMed] [Google Scholar]
- 101.Khalil RA, Crews JK, Novak J, Kassab S, Granger JP. Enhanced vascular reactivity during inhibition of nitric oxide synthesis in pregnant rats. Hypertension. 1998;31:1065–1069. doi: 10.1161/01.hyp.31.5.1065. [DOI] [PubMed] [Google Scholar]
- 102.Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol. 2003;284:L316–L323. doi: 10.1152/ajplung.00228.2002. [DOI] [PubMed] [Google Scholar]
- 103.Suzuki Y, Hattori T, Kajikuri J, Yamamoto T, Suzumori K, Itoh T. Reduced function of endothelial prostacyclin in human omental resistance arteries in pre-eclampsia. J Physiol. 2002;545:269–277. doi: 10.1113/jphysiol.2002.022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bowen RS, Zhang Y, Gu Y, Lewis DF, Wang Y. Increased phospholipase A2 and thromboxane but not prostacyclin production by placental trophoblast cells from normal and preeclamptic pregnancies cultured under hypoxia condition. Placenta. 2005;26(5):402–409. doi: 10.1016/j.placenta.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 105.Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111(6):796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- 106.Fulep EE, Vedernikov YP, Saade GR, Garfield RE. The role of endothelium-derived hyperpolarizing factor in the regulation of the uterine circulation in pregnant rats. Am J Obstet Gynecol. 2001;185:638–642. doi: 10.1067/mob.2001.117665. [DOI] [PubMed] [Google Scholar]
- 107.Petersson J, Zygmunt PM, Jonsson P, Hogestatt ED. Characterization of endothelium- dependent relaxation in guinea pig basilar artery - effect of hypoxia and role of cytochrome P450 mono-oxygenase. J Vasc Res. 1998;35:285–294. doi: 10.1159/000025595. [DOI] [PubMed] [Google Scholar]
- 108.Resta TC, Walker BR. Chronic hypoxia selectively augments endothelium-dependent pulmonary arterial vasodilation. Am J Physiol. 1996;270:H888–H896. doi: 10.1152/ajpheart.1996.270.3.H888. [DOI] [PubMed] [Google Scholar]
- 109.Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. Differential mechanisms of endothelium-dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre-eclampsia. Clin Sci (Lond) 2002;103:67–73. doi: 10.1042/cs1030067. [DOI] [PubMed] [Google Scholar]
- 110.Luksha L, Nisell H, Luksha N, Kublickas M, Hultenby K, Kublickiene K. Endothelium-derived hyperpolarizing factor in preeclampsia: heterogeneous contribution, mechanisms, and morphological prerequisites. Am J Physiol Regul Integr Comp Physiol. 2008;294(2):R510–R519. doi: 10.1152/ajpregu.00458.2007. [DOI] [PubMed] [Google Scholar]
- 111.Niwano K, Arai M, Tomaru K, Uchiyama T, Ohyama Y, Kurabayashi M. Transcriptional stimulation of the eNOS gene by the stable prostacyclin analogue beraprost is mediated through cAMP-responsive element in vascular endothelial cells: close link between PGI2 signal and NO pathways. Circ Res. 2003;93(6):523–30. doi: 10.1161/01.RES.0000091336.55487.F7. [DOI] [PubMed] [Google Scholar]
- 112.Orie NN, Fry CH, Clapp LH. Evidence that inward rectifier K+ channels mediate relaxation by the PGI2 receptor agonist cicaprost via a cyclic AMP-independent mechanism. Cardiovasc Res. 2006;69:107–115. doi: 10.1016/j.cardiores.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 113.Bernardi F, Constantino L, Machado R, Petronilho F, Dal-Pizzol F. Plasma nitric oxide, endothelin-1, arginase and superoxide dismutase in pre-eclamptic women. J Obstet Gynaecol Res. 2008;34(6):957–963. doi: 10.1111/j.1447-0756.2008.00860.x. [DOI] [PubMed] [Google Scholar]
- 114.Kourembanas S, McQuillan LP, Leung GK, Faller DV. Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J Clin Invest. 1993;92:99–104. doi: 10.1172/JCI116604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taylor RN, Varma M, Teng NN, Roberts JM. Women with preeclampsia have higher plasma endothelin levels than women with normal pregnancies. J Clin Endocrinol Metab. 1990;71:1675–1677. doi: 10.1210/jcem-71-6-1675. [DOI] [PubMed] [Google Scholar]
- 116.Yao L, Kobori H, Rahman M, Seth DM, Shokoji T, Fan Y, Zhang GX, Kimura S, Abe Y, Nishiyama A. Olmesartan improves endothelin-induced hypertension and oxidative stress in rats. Hypertens Res. 2004;27(7):493–500. doi: 10.1291/hypres.27.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Giardina JB, Green GM, Rinewalt AN, Granger JP, Khalil RA. Role of endothelin B receptors in enhancing endothelium-dependent nitric oxide-mediated vascular relaxation during high salt diet. Hypertension. 2001;37:516–23. doi: 10.1161/01.hyp.37.2.516. [DOI] [PubMed] [Google Scholar]
- 118.Conrad KP, Gandley RE, Ogawa T, Nakanishi S, Danielson LA. Endothelin mediates renal vasodilation and hyperfiltration during pregnancy in chronically instrumented conscious rats. Am J Physiol. 1999;276:F767–F776. doi: 10.1152/ajprenal.1999.276.5.F767. [DOI] [PubMed] [Google Scholar]
- 119.Gandley RE, Conrad KP, McLaughlin MK. Endothelin and nitric oxide mediate reduced myogenic reactivity of small renal arteries from pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1–R7. doi: 10.1152/ajpregu.2001.280.1.R1. [DOI] [PubMed] [Google Scholar]
- 120.Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L284–L290. doi: 10.1152/ajplung.00449.2004. [DOI] [PubMed] [Google Scholar]
- 121.Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Med Sci Monit. 2005;11:RA194–RA205. [PubMed] [Google Scholar]
- 122.Chassagne C, Eddahibi S, Adamy C, Rideau D, Marotte F, Dubois-Randé JL, Adnot S, Samuel JL, Teiger E. Modulation of angiotensin II receptor expression during development and regression of hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol. 2000;22(3):323–332. doi: 10.1165/ajrcmb.22.3.3701. [DOI] [PubMed] [Google Scholar]
- 123.Merrill DC, Karoly M, Chen K, Ferrario CM, Brosnihan KB. Angiotensin-(1–7) in normal and preeclamptic pregnancy. Endocrine. 2002;18:239–245. doi: 10.1385/ENDO:18:3:239. [DOI] [PubMed] [Google Scholar]
- 124.Wesselman JP, De Mey JG. Angiotensin and cytoskeletal proteins: role in vascular remodeling. Curr Hypertens Rep. 2002;4:63–70. doi: 10.1007/s11906-002-0055-9. [DOI] [PubMed] [Google Scholar]
- 125.Mizutani S, Tomoda Y. Effects of placental proteases on maternal and fetal blood pressure in normal pregnancy and preeclampsia. Am J Hypertens. 1996;9(6):591–597. doi: 10.1016/0895-7061(96)00016-7. [DOI] [PubMed] [Google Scholar]
- 126.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101(20):2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 127.Wijayagunawardane MP, Gabler C, Killian G, Miyamoto A. Tumor necrosis factor alpha in the bovine oviduct during the estrous cycle: messenger RNA expression and effect on secretion of prostaglandins, endothelin-1, and angiotensin II. Biol Reprod. 2003;69:1341–1346. doi: 10.1095/biolreprod.103.017327. [DOI] [PubMed] [Google Scholar]
- 128.Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Böhm M, Nickenig G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. 2004;94(4):534–541. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- 129.Cingolani HE, Villa-Abrille MC, Cornelli M, Nolly A, Ennis IL, Garciarena C, Suburo AM, Torbidoni V, Correa MV, Camiliónde Hurtado MC, Aiello EA. The positive inotropic effect of angiotensin II: role of endothelin-1 and reactive oxygen species. Hypertension. 2006;47(4):727–734. doi: 10.1161/01.HYP.0000208302.62399.68. [DOI] [PubMed] [Google Scholar]
- 130.Askari B, Ferrerib NR. Regulation of prostacyclin synthesis by angiotensin II and TNF-α in vascular smooth muscle. Prostaglandins. 2001;63:175–187. doi: 10.1016/s0090-6980(01)00098-3. [DOI] [PubMed] [Google Scholar]
- 131.Wang D, Chabrashvili T, Borrego L, Aslam S, Umans JG. Angiotensin II infusion alters vascular function in mouse resistance vessels: roles of superoxide and endothelium. J Vasc Res. 2006;43:109–119. doi: 10.1159/000089969. [DOI] [PubMed] [Google Scholar]
- 132.Goto K, Fujii K, Kansui Y, Iida M. Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: Response to chronic treatment with renin-angiotensin system inhibitors. Clin Exp Pharmacol Physiol. 2004;31:650–655. doi: 10.1111/j.1440-1681.2004.04054.x. [DOI] [PubMed] [Google Scholar]
- 133.Deja MA, Gołba KS, Widenka K, Mrozek R, Biernat J, Kolowca M, Malinowski M, Woś S. Angiotensin-converting enzyme inhibitors reveal non-NO-, non-prostacycline-mediated endothelium-dependent relaxation in internal thoracic artery of hypertensive patients. Int J Cardiol. 2005;102(3):455–460. doi: 10.1016/j.ijcard.2004.05.050. [DOI] [PubMed] [Google Scholar]
- 134.Wang Y, Walsh SW, Kay HH. Placental lipid peroxides and thromboxane are increased and prostacyclin is decreased in women with preeclampsia. Am J Obstet Gynecol. 1999;167:946–949. doi: 10.1016/s0002-9378(12)80017-2. [DOI] [PubMed] [Google Scholar]
- 135.Llinás MT, Alexander BT, Seedek M, Abram SR, Crell A, Granger JP. Enhanced thromboxane synthesis during chronic reductions in uterine perfusion pressure in pregnant rats. Am J Hypertens. 2002;15(9):793–797. doi: 10.1016/s0895-7061(02)02975-8. [DOI] [PubMed] [Google Scholar]
- 136.Coomarasamy A, Papaioannou S, Gee H, Khan KS. Aspirin for the prevention of preeclampsia in women with abnormal uterine artery doppler: a meta-analysis. Obstet Gynecol. 2001;98:861–866. doi: 10.1016/s0029-7844(01)01569-1. [DOI] [PubMed] [Google Scholar]
- 137.Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol. 2003;101(6):1319–1332. doi: 10.1016/s0029-7844(03)00169-8. [DOI] [PubMed] [Google Scholar]
- 138.de Berrazueta JR. The role of calcium in the regulation of normal vascular tone and in arterial hypertension. Rev Esp Cardiol. 1999;52(3):25–33. [PubMed] [Google Scholar]
- 139.Wimalasundera RC, Thom SA, Regan L, Hughes AD. Effects of vasoactive agents on intracellular calcium and force in myometrial and subcutaneous resistance arteries isolated from preeclamptic, pregnant, and nonpregnant woman. Am J Obstet Gynecol. 2005;192(2):625–632. doi: 10.1016/j.ajog.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 140.Magness RR, Rosenfeld CR, Carr BR. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am J Physiol. 1991;260(3):E464–E470. doi: 10.1152/ajpendo.1991.260.3.E464. [DOI] [PubMed] [Google Scholar]
- 141.Kanashiro CA, Cockrell KL, Alexander BT, Granger JP, Khalil RA. Pregnancy-associated reduction in vascular protein kinase C activity rebounds during inhibition of NO synthesis. Am J Physiol Regul Integr Comp Physiol. 2000;278(2):R295–R303. doi: 10.1152/ajpregu.2000.278.2.R295. [DOI] [PubMed] [Google Scholar]
- 142.Kanashiro CA, Alexander BT, Granger JP, Khalil RA. Ca2+-insensitive vascular protein kinase C during pregnancy and NOS inhibition. Hypertension. 1999;34(4 Pt 2):924–930. doi: 10.1161/01.hyp.34.4.924. [DOI] [PubMed] [Google Scholar]
- 143.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76(4):967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- 144.Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005;70(11):1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.VanWijk MJ, Boer K, van der Meulen ET, Bleker OP, Spaan JA, VanBavel E. Resistance artery smooth muscle function in pregnancy and preeclampsia. Am J Obstet Gynecol. 2002;186(1):148–154. doi: 10.1067/mob.2002.119184. [DOI] [PubMed] [Google Scholar]
- 146.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hu E. Recent patents on Rho signaling pathway as therapeutic target for cardiovascular diseases. Recent Patents Cardiovasc Drug Discov. 2006;1(3):249–263. doi: 10.2174/157489006778777034. [DOI] [PubMed] [Google Scholar]
- 148.Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, Kitamoto S, Usui M, Kaibuchi K, Shimokawa H, Takeshita A. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. 2002;39(2):245–250. doi: 10.1161/hy0202.103271. [DOI] [PubMed] [Google Scholar]
- 149.Friel AM, Sexton DJ, O’reilly MW, Smith TJ, Morrison JJ. Rho A/Rho kinase: human umbilical artery mRNA expression in normal and pre eclamptic pregnancies and functional role in isoprostane-induced vasoconstriction. Reproduction. 2006;132(1):169–76. doi: 10.1530/rep.1.01088. [DOI] [PubMed] [Google Scholar]
- 150.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 151.Khalil RA, Menice CB, Wang CL, Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ Res. 1995;76(6):1101–1108. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- 152.D’Angelo G, Graceffa P, Wang CA, Wrangle J, Adam LP. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J Biol Chem. 1999;274(42):30115–30121. doi: 10.1074/jbc.274.42.30115. [DOI] [PubMed] [Google Scholar]
- 153.Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am J Physiol Cell Physiol. 2000;278(4):C718–C726. doi: 10.1152/ajpcell.2000.278.4.C718. [DOI] [PubMed] [Google Scholar]
- 154.Kyaw M, Yoshizumi M, Tsuchiya K, Izawa Y, Kanematsu Y, Tamaki T. Atheroprotective effects of antioxidants through inhibition of mitogen-activated protein kinases. Acta Pharmacol Sin. 2004;25(8):977–985. [PubMed] [Google Scholar]
- 155.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290(3):H935–H940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 156.Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension. 2004;44(3):264–270. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- 157.Brilla CG, Zhou G, Rupp H, Maisch B, Weber KT. Role of angiotensin II and prostaglandin E2 in regulating cardiac fibroblast collagen turnover. Am J Cardiol. 1995;76(13):8D–13D. doi: 10.1016/s0002-9149(99)80485-8. [DOI] [PubMed] [Google Scholar]
- 158.Ergul A, Portik-Dobos V, Giulumian AD, Molero MM, Fuchs LC. Stress upregulates arterial matrix metalloproteinase expression and activity via endothelin A receptor activation. Am J Physiol Heart Circ Physiol. 2003;285(5):H2225–H2232. doi: 10.1152/ajpheart.00133.2003. [DOI] [PubMed] [Google Scholar]
- 159.Salani D, Taraboletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157(5):1703–1711. doi: 10.1016/S0002-9440(10)64807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abalos E, Duley L, Steyn DW, Henderson-Smart DJ. Antihypertensive drug therapy for mild to moderate hypertension during pregnancy. Cochrane Database Syst Rev. 2007;(1):CD002252. doi: 10.1002/14651858.CD002252.pub2. [DOI] [PubMed] [Google Scholar]