

Abstract
Background
Reduced cardiac output is traditionally believed to be the main determinant of worsening renal function (WRF) in advanced decompensated heart failure (ADHF).
Objective
To determine if venous congestion, rather than impairment of cardiac output, is primarily associated with the development of WRF in ADHF.
Methods
A total of 145 consecutive patients admitted with ADHF treated with intensive medical therapy guided by pulmonary artery catheter were studied. WRF was defined as an increase of serum creatinine ≥0.3 mg/dl during hospitalization.
Results
In the study cohort (age 57 ±14 years, cardiac index 1.9 ±0.6 l/kg.m2, LVEF 20 ±8%, serum creatinine 1.7 ±0.9 mg/dl), 58 patients (40%) developed WRF. Patients who developed WRF had a higher central venous pressure on admission (CVP, 18 ±7 versus 12 ±6 mmHg, p<0.001) and after intensive medical therapy (11 ±8 versus 8 ±5 mmHg, p=0.04). The development of WRF occurred less frequently in patients that achieved a CVP <8 mmHg (p=0.01). Furthermore, the ability of CVP to stratify risk for development of WRF was apparent across the spectrum of systemic blood pressure, pulmonary capillary wedge pressure, cardiac index, and estimated glomerular filtration rates.
Conclusions
Venous congestion is the most important hemodynamic factor driving WRF in decompensated patients with advanced heart failure.
Keywords: worsening renal function, venous congestion, cardiac index, decompensated heart failure
INTRODUCTION
The pathophysiology of the cardio-renal interaction in the setting of advanced decompensated heart failure (ADHF) is poorly understood. It is commonly observed that coexisting renal dysfunction may complicate the treatment course of heart failure, and the use of intravenous loop diuretics often alleviate congestion at the cost of worsening renal function (WRF) (1,2). WRF during treatment of ADHF typically occurs within days of hospitalization and is a strong independent predictor of adverse outcomes (3,4,5).
Traditionally, WRF has been attributed to hypoperfusion of the kidney due to progressive impairment of cardiac output or intravascular volume depletion secondary to overzealous use of diuretics (6). Although the majority of patients hospitalized with ADHF also present with increased central or peripheral congestion, the presence of venous congestion has been considered a secondary phenomenon due to the “backward failure” caused by impaired cardiac output. Nevertheless, experimental animal data as far back as the 1930’s have demonstrated that temporary isolated elevation of central venous pressure (CVP) can be transmitted back to the renal veins, resulting in direct impairment of renal function (7,8). However, human data regarding the differential contributions of venous congestion and cardiac output in the development of WRF during ADHF are lacking.
The primary aim of this study is to test the hypothesis that WRF is more dependent on venous congestion rather than on impairment of cardiac output in patients admitted with ADHF. The secondary aim is to investigate if effective reduction of CVP with intensive medical therapy can prevent the development of WRF.
METHODS
Subject Population
We enrolled consecutive subjects, ≥18 year, with ADHF including New York Heart Association class III–IV symptoms, who underwent intensive medical therapy guided by pulmonary artery catheter at the Cleveland Clinic in a dedicated heart failure intensive care unit between January 1, 2006 and June 30, 2007. Subjects who met the following inclusion criteria at the time of admission were enrolled in the study: 1) left ventricular ejection fraction < 30%; 2) cardiac index (CI) ≤2.4 l/min/m2; and 3) pulmonary capillary wedge pressure (PCWP) > 18 mmHg and/or central venous pressure (CVP) > 8 mmHg. Exclusion criteria included: 1) mechanical ventilation; 2) renal replacement therapy; 3) intravenous inotropic support on admission; 4) congenital heart disease; 5) recipients of heart transplantation. Institutional Review Board approval of this research project was obtained, and informed consent was obtained for hospitalization, treatment and documented in the medical records, according to protocol and Cleveland Clinic policy.
Intensive Medical Therapy
The hemodynamic goals and pharmacologic approach of intravenous therapy in the dedicated heart failure intensive care unit have been previously described (9). Briefly, optimal hemodynamic response was defined as a decrease in PCWP to ≤ 18 mmHg, decrease in CVP to ≤ 8 mmHg, and improvement in CI to ≥ 2.4 l/min/m2, all while maintaining mean arterial pressure > 65–70 mmHg. In order to achieve these hemodynamic goals, all subjects were treated according to protocols developed in our intensive care unit with intravenous or oral loop diuretics in combination with intravenous vasodilators (and/or inotropic agents), while continuing and optimizing evidence-based therapies (angiotensin-converting enzyme inhibitors, beta-blockers and spironolactone) as tolerated.
Data Collection and Renal Assessment
Two experienced heart failure cardiologists manually collected hemodynamic data, demographic characteristics, treatment, and echocardiographical data. Sequential serum creatinine and blood urea nitrogen values were recorded on admission and daily throughout the hospitalization period including the day of discharge. We defined a strict definition on the development of WRF as an increase in serum creatinine of ≥ 0.3 mg/dl during hospitalization, consistent with several previous investigations (4,5,10). It takes into account any significant renal deterioration during the treatment period in the setting of low cardiac output and congestion as defined by the inclusion criteria. Glomerular filtration rate (GFR) in ml/min was estimated daily using the four-variable Modification of Diet in Renal Disease equation (11). Normal or mild renal insufficiency was defined as GFR ≥60 ml/min/1.73m2. Moderate renal insufficiency was defined as GFR 30–59 ml/min/1.73m2 and severe renal insufficiency as GFR <30 ml/min/1.73m2.
Hemodynamic Assessment
Complete hemodynamic assessment was collected in all subjects before the start of intensive medical therapy, and again before removing the pulmonary artery catheter. The CVP and PCWP were assessed at end-expiration with a balloon-tipped catheter at steady state with the subject in a supine position. CI was determined by calculation using the Fick equation through sampling of a mixed central venous blood gas taken in the pulmonary artery while assuming standard metabolic rates. The systemic blood pressure was measured non-invasively by an automatic cuff sphygmomanometer. Renal perfusion pressure on admission was assessed as the difference between mean arterial pressure and central venous pressure.
Statistical Analysis
All data were expressed as mean ±standard deviation for continuous data (median and inter-quartile range [IQR] for non-parametric data), and as a ratio for categorical data. Univariate comparisons of these variables were performed between baseline and follow-up variables and between subjects who developed WRF versus those who did not. A paired and unpaired t-test for continuous data and chi-square, Pearson’s correlation and Fisher’s exact test for categorical data was used for appropriate comparisons. The predictive value of CVP and CI as continuous variables to predict WRF was assessed using a receiver operating characteristic curve analysis. Separate c-statistics for CVP and CI from logistic regression models were calculated and a set of 300 bootstrapped (with replacement) samples were generated to compute the difference and standard error. The difference between the c-statistics was bias corrected, and a one-sample t-test was performed to determine if the difference was equal to zero. Stepwise multivariate linear regression analysis was used to determine the independent relationships between hemodynamic variables, baseline renal function and hemoglobin with WRF. Statistical significance was set at a two-tailed probability level < 0.05. Statistical analyses were performed using SPSS for Windows, release 13.0 (SPSS Inc., Chicago IL) and SAS version 8.2 (Cary, NC). The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
RESULTS
Subject characteristics
A total of 145 subjects, mean LVEF 20 ±8%, were included in this observational prospective study. Patient characteristics on admission are listed in Table 1, which were comparable between subjects who developed WRF versus those who did not (except for serum creatinine, blood urea nitrogen, and hemoglobin at admission). The percentage of patients with moderate to severe right ventricular dysfunction (60%) was similar between the two groups. Plasma B-type natriuretic peptide measurements on admission were available in 40% of subjects, and were comparable between patients with and without incident WRF (median [IQR]: 1,100 [497, 1,921] versus 874 [333, 1,430] pg/ml, p=ns).
Table 1.
Baseline patient characteristics and medication use.
| Patients with worsening renal function (n=58) | Patients without worsening renal function (n=87) | p value | |
|---|---|---|---|
| Age | 59 ±14 | 56 ±14 | ns |
| NYHA class III/IV (%) | 9/91 | 10/90 | ns |
| Ischemic Etiology (%) | 54 | 50 | ns |
| Body mass index (kg/m2) | 28 ±3 | 29 ±4 | ns |
| Male Gender (%) | 74 | 73 | ns |
| Caucasian Race (%) | 78 | 76 | ns |
| Medical history (%) | |||
| Smoking history | 49 | 51 | ns |
| Diabetes | 44 | 34 | ns |
| Hypertension | 48 | 40 | ns |
| Hyperlipidemia | 60 | 59 | ns |
| ICD/CRT-D | 38/29 | 42/27 | ns |
| Labs on admission | |||
| Hemoglobin (g/dl) | 11.5 ±.2.5 | 13.0 ±1.5 | 0.05 |
| Creatinine (mg/dl) | 1.9 ±0.9 | 1.5 ±0.8 | 0.007 |
| GFR (ml/min/1.73.m2) | 48 ±19 | 56 ±25 | 0.05 |
| BUN (mg/dl) | 58 ±25 | 36 ±20 | <0.001 |
| Sodium (mmol/l) | 134 ±6 | 134 ±5 | ns |
| BNP (pg/ml) | 1,559 ±1340 | 1,157 ±1073 | ns |
| Oral Medication on Admission (%) | |||
| Aspirin/coumadin | 44 | 48 | ns |
| ACE-inhibitor/ARB | 49 | 50 | ns |
| Digoxin | 38 | 43 | ns |
| Beta-blockers | 56 | 62 | ns |
| Spironolactone | 27 | 47 | 0.03 |
| Loop diuretics | 80 | 86 | ns |
| Hydralazine | 23 | 17 | ns |
| Isosorbide dinitrate | 27 | 22 | ns |
| Statin | 57 | 54 | ns |
| Amiodarone | 22 | 19 | ns |
| Medication During PAC-Guided Therapy (%) | |||
| IV or PO furosemide | 85 | 86 | ns |
| IV vasodilators | 51 | 56 | ns |
| Milrinone | 34 | 30 | ns |
| Dobutamine | 30 | 27 | ns |
NYHA indicates New York Heart Association functional class, ICD: implantable cardiac defibrillator, CRT-D: cardiac resynchronization therapy with defibrillator, GFR: estimated glomerular filtration rate, BUN: blood urea nitrogen, ACE: angiotensin converting enzyme, ARB: angiotensin receptor blocker. Values are mean ± SD or n (%).
The mean time to develop WRF was 1.0 ±1.5 day. Mean duration of pulmonary artery catheter guided therapy was 3.5 ±1.5 days, and mean total length of stay was 9 ±9 days, similar between those with or without incident WRF. On admission, 19% of the study population had severe renal insufficiency, 45% had moderate renal insufficiency, and 36% had normal or mild renal insufficiency. Overall, 53% of patients who developed WRF during admission demonstrated serum creatinine level at discharge to be less than the peak serum creatinine level
A statistically significant correlation was observed between baseline CI and baseline renal function expressed by serum creatinine (r = 0.32, p=0.001) or GFR (r = −0.3, p=0.002). However, there was no correlation between baseline CI and baseline CVP. Finally, no correlation between baseline CVP and baseline renal function could be found.
Incidence and renal predictors of worsening renal function
Overall, 58 subjects (40%) developed WRF during their hospitalization, predominantly within the first 5 days of hospitalization. The development of WRF was associated with a higher peak of serum creatinine (2.5 ±1.1 versus 1.5 ±0.8 mg/dL, p<0.001) during hospitalization. Subjects who developed WRF were more likely to have severe renal insufficiency at baseline (p=0.05), and had higher serum creatinine both at baseline (1.9 ±0.9 versus 1.5±0.8 mg/dL, p=0.007) and at discharge (2.2 ±1.1 versus 1.4 ±0.7 mg/dL, p<0.001).
Impact of medication on development of worsening renal function
Subjects who developed WRF versus those who did not had comparable baseline medication use on admission, with the exception of lower spironolactone utilization (Table 1). Overall, no statistically significant differences in medication use during pulmonary artery catheter guided therapy were observed. Mean dose of furosemide during intensive medical therapy guided by pulmonary artery catheter was similar among patients who developed WRF or not (117±130 mg/day and 116±81 mg/day, p=ns). Half of the patients in both groups received furosemide through continuous parental infusion.
Baseline hemodynamic predictors of incident worsening renal function
Table 2 illustrates the baseline hemodynamic measurements, stratified by the presence or absence of incident WRF. All subjects showed signs of impaired hemodynamics with impaired CI and elevated right- and left-sided filling pressures at baseline. Heart rate, systolic arterial blood pressure, PCWP, and systolic pulmonary artery pressure at baseline were comparable (p=ns) between the two cohorts and were not predictive for WRF.
Table 2.
Hemodynamic variables on admission and time of pulmonary artery catheter removal in all patients and stratified according to those who developed worsening renal function (n=58) and those who did not (n=87).
| All patients (n=145) |
Patients with worsening renal function (n=58) |
Patients without worsening renal function (n=87) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | p-value | Baseline | Follow-up | p-value | Baseline | Follow-up | p-value | |
| HR (bpm) | 88 ±40 | 89 ±18 | ns | 86 ±22′ | 90 ±16′ | Ns | 89 ±46 | 88 ±19 | ns |
| SBP (mmHg) | 109 ±18 | 109 ±18 | ns | 111 ±21′ | 110 ±25′ | Ns | 108 ±15 | 109 ±15 | ns |
| CVP (mmHg) | 14 ±7 | 9 ±6 | < 0.001 | 18 ±7* | 11 ±8** | < 0.001 | 12 ±6 | 8 ±5 | < 0.001 |
| SPA (mmHg) | 55 ±15 | 46 ±7 | < 0.001 | 57 ±13′ | 49 ±15′ | < 0.001 | 54 ±16 | 46 ±12 | < 0.001 |
| PCWP (mmHg) | 24 ±7 | 18 ±5 | < 0.001 | 25 ±7′ | 19 ±5′ | < 0.001 | 24 ±7 | 18 ±5 | < 0.001 |
| CI (l/min/m2) | 1.9 ±0.6 | 2.5 ±0.6 | < 0.001 | 2.0±0.8† | 2.7±0.7‡ | < 0.001 | 1.8 ±0.4 | 2.4 ±0.5 | < 0.001 |
HR: heart rate, SBP: systolic arterial blood pressure, CVP: central venous pressure, SPA: systolic pulmonary artery pressure, PCWP: pulmonary capillary wedge pressure, CI: cardiac index.
‘p = ns,
p < 0.001,
p = 0.01,
p = 0.008,
p = 0.01 between patients who did and did not develop worsening renal function at same moment in time.
There was an incremental risk in WRF with increasing categories of baseline CVP, with 75% of subjects presenting with a baseline CVP >24 mmHg developing WRF (Figure 1). Furthermore, the mean baseline CVP was statistically higher in subjects who developed WRF versus those with did not (18 ±7 versus 12 ±6 mmHg, p<0.001). In addition, a significant correlation between admission CVP and severity of WRF was found (r = 0.4, p<0.0001). Estimated renal perfusion pressure on admission was similar among patients who did and did not develop WRF (63±15 vs 65±12 mmHg, p = 0.2).
Figure 1. Prevalence of worsening renal function during hospitalization according to categories of admission central venous pressure, cardiac index, systolic blood pressure and pulmonary capillary wedge pressure.
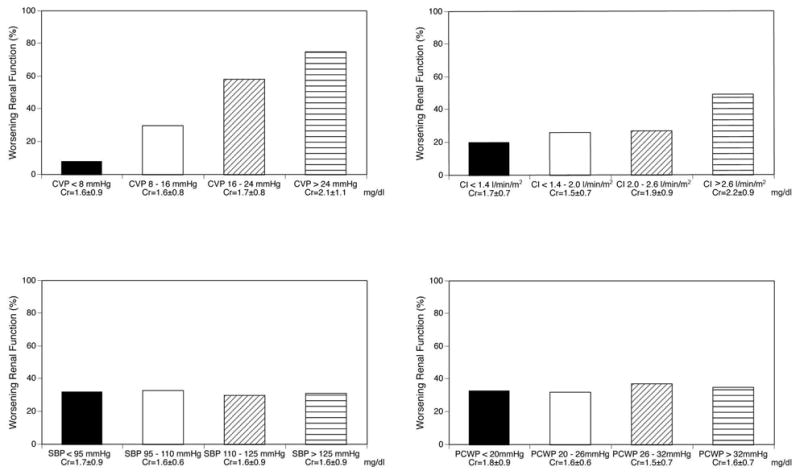
Abbreviations: CVP = central venous pressure, Cr = serum creatinine, CI = cardiac index, SBP = systolic blood pressure, PCWP = pulmonary capillary wedge pressure.
The mean baseline CI was significantly higher (rather than lower) in subjects who developed WRF versus those who did not (2.0 ±0.8 versus 1.8 ±0.4 l/min/m2, p=0.008). However, the pattern of change in GFR during hospitalization was similar between those with CI above and below mean admission CI, indicating that changes in GFR were not related to baseline CI. In addition, using ROC curve analysis, we observed that baseline CVP (0.734, p < 0.0001) but not baseline CI (0.552, p = 0.6) predicted the development of WRF (Figure 2, difference p = 0.012). In a separate ROC analysis (not shown), baseline CVP remained a predictor of WRF when patients were categorized according to the presence or absence of diabetes mellitus, hypertension or significant baseline renal dysfunction. Finally, another sub-analysis was performed in patients without severe renal insufficiency (GFR >30 ml/min/1.73.m2). In this subset, patients who developed WRF still had higher admission CVP (17 ±4 versus 12 ±5 mmHg, p=0.007) but similar admission CI (1.9 ±0.4 versus 1.8 ±0.5 l/min/m2, p=ns).
Figure 2.
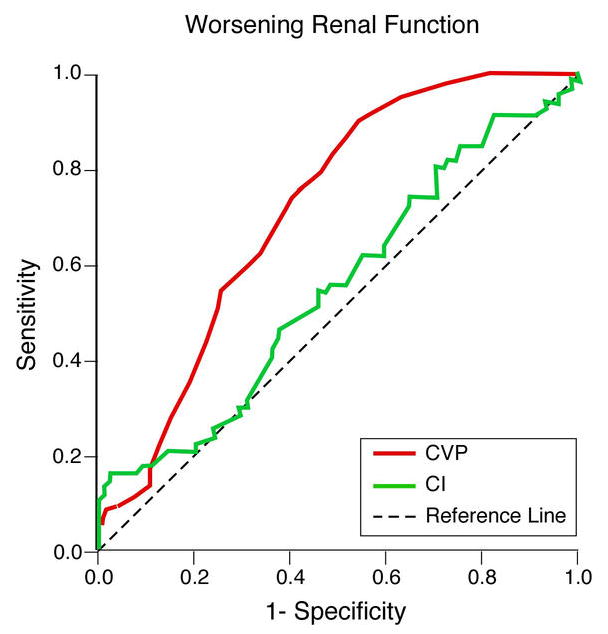
ROC curves for central venous pressure (CVP) and cardiac index (CI) on admission for worsening renal function development.
Impact of hemodynamic changes on incident worsening renal function
Table 2 also compares the hemodynamic measurements from baseline to follow-up, stratified by the presence or absence of incident WRF. All hemodynamic alterations demonstrated significant improvements following intensive medical therapy as expected (all p<0.001). Heart rate, systolic arterial blood pressure, PCWP, and systolic pulmonary artery pressure at the time of pulmonary artery catheter removal remained comparable (p=ns) between the two cohorts.
Follow-up hemodynamic predictors of incident worsening renal function
At follow-up, the mean CI remained significantly higher (2.7 ±0.7 versus 2.4 ±0.5 l/min/m2, p=0.01) and the CVP significantly higher (11 ±8 versus 8 ±5 mmHg, p=0.04) in subjects who developed WRF versus those who did not. In particular, a persistently elevated CVP >8 mmHg at the time of PAC removal was associated with greater incidence of WRF (51% versus 18 %, p=0.01). Overall discharge CVP also correlated with the severity of WRF (r = 0.3, p=0.007). Finally, discharge CVP rather than discharge CI was associated with renal impairment (lower GFR) as illustrated in Figure 3.
Figure 3. Relative contributions of central venous pressure (CVP) and cardiac index (CI) to glomerular filtration rate (GFR) at time of pulmonary artery catheter removal.
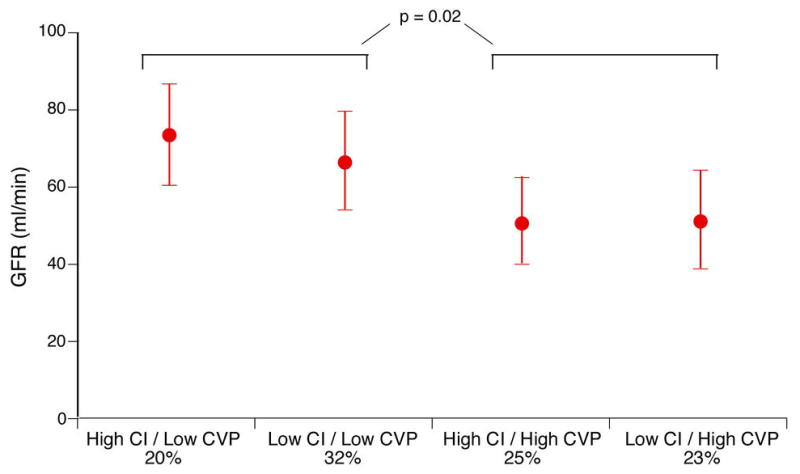
Error bars represent 95% confidence interval. Cut-off values for CI = 2.4 l/min.m2 and CVP = 8 mmHg.
The ability of CVP on admission (p=0.01) or at time of PAC removal (p=0.03) to stratify risk to develop WRF was apparent across the spectrum of heart rate, PCWP, systolic blood pressure, systolic pulmonary artery pressure, CI, serum creatinine, and hemoglobin in multivariable analysis.
DISCUSSION
There have been numerous contemporary reports describing the natural history of the development of WRF in the setting of decompensated heart failure. However, the majority lacked careful cardiac and hemodynamic profiling during the clinical course of WRF. Based on early work, WRF is often attributed to hypoperfusion of the kidney due to progressive impairment of cardiac output or intravascular volume depletion secondary to overzealous use of diuretics (6). We observed in our patient population with low-output decompensated heart failure that besides the presence of intrinsic renal insufficiency, venous congestion (both with elevated CVP on admission as well as insufficient reduction of CVP during hospitalization) was the strongest hemodynamic determinant for the development of WRF. In contrast, impaired CI on admission and improvement in CI following intensive medical therapy had limited contribution to WRF. These observations provide important clinical confirmation of experimental data that preservation of cardiac output without relieving venous congestion may not necessarily avert the development of WRF. While many of these findings may seem intuitive to the experienced clinician, the concept of “congestive kidney failure” is of high clinical value with the contemporary epidemic proportions of ADHF where cardiac insufficiency (rather than venous congestion) is often considered the core lesion.
The pathophysiology of WRF in the setting of ADHF is complex and multifactorial. The term “cardio-renal syndrome” is often used to describe progressive renal deterioration with heart failure therapy in an aggressive attempt to relieve congestive signs and symptoms. We chose to use the term “worsening renal function”, as there remains much uncertainty regarding the precise definition of the cardio-renal syndrome. Using a clinical surrogate of rise in serum creatinine levels, previous reports have suggested that WRF occurs in one third of patients admitted with ADHF (4,5,12). We found this incidence to be even higher (approaching 40%) in a “cold and wet” patient population. While the initiation or maintenance of certain classes of drugs like angiotensin-converting enzyme inhibitors and loop diuretics have been linked to WRF, we did not find any difference in their usage at admission or during hospitalization to account for the occurrence of worsening renal function (2,13,14). The lower rates of spironolactone use in those developing WRF is likely due to the relative contraindication of the drug in patients with intrinsic renal diseases.
In patients with severe renal insufficiency at baseline, almost 60% developed WRF. Indeed, the highest quartile of baseline CVP and CI both had the highest mean serum creatinine and corresponding highest rates of WRF. This indicates that the underlying intrinsic kidney disease remains an important determinant of the “reserve” available for the kidneys to relieve congestion and to respond to the insult posed by ADHF and the aggressive diuresis and natriuresis necessary during treatment of ADHF. Naturally, this raises the question as to whether treatment primarily directed with the aim of “renal preservation” should be administered prophylatically, especially in this extraordinary high-risk group.
WRF occurs during the initial days following treatment for ADHF during hospitalization. As a result, the most commonly assumed cause of worsening renal function has been hypoperfusion of the kidney secondary to low-output or hypotension (leading to pre-renal hypoperfusion or impaired renal “preload”) (6). In our patient population, we observed that systemic blood pressures were similar between those with versus without WRF, consistent with previous reports (2). Also during intensive medical therapy, systolic blood pressures were carefully monitored and targeted as drugs were being titrated to prevent overzealous hypotension. Although we did not directly assessed regional renal perfusion, the persistently elevated intracardiac pressures in our patient population (with a mean PCWP in the range of 18–19 mmHg) suggested that the overall vasculature was unlikely to be “under-filled.” In particular, estimated renal perfusion pressures were similar between those with versus without WRF. Clearly, judicious lowering of filling pressures is still of utmost importance to prevent hypoperfusion and pre-renal azotemia, and there are still indicators that careful monitoring can be helpful in vulnerable patients. For example, the ESCAPE trial demonstrated that renal function did not worsen when treatment was directed at lowering invasively measured CVP and PCWP, while it did worsen in the treatment arm guided by clinical assessment alone (15).
Our data also demonstrate that progressive or persistent impairment of cardiac output may not be the primary culprit in the development of WRF during the treatment for ADHF. Patients who developed WRF did not have a lower CI on admission and at discharge when compared to those without WRF. Furthermore, the patterns of change in GFR were similar between those with a different degree of CI impairment, independent of inotropic usage. However, this is not to imply that impairment of CI itself does not contribute to WRF, as we acknowledge that patients with progressive pump failure or cardiogenic shock may progress to renal impairment as a result of impaired organ perfusion or indirectly through “backward failure” and venous congestion. Instead, our data indicate that in the setting of hemodynamic alterations of ADHF on admission and following treatment, the relative contributions of CI may be less apparent than historically assumed. Thus, even in this advanced heart failure population with relatively low-output cardiac failure and marginal blood pressures, routine use of inotropic therapy may not necessarily relieve or prevent WRF.
Our observation suggests that the strongest hemodynamic determinant of development of WRF is the presence of venous congestion as measured by elevated CVP, both on admission and at follow-up. There appears to be a near-linear relationship since if the baseline CVP reached >16 or >24 mmHg, we observed a sharp rise in the incidence of WRF approaching 59% and 75%, respectively. During treatment for ADHF, persistent venous congestion also posed a very high risk for the development of WRF. Clearly, this could simply be interpreted as a “sicker” patient population with more advanced disease states that were reflected by higher CVP. However, common cardiovascular measures of disease severity (including systolic blood pressure, serum sodium, plasma B-type natriuretic peptide, PCWP, systolic pulmonary arterial pressure, and dosage of loop diuretics) were similar between those with versus without WRF.
The concept of venous congestion being transmitted to the renal veins and kidneys leading to renal dysfunction is supported by a substantial amount of literature as early as in the 1930s. In an experimental model that iatrogenically induced hypervolemia, an increase in renal vein pressure directly led to renal insufficiency independent of cardiac output or renal blood flow (7,8). Importantly, this was also shown to be a reversible phenomenon as lowering of renal vein pressure immediately improved urine output and GFR (7,8). Other studies indicated that temporary renal vein compression resulted in reduced sodium excretion, reduced GFR and reduced renal blood flow (16–18). Increased CVP also causes an increase in renal interstitial pressure, which might lead to a hypoxic state of the renal parenchyma similar to the mechanism by which hepatic congestion leads to liver dysfunction in heart failure (19–25). In addition, our group recently provided some mechanistic data to suggest the contributions of raised intra-abdominal pressure caused by visceral edema or ascites in this pathophysiology (26). On the other hand, prolonged increases in plasma volume or CVP will attenuate several vascular reflexes leading to an impaired arterial responsiveness, thereby further impairing the effective renal blood flow (27–32). Increased CVP has also been associated with reduced GFR in patients with primary pulmonary hypertension and relatively preserved cardiac outputs (33). Finally, a recent sub-analysis of the ESCAPE trial also suggested that incident WRF was related to CVP (34).
It is conceivable that in the setting of ADHF, the development of “congestive kidney failure” led by elevated renal venous pressure from venous congestion (increased renal afterload) and increased renal interstitial pressure (intrinsic renal compromise) might be under-appreciated mechanisms by which WRF develops. These findings may therefore help to explain why extra-renal strategies primarily aim to relieve venous congestion (such as ultrafiltration) may be effective in alleviating “congestive kidney failure” in selected cases of heart failure rather than those augmenting cardiac output or forward perfusion. We believe that this is an important conceptual shift with broad implications, implying that the search for future ADHF therapies should focus on strategies that allow safe and optimal reduction of venous congestion to prevent such a devastating complication.
Study Limitations
There are several limitations in our study, including the lack of serial weight assessments and direct measurements on glomerular filtration. There were no direct physiological measurements of renal hemodynamics or regional intravascular volume to fully explain the complex underlying pathophysiology, although CI has been considered a reasonable surrogate for renal blood flow under the circumstances. To analyze CI, a standard resting metabolic rate was assumed but overall CI assessed by Fick was comparable with those assessed by thermodilution. Although differences in hemoglobin concentration might have also contributed to differences in absolute oxygen delivery to the kidney, arterio-venous oxygen differences on admission could be retrieved in 50% of patients, and were found to be similar between patients with and without WRF. The relatively low admission rates of neurohormonal antagonists and difference in spironolactone use were probably secondary to the underlying kidney disease, severity of the heart failure, and the withholding of medications due to intolerance related to their “cold and wet” conditions. Finally, though invasive measurements were used in our protocol, it is not the intention of these data to imply the need for invasive monitoring, but solely to understand the hemodynamic contributors of WRF in ADHF.
CONCLUSION
In our cohort of patients with advanced heart failure admitted for decompensation, WRF is commonly observed despite hemodynamic improvements with intensive medical therapy. Our data imply that apart from intrinsic renal insufficiency, the presence of venous congestion, rather than reduced cardiac output, may be the primary hemodynamic factor driving WRF in this patient population.
Acknowledgments
Dr. Tang is supported in part by the National Institutes of Health, National Center for Research Resources, CTSA 1UL1RR024989, Cleveland, Ohio
ABBREVIATIONS
- ADHF
advanced decompensated heart failure
- WRF
worsening renal function
- CI
cardiac index
- CVP
central venous pressure
- GFR
glomerular filtration rate
- LVEF
left ventricular ejection fraction
- PCWP
pulmonary capillary wedge pressure
Footnotes
Disclosure: There are no financial conflicts of interest and this project did not receive funding support.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111:1487–91. doi: 10.1161/01.CIR.0000159340.93220.E4. [DOI] [PubMed] [Google Scholar]
- 2.Heywood JT. The cardiorenal syndrome: lessons from the ADHERE database and treatment options. Heart Fail Rev. 2004;9:195–201. doi: 10.1007/s10741-005-6129-4. [DOI] [PubMed] [Google Scholar]
- 3.Hillege HL, Girbes AR, de Kam PJ, et al. Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation. 2000;102:203–10. doi: 10.1161/01.cir.102.2.203. [DOI] [PubMed] [Google Scholar]
- 4.Krumholz HM, Chen YT, Vaccarino V, et al. Correlates and impact on outcomes of worsening renal function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85:1110–3. doi: 10.1016/s0002-9149(00)00705-0. [DOI] [PubMed] [Google Scholar]
- 5.Forman DE, Butler J, Wang Y, et al. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61–7. doi: 10.1016/j.jacc.2003.07.031. [DOI] [PubMed] [Google Scholar]
- 6.Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 1990;39:10–24. doi: 10.2165/00003495-199000394-00004. [DOI] [PubMed] [Google Scholar]
- 7.Firth JF, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet. 1988;1:1033–5. doi: 10.1016/s0140-6736(88)91851-x. [DOI] [PubMed] [Google Scholar]
- 8.Winton FR. The influence of venous pressure on the isolated mammalian kidney. J Physiol. 1931;72:49–61. doi: 10.1113/jphysiol.1931.sp002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steimle AE, Stevenson LW, Chelimsky-Fallick C, et al. Sustained hemodynamic efficacy of therapy tailored to reduce filling pressures in survivors with advanced heart failure. Circulation. 1997;19:1165–72. doi: 10.1161/01.cir.96.4.1165. [DOI] [PubMed] [Google Scholar]
- 10.Gottlieb SS, Abraham W, Butler J, et al. The prognostic Importance of Different definitions of Worsening Renal Function in Congestive Heart Failure. J Card Fail. 2002;8:136–41. doi: 10.1054/jcaf.2002.125289. [DOI] [PubMed] [Google Scholar]
- 11.Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–70. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 12.Smith GL, Lichtman JH, Bracken MB, et al. Renal impairment and outcomes in heart failure: systematic review and meta-analysis. J Am Coll Cardiol. 2006;47:1987–97. doi: 10.1016/j.jacc.2005.11.084. [DOI] [PubMed] [Google Scholar]
- 13.Oster JR, Materson BJ. Renal and electrolyte complications of congestive heart failure and effects of treatment with angiotensin-converting enzyme inhibitors. Arch Intern Med. 1992;152:704–10. [PubMed] [Google Scholar]
- 14.Packer M, Lee WH, Medina N, Yushak M, Kessler PD. Functional renal insufficiency during long-term therapy with captopril and enalapril in severe chronic heart failure. Ann Intern Med. 1987;106:346–54. doi: 10.7326/0003-4819-106-3-346. [DOI] [PubMed] [Google Scholar]
- 15.Binanay C, Califf RM, Hasselblad V, et al. ESCAPE Investigators and ESCAPE Study Coordinators. JAMA. 2005;294:1625–33. doi: 10.1001/jama.294.13.1625. [DOI] [PubMed] [Google Scholar]
- 16.Burnett JC, Haas JA, Knox FG. Segmental analysis of sodium reabsorption during renal vein constriction. Am J Physiol. 1982;243:19–22. doi: 10.1152/ajprenal.1982.243.1.F19. [DOI] [PubMed] [Google Scholar]
- 17.Wathen RL, Selkurt EE. Intrarenal regulatory factors of salt excretion during renal venous pressure elevation. Am J Physiol. 1969;216:1517–24. doi: 10.1152/ajplegacy.1969.216.6.1517. [DOI] [PubMed] [Google Scholar]
- 18.Burnett JC, Knox FR. Renal interstitial pressure and sodium excretion during renal vein constriction. Am J Physiol. 1980;238:279–82. doi: 10.1152/ajprenal.1980.238.4.F279. [DOI] [PubMed] [Google Scholar]
- 19.Maxwell MH, Breed ES, Schwartz IL. Renal venous pressure in chronic congestive heart failure. J Clin Invest. 1950;29:342–48. doi: 10.1172/JCI102263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiksen-Olsen MJ, Romero JC. Renal effects of prostaglandin inhibition during increases in renal venous pressure. Am J Physiol. 1991;260:525–29. doi: 10.1152/ajprenal.1991.260.4.F525. [DOI] [PubMed] [Google Scholar]
- 21.Fiksen-Olsen MJ, Strick DM, Hawley H, Romero JC. Renal effects of angiotensin II inhibition during increases in renal venous pressure. Hypertension. 1992;19:137–41. doi: 10.1161/01.hyp.19.2_suppl.ii137. [DOI] [PubMed] [Google Scholar]
- 22.Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis: clinical presentation and pathogenesis. Am J Med. 2000;109:109–13. doi: 10.1016/s0002-9343(00)00461-7. [DOI] [PubMed] [Google Scholar]
- 23.Badalamenti S, Graziani G, Salerno F, Ponticelli C. Hepatorenal syndrome. New perspectives in pathogenesis and treatment. Arch Intern Med. 1993;153:1957–67. doi: 10.1001/archinte.153.17.1957. [DOI] [PubMed] [Google Scholar]
- 24.Castells A, Salo J, Planas R, et al. Impact of shunt surgery for variceal bleeding in the natural history of ascites in cirrhosis: a retrospective study. Hepatology. 1994;20:584–91. [PubMed] [Google Scholar]
- 25.Hamza S, Kaufman S. Effect of mesenteric vascular congestion on reflex control of renal blood flow. Am J Physiol Regul Integr Comp Physiol. 2007;293:1917–22. doi: 10.1152/ajpregu.00180.2007. [DOI] [PubMed] [Google Scholar]
- 26.Mullens W, Abrahams Z, Skouri H, et al. Elevated Intra-Abdominal Pressure in Acute Decompensated Heart Failure: A Potential Contributor to Worsening Renal Function? J Am Coll Cardiol. 2008;51:300–6. doi: 10.1016/j.jacc.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 27.Charkoudian N, Martin EA, Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Influence of increased central venous pressure on baroreflex control of sympathetic activity in humans. Am J Physiol Heart Circ Physiol. 2004;287:1658–62. doi: 10.1152/ajpheart.00265.2004. [DOI] [PubMed] [Google Scholar]
- 28.Creager MA, Creager SJ. Arterial baroreflex regulation of blood pressure in patients with congestive heart failure. J Am Coll Cardiol. 1994;23:401–5. doi: 10.1016/0735-1097(94)90427-8. [DOI] [PubMed] [Google Scholar]
- 29.Shi X, Foresman BH, Raven PB. Interaction of central venous pressure, intramuscular pressure, and carotid baroreflex function. Am J Physiol Heart Circ Physiol. 1997;272:1359–63. doi: 10.1152/ajpheart.1997.272.3.H1359. [DOI] [PubMed] [Google Scholar]
- 30.Cody RJ, Ljungman S, Covit AB, et al. Regulation of glomerular filtration rate in chronic congestive heart failure patients. Kidney Int. 1988;34:361–67. doi: 10.1038/ki.1988.189. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg TT, Richmond WH, Stocking RA, Gupta PD, Meehan JP, Henry JP. Impaired atrial receptor responses in dogs with heart failure due to tricuspid insufficiency and pulmonary artery stenosis. Circ Res. 1973;32:424–33. doi: 10.1161/01.res.32.4.424. [DOI] [PubMed] [Google Scholar]
- 32.Gauer OH, Henry JP. Neurohormonal control of plasma volume. Int Rev Physiol. 1976;9:145–90. [PubMed] [Google Scholar]
- 33.Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872–8. doi: 10.1016/j.ejheart.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Nohria A, Hasselblad V, Stebbins A, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51:1268–74. doi: 10.1016/j.jacc.2007.08.072. [DOI] [PubMed] [Google Scholar]