

Abstract
Proinflammatory cytokines play a crucial role in the pathogenesis of type 1 diabetes mellitus. One of the cytokine-regulated pathways mediating inflammation in this autoimmune disease is the arachidonic acid metabolism pathway, comprising both the induction of cyclooxygenases and the production of different prostaglandins. Cytokine toxicity is mediated in many cell types, including pancreatic β cells through this pathway. Interestingly, some cell types have been shown to be insensitive to such toxicity, and this correlated with a high expression of prostacyclin synthase (PGIS). Using insulin-producing RINm5F cells as a model for pancreatic β cells, PGIS was overexpressed and exhibited a large protective effect against cytokine toxicity. This protective effect of PGIS against cytokine toxicity correlated with a decreased activation of the transcription factor NFκB and the inducible NO synthase promoter as well as a reduced inducible NO synthase protein expression and nitrite production. A reduction in the cytokine-stimulated endoplasmic reticulum and mitochondrial stress was also found in the PGIS-overexpressing cells. Moreover, cytokine-induced caspase-3 activation and reduction of glucose oxidation and cell proliferation were suppressed. Thus, PGIS overexpression apparently protects insulin-producing cells against cytokine toxicity via suppression of endoplasmic reticulum and mitochondrial stress-mediated cell death pathways.
Keywords: Apoptosis, Diabetes, ER Stress, Mitochondrial Apoptosis, Prostaglandins, NFκB, Cytokines, Insulin-producing Cells, Nitric Oxide, Prostacyclin
Introduction
Inflammation is an essential element in the pathogenesis of type 1 diabetes mellitus (1–3). Proinflammatory cytokines induce cell death in insulin-producing cells through complex signaling pathways, involving nitrosative, oxidative, and ER and mitochondrial stress (4–9). The mechanisms of action of these cytokines have been extensively studied, but many details are still unclear. On the other hand, research efforts focus on potential protective strategies that can counteract proinflammatory cytokine-induced β cell dysfunction and death.
Among the complex mechanisms that initiate the inflammatory response are the activation of cyclooxygenases (COX)2 and the synthesis and activity of arachidonic acid metabolites. COX enzymes (constitutive COX-1 and inducible COX-2) convert arachidonic acid into an unstable endoperoxide intermediate, which is subsequently converted into several related products, including the prostaglandins PGD2, PGE2, and PGF2α, prostacyclin, and thromboxane A2. Cyclooxygenases are well expressed in insulin-producing cells, and the inducible form (COX-2) is activated in islet cells by cytokines (10–15). COX-2 has been shown to generate free radicals in the process of arachidonic acid metabolism (12, 13, 16, 17), and this mechanism has been considered to be responsible for the deleterious effects of COX-2 activation in pancreatic islets (2). Some products of arachidonic acid metabolism produced by COX-2, like prostaglandin E2, have been shown to mediate inhibition of insulin secretion by IL-1β in insulin-producing cells (18). PGJ2 has also been considered to cause β cell apoptosis (19).
Interestingly, it has been reported that a lack of toxicity caused by COX-2 induction in some cell types is accompanied by a high expression of prostacyclin synthase (PGIS), the heme-thiolate enzyme converting prostaglandin H2 into prostacyclin (PGI2) (20), and this promotes cell survival (20). The action of PGI2 is mediated by specific cell surface receptors called IP receptors (20) or, as recently shown, by the nuclear receptors PPARs (21).
PGIS deficiency plays a major role in various diseases including arterial intima hyperplasia, pulmonary hypertension, and tissue infarction. Enhanced PGIS expression with or without a concurrent COX-1 overexpression has been shown to control these disease processes (22). PGI2 produced from PGIS is generally considered to be released into the extracellular matrix. On the other hand, it has been hypothesized that endogenously produced PGI2 at the nuclear membrane enters the nucleus and binds and activates PPARδ to exert its cytoprotective action via induction of 14-3-3 protein expression, which in turn prevents apoptosis (23). It has been shown that inhibition of IL-1β-induced COX-2 gene expression by sodium salicylate enhances the insulin-producing islet cell function (24). It has also been reported in several studies that administration of prostacyclin analogues such as isocarbacyclin, iloprost, or beraprost to isolated pancreatic β cells and islets exerts protective effects (25–29). The mechanisms underlying these effects, however, are unknown.
So far very little is known about the functional role of PGIS in insulin-producing cells. The present study demonstrates an extremely low endogenous level of PGIS in insulin-producing cells and reports that overexpression of PGIS can efficiently prevent cytokine-induced cell death by prevention of NO formation as well as ER and mitochondrial stress in insulin-producing cells.
EXPERIMENTAL PROCEDURES
Chemicals
The cytokines were obtained from PromoCell (Heidelberg, Germany). Biotherm™ Taq polymerase from GeneCraft (Münster, Germany). Hybond N nylon membranes, the ECL detection system, and autoradiography films were from Amersham Biosciences (Freiburg, Germany). U51605 was from Biozol (Eching, Germany). Lentiviral particles with rat GADD 153 small hairpin RNA and lentiviral particles with rat caspase-12 small hairpin RNA were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). All other reagents were from Sigma.
Cell Culture
Insulin-producing RINm5F cells were cultured as described (30, 31) in RPMI medium, supplemented with 10 mm glucose, 10% (v/v) fetal calf serum, penicillin, and streptomycin in a humidified atmosphere at 37 °C and 5% CO2.
Overexpression of PGIS in Insulin-producing Cells
Human prostacyclin synthase cDNA (pcDNA3.1-hPGIS vector, a kind gift from Dr. Rolf Müller, Marburg, Germany) was stably overexpressed in insulin-producing RINm5F cells using the Lipofectamine transfection method. Positive clones were selected against G418, and the PGIS expression levels were confirmed by real time PCR measurements and Western blotting.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide Cell Viability Assay
Viability of the cells was determined after a 72-h incubation with cytokines using a microplate-based 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (32). Viability was expressed as a percentage of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide absorbance at 550 nm in the absence of the test compounds.
Proliferation Assay
The proliferation rate of RINm5F cells was quantified by a colorimetric method using cell proliferation bromodeoxyuridine-enzyme-linked immunosorbent assay (Roche Applied Science). The cells were seeded at a concentration of 5, 000 cells/well in 96-well microtiter plates and allowed to attach for 24 h. Thereafter cells were incubated with cytokines for 72 h. The proliferation assay was performed as described earlier (33). The absorbance at 450 nm (reference wavelength, 650 nm) was measured. The data are expressed as percentages of untreated cells.
Reporter Gene Assays
For the NFκB enhancer element, activator protein 1, and iNOS promoter activity studies, 2 × 104 cells/well were seeded in 96-well plates 24 h before transient transfection was performed (jetPEI™ transfection reagent) and 48 h before addition of test compounds for 6 h. The pSEAP constructs were used as described before (34). Secreted alkaline phosphatase expression was measured using the Phospha-Light™ system kit (Applera, Darmstadt, Germany).
Caspase-3 Activity Assay
Activation of caspase 3 was analyzed after 24 h of incubation with cytokines. The fluorimetric method of cleavage of the DEVD was employed as described earlier (35). The results are presented as percentages of untreated cells.
Caspase-9 and -12 Activity Assay
Activation of caspase-9 and -12 was quantified in control and RIN-PGIS cells after 24 h of exposure to cytokines using a Green Caspase-9 or -12 staining kit (PromoCell, Heidelberg, Germany) according to the instruction manual. After staining and washing, the cell suspensions were promptly read in the CyFlow ML cytometer (Partec, Münster, Germany). A total of 20,000 events were acquired. Nonlabeled cells were used as a negative control and for the determination of the gates. The data were analyzed by FlowJo software (Tree Star, Ashland, OR). The data are expressed as percentages of the caspase-9- or -12-positive cells without exposure to cytokines.
RNA Isolation and cDNA Preparation
Total RNA from insulin-producing cell clones was obtained using NucleoSpin RNA columns (Marcherey-Nagel, Düren, Germany). The quality of the total RNA was verified by agarose gel electrophoresis. RNA was quantified spectrophotometrically at 260/280 nm. Thereafter, 2 μg of RNA were reverse transcribed into complementary DNA using a random hexamer primer and a reverse transcriptase.
Real Time Reverse Transcription-PCR
The QuantiTect SYBR Green™ technology (Qiagen), which uses a fluorescent dye that binds only double-stranded DNA, was employed. The reactions were performed using the DNA Engine Opticon™ sequence detection system (Biozym Diagnostik, Hess. Oldendorf, Germany). A total volume of 25 μl was used for the PCRs. The samples were first denatured at 94 °C for 2 min followed by up to 30 PCR cycles. Each PCR cycle comprised a melting at 94 °C for 30 s, an annealing at 62 °C (PGIS) or 58 °C (CHOP) for 30 s, and an extension at 72 °C for 30 s. Each PCR amplification was performed in triplicate. The optimal parameters for the PCRs were empirically defined. The purity of the amplified PCR products was verified by melting curves. The primer sequences were as follows: for rCHOP: forward, CCAGCAGAGGTCACAAGCAC, and reverse, CGCACTGACCACTCTGTTTC; for hPGIS: forward, CCTGGTTGGGGTATGCCTTG, and reverse, TGTGGGTCCAGGAGAACGGT; for rPGIS: forward, CTGGGCCATGCCTTGGAGTT, and reverse, AGCCGTGTACGCAGATCCCA; and for rat β-actin: forward, GAACACGGCATTGTAACCAACTGG, and reverse, GGCCACACGCAGCTCATTGTA. The values for rPGIS, hPGIS, and rCHOP were normalized to β-actin.
iNOS, P-IKK, and PGIS Western Blot Analyses
The cells were plated at a density of 2 × 106/90-mm plastic dish and grown to confluence within 2 days. Thereafter the cells were exposed to the desired concentration of proinflammatory cytokines for 24 h (iNOS) or for 30 min (P-IKK). Concentrations of the cytokines were: IL-1β (600 units/ml) or a cytokine mixture containing IL-1β (60 units/ml) + TNFα (185 units/ml) + IFNγ (14 units/ml). RINm5F cells were homogenized in ice-cold phosphate-buffered saline using short bursts (Braun-Sonic 125 Homogenisator, Quigley-Rochester Inc., Rochester, NY) (iNOS and PGIS). The cytosolic extracts for analysis of P-IKK were obtained as described earlier (36). Protein content was determined by a BCA assay (Pierce). For the assays, 40 μg of total protein was resolved in SDS-polyacrylamide gel electrophoresis and then electroblotted onto membranes. Immunodetection was performed using specific primary antibodies against iNOS (NOS2 rabbit polyclonal IgG), against β-actin, or against P-IKKα/β Ser176/177 (all from Santa Cruz Biotechnology) or hPGIS (mouse polyclonal IgG, Biozol, Eching, Germany) followed by exposition to secondary peroxidase-conjugated AffiniPure donkey anti-rabbit IgG (H+L) or anti-mouse IgG (Dianova, Hamburg, Germany). The hybrids were visualized through chemiluminescence using the ECL detection system after a short exposure (2–3 min) to autoradiograms.
Nitrite Measurements
Nitrite accumulation after 72 h of cytokine exposure was determined spectrophotometrically at 562 nm by the Griess reaction as described earlier (37).
Glucose Oxidation
Glucose oxidation in the different cell clones after a 72-h incubation with IL-1β (600 units/ml) or a cytokine mixture containing IL-1β (60 units/ml) + TNFα (185 units/ml) + IFNγ (14 units/ml) was determined at 10 mmol/liter [U-14C]glucose (1 Ci/mol) as described earlier (33, 38).
6-Keto-PGF1α Measurements
The concentration of a stable product of prostacyclin metabolism was measured by a 6-keto-PGF1α EIA kit from Biozol (Eching, Germany) according to the manufacturer's protocol.
Data Analysis
All of the data are expressed as the means ± S.E. Statistical analyses were performed using the Prism analysis program (Graphpad, San Diego, CA).
RESULTS
Stable Overexpression of PGIS in Insulin-producing Cells
The endogenous level of prostacyclin synthase expression in insulin-producing RINm5F cells was virtually undetectable, so this insulin-producing cell line is particularly suited as a model for studies on the effects of PGIS overexpression. Therefore cDNA for human PGIS was introduced, and several positive clones were obtained. To exclude a possible influence of clonal variation on the results, for further analyses two clones with medium expression levels (K1 and K2) and two clones with high expression levels (K3 and K4) of PGIS were selected (Fig. 1). The PGIS expression was estimated on the RNA and protein level as well as by measurements of a stable product of prostacyclin metabolism 6-keto-PGF1α (Fig. 1, and concentrations of 6-keto-PGF1α in cell clones: RINm5F, not detected; RIN-PGIS K1, 46 ± 12; RIN-PGIS K2, 210 ± 65; RIN-PGIS K3, 637 ± 27, RIN-PGIS K4, 670 ± 82 pg/ml/mg cellular protein). Insulin-producing RINm5F cells transfected with the empty pcDNA3 vector were used as control cells. A decrease of 35% in 6-keto-PGF1α formation after exposure to cytokines was observed in the cells overexpressing PGIS (supplemental Table S1). But the concentration of a stable PGI2 product in the cells overexpressing PGIS and exposed to cytokines remained significantly higher than in the control cells, in which virtually no 6-keto-PGF1α could be detected (supplemental Table S1).
FIGURE 1.

Prostacyclin synthase expression in insulin-producing RINm5F cell clones. The cells were stably transfected with either the empty vector (control cells) or with the pcDNA3-hPGIS. A, gene expression of hPGIS was measured by quantitative real time reverse transcription-PCR. B, protein expression. *, p < 0.05 versus control cells, analysis of variance followed by Bonferroni. The basal expression of rat PGIS in the control as well as in all clones overexpressing PGIS was negligible (data not shown).
Effects of PGIS Overexpression on Cytokine Toxicity
A 72-h incubation of insulin-producing RINm5F control cells with cytokines caused ∼30 or 60% of cell viability loss after exposure to IL-1β (600 units/ml) or to a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ), respectively (Table 1). A significant protection through overexpression of PGIS was observed in all of the analyzed clones, with the greatest effect in the clones with the highest PGIS expression levels (Table 1). In the case of IL-1β, a 50% reduction of the toxic effect was seen in the RIN-PGIS clones K1 and K2 when compared with control cells and virtually full protection in the case of the RIN-PGIS clones K3 and K4 (Table 1). Similar protective effects, although somewhat less pronounced, were observed after incubation with the cytokine mixture (Table 1). The use of a specific PGIS inhibitor (U51605) at a concentration of 3 μm abrogated the protective effect of PGIS overexpression (Table 1). The PGIS inhibitor caused an ∼33% reduction in the formation of 6-keto-PGF1α. Higher concentrations of this inhibitor were toxic to the cells (data not shown). The reduced PGIS activity in the RIN-PGIS cells treated with cytokines and the PGIS inhibitor resulted in a reduction of the protective effect of PGIS overexpression against cytokines (∼20% reduction of the beneficial effect; Table 1).
TABLE 1.
Effects of PGIS overexpression in RINm5F insulin-producing cells on cell viability after exposure to IL-1β alone or a cytokine mixture in the absence or presence of a PGIS inhibitor (U51605)
RINm5F insulin-producing cells overexpressing PGIS as well as control cells were incubated with IL-1β (600 units/ml) alone or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, 14 units/ml IFNγ) for 72 h in the absence or presence of 3 μm PGIS inhibitor (U51605). The viability of the cells was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and expressed as a percentage of the untreated cells. The data are the means ± S.E. with the numbers of experiments in parentheses, each measured in at least three repetitions (analysis of variance followed by Bonferroni).
| RINm5F | RIN-PGIS K1 | RIN-PGIS K2 | RIN-PGIS K3 | RIN-PGIS K4 | |
|---|---|---|---|---|---|
| Control | 100 ± 3 (8) | 100 ± 5 (4) | 100 ± 4 (4)f | 100 ± 4 (4) | 100 ± 1 (8) |
| IL-1β | 67 ± 3 (8)a | 84 ± 7 (4)a,c | 83 ± 1 (4)a,c | 92 ± 1 (4)c | 91 ± 1 (8)c |
| Mix | 40 ± 3 (8)a | 67 ± 5 (4)a,d | 77 ± 1 (4)a,d | 83 ± 1 (4)a,d | 85 ± 1 (8)a,d |
| U51605 | 98 ± 2 (4) | 97 ± 2 (4) | 97 ± 3 (4) | 99 ± 3 (4) | 98 ± 3 (4) |
| IL-1β + U51605 | 60 ± 3 (4)b | 76 ± 1 (4)e | 77 ± 2 (4)b | 79 ± 1 (4)b,g | 85 ± 1 (4)b,g |
| Mix + U51605 | 44 ± 2 (4)b | 57 ± 2 (4)e,f | 68 ± 2 (4)b | 70 ± 2 (4)b,f | 80 ± 2 (4)b |
Effects of PGIS Overexpression on Proliferation Rate
The proliferation rate in untreated RIN-PGIS cell clones was slightly higher compared with control RINm5F cells (after 72 h under control condition: RINm5F, 100 ± 3%; RIN-PGIS K1, 125 ± 6%; RIN-PGIS K2, 118 ± 6%; RIN-PGIS K3, 119 ± 3%; RIN-PGIS K4, 114 ± 4%; n = 4, expressed as percentages of proliferation of RINm5F control cells). A 72-h exposure of insulin-producing RINm5F control as well as PGIS-overexpressing cell clones to IL-1β alone did not cause a significant decrease in the cell proliferation rate (Table 2). The incubation of insulin-producing RINm5F control cells with the cytokine mixture led to a 45% reduction in the proliferation rate (Table 2). Interestingly, a significant protective effect of PGIS overexpression against the cytokine mixture-induced decrease in the proliferation rate was observed in all clones overexpressing PGIS (Table 2). The protection correlated well with the degree of the PGIS expression and was most pronounced in the RIN-PGIS K4 clone with the highest PGIS expression (Table 2). Indeed, in this clone, no significant reduction in the proliferation rate could be detected after incubation with the cytokine mixture (Table 2). Because the largest protective effects on the cell viability as well as the proliferation rate were seen in the case of the RIN-PGIS K4 clone, all of the subsequent experiments were performed with this clone.
TABLE 2.
Effects of PGIS overexpression in RINm5F insulin-producing cells on cell proliferation after exposure to IL-1β alone or a cytokine mixture
RINm5F insulin-producing cells overexpressing PGIS as well as control cells were incubated with IL-1β (600 units/ml) alone or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNF-α, and 14 units/ml IFNγ) for 72 h. The proliferation rate of the cells was determined by the bromodeoxyuridine-enzyme-linked immunosorbent assay and expressed as a percentage of the untreated cells. The data are the means ± S.E. with the numbers of experiments in parentheses, each measured in at least three repetitions (analysis of variance followed by Bonferroni).
| RINm5F cell clone | Cell proliferation |
||
|---|---|---|---|
| Untreated | IL-1β | Mix | |
| RINm5F | 100 ± 3 (4) | 88 ± 3 (4) | 55 ± 3 (4)a |
| RIN-PGIS K1 | 100 ± 5 (4) | 91 ± 6 (4) | 73 ± 6 (4)a |
| RIN-PGIS K2 | 100 ± 4 (4) | 93 ± 3 (4) | 75 ± 4 (4)a |
| RIN-PGIS K3 | 100 ± 4 (4) | 97 ± 4 (4) | 80 ± 4 (4)a,b |
| RIN-PGIS K4 | 100 ± 1 (4) | 102 ± 4 (4) | 84 ± 5 (4)b |
a p < 0.05 versus untreated.
b p < 0.05 cytokine mixture versus control clone.
Effects of PGIS Overexpression on Cytokine-stimulated Caspase-3, -9, and -12 Activation
Exposure of insulin-producing RINm5F control cells to IL-1β for 24 h induced caspase-12 and caspase-3 activation (Fig. 2). The increase in the activity of both caspases could also be detected after incubation of the control cells with the cytokine mixture; however, this induction was lower than in the case of IL-1β (4-fold increase after cytokine mixture versus 10-fold increase after IL-1β; Fig. 2) in the case of caspase-3. The activation of caspase-3 was almost completely abolished by overexpression of PGIS in the RIN-PGIS K4 clone (Fig. 2). Camptothecin (0.5 μm) induced caspase-3 activation in both control and PGIS K4 clones, indicating that the protective effect was specific for cytokine-mediated toxicity. PGIS overexpression completely blocked cytokine-induced caspase-12 activation but not the camptothecin-induced caspase-12 activation (Fig. 2). In the case of caspase-9 activation, PGIS overexpression diminished cytokine-induced activation but did not prevent the activation induced by camptothecin (Fig. 2).
FIGURE 2.

Effects of PGIS overexpression in insulin-producing RINm5F cells on cytokine-stimulated caspase-3, -9, and -12 activity. RINm5F insulin-producing cells were incubated with IL-1β (600 units/ml) or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ), or camptothecin (0.5 μm) for 24 h. After cell lysis and extraction of the total intracellular protein, the activity of caspase-3 was measured using the DEVD cleavage method. Caspase-9 and -12 activation was measured using flow cytometry. The data are mean values from four independent experiments, each measured in at least two repetitions. *, p < 0.05 versus untreated; #, p < 0.05 versus control clone treated in the same way; analysis of variance followed by Bonferroni.
Effects of PGIS Overexpression on CHOP Expression after Exposure to Cytokines
Because a complete prevention of caspase-12 activation in PGIS-overexpressing cells was observed, which is thought to be induced by ER stress, CHOP gene expression was analyzed in untreated and cytokine-treated control and PGIS-overexpressing cells. In RINm5F control cells a 2-fold induction after IL-1β and a 3-fold induction after a cytokine mix was observed. This cytokine-stimulated CHOP gene expression was almost completely prevented by the overexpression of PGIS (Fig. 3). In contrast to PGIS overexpression, the reduction of ER stress (by a use of small hairpin RNA-CHOP ∼65% or small hairpin RNA-caspase-12 ∼60% reduction) in control experiments resulted only in a weak protection against cytokine toxicity (supplemental Table S2).
FIGURE 3.

Effects of PGIS overexpression in insulin-producing RINm5F cells on cytokine-induced CHOP gene expression. The cells were incubated with IL-1β (600 units/ml) or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ) for 24 h. Gene expression of CHOP was measured by quantitative real time reverse transcription-PCR. *, p < 0.05 versus untreated cells. Analysis of variance was followed by Bonferroni.
Effects of PGIS Overexpression on Cytokine-induced NFκB and Activator Protein 1 Activation
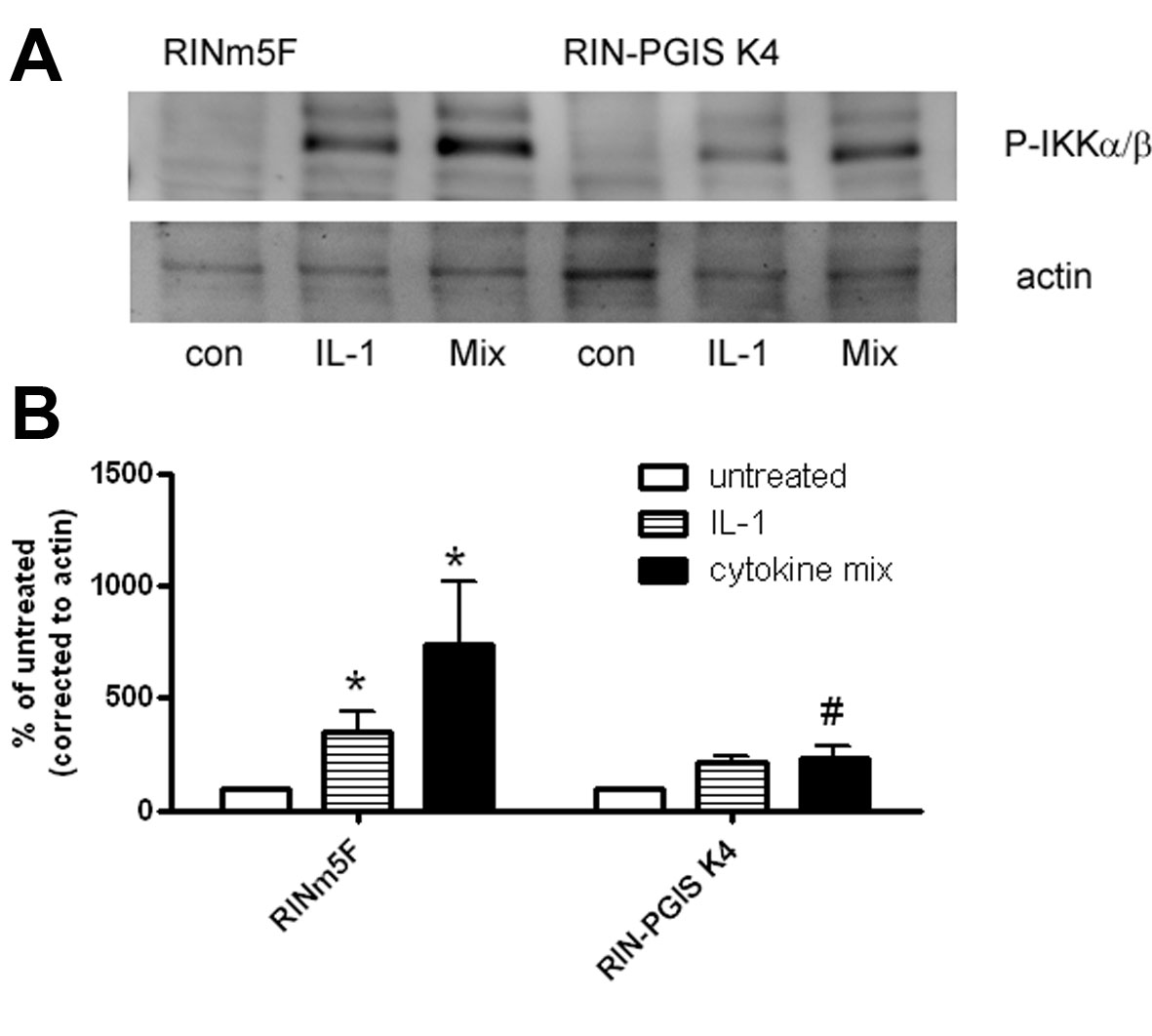
As expected, incubation of insulin-producing RINm5F control cells with cytokines stimulated NFκB activation (2-fold increase; Table 3). In contrast, there was no induction of NFκB activation in the RIN-PGIS K4 clone (Table 3). The prevention of NFκB activation in RIN-PGIS K4 clone went along with a reduction of IKK phosphorylation by cytokines. This effect was particularly prominent in the case of the cytokine mixture (supplemental Fig. S1). On the other hand, cytokine-induced activator protein 1 activation in both control and PGIS K4 clones did not differ significantly (Table 3).
TABLE 3.
Effects of PGIS overexpression on cytokine-induced NFκB, activator protein 1, and iNOS promoter activation and nitrite production in RINm5F insulin-producing cell clones
For estimation of transcription factor and iNOS promoter activation, RINm5F insulin-producing cells were transfected 24 h prior to cytokine incubation and then treated with IL-1β (600 units/ml) alone or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNF-α, 14 units/ml IFNγ) for 6 h. Thereafter the medium was collected, and a SEAP-reporter gene assay was performed. For nitrite measurements, RINm5F insulin-producing cells were incubated with IL-1β (600 units/ml) alone or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNF-α, and 14 units/ml IFNγ) for 72 h. Thereafter the medium was collected, and the nitrite concentration was measured. The numbers of experiments are given in parentheses. The data are mean values for the indicated numbers (n) of independent experiments, each measured in at least three repetitions (analysis of variance followed by Bonferroni).
| RINm5F cell clone | RINm5F | RIN-PGIS K4 |
|---|---|---|
| NFκB (% of untreated) | ||
| Untreated | 100 ± 2 (4) | 100 ± 1 (4) |
| IL-1β | 217 ± 23 (4)a | 111 ± 5 (4)b |
| Mix | 190 ± 19 (4)a | 119 ± 15 (4)c |
| AP-1 (% of untreated) | ||
| Untreated | 100 ± 1 (6) | 100 ± 1 (6) |
| IL-1β | 125 ± 6 (6) | 110 ± 20 (6) |
| Mix | 129 ± 10 (6) | 94 ± 18 (6) |
| iNOS promoter (% of untreated) | ||
| Untreated | 100 ± 1 (4) | 100 ± 3 (4) |
| IL-1β | 422 ± 49 (4)a | 144 ± 9 (4)b |
| Mix | 531 ± 91 (4)a | 182 ± 19 (4)c |
| Nitrite (pmol/μg protein) | ||
| Untreated | 0 ± 0 (6) | 0 ± 0 (6) |
| IL-1β | 3.9 ± 0.5 (6)a | 0 ± 0 (6)b |
| Mix | 5.5 ± 0.7 (6)a | 0 ± 0 (6)c |
a p < 0.05 versus untreated.
b p < 0.05 IL-1β versus control clone.
c p < 0.05 cytokine mixture versus control clone.
Effects of PGIS Overexpression on Cytokine-induced iNOS Promoter Activation, iNOS Protein Expression, and Nitrite Production
Cytokines also caused iNOS promoter activation, and again, as in the case of NFκB, this induction was significantly lower in the RIN-PGIS K4 clone than in the control clone (Table 3). Consequently, in the insulin-producing RINm5F control cells, cytokine exposure led to a very pronounced protein expression of the iNOS, whereas in the RIN-PGIS K4 clone, this protein was hardly detectable (Fig. 4). These results were confirmed through nitrite measurements, revealing high concentrations of nitrite in the control cells and virtually no nitrite in the RIN-PGIS K4 clone after incubation with cytokines (Table 3).
FIGURE 4.
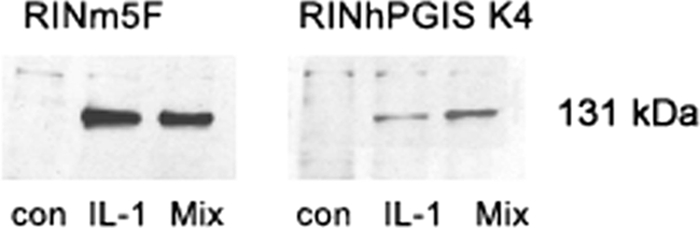
Effects of PGIS overexpression in insulin-producing RINm5F cells on cytokine-stimulated iNOS protein expression. The cells were incubated with IL-1β (600 units/ml) or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ) for 24 h, and thereafter the samples were collected for Western-blotting. A representative blot from four independent experiments is shown. con, control. Mix, cytokine mixture.
Effects of PGIS Overexpression on Glucose Oxidation after Exposure to Cytokines
Incubation of control insulin-producing RINm5F cells with IL-1β caused a decrease of the glucose oxidation rate by ∼40% (Table 4). A 60% reduction of the glucose oxidation rate was observed after exposure of the control cells to the cytokine mixture (Table 4). The cytokines did not reduce glucose oxidation in the RIN-PGIS K4 clone; rather the opposite effect was observed, namely a pronounced increase above 100% of the glucose oxidation rate in the RIN-PGIS K4 clone after cytokine treatment (Table 4).
TABLE 4.
Effects of PGIS overexpression in RINm5F insulin-producing cells on the glucose oxidation rate after exposure to IL-1β alone or a cytokine mix
RINm5F insulin-producing cells overexpressing PGIS as well as control cells were incubated with IL-1β (600 units/ml) alone or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNF-α, and 14 units/ml IFNγ) for 72 h. Glucose oxidation rates at 10 mmol/liter [U-14C]glucose (1Ci/mol) were expressed as a percentage of untreated cells. Glucose oxidation rates under control conditions were identical in the control cells (0.09 ± 0.005 mmol/g of DNA/h) and in the RIN-PGIS K4 clone (0.12 ± 0.003 mmol/g of DNA/h). The data are the means ± S.E. with the numbers of experiments in parentheses, each measured in two repetitions (analysis of variance followed by Bonferroni).
| RINm5F cell clone | Glucose oxidation rate |
||
|---|---|---|---|
| Untreated | IL-1β | Cytokine mixture | |
| RINm5F | 100 ± 3 (8) | 63 ± 19 (7)a | 34 ± 7 (8)a |
| RIN-PGIS K4 | 100 ± 1 (8) | 286 ± 57 (6)a,b | 376 ± 41 (7)a,c |
a p < 0.05 versus untreated.
b p < 0.05 IL-1β versus control clone.
c p < 0.05 cytokine mixture versus control clone.
DISCUSSION
It is generally accepted that proinflammatory cytokines are major effectors of programmed cell death during the development of type 1 diabetes mellitus (4, 6, 8, 39–44). The mechanisms of action are only partially understood, but it is assumed that increased nitrosative, oxidative, mitochondrial, and ER stresses (4, 30, 33, 35, 45–49) play a crucial role.
Arachidonic acid metabolism is a crucial pathway induced in many diseases with an inflammatory background. It is known that proinflammatory cytokines induce the expression of COX-2, the enzyme that converts arachidonic acid into an unstable endoperoxide intermediate, which is subsequently converted into several related products, including prostaglandins and thromboxanes. This induction is thought to cause intracellular generation of reactive oxygen species. Interestingly, it has been shown in some cell types that COX-2 induction has no deleterious effects on cell viability and function, when the level of PGIS, the synthase producing prostacyclin, is sufficiently high (20). It appears therefore that high levels of prostacyclin synthase block this pro-inflammatory pathway, thereby exhibiting anti-inflammatory properties. It has been shown that prostacyclin analogues, such as beraprost, have a great cytoprotective potential in isolated pancreatic islets (25–29). For example beraprost improved islet yield and viability in canine islet cryopreservation (25) and protected islets during collagenase digestion (27). Moreover, a low dose of another PGI2 analogue, namely iloprost, improved insulin action in aged healthy subjects and T2DM patients (50). Very little is known, however, about the role of prostacyclin in insulin-producing cells, especially with respect to the molecular mechanisms involved in its protective action.
In the present study we detected an extremely low endogenous level of PGIS in insulin-producing cells. Overexpression of PGIS in insulin-producing RINm5F cells provided excellent protection against cytokine toxicity. This protection resulted from the reduced NO formation and, subsequently, the reduced ER as well as mitochondrial stress and could be correlated with the prevention of caspase-3 activation after exposure to cytokines.
It is generally accepted that reduction of nitrosative stress can prevent β cell death (51, 52), and this is accompanied by a decrease in cytokine-stimulated caspase-3 activation (35). The present study reveals that PGIS overexpression decreases NFκB activation and blocks the NO pathway in PGIS-overexpressing insulin-producing cells, which is in line with previous reports about anti-inflammatory properties of PGJ2 in β cells (53, 54).
It has been shown in some cell types that so-called cyclopentenone prostaglandins are able to inhibit the activity of IκB kinase (IKK), reduce cytokine-stimulated NFκB activation, and therefore prevent NFκB translocation to the nucleus (55, 56). PGIS overexpression reduced cytokine-induced IKK phosphorylation also in insulin-producing cells, which can explain the reduced cytokine-stimulated NFκB activation in the PGIS-overexpressing cells.
Cytokine toxicity in insulin-producing cells is mostly related to the NFκB-induced up-regulation of iNOS (57–59). From our study it becomes clear that in insulin-producing cells overexpressing PGIS, the reduced nitrosative stress can be related to the prevention of the cytokine-induced NFκB activation. It is known from studies in astrocytes and microglia that anti-inflammatory cyclopentenone prostaglandins are potent inhibitors of iNOS induction (60). It is also generally accepted that a blockade of the NFκB and iNOS in insulin-producing cells protects these cells against cytokine toxicity (35, 49, 52, 61).
The prevention of NFκB activation or NO formation in insulin-producing cells has been reported to lower the cytokine-stimulated ER stress response (62, 63). Interestingly, the present results demonstrate that overexpression of PGIS can down-regulate CHOP, the main inflammatory player in ER stress, as documented by decreased CHOP gene expression and the ER-associated caspase-12 activation via inhibition of the cytokine-stimulated iNOS induction. This is at variance from another prostaglandin, PGJ2, which also attenuates cytokine signaling, but has been reported to stimulate the ER stress response (64). It is important to state that ER stress is activated by cytokines in insulin-producing cells (for review see Ref. 65), but it is only partially responsible for cytokine toxicity in these cells as shown by our experiments (supplemental Table S2) and by others (63, 66).
PGIS overexpression blocked almost completely cytokine toxicity, whereas a down-regulation of either CHOP or caspase-12 did not prevent cytokine toxicity. The beneficial effects of PGIS overexpression come not only from a reduced ER stress but also from reduced mitochondrial stress. This is an additional proof that cytokine toxicity in insulin-producing cells results in the induction of many different cell death pathways.
Interestingly, cytokines affected neither cell viability nor the proliferation rate of insulin-producing cells highly overexpressing PGIS, which indicates that high levels of prostacyclin can provide protection not only by decreasing the cell death rate but also by maintaining the proliferative capacity. These observations are in line with an earlier report in which PGI2 levels were correlated with the blastocyst development, and an PGIS inhibitor was shown to reduce the number of blastomeres (67). It has also been reported that endogenous prostacyclin reduces apoptosis and enhances embryo viability in mice (68). In addition, the PGIS gene transfer approach has been successfully used to stimulate wound healing in skin diseases (69), suggesting an important role for prostacyclin in cell proliferation and survival.
Our study also provides additional evidence, namely improved glucose oxidation capacity upon exposure to cytokines, for the excellent metabolic status of the cells expressing a high level of prostacyclin synthase. Similar observations have been made before in patients treated with a PGI2 analogue in whom improved oxidative and nonoxidative glucose metabolism has been reported (50, 70, 71).
The mechanism by which caspase-3 activation is prevented in PGIS-overexpressing cells probably involves not only inhibition of caspase-9 and caspase-12 activation but also some other pathways. It has been shown in endothelial cells that endogenously produced PGI2 binds and activates PPARδ, which subsequently activates the 14-3-3ϵ promoter (23). 14-3-3ϵ is a member of the 14-3-3 family, which binds phosphorylated Bad and sequesters Bad in the cytosol. The up-regulation of 14-3-3ϵ in endothelial cells amplifies Bad binding and reduces Bad translocation to mitochondria, whereby it inhibits cytochrome c release and caspase-3 activation. Whether a similar mechanism might play a role in the reduced cytokine-induced caspase-3 activation also in insulin-producing cells overexpressing PGIS remains to be elucidated.
Our results have demonstrated an extremely low endogenous PGIS expression in insulin-producing cells, which can provide the explanation for the destructive action of COX-2 on one hand and the protective effects of PGI2 analogues on the other hand. Overexpression of PGIS could prevent the toxic effects of cytokines in insulin-producing cells. It is evident that the suppression of the cytokine-stimulated NFκB-iNOS pathway preventing ER and mitochondrial stress and caspase-3 activation together with a stimulatory effect on cell proliferation are of crucial importance for this effect. The results of the present study provide further evidence for the inflammatory nature of cytokine-mediated β cell toxicity.
Supplementary Material
Acknowledgments
We are grateful to Dr. Rolf Müller (Marburg, Germany) for the pcDNA3-hPGIS vector. The excellent technical assistance of C. Heinrichs, M. Funck, and J. Kresse is acknowledged.
This work was supported by European Union Grant STREP SaveBeta LSHM-CT-2006-036903 in Framework Programme 6 of the European Community) (to S. L.) and by a grant from the German Foundation Das Zuckerkranke Kind (to E. G.-C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Fig. S1.
- COX
- cyclooxygenase
- IFNγ
- interferon γ
- IKK
- IκB kinase
- IL-1β
- interleukin 1β
- iNOS
- inducible NO synthase
- NFκB
- nuclear factor κB
- PGI2
- prostaglandin I2 (prostacyclin)
- PG
- prostaglandin
- PGIS
- prostacyclin synthase
- PPAR
- peroxisome proliferator-activated receptor
- TNF
- tumor necrosis factor
- ER
- endoplasmic reticulum
- IL
- interleukin.
REFERENCES
- 1.Bach J. F. (1994) Endocr. Rev. 15, 516–542 [DOI] [PubMed] [Google Scholar]
- 2.McDaniel M. L., Kwon G., Hill J. R., Marshall C. A., Corbett J. A. (1996) Proc. Soc. Exp. Biol. Med. 211, 24–32 [DOI] [PubMed] [Google Scholar]
- 3.Yang Z., Chen M., Ellett J. D., Carter J. D., Brayman K. L., Nadler J. L. (2005) Am. J. Transplant 5, 475–483 [DOI] [PubMed] [Google Scholar]
- 4.Cnop M., Welsh N., Jonas J. C., Jörns A., Lenzen S., Eizirik D. L. (2005) Diabetes 54, (Suppl. 2) S97–S107 [DOI] [PubMed] [Google Scholar]
- 5.Eizirik D. L. (1996) Horm. Metab. Res. 28, 302–305 [DOI] [PubMed] [Google Scholar]
- 6.Eizirik D. L., Mandrup-Poulsen T. (2001) Diabetologia 44, 2115–2133 [DOI] [PubMed] [Google Scholar]
- 7.Lenzen S. (2008) Biochem. Soc. Trans. 36, 343–347 [DOI] [PubMed] [Google Scholar]
- 8.Mandrup-Poulsen T. (1996) Diabetologia 39, 1005–1029 [DOI] [PubMed] [Google Scholar]
- 9.Mandrup-Poulsen T. (2003) Ann. N.Y. Acad. Sci. 1005, 32–42 [DOI] [PubMed] [Google Scholar]
- 10.Sorli C. H., Zhang H. J., Armstrong M. B., Rajotte R. V., Maclouf J., Robertson R. P. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 1788–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson R. P. (1998) Diabetes 47, 1379–1383 [DOI] [PubMed] [Google Scholar]
- 12.Jobin C., Morteau O., Han D. S., Balfour Sartor R. (1998) Immunology 95, 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabatabaie T., Vasquez-Weldon A., Moore D. R., Kotake Y. (2003) Diabetes 52, 1994–1999 [DOI] [PubMed] [Google Scholar]
- 14.Heitmeier M. R., Kelly C. B., Ensor N. J., Gibson K. A., Mullis K. G., Corbett J. A., Maziasz T. J. (2004) J. Biol. Chem. 279, 53145–53151 [DOI] [PubMed] [Google Scholar]
- 15.Kwon G., Corbett J. A., Hauser S., Hill J. R., Turk J., McDaniel M. L. (1998) Diabetes 47, 583–591 [DOI] [PubMed] [Google Scholar]
- 16.Marnett L. J. (2000) Ernst Schering Research Foundation Workshop 31, 65–83 [DOI] [PubMed] [Google Scholar]
- 17.Marnett L. J., Panthananickal A., Reed G. A. (1982) Drug Metab. Rev. 13, 235–247 [DOI] [PubMed] [Google Scholar]
- 18.Tran P. O., Gleason C. E., Poitout V., Robertson R. P. (1999) J. Biol. Chem. 274, 31245–31248 [DOI] [PubMed] [Google Scholar]
- 19.Chambers K. T., Weber S. M., Corbett J. A. (2007) Am. J. Physiol. Endocrinol. Metab. 292, E1052–E1061 [DOI] [PubMed] [Google Scholar]
- 20.Siegle I., Klein T., Zou M. H., Fritz P., Kömhoff M. (2000) J. Histochem. Cytochem. 48, 631–641 [DOI] [PubMed] [Google Scholar]
- 21.Hatae T., Wada M., Yokoyama C., Shimonishi M., Tanabe T. (2001) J. Biol. Chem. 276, 46260–46267 [DOI] [PubMed] [Google Scholar]
- 22.Wu K. K., Liou J. Y. (2005) Biochem. Biophys. Res. Commun. 338, 45–52 [DOI] [PubMed] [Google Scholar]
- 23.Liou J. Y., Lee S., Ghelani D., Matijevic-Aleksic N., Wu K. K. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 1481–1487 [DOI] [PubMed] [Google Scholar]
- 24.Tran P. O., Gleason C. E., Robertson R. P. (2002) Diabetes 51, 1772–1778 [DOI] [PubMed] [Google Scholar]
- 25.Arita S., Kasraie A., Une S., Ohtsuka S., Kawahara T., Shevlin L., Matusda N., Amirmoazzami S., Mullen Y. (1997) Transplant. Proc. 29, 1977. [DOI] [PubMed] [Google Scholar]
- 26.Arita S., Kasraie A., Une S., Ohtsuka S., Kawahara T., Smith C. V., Mullen Y. (1999) Pancreas 19, 289–296 [DOI] [PubMed] [Google Scholar]
- 27.Arita S., Une S., Ohtsuka S., Kawahara T., Kasraie A., Shevlin L., Mullen Y. (1998) Transplant. Proc. 30, 366. [DOI] [PubMed] [Google Scholar]
- 28.Arita S., Une S., Ohtsuka S., Kawahara T., Kasraie A., Smith C. V., Mullen Y. (2001) Pancreas 23, 62–67 [DOI] [PubMed] [Google Scholar]
- 29.Yeğen C., Aktan A. O., Döşlüoğlu H. H., Ercan S., Yalin R. (1994) Prostaglandins Leukot. Essent. Fatty Acids 51, 257–262 [DOI] [PubMed] [Google Scholar]
- 30.Lortz S., Tiedge M., Nachtwey T., Karlsen A. E., Nerup J., Lenzen S. (2000) Diabetes 49, 1123–1130 [DOI] [PubMed] [Google Scholar]
- 31.Elsner M., Tiedge M., Guldbakke B., Munday R., Lenzen S. (2002) Diabetologia 45, 1542–1549 [DOI] [PubMed] [Google Scholar]
- 32.Mosmann T. (1983) J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 33.Gurgul E., Lortz S., Tiedge M., Jörns A., Lenzen S. (2004) Diabetes 53, 2271–2280 [DOI] [PubMed] [Google Scholar]
- 34.Azevedo-Martins A. K., Lortz S., Lenzen S., Curi R., Eizirik D. L., Tiedge M. (2003) Diabetes 52, 93–101 [DOI] [PubMed] [Google Scholar]
- 35.Souza K. L., Gurgul-Convey E., Elsner M., Lenzen S. (2008) J. Endocrinol. 197, 139–150 [DOI] [PubMed] [Google Scholar]
- 36.Schreiber E., Matthias P., Müller M. M., Schaffner W. (1989) Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiedge M., Lortz S., Munday R., Lenzen S. (1999) Diabetologia 42, 849–855 [DOI] [PubMed] [Google Scholar]
- 38.Lenzen S., Panten U. (1980) Biochem. J. 186, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eizirik D. L. (1988) Acta Endocrinol. 119, 321–325 [DOI] [PubMed] [Google Scholar]
- 40.Eizirik D. L. (1991) Autoimmunity 10, 107–113 [DOI] [PubMed] [Google Scholar]
- 41.Eizirik D. L., Bendtzen K., Sandler S. (1991) Endocrinology 128, 1611–1616 [DOI] [PubMed] [Google Scholar]
- 42.Eizirik D. L., Sandler S., Welsh N., Cetkovic-Cvrlje M., Nieman A., Geller D. A., Pipeleers D. G., Bendtzen K., Hellerström C. (1994) J. Clin. Invest. 93, 1968–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rønn S. G., Billestrup N., Mandrup-Poulsen T. (2007) Diabetes 56, 541–548 [DOI] [PubMed] [Google Scholar]
- 44.Sandler S., Eizirik D. L., Svensson C., Strandell E., Welsh M., Welsh N. (1992) Lakartidningen 89, 1777–1779 [PubMed] [Google Scholar]
- 45.Bast A., Wolf G., Oberbäumer I., Walther R. (2002) Diabetologia 45, 867–876 [DOI] [PubMed] [Google Scholar]
- 46.Lortz S., Gurgul-Convey E., Lenzen S., Tiedge M. (2005) Diabetologia 48, 1541–1548 [DOI] [PubMed] [Google Scholar]
- 47.Lortz S., Tiedge M. (2003) Free Radic. Biol. Med. 34, 683–688 [DOI] [PubMed] [Google Scholar]
- 48.Størling J., Binzer J., Andersson A. K., Züllig R. A., Tonnesen M., Lehmann R., Spinas G. A., Sandler S., Billestrup N., Mandrup-Poulsen T. (2005) Diabetologia 48, 2039–2050 [DOI] [PubMed] [Google Scholar]
- 49.Suarez-Pinzon W. L., Mabley J. G., Strynadka K., Power R. F., Szabó C., Rabinovitch A. (2001) J. Autoimmun. 16, 449–455 [DOI] [PubMed] [Google Scholar]
- 50.Paolisso G., Di Maro G., D'Amore A., Passariello N., Gambardella A., Varricchio M., D'Onofrio F. (1995) Diabetes Care 18, 200–205 [DOI] [PubMed] [Google Scholar]
- 51.Eldor R., Yeffet A., Baum K., Doviner V., Amar D., Ben-Neriah Y., Christofori G., Peled A., Carel J. C., Boitard C., Klein T., Serup P., Eizirik D. L., Melloul D. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5072–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flodström M., Tyrberg B., Eizirik D. L., Sandler S. (1999) Diabetes 48, 706–713 [DOI] [PubMed] [Google Scholar]
- 53.Maggi L. B., Jr., Sadeghi H., Weigand C., Scarim A. L., Heitmeier M. R., Corbett J. A. (2000) Diabetes 49, 346–355 [DOI] [PubMed] [Google Scholar]
- 54.Weber S. M., Scarim A. L., Corbett J. A. (2003) Am. J. Physiol. Endocrinol. Metab. 284, E883–E891 [DOI] [PubMed] [Google Scholar]
- 55.Rossi A., Kapahi P., Natoli G., Takahashi T., Chen Y., Karin M., Santoro M. G. (2000) Nature 403, 103–108 [DOI] [PubMed] [Google Scholar]
- 56.Rossi A., Elia G., Santoro M. G. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 746–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darville M. I., Eizirik D. L. (1998) Diabetologia 41, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 58.Eizirik D. L., Leijerstam F. (1994) Diabetes Metab. 20, 116–122 [PubMed] [Google Scholar]
- 59.Flodström M., Welsh N., Eizirik D. L. (1996) FEBS Lett. 385, 4–6 [DOI] [PubMed] [Google Scholar]
- 60.Petrova T. V., Akama K. T., Van Eldik L. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 4668–4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ortis F., Pirot P., Naamane N., Kreins A. Y., Rasschaert J., Moore F., Théâtre E., Verhaeghe C., Magnusson N. E., Chariot A., Orntoft T. F., Eizirik D. L. (2008) Diabetologia 51, 1213–1225 [DOI] [PubMed] [Google Scholar]
- 62.Tonnesen M. F., Grunnet L. G., Friberg J., Cardozo A. K., Billestrup N., Eizirik D. L., Størling J., Mandrup-Poulsen T. (2009) Endocrinology 150, 4094–4103 [DOI] [PubMed] [Google Scholar]
- 63.Chambers K. T., Unverferth J. A., Weber S. M., Wek R. C., Urano F., Corbett J. A. (2008) Diabetes 57, 124–132 [DOI] [PubMed] [Google Scholar]
- 64.Weber S. M., Chambers K. T., Bensch K. G., Scarim A. L., Corbett J. A. (2004) Am. J. Physiol. Endocrinol. Metab. 287, E1171–E1177 [DOI] [PubMed] [Google Scholar]
- 65.Eizirik D. L., Cardozo A. K., Cnop M. (2008) Endocr. Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 66.Scarim A. L., Heitmeier M. R., Corbett J. A. (1997) Endocrinology 138, 5301–5307 [DOI] [PubMed] [Google Scholar]
- 67.Pakrasi P. L., Jain A. K. (2007) Life Sci. 80, 1503–1507 [DOI] [PubMed] [Google Scholar]
- 68.Pakrasi P. L., Jain A. K. (2008) Prostaglandins Leukot. Essent. Fatty Acids 79, 27–33 [DOI] [PubMed] [Google Scholar]
- 69.Kunugiza Y., Tomita N., Taniyama Y., Tomita T., Osako M. K., Tamai K., Tanabe T., Kaneda Y., Yoshikawa H., Morishita R. (2006) Gene Ther. 13, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 70.Paolisso G., Gambardella A., Saccomanno F., Varricchio G., D'Amore A., Varricchio M. (1995) Eur. J. Clin. Pharmacol. 48, 333–338 [DOI] [PubMed] [Google Scholar]
- 71.Scheeren T., Susanto F., Reinauer H., Tarnow J., Radermacher P. (1994) J. Crit. Care 9, 175–184 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}