

Abstract
The histone acetyltransferase TIP60, a frequent target of monoallelic loss in human carcinomas, can acetylate many substrates, including histones and p53, and thus promote apoptosis following UV radiation. Here we showed that TIP60 is autoacetylated in response to UV damage, which is critically important for TIP60 activation. Mechanistically we demonstrated that TIP60 autoacetylation leads to the dissociation of TIP60 oligomer and enhances its interaction with substrates. Moreover, we identified SIRT1 that specifically deacetylates TIP60 and negatively regulates TIP60 activity in vivo. Taken together, our data reveal TIP60 autoacetylation as a key step in the control of its histone acetyltransferase activity and function in response to DNA damage.
Keywords: Apoptosis, DNA Damage, Protein Acylation, Signal Transduction, Tumor Suppressor
Introduction
TIP60 is a histone acetyltransferase that serves to integrate diverse signaling pathways involved in a variety of cellular functions (1–3). It is known that TIP60 can acetylate core histones and thereby regulate chromatin remodeling and gene transcription (4). TIP60 also has a number of non-histone substrates. For example, suppression of TIP60 results in defective double-stranded DNA break repair and failure to induce apoptosis following IR or UV radiation (5–7). This function of TIP60 in DNA damage-induced apoptosis is likely to be mediated through its ability to acetylate substrates, such as p53 and ATM, following DNA damage (7–9). Given the diverse and important roles of TIP60 in the cell, it is not surprising that the TIP60 gene is a frequent target for monoallelic loss in many human carcinomas, including lymphomas and head-and-neck and mammary carcinomas (10).
One of the critical substrates of TIP60 involved in its tumor suppression function is likely to be p53. Posttranslational modifications, such as phosphorylation and acetylation, of p53 are important modes to activate p53 activity in response to DNA damage and other cellular stresses (11). Until now, eight acetylation sites of p53 have been identified, and substitution of these residues completely abolishes p53-dependent cell cycle arrest and apoptosis (11, 12). Among these acetylation sites, the CREB-binding protein/p300-mediated p53 acetylation at lysine 164 and the six C-terminal lysine residues enhances p53 activation and stabilization in response to stress (13). The TIP60 (and hMOF) mediates p53 acetylation at lysine 120, which locates within the DNA-binding domain of p53 (8, 9), enhances the binding of p53 to promoters of pro-apoptotic genes, such as BAX and PUMA, and therefore plays a critical role in p53 mediated apoptosis in response to DNA damage (8, 9).
As an important enzyme involved in multiple cellular processes, TIP60 activity should be tightly regulated, since even 2-fold reduction of its activity would result in tumorigenesis and its excessive activation would result in apoptosis (8–10). However, it is still unclear as how TIP60 activity is regulated in vivo, especially in response to DNA damage. In this study, we showed that TIP60 autoacetylates in response to UV and this autoacetylation is important for TIP60 activation in response to DNA damage.
EXPERIMENTAL PROCEDURES
Cell Culture and Plasmids
293T, U2OS and HCT116 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in a humidified incubator with 5% CO2 (v/v). TIP60, p53 or SIRT1 cDNA was subcloned into pDONR201 entry vector and then transferred to the destination vectors containing N-terminal triple-epitope S protein, FLAG, and streptavidin-binding peptide (SFB) tag, Myc-epitope tag, GST tag, or MBP tag using gateway technology (Invitrogen). All point mutants were generated by site-directed mutagenesis (Stratagene) and verified by sequencing. Transfections were performed using Fugene 6 or Lipofectamine according to manufacturer's instructions.
Antibodies
Rabbit polyclonal anti-SIRT1 antibody was described previously (14). Monoclonal anti-Flag M2 and anti-β-actin antibodies were purchased from Sigma. Anti-Ac-Lysine polyclonal antibody was purchased from Calbiochem. The anti-MBP antibody was purchased from Millipore. TIP60 Antibody was a kind gift of Dr. Bruno Amati at European Institute of Oncology. Ac-Lys120-p53 monoclonal antibody was purchased from MBL.
Co-immunoprecipitation and Western Blotting
Cells were lysed with NTEN buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40) containing 20 mm NaF and 1 μg/ml pepstatin A and aprotonin on ice for 20 min. After removal of cell debris by centrifugation, the soluble fractions were collected and incubated with either protein A-agarose beads coupled with anti-TIP60, SIRT1 antibodies, or S protein-Sepharose (Amersham Biosciences) for 3 h at 4 °C. The precipitates were then washed four times with NTEN buffer and boiled in 2× SDS loading buffer. Samples were resolved on SDS-PAGE and transferred to polyvinylidine difluoride membrane, and immunoblottings were carried out with antibodies as indicated.
Pull-down Assay
GST2 or MBP fusion proteins were expressed in Escherichia coli and purified as described previously (15). GST fusion proteins were immobilized on glutathione-Sepharose 4B beads and incubated with lysates prepared from cells transiently transfected with plasmids encoding the indicated constructs. The samples were subjected to SDS-PAGE and analyzed by Western blotting.
RNA Interference
Briefly, cells were transfected twice at 24-h intervals with the indicated small interfering RNAs (siRNAs) using oligofectamine (Invitrogen) according to the manufacturer's instructions. siRNAs against human SIRT1 used were described previously (14). The TIP60 siRNA sequence is GGACAGCUCUGAUGGAAUA.
Acetylation and Deacetylation Assays in Vitro
Acetylation and deacetylation assays in vitro were performed as described before (14, 16). 200 ng of bacterially expressed MBP-TIP60 or MBP-TIP60 mutants were separately incubated with the indicated substrates with 5 mm acetyl-CoA in HAT buffer at 30 °C for the indicated time period. The reaction mixtures were resolved on SDS-PAGE and analyzed by immunoblotting or autoradiography.
RESULTS
TIP60 Autoacetylates in Vivo
Intra- or intermolecular posttranslational modification is a key mechanism for regulating protein trafficking, localization, stability, and activity. For example, autophosphorylation of ATM is believed to be an important step during its activation following DNA damage (17). Likewise, autoacetylation of p300 induces a conformational change in the catalytic domain of p300, which serves as a switch that allows the dissociation of p300, enhances TFIID binding, and activates transcription (18). Moreover, autoubiquitination of MDM2 followed by proteasome-mediated degradation is important at the early stage of DNA damage response because it permits the initial increase of p53 protein level following DNA damage (19). Here, we decided to explore whether a similar mechanism is involved in TIP60 activation in response to DNA damage.
To determine whether TIP60 could be acetylated in vivo, 293T cells were transfected with plasmids encoding SFB-tagged TIP60 or control plasmids encoding SFB-tagged DDB2. These overexpressed proteins were immunoprecipated by S protein-Sepharose and immunoblotted with anti-FLAG or Ac-lysine antibodies. As shown in Fig. 1A, SFB-TIP60, but not SFB-DDB2, could be acetylated in vivo. Further, acetylation of endogenous TIP60 was detected in extracts prepared from U2OS cells transfected with control siRNA but not from those prepared from cells transfected with siRNA specifically targeting TIP60 (Fig. 1B), indicating that endogenous TIP60 is acetylated in vivo. To determine whether this is due to TIP60 autoacetylation, we used wild type or HAT mutant (Q377E/G380E) (7) of TIP60. We first confirmed the HAT domain-inactive mutant of TIP60 using in vitro acetylation assays. Recombinant MBP protein and MBP protein fused with wild type or HAT mutant of TIP60 were purified from E. coli, incubated with acetyl-CoA, and then immunoblotted with Ac-lysine antibody. Wild type TIP60, but not its HAT mutant, was strongly acetylated (Fig. 1C), suggesting that TIP60 can autoacetylate in vitro. Similarly, when we expressed these TIP60 constructs in vivo, we could easily detect acetylation of wild type TIP60 but not that of TIP60 HAT mutant (Fig. 1D). Together, these results suggest that TIP60 can autoacetylate both in vivo and in vitro.
FIGURE 1.

TIP60 autoacetylates itself in vivo and in vitro. A, ectopically expressed SFB-TIP60 was acetylated in vivo. 293T cells were transfected with plasmids encoding SFB-TIP60 or SFB-DDB2. Cell extracts were prepared, and SFB-tagged proteins were precipitated (IP) using S protein-Sepharose and immunoblotted (WB) with anti-FLAG and anti-Ac-lysine antibodies. B, endogenous TIP60 was acetylated in vivo. Extracts prepared from U2OS cells transfected with control or TIP60 siRNAs were immunoprecipitated using anti-TIP60 antibody and immunoblotted with anti-Ac-lysine antibody. C, TIP60 could be autoacetylated in vitro. Recombinant MBP-TIP60 was incubated in HAT buffer with or without acetyl-CoA for 45 min and subjected to immunoblotting with anti-Ac-lysine antibody. D, TIP60 HAT mutant (TIP60-MT) failed to be acetylated in vivo. 293T cells were transfected with plasmids encoding wild type or HAT mutant (TIP60-MT) of TIP60. Cell extracts were prepared, immunoprecipitated using S protein Sepharose beads, and blotted with anti-Ac-lysine antibody and anti-FLAG antibodies.
Acetylation Status of TIP60 Correlates with TIP60 Acetylase Activity
Interestingly, the in vivo acetylation of SFB-tagged TIP60 is quite low in cells stably expressing this construct (Fig. 2A, left lane). In order to increase its acetylation level, we treated cells with trichostatin A, which specifically inhibits the class I and II HDAC family of deacetylase (20), or with nicotinamide, an inhibitor of the NAD-dependent SIRT deacetylase family (16). We found that treatment with trichostatin A had no apparent effect on the acetylation level of TIP60 (Fig. 2A). However, TIP60 acetylation level increased significantly in cells treated with nicotinamide, suggesting that one or more of the SIRT family deacetylases is responsible for TIP60 deacetylation (Fig. 2A).
FIGURE 2.

Autoacetylation activates TIP60 by dissociating TIP60 oligomer and enhances substrate binding. A, nicotinamide but not trichostatin A increased TIP60 acetylation in vivo. U2OS cells stably expressing SFB-TIP60 were treated with varying concentrations of trichostatin A (10–50 μm) for 12 h or nicotinamide (100 μm to 1 mm) for 6 h. Cell extracts were immunoprecipitated (IP) using S protein-Sepharose beads and immunoblotted (WB) with anti-FLAG antibody and anti-Ac-lysine antibodies. B, SFB-TIP60 was purified from nicotinamide-treated or -untreated 293T cells and incubated with GST-p53 and acetyl-CoA in HAT buffer for the indicated times. Protein levels and their acetylation status were evaluated by immunoblotting using antibodies as indicated. C, reactions similar to those described in B were carried out using 14C-labeled acetyl-CoA and 50 ng of calf thymus histones (instead of GST-p53) as substrate. Proteins were resolved on SDS-PAGE and analyzed by autoradiography or Coomassie staining as indicated. D, acetylated TIP60 binds to GST-p53 more efficiently than unacetylated TIP60. SFB-TIP60 was purified from nicotinamide-treated or -untreated 293T cells and then incubated with beads coated with GST-p53. A pull-down assay was performed and assessed by immunoblotting as indicated. E, TIP60 forms oligomer in vivo. 293T cells were transfected with plasmids encoding SFB-TIP60 together with plasmids encoding Myc-TIP60. Cell lysates were precipitated by S protein-Sepharose beads and blotted with the indicated antibodies. F, TIP60 forms oligomer in vitro. Beads coated with GST-TIP60 were incubated with bacterially expressed and purified MBP-TIP60. After extensive wash, proteins were eluted from beads and analyzed by Western blotting using anti-MBP antibody. The arrow indicates the position of full-length MBP-TIP60. The faster migrating bands are degradation products of MBP-TIP60. G, autoacetylation dissociates TIP60 oligomer. 293T cells were transfected with plasmids encoding Myc-tagged TIP60 together with plasmids encoding SFB-TIP60. Cell extracts were immunoprecipitated and immunoblotted using the indicated antibodies.
To determine whether acetylation of TIP60 affects its HAT activity, SFB-TIP60 was purified from untreated or nicotinamide-treated U2OS cells, and in vitro acetylation assays were performed using recombinant GST-p53 as substrate. As shown in Fig. 2B, TIP60 isolated from nicotinamide-treated cells acetylated GST-p53 more efficiently at early time points (5 or 10 min) than TIP60 isolated from control cells. This acetylase activity of TIP60 toward its substrate p53 correlates well with its own acetylation status, suggesting that acetylated TIP60 is more active than unacetylated TIP60. However, after 30 min of incubation, both nicotinamide-treated and untreated TIP60 were acetylated at similar levels due to its autoacetylation in vitro. As a consequence, their abilities to acetylate GST-p53 were comparable (Fig. 2B, lanes 5 and 6).
We also used calf thymus histones as substrates for these in vitro assays. Similarly, TIP60 isolated from nicotinamide-treated cells exhibited enhanced histone acetylation activity, which correlates well with its acetylation status (Fig. 2C). These results suggest that the acetylation level of TIP60 is important for regulating TIP60 activity.
Autoacetylation Activates TIP60 by Dissociating TIP60 Oligomer and Enhancing Substrate Access
There are several potential mechanisms for how autoacetylation could affect recognition of substrates. It is possible that autoacetylation of TIP60 could induce a conformational change in its catalytic domain, similar to that reported for p300 (18), and thus increase its activity. This may be in part mediated by an enhanced binding of TIP60 to its substrates. To test this possibility, we isolated differentially acetylated TIP60 from control cells or cells treated with nicotinamide. As shown in Fig. 2D, acetylated SFB-TIP60 bound strongly to immobilized GST-p53 but not to GST alone, whereas weakly acetylated SFB-TIP60 barely interacted with GST-p53, indicating that indeed acetylation of TIP60 promotes the binding of TIP60 to its substrates.
Another non-exclusive explanation is that autoacetylation may lead to the dissociation of potential TIP60 inhibitors and thus permit TIP60 activation. Such inhibitors can be TIP60 itself or other TIP60-associated proteins. We first checked whether TIP60 could form oligomers. Myc-tagged TIP60 was co-immunoprecipitated with SFB-tagged TIP60, suggesting that TIP60 was able to interact with itself in vivo (Fig. 2E). To further examine whether or not this interaction is direct, a pull-down assay was performed using GST-TIP60 and MBP-TIP60 purified from bacteria. As shown in Fig. 2F, GST-TIP60 specifically associated with MBP-TIP60 but not MBP, indicating that TIP60 interacts directly with itself in vitro.
Next we asked whether autoacetylation of TIP60 would affect its oligomer formation. To examine this possibility, cells were transfected with plasmids encoding wild type Myc-TIP60 together with plasmids encoding corresponding wild type or HAT mutant of SFB-TIP60. Both wild type and HAT mutant of Myc-tagged TIP60 could be co-immunoprecipitated with their corresponding SFB-tagged TIP60 partners (Fig. 2G). Interestingly, the association between wild type TIP60s was significantly reduced in nicotinamide-treated cells compared with that observed in control cells (Fig. 2G). This was accompanied by increased TIP60 acetylation isolated from cells treated with nicotinamide (Fig. 2G). In comparison, the association between HAT mutants of TIP60 was always stronger and was not affected by nicotinamide treatment (Fig. 2G). Together, these data indicate that TIP60 may normally exist as oligomers in the cell. Its autoacetylation would dissociate this oligomer formation, increase the binding of TIP60 to its substrates, and thus enhance TIP60 acetylase activity.
UV Radiation Augments TIP60 Autoacetylation
In order to assess the physiological relevance of TIP60 autoacetylation, we explored whether TIP60 acetylation would change in response to DNA damage. U2OS cells stably expressing SBP-tagged TIP60 were mock-treated or UV-irradiated. UV irradiation resulted in robust autoacetylation of TIP60 in a time-dependent and dose-dependent manner (Fig. 3, A and B). Caffeine, which inhibits the catalytic activity of both ATM and ATR, efficiently inhibited autoaceylation of TIP60 (Fig. 3C), suggesting that indeed DNA damage could signal to TIP60 and control TIP60 autoacetylation.
FIGURE 3.
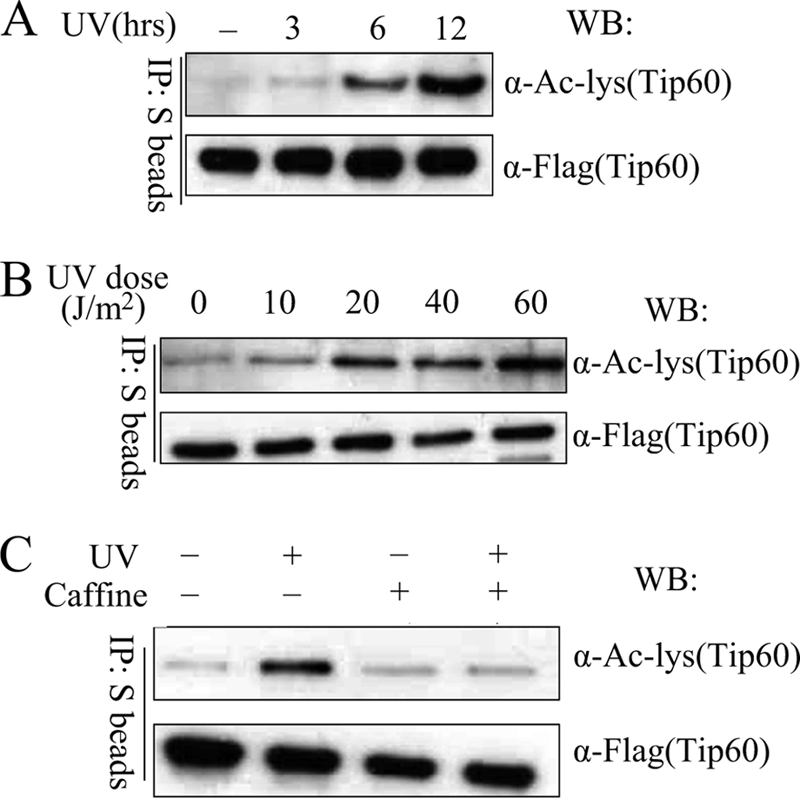
UV radiation augments TIP60 autoacetylation. A, autoacetylation of TIP60 is induced by UV radiation. U2OS cells stably expressing TIP60 were irradiated with 50 J/m2 UV and then incubated for the indicated period of time. Cells were harvested, lysed, and subjected to precipitation (IP) and immunoblotting (WB) as indicated. B, dose-dependent induction of TIP60 autoacetylation following UV radiation. U2OS cells stably expressing TIP60 were UV-irradiated with the indicated dosages. Cells were collected at 12 h. Precipitation and immunoblotting were performed as indicated. C, caffeine blocks UV-induced TIP60 autoacetylation. U2OS cells stably expressing TIP60 were preincubated with 1 mm caffeine for 1 h before they were irradiated (40 J/m2). Precipitation and immunoblotting were performed as outlined in B.
The time course experiment performed above showed that autoacetylation of TIP60 is not a rapid event following UV-induced DNA damage. We speculate that this may indicate that autoacetylation of TIP60 occurs late in cellular DNA damage response and may participate in late events like cell cycle arrest or apoptosis.
SIRT1 Is a Negative Regulator of TIP60
The significant increase of TIP60 acetylation following nicotinamide treatment (Fig. 2A) suggests that a major mechanism for regulating TIP60 acetylation is mediated through TIP60 deacetylation. Therefore, we decided to examine whether one or more members of the SIRT family of deacetylases would participate in deacetylating TIP60. Interestingly, when we examined the interaction between TIP60 and SIRTs, we found that only SIRT1, and not any other SIRTs, co-immunoprecipitated strongly with TIP60 (Fig. 4A). In addition, only overexpression of SIRT1 led to a decreased TIP60 acetylation level (Fig. 4A), indicating a specific functional interaction between SIRT1 and TIP60. Indeed, we confirmed the interaction between endogenous SIRT1 with endogenous or overexpressed TIP60 (Fig. 4B).
FIGURE 4.

SIRT1 binds to acetylated TIP60. A, TIP60 specifically interacts with SIRT1 in vivo. 293T cells were transfected with plasmids encoding each of the seven Myc-tagged SIRTs (SIRT1 to -7), together with plasmids encoding SFB-TIP60. Cell extracts were precipitated (IP) using S protein-Sepharose beads. The precipitates were analyzed by immunoblotting (WB) using antibodies as indicated. B, a co-immunoprecipitation assay was performed to verify the interaction between TIP60 and SIRT1. C, SIRT1 failed to bind to unacetylated TIP60. 293T cells were transfected with plasmids encoding wild type or the HAT mutant of SFB-TIP60. Cell extracts were precipitated with S protein-Sepharose beads and immunoblotted using antibodies as indicated. D, recombinant MBP-TIP60 was autoacetylated to different levels in vitro before they were used in a pull-down assay and incubated with U2OS cell extracts. Inputs and MBP-TIP60-associated SIRT1 were analyzed by immunoblotting using indicated antibodies.
When we used wild type or HAT domain mutant of TIP60, we found that endogenous SIRT1 associated readily with wild type but not the HAT mutant of TIP60 (Fig. 4C). This raises the question of whether or not SIRT1 only binds strongly to TIP60 when TIP60 is acetylated. In order to test this directly, we generated MBP-TIP60 with variable acetylation levels by incubating it in acetylation reactions for different times. Indeed, we observed an increased interaction between SIRT1 and TIP60 when TIP60 was acetylated (Fig. 4D). In order to determine whether TIP60-SIRT1 association is regulated by DNA damage, co-immunoprecipitation experiments were performed in mock-treated cells or UV-irradiated cells. As shown in supplemental Fig. 1, UV irradiation greatly enhanced TIP60 acetylation and likewise augmented TIP60-SIRT1 interaction. These data imply that SIRT1 is a negative regulator of TIP60. When TIP60 is acetylated, SIRT1 would bind to and deacetylate TIP60.
SIRT1 Deacetylates TIP60 and Negatively Regulates TIP60 Activity in Vivo
Next we further examined the functional interaction between TIP60 and SIRT1. Because endogenous TIP60 migrates with immunoglobin heavy chains, we chose to use U2OS cells stably expressing SFB-tagged TIP60. As shown in Fig. 5A, RNA interference-mediated knockdown of SIRT1 significantly increased the acetylation level of TIP60, suggesting that endogenous SIRT1 is involved in deacetylating TIP60 in vivo. To confirm directly that it is SIRT1, and not any SIRT1-regulated events, that is involved in TIP60 deacetylation, we performed in vitro deacetylation experiments and showed that only wild type and not the deacetylase-defective mutant (H363Y) of SIRT1 could specifically deacetylate TIP60 in vitro (Fig. 5B). In addition, these in vitro deacetylation reactions require NAD and can be inhibited by nicotinamide (Fig. 5B), further suggesting that SIRT1 is the enzyme that directly deacetylates TIP60 in vitro. Taken together, these data demonstrate that SIRT1 can specifically deacetylate TIP60 in vivo and in vitro.
FIGURE 5.

SIRT1 deacetylates TIP60 and negatively regulates TIP60 activity. A, SIRT1 deacetylates TIP60 in vivo. U2OS cells stably expressing SFB-TIP60 was transfected with SIRT1-specific siRNA or control siRNA. Cells were harvested and subjected to immunoblotting analysis with the indicated antibodies. B, SIRT1 deacetylates TIP60 in vitro. Wild type or deacetylase-defective mutant (H363Y) of SIRT1 was expressed in 293T cells and purified by S protein-Sepharose beads. The purified SIRT1s were then incubated with acetylated MBP-TIP60 without or with the addition of NAD+ or nicotinamide in the reactions. The mixtures were analyzed by immunoblotting using the indicated antibodies. C, SIRT1 failed to deacetylate Lys120 of p53 in vitro. Recombinant MBP-SIRT1 was purified from E. coli and incubated with acetylated SFB-p53. The proteins were immunoblotted with the indicated antibodies. D, SIRT1 negatively regulated HAT activity of TIP60. SFB-TIP60 purified from U2OS cells was incubated with GST-p53 together with wild type or catalytic inactive H363Y mutant of MBP-SIRT1 in HAT buffer for 45 min. The mixtures were analyzed by immunoblotting with the indicated antibodies. E and F, HCT116 cells were transfected with TIP60, SIRT1, or control siRNAs. 72 h after transfection, cells were UV-irradiated (30 J/m2) and harvested 8 h (E) 16 h (F) later. Western blotting was performed using the indicated antibodies (E). The cell cycle profile was determined by FACS analysis, and the percentage of cells in the sub-G1 fraction is indicated (F). G, a proposed model indicating how acetylation of TIP60 regulates its HAT activity. During normal cell proliferation, most TIP60 proteins are unacetylated and form inactive oligomers. In response to DNA damage, TIP60 autoacetylates itself, and the inactive oligomer of TIP60 dissociates. Activated TIP60 can now acetylate p53 at the Lys120 site, which promotes apoptosis. SIRT1 on the other hand, deacetylates TIP60, thereby regulating its HAT activity and keeping the levels of activated TIP60 in check. IP, immunoprecipitation; WB, Western blotting.
The major downstream substrate of TIP60 involved in DNA damage response is likely to be p53. TIP60 specifically acetylates p53 at the Lys120 site, which is critically important for activating apoptosis following DNA damage (8, 9). Thus, we would like to further examine whether or not SIRT1 would regulate TIP60 acetylation and thus influence p53 activity. However, early studies have already shown that SIRT1 can directly deacetylate p53 (e.g. at the Lys382 residue) in vitro and in vivo (21). Therefore, we first wanted to examine whether or not SIRT1 would directly deacetylate p53 at the Lys120 site, which is the major site acetylated by TIP60. As shown in Fig. 5C, although the total acetylation level of p53 decreased following incubation with SIRT1, acetylation of p53 at the Lys120 site did not change. This result indicates that although SIRT1 can deacetylate p53 as previously reported (16, 21), SIRT1 does not deacetylate p53 at the Lys120 site.
Having established that the Lys120 site of p53 is not the SIRT1 substrate, we went on and tested whether SIRT1 could regulate p53 acetylation at the Lys120 site via TIP60. As expected, TIP60 specifically acetylated p53 at the Lys120 site in vitro (Fig. 5D). Interestingly, SIRT1 inhibited the autoacetylation of TIP60 and also specifically inhibited the ability of TIP60 to acetylate p53 at the Lys120 site (Fig. 5D). As a control, the SIRT1 catalytic inactive mutant did not have any effect in this assay (Fig. 5D). Together, these data showed that SIRT1 negatively regulates TIP60 activity in vitro.
In order to provide direct evidence to support the theory that SIRT1 deacetylates TIP60 and negatively regulates TIP60 activity in vivo, we used siRNAs to specifically down-regulate SIRT1 in the cell. Reduction of SIRT1 in U2OS cells resulted in enhanced TIP60 acetylation before and after UV radiation, which subsequently led to increased p53 acetylation at the Lys120 site (supplemental Fig. 2). These data suggest that SIRT1 negatively regulates TIP60 and therefore affects p53 Lys120 acetylation in vivo.
SIRT1 Represses UV-induced Apoptosis Partly through Negatively Regulating TIP60
Because acetylation of p53 at the Lys120 site by TIP60 is known to play a role in apoptosis following DNA damage (8, 9), we explored whether regulation of TIP60 activity by SIRT1 would play a role in this process. We depleted TIP60, SIRT1, or both in p53-proficient HCT116 cells (Fig. 5E) and examined UV-induced apoptosis. UV-induced apoptosis, as measured by poly-ADP-ribose polymerase-1 (PARP1) cleavage (Fig. 5E) or by sub-G1 cells (Fig. 5F), increased significantly following SIRT1 depletion, indicating that SIRT1 plays a negative role in promoting apoptosis following DNA damage. The removal of TIP60 in SIRT1 depletion cells abolished the effects of SIRT1 (Fig. 5, E and F), indicating that SIRT1 and TIP60 antagonize each other in this process and that SIRT1 negatively regulates UV-induced apoptosis at least partially through its regulation of TIP60.
DISCUSSION
Recent studies suggest that TIP60 participates in many cellular processes, including signal transduction, DNA damage repair, cell cycle and checkpoint control, and apoptosis (1, 3, 22). In this study, we have uncovered a regulatory mechanism of TIP60. We showed that TIP60 autoacetylation is important for TIP60 activation following DNA damage and that this activation is negatively regulated by SIRT1 in vivo. Thus, our study has not only provided new insights into the regulation of TIP60 activity but also revealed a new substrate of SIRT1.
Protein acetylation, similar to phosphorylation, is emerging as an important player in regulating protein activity and signal transduction in response to DNA damage (23, 24). TIP60 is known to acetylate and activate several key DNA damage-responsive proteins, such as H2AX, ATM, and p53 (7–9, 24). Interestingly, although H2AX and ATM are acetylated rapidly (<30 min) in response to DNA damage (25, 26), the TIP60-dependent acetylation of p53 occurs relative later (about 6 h after UV irradiation) (8), suggesting that p53 acetylation may somehow be regulated differently. In our study, we showed that similar to p53 acetylation of the Lys120 site, TIP60 autoacetylation took place late after UV damage, suggesting that autoacetylation of TIP60 enhances TIP60 activity, which may not be critical for other substrates but is important for regulating p53 activity.
Acetylation of p53 at the Lys120 site is a key factor in deciding cell cycle arrest versus apoptosis (8, 9). In the presence of a lower amount of DNA damage, p53 is mainly deacetylated at the Lys120 site and induces cell cycle arrest through up-regulation of p21 and other cell cycle inhbitors (8, 9). Increasing amounts of DNA damage lead to increased p53 acetylation at the Lys120 site, which results in decreased p53 affinity for the p21 promoter and enhanced activation of proapoptotic gene transcription, thereby turning the balance of p53-responsive genes toward apoptosis (8, 9). This hypothesis is entirely consistent with the data we provided in this study. As we showed in Fig. 3, only relatively high doses of UV irradiation would induce TIP60 autoaceylation, suggesting that high doses of UV irradiation are required to activate TIP60 and enhance TIP60 activity, which subsequently acetylates p53 at the Lys120 site to promote apoptosis.
Our study also provides a mechanism for how autoacetylation of TIP60 would enhance its HAT activity. We showed that TIP60 could form oligomers in vivo, which dissociate after TIP60 autoacetylation. This dissociation enables TIP60 to bind to its substrates like p53 more efficiently and thus promote substrate acetylation. In order to directly test that autoacetylation of TIP60 is critical for its activation, we need to identify all of the autoacetylation sites on TIP60. Our initial attempts have led to the identification of four acetylation sites on TIP60 (Lys76, Lys80, Lys189, and Lys327) by mass spectrometry analysis (data not shown). We are currently investigating whether these acetylation events are important for TIP60 activation and also improving our methodology to identify additional autoacetylation sites.
Our study has also allowed us to discover SIRT1 as a negative regulator of TIP60. SIRT1 is known to promote cell survival through deacetylating and negatively regulating many substrates, including p53 and FOXO (16, 27, 28). We have added one more substrate, TIP60, to this growing list. We showed that SIRT1 does not directly deacetylate Lys120 of p53. Instead, SIRT1 negatively regulates TIP60 activation and thus inhibits p53 acetylation at Lys120. Such a complex network of regulations suggests that the balance of TIP60 (and p53) activity is critically important in vivo. This is supported by the observations that loss of one allele of TIP60 or p53 would greatly promote tumorigenesis (10).
SIRT1 is known to deacetylate several substrates involved in DNA damage response. For example, the acetylation level of NBS1 is tightly regulated by SIRT1, and the deacetylation of NBS1 by SIRT1 plays a key role in the dynamic regulation of DNA damage response (29). Interestingly, TIP60 may also play a role in this process because depletion of TIP60 inhibits NBS1 recruitment to the DNA damage site (30). Recently, the acetylation of H2AX, another TIP60 substrate, has also been shown to be negatively regulated by SIRT1 (31). These data suggest that besides the indirect mechanism we have proposed here, SIRT1 could also work directly on certain TIP60 substrates and thus counteract TIP60 functions in vivo. Such a complex relationship between SIRT1 and TIP60 in DNA damage response warrants further investigation.
In summary, our study has revealed a new regulatory mechanism of controlling TIP60 activity and function in response to UV radiation. Our data strongly suggest that TIP60 autoacetylation is important for its activation following UV damage. This activity of TIP60 is negatively regulated by SIRT1, which permits a fine balance to control p53 activity and outcomes following DNA damage.
Supplementary Material
Acknowledgments
We thank all colleagues in the Chen laboratory, especially Dr. Gargi Ghosal, for proofreading of the manuscript. We also thank Dr. Bruno Amati at the European Institute of Oncology for sharing the TIP60 antibody.
This work was supported, in whole or in part, by National Institutes of Health Grants CA089239, CA092312, and CA100109 (to J. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- GST
- glutathione S-transferase
- MBP
- maltose-binding protein
- HAT
- histone acetyltransferase
- siRNA
- small interfering RNA
- SFB
- S protein, FLAG, and streptavidin-binding peptide.
REFERENCES
- 1.Sapountzi V., Logan I. R., Robson C. N. (2006) Int. J. Biochem. Cell Biol. 38, 1496–1509 [DOI] [PubMed] [Google Scholar]
- 2.Squatrito M., Gorrini C., Amati B. (2006) Trends Cell Biol. 16, 433–442 [DOI] [PubMed] [Google Scholar]
- 3.Sun Y., Jiang X., Xu Y., Ayrapetov M. K., Moreau L. A., Whetstine J. R., Price B. D. (2009) Nat. Cell Biol. 11, 1279–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimura A., Horikoshi M. (1998) Genes Cells 3, 789–800 [DOI] [PubMed] [Google Scholar]
- 5.Ikura T., Ogryzko V. V., Grigoriev M., Groisman R., Wang J., Horikoshi M., Scully R., Qin J., Nakatani Y. (2000) Cell 102, 463–473 [DOI] [PubMed] [Google Scholar]
- 6.Kranz D., Dohmesen C., Dobbelstein M. (2008) J. Cell Biol. 182, 197–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y., Jiang X., Chen S., Fernandes N., Price B. D. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sykes S. M., Mellert H. S., Holbert M. A., Li K., Marmorstein R., Lane W. S., McMahon S. B. (2006) Mol. Cell 24, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Y., Luo J., Zhang W., Gu W. (2006) Mol. Cell 24, 827–839 [DOI] [PubMed] [Google Scholar]
- 10.Gorrini C., Squatrito M., Luise C., Syed N., Perna D., Wark L., Martinato F., Sardella D., Verrecchia A., Bennett S., Confalonieri S., Cesaroni M., Marchesi F., Gasco M., Scanziani E., Capra M., Mai S., Nuciforo P., Crook T., Lough J., Amati B. (2007) Nature 448, 1063–1067 [DOI] [PubMed] [Google Scholar]
- 11.Kruse J. P., Gu W. (2009) Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter S., Vousden K. H. (2009) Curr. Opin. Genet. Dev. 19, 18–24 [DOI] [PubMed] [Google Scholar]
- 13.Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. (2008) Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J. E., Chen J., Lou Z. (2008) Nature 451, 583–586 [DOI] [PubMed] [Google Scholar]
- 15.Hofer B., Backhaus S., Timmis K. N. (1994) Gene 144, 9–16 [DOI] [PubMed] [Google Scholar]
- 16.Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 17.Bakkenist C. J., Kastan M. B. (2003) Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 18.Black J. C., Choi J. E., Lombardo S. R., Carey M. (2006) Mol. Cell 23, 809–818 [DOI] [PubMed] [Google Scholar]
- 19.Stommel J. M., Wahl G. M. (2004) EMBO J. 23, 1547–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laherty C. D., Yang W. M., Sun J. M., Davie J. R., Seto E., Eisenman R. N. (1997) Cell 89, 349–356 [DOI] [PubMed] [Google Scholar]
- 21.Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 22.Fazzio T. G., Huff J. T., Panning B. (2008) Cell 134, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huen M. S., Chen J. (2008) Cell Res. 18, 8–16 [DOI] [PubMed] [Google Scholar]
- 24.van Attikum H., Gasser S. M. (2009) Trends Cell Biol. 19, 207–217 [DOI] [PubMed] [Google Scholar]
- 25.Ikura T., Tashiro S., Kakino A., Shima H., Jacob N., Amunugama R., Yoder K., Izumi S., Kuraoka I., Tanaka K., Kimura H., Ikura M., Nishikubo S., Ito T., Muto A., Miyagawa K., Takeda S., Fishel R., Igarashi K., Kamiya K. (2007) Mol. Cell Biol. 27, 7028–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y., Xu Y., Roy K., Price B. D. (2007) Mol. Cell Biol. 27, 8502–8509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., Alt F. W., Greenberg M. E. (2004) Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 28.Giannakou M. E., Partridge L. (2004) Trends Cell Biol. 14, 408–412 [DOI] [PubMed] [Google Scholar]
- 29.Yuan Z., Zhang X., Sengupta N., Lane W. S., Seto E. (2007) Mol. Cell 27, 149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chailleux C., Tyteca S., Papin C., Boudsocq F., Puget N., Courilleau C., Grigoriev M., Canitrot Y., Trouche D. (2010) Biochem. J. 426, 365–371 [DOI] [PubMed] [Google Scholar]
- 31.Yamagata K., Kitabayashi I. (2009) Biochem. Biophys. Res. Commun. 390, 1355–1360 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.