

Abstract
In this study, we demonstrate that human cardiomyocytes (AC16) produce reactive oxygen species (ROS) and inflammatory cytokines in response to Trypanosoma cruzi. ROS were primarily produced by mitochondria, some of which diffused to cytosol of infected cardiomyocytes. These ROS resulted in an increase in 8-hydroxyguanine lesions and DNA fragmentation that signaled PARP-1 activation evidenced by poly(ADP-ribose) (PAR) modification of PARP-1 and other proteins in infected cardiomyocytes. Phenyl-α-tert-butylnitrone blocked the mitochondrial ROS (mtROS) formation, DNA damage, and PARP-1 activation in infected cardiomyocytes. Further inhibition studies demonstrated that ROS and PARP-1 signaled TNF-α and IL-1β expression in infected cardiomyocytes. ROS directly signaled the nuclear translocation of RelA (p65), NF-κB activation, and cytokine gene expression. PARP-1 exhibited no direct interaction with p65 and did not signal its translocation to nuclei in infected cardiomyocytes. Instead, PARP-1 contributed to PAR modification of p65-interacting nuclear proteins and assembly of the NF-κB transcription complex. PJ34 (PARP-1 inhibitor) also prevented mitochondrial poly(ADP-ribosyl)ation (PARylation) and ROS formation. We conclude that T. cruzi-mediated mtROS provide primary stimulus for PARP-1-NF-κB activation and cytokine gene expression in infected cardiomyocytes. PAR modification of mitochondrial membranes then results in a feedback cycle of mtROS formation and DNA damage/PARP-1 activation. ROS, either through direct modulation of cytosolic NF-κB, or via PARP-1-dependent PAR modification of p65-interacting nuclear proteins, contributes to cytokine gene expression. Our results demonstrate a link between ROS and inflammatory responses in cardiomyocytes infected by T. cruzi and provide a clue to the pathomechanism of sustained inflammation in Chagas disease.
Keywords: Oxygen/Radicals, Cytokines Induction, Mitochondria, NF-κB, Parasite, PARP-1, Trypanosoma cruzi, Cardiomyocytes
Introduction
Trypanosoma cruzi, a hemoflagellate protozoan parasite, accounts for ∼16–18 million cases in Latin America (1). Upon infection, asynchronous cycles of parasite multiplication and host cell destruction result in nonspecific clinical symptoms. Most patients then remain seropositive and clinically asymptomatic. Several years later, 30–40% of the patients develop chronic Chagas disease with symptomatic arrhythmia, thromboembolic events, and heart failure (2).
The inflammatory response in the myocardium is an important aspect of the pathogenesis of chagasic heart disease. Acute infection by T. cruzi results in intense inflammatory reaction consisting primarily of leukocytes, including eosinophils and macrophages, accompanied by increased expression of inflammatory mediators such as cytokines, chemokines, and nitric oxide synthase in the heart (3, 4). During the chronic phase, diffused inflammatory infiltrate persists in the myocardium (reviewed in Refs. 5, 6). Besides infiltration of inflammatory infiltrate, cardiomyocytes are also reported to produce cytokines in response to T. cruzi infection (7), although the signaling events are not known.
Chagasic hearts are also exposed to oxidative stress (reviewed in Ref. 8). Others and we have evaluated the antioxidant/oxidant status in chagasic experimental models and human patients. We have documented that an inefficient antioxidant defense in the myocardium of infected rodents with progressive disease development is associated with increased level of ROS,2 decreased activity of superoxide dismutase and insensitivity of glutathione defense to oxidative stress (9). A substantial increase in glutathione disulfide contents and decline in superoxide dismutase and glutathione peroxidase activities was also noted in peripheral blood of chagasic animals and seropositive patients (10–12). Although inflammatory infiltrate constituted by activated neutrophils and macrophages contributes to oxidative stress in acute hearts (13, 14), we have found that T. cruzi alters the mitochondrial membrane potential and initiates a feedback cycle of mitochondrial leakage of electrons to molecular oxygen and ROS production in the cardiomyocytes and chagasic myocardium (15, 16).
ROS cytotoxicity triggers protein, lipid, and DNA oxidation. ROS induces single strand breaks in DNA, and oxidation of guanine to 8-oxoguanine (8-oxoG) signals the activation of members of the poly(ADP-ribose) polymerase (PARP) family that facilitate DNA repair (17). PARP-1 accounts for >85% of the PARP activity in most cellular systems, and it catalyzes cleavage of NAD+ into nicotinamide and ADP-ribose and uses the latter to synthesize long branching PAR polymers (17). The basal level activation of PARP-1 by mild genotoxic stimuli causes PARylation of histone proteins (e.g. H1 and H2B) that mediates relaxation of the chromatin superstructure and recruitment of DNA-break repair enzymes (18, 19), resulting in DNA repair and cell survival. PARP-1 is also suggested to mediate transcriptional regulation of NF-κB and AP-1 via noncovalent protein-protein interaction or by active PARylation of signaling molecules, although the mechanistic details of PARP interaction with NF-κB are yet to be elucidated.
In this study, we determined whether ROS signals PARP-1 activation in T. cruzi-infected cardiomyocytes and if PARP-1 up-regulates the transcriptional activity of NF-κB and production of pro-inflammatory cytokines in cardiomyocytes. Our results demonstrate a link between ROS and inflammatory responses in cardiomyocytes infected by T. cruzi and provide clues to the pathomechanism of sustained inflammation in Chagas disease.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Monoclonal antibody against poly ADP-ribose (PAR) (ALX-804-220) was purchased from Alexis (San Diego, CA). Monoclonal antibodies against PARP-1 (B-10, sc-74470) and NF-κB p65 (F-6, sc-8008), and polyclonal antibodies against PARP-1 (H-300, sc-25780) and Lamin A/C (H-110, sc-20681), were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody against 8-oxoG (MAB3560) was purchased from Millipore (Billerica, MA). PJ34 (P4365, PARP-1 inhibitor), PBN (B7263, free-radical spin trap), 3-methyl-1,6,8-trihydroxyanthraquinone (emodin, E7881, NF-κB inhibitor), and other chemicals were of highest purity and purchased from Sigma-Aldrich.
Cell Culture and Infection
Generation of a human cardiomyocyte cell line, AC16, has been described previously (20). AC16 cardiomyocytes were cultured and maintained in Dulbecco's modified Eagle's medium/F-12 medium with 12.5% fetal bovine serum. T. cruzi trypomastigotes (SylvioX10/4 strain) were propagated in C2C12 cells in RPMI 1640 medium with 5% fetal bovine serum. Cardiomyocytes seeded in 6-well plates (5 × 104/well) or Nunc® Lab-Tek II chamber slides (104/well) were infected with T. cruzi trypomastigotes (cell:parasite ratio, 1:3) and then incubated for 0–48 h. In some experiments, cells were incubated with addition of PJ34 (10 μm) or PBN (1–500 μm) in the media.
Fluorescence Microscopy
AC16 cardiomyocytes were cultured in Nunc® chamber slides and infected with parasites in the presence or absence of PBN or PJ34 for 12 h. The cells were washed thrice to remove free parasites and labeled at 37 °C for 30 min with the following fluorescent probes: 200 nm MitoTracker Red (Ex581 nm/Em644 nm) or MitoTracker Green (Ex490 nm/Em516 nm), 10 μm H2DCFDA (detects intracellular/intramitochondrial ROS, Ex498 nm/Em598 nm), 5 μm MitoSOX Red (specifically detects mitochondrial O2˙̄, Ex510 nm/Em580 nm), 0.1 μm dihydroethidium (DHE, oxidized by cytosolic ROS to fluorescent ethidium that binds nuclear DNA, Ex518 nm/Em605 nm), and 10 μm JC-1 (a sensitive marker of mitochondrial membrane potential (Δψm), Ex485 nm/Em535 nm for green monomers and Ex560 nm/Em595 nm for red aggregates). For immunofluorescent detection of PAR polymers, AC16 cells were labeled with MitoTracker Red (as above), washed to remove unbound probe, and fixed with ice-cold methanol for 20 min. After blocking with 2% fetal bovine serum for 20 min, cells were incubated for 1 h each with anti-PAR antibody (1:100 dilution) and fluorescein isothiocyanate-conjugated secondary antibody. Fluorescence was detected on an Olympus BX-15 fluorescence microscope equipped with a digital camera (magnification, 40× or 60×) (16). All fluorescent probes were purchased from Invitrogen/Molecular Probes.
Immunocytochemistry of 8-oxoG
AC16 cells (wt and infected) were incubated for 12 h in the presence and absence of 500 μm PBN. Cells were fixed and blocked as above and incubated for 1 h each with anti-8-oxoG antibody (1:100 dilution) and horseradish peroxidase-conjugated goat anti-mouse secondary antibody. Color was developed with diaminobenzidine tetrahydrochloride substrate, and images were captured as above (magnification, 40×).
Comet Assay
We utilized a Trevigen Comet Assay kit to detect the DNA damage and fragmentation according to protocols provided by the manufacturer. Briefly, wt and infected cardiomyocytes (±PBN) were harvested, and washed with ice-cold phosphate-buffered saline. Pellets (1 × 105 cells) were suspended in low melting point agarose (1:10 ratio, v/v) and immediately transferred to slides. After agarose was polymerized, slides were incubated in lysis solution at 4 °C for 60 min, and alkaline solution (300 mm NaOH, 1 mm EDTA, pH 13) for 30 min. Electrophoresis was performed in alkaline solution at 25 V (1 V/cm, 300 mA), 4 °C for 40 min. Slides were then washed twice with H2O, once with 70% ethanol, and air-dried and stained with SYBR Green I. Fluorescence was examined as above, and tail moment, indicative of DNA fragmentation, was calculated for at least 50 nuclei per sample using the Comet scoring software (TriTek, Sumerduck, VA).
Cytosolic and Nuclear Fractions
AC16 cardiomyocytes (wt and infected, 2 × 106/ml) were incubated on ice for 30 min in buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride) containing 0.625% Nonidet P-40. Cell lysates were centrifuged at 4 °C, 10,000 × g for 1 min, and supernatants were stored as cytosolic fraction. Pellets were washed with buffer A, suspended in buffer B (20 mm HEPES, pH 7.9, 0.4 m NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride), centrifuged at 4 °C, 13,000 × g for 5 min, and the resultant supernatant (nuclear extract) stored at −20 °C (21).
Immunoprecipitation and Immunoblotting
AC16 cardiomyocytes (wt and infected, ± inhibitors) were incubated on ice for 30 min in lysis buffer (2 × 107 cells/ml) constituted of 50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Nonidet P-40, 2.5 mm KH2PO4, 1 mm each of glycerophosphate, NaF, and Na3VO4, and proteinase inhibitor mixture (Sigma). Cell lysates were centrifuged at 4 °C, 13,000 × g for 15 min, and the resultant supernatants were pre-adsorbed with protein G-Sepharose (Amersham Biosciences). The pre-cleared supernatants were incubated at 4 °C with monoclonal anti-PARP-1 or anti-p65 antibody (1:100 dilution) for 12 h and protein G-Sepharose beads for 2 h. The antigen-antibody-protein G-Sepharose complex was precipitated by centrifugation at 13,000 × g for 1 min, and proteins were resolved on 10% gradient gels, and transferred to polyvinylidene difluoride membrane (Bio-Rad). Membranes were probed with specific antibodies against PAR, PARP-1, or p65, and chemiluminescence was detected by using an ECL Plus system (Amersham Biosciences).
Transient Transfection and Luciferase Activity Assay
The luciferase reporter plasmid pNF-κB-luc with 5× NF-κB binding site (TGGGGACTTTCCGC)5 was purchased from Stratagene (La Jolla, CA). The pREP7-Rluc containing Renilla luciferase (positive control) was kindly provided by Dr. Keji Zhao (National Institutes of Health, Bethesda, MD). Cardiomyocytes were seeded in 6-well plates and incubated overnight in antibiotic-free Opti-MEM medium. Cells were transfected with pNF-κB-luc (3 μg) and pREP7-Rluc (100 ng) using Lipofectamine 2000 (Invitrogen) according to instructions provided by the manufacturer. After 6 h of incubation with plasmids and Lipofectamine 2000, cells were replenished with complete medium for 3–5 h to promote recovery, and then used for infection and/or treatment with inhibitors (as above). The relative NF-κB transcriptional activity was detected using a Dual Luciferase Assay System (Promega, Madison, WI) and normalized to Renilla luciferase activity.
Reverse Transcription and Real-time RT-PCR
Total RNA from infected and/or treated cardiomyocytes was extracted using TRIzol reagent (Invitrogen), the contaminating DNA was removed with a DNA-free kit (Ambion), and 2 μg of purified RNA was transcribed to cDNA. The cDNA was used as template, and real-time PCR was performed with an ABI Prism 7000 Sequence Detection System using the SYBR Green I master mix (Applied Biosystems, Foster City, CA) (22). Specific oligonucleotides were used to amplify mRNAs for β-actin (5′-ATGCCAGGGTACATGGTGGT-3′ and 5′-TCGTGCGTGACATTAAGGAG-3′), TNF-α (5′-TCCTTCAGACACCCTCAACC-3′ and 5′-AGGCCCCAGTTTGAATTCTT-3′), and IL-1β (5′-AAACAGATGAAGTGCTCCTTCCAGG-3′ and 5′-TGGAGAACACCACTTGTTGCTCCA-3′).
Small Interfering RNA Transfection
Silencing of gene PARP-1 was performed by using small interfering RNA directed against human PARP-1 (ID: 111005) or a negative control purchased from Ambion (Austin, TX). Cells were seeded at 30–50% confluence and transfected with small interfering RNA by using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. All the experiments were performed 48 h after transfection.
Statistical Analysis
All experiments were performed at least thrice for each determination. Data were expressed as mean ± S.D. and analyzed using one-way analysis of variance. The level of significance was accepted at p < 0.01.
RESULTS
Human AC16 cardiomyocytes were infected with T. cruzi (cell:parasite ratio, 1:3) for 0–24 h, and intracellular ROS were detected using an H2DCFDA probe. We noted a significant increase in dichlorofluorescein fluorescence beginning 6 h pi, the maximum level of fluorescence being detected at 12 h pi (Fig. 1A). Overlay images of cells labeled with H2DCFDA and MitoTracker Red, or with MitoSOX Red (detects mitochondrial O2˙̄) and MitoTracker Green (Fig. 1, B and C, compare panels c and f) suggested that mitochondria are the primary site of ROS production in infected cardiomyocytes. A substantial increase in DHE fluorescence in infected cells (Fig. 1D) suggested that at least some of the mtROS were diffused to cytosol, because the cytosolic ROS oxidizes DHE and the later binds to nuclear DNA, leading to nuclear red fluorescence, observed during 3–12 h pi.
FIGURE 1.

T. cruzi-induced ROS production in AC16 cardiomyocytes. A, AC16 cardiomyocytes were infected with T. cruzi (cell:parasite ratio, 1:3). Shown are representative fluorescent micrographs of AC16 cells infected for 0–24 h and stained with H2DCFDA (detects intracellular ROS, green). B, normal (panels a–c) and infected (panels d–f) cardiomyocytes at 12 h pi were stained with H2DCFDA (panels a and d) and MitoTracker Red (red, localizes to mitochondria, panels b and e). Overlay images (panels c and f, yellow) indicated mitochondria are the major source of intracellular ROS in infected cardiomyocytes. C, overlay images (panels c and f, yellow) of cardiomyocytes stained with MitoSOX Red (red, detects mtROS, panels a and d) and MitoTracker Green (green, localizes to mitochondria, panels b and e) validates mitochondrial production of ROS in infected cardiomyocytes at 12 h pi (panels d–f). D, dihydroethidium (DHE) is oxidized by cytosolic ROS to fluorescent ethidium bromide that intercalates DNA yielding a bright red nuclear fluorescence. A gradual increase in DHE fluorescence during 0–12 h pi indicates that mtROS are diffused to cytosol in infected cardiomyocytes.
DNA is ROS-sensitive. The low redox potential of guanine facilitates electron transfer from guanine to nearby base radicals, leaving guanine with an unpaired electron, commonly resolved as 8-oxoG lesions. Immunostaining with anti-8-oxoG antibody showed a significant increase in nuclear 8-oxoG lesions in infected cardiomyocytes (Fig. 2A). Likewise, the Comet assay showed >10-fold increase in tail moments, indicative of nuclear DNA fragmentation, in infected cells as compared with that noted in normal controls (Fig. 2, B and C). Treatment of infected cardiomyocytes with PBN resulted in a significant decline in intracellular ROS level (Fig. 2D), and subsequently, the 8-oxoG lesions (Fig. 2A) and DNA fragmentation (Fig. 2, B and C) were decreased in PBN-treated/infected cardiomyocytes in a dose-dependent manner. These data showed that intracellular ROS caused DNA oxidative damage and DNA fragmentation in cardiomyocytes infected by T. cruzi.
FIGURE 2.

ROS elicit DNA oxidative damage and DNA fragmentation in T. cruzi-infected cardiomyocytes. AC16 cells (wt and infected) were treated with 500 μm PBN for 12 h. A, cardiomyocytes were fixed and permeabilized, and immunostaining was performed with anti-8-oxoG antibody (brown nuclei). B, a Comet assay was performed to detect DNA fragmentation. Shown are representative micrographs of SYBR-green-stained nuclear DNA and Comet tails in normal, infected, and PBN-treated/infected cardiomyocytes. C, quantitation of tail moment, indicative of DNA fragmentation, was performed as described under “Experimental Procedures.” Data (mean ± S.D.) are representative of three independent experiments (p < 0.01; *, normal versus T. cruzi-infected; #, infected/PBN-treated versus infected/untreated). D, PBN scavenges ROS in infected cardiomyocytes. AC16 cardiomyocytes (normal and infected) were incubated for 12 h (± PBN), labeled with 10 μm H2DCFDA (green), and ROS-specific signal detected by fluorescence microscopy.
The nuclear enzyme PARP-1 is a sensor of DNA damage. We monitored PARP-1 activity by determination of poly(ADP-ribosyl) (PAR) level. Immunoblotting with anti-PAR antibody showed PAR modification of several proteins was distinctly enhanced in infected cardiomyocytes during 6–24 h pi (Fig. 3A). At 48 h pi, the extent of PARylation of proteins was decreased that correlated with a decline in cell survival. No PARylation of host proteins was detectable at 1 and 3 h pi (data not shown). Increased PARylation of host proteins in infected cardiomyocytes was inhibited by PJ34 (PARP-1 inhibitor, Fig. 3B). Normal cardiomyocytes incubated with H2O2 exhibited a similar pattern of increased protein PARylation as was noted in T. cruzi-infected cells (Fig. 3B) indicating that ROS plays an upstream role in PARP-1 activation. To demonstrate such is the case, we incubated infected cardiomyocytes with PBN and examined protein PARylation level. PBN treatment decreased the extent of PARylation of host proteins in a dose-dependent manner (Fig. 3C), thus, suggesting that ROS signals PARP-1-dependent protein PARylation in infected cardiomyocytes. Immunoprecipitation of PARP-1 followed by immunoblotting with anti-PARP-1 and anti-PAR antibodies showed that PARP-1 protein level was not increased, yet PARylation of PARP-1 was enhanced by 2-fold in infected cardiomyocytes (Fig. 3, D and E). The PARylation of PARP-1 was normalized to control level when infected cardiomyocytes were incubated with 10 μm PJ34 or 500 μm PBN (Fig. 3, D and E). Together, these results demonstrated that (a) ROS stimulates PARP-1 activation in T. cruzi-infected cardiomyocytes and (b) PARP-1 activation contributes to PARylation of self and other proteins in infected cardiomyocytes.
FIGURE 3.

ROS stimulates PARP-1 activation in cardiomyocytes infected by T. cruzi. A, PAR modification. AC16 cells were infected with T. cruzi for 0–48 h. Cell lysates (40 μg of protein) were resolved by SDS-PAGE, and protein PAR modification was detected using anti-PAR antibody. B and C, enhanced PAR modification of cellular proteins is dependent upon PARP-1 and ROS activation. AC16 cells (wt and infected) were incubated with 10 μm PJ34 (PARP-1 inhibitor), 50 μm H2O2, or 0–500 μm PBN for 12 h. Cell lysates (40 μg of protein) were resolved by SDS-PAGE, and Western blotting was performed with anti-PAR antibody. D and E, PARP-1 is PAR-modified in a ROS-dependent manner. AC16 cells were infected with T. cruzi for 12 h (±10 μm PJ34 or 500 μm PBN). Cell lysates were subjected to immunoprecipitation (IP) with anti-PARP antibody, and immunoblotting (IB) was performed with anti-PARP-1 and anti-PAR-1 antibodies. Densitometric analysis of PAR-modified PARP-1, normalized to β-actin signal, is shown in E. Data (mean ± S.D.) are representative of three independent experiments (p < 0.01; *, normal versus T. cruzi-infected; #, infected/PBN-treated versus infected/untreated). All membranes were re-probed with β-actin antibody to validate equal loading of samples.
Next, we determined if ROS or PARP-1 contribute to inflammatory responses in cardiomyocytes infected by T. cruzi. Real-time PCR analysis showed that mRNA level for TNF-α and IL-1β cytokines was increased in infected cardiomyocytes, beginning at 3 h pi. The maximal increase in mRNA level for TNF-α (5-fold increase) and IL-1β (7-fold increase) was observed at 12 h pi (Fig. 4A). Treatment with PJ34 inhibited the increased TNF-α and IL-1β mRNA levels in infected cardiomyocytes, thus, suggesting that PARP-1 activity is an important regulator of cytokine gene expression in cardiomyocytes (Fig. 4B). A substantial increase in TNF-α and IL-1β mRNA levels in normal cardiomyocytes treated with H2O2 (Fig. 4B) and inhibition of their expression in infected cardiomyocytes treated with PBN (Fig. 4C) suggested that ROS, either directly or as an upstream regulator of PARP-1, contributes to cytokine gene expression in cardiomyocytes infected by T. cruzi. Emodin (NF-κB inhibitor) blocked the enhanced levels of TNF-α and IL-1β mRNAs in infected cardiomyocytes (Fig. 4C), suggesting that ROS and PARP-1 signal cytokine gene expression through NF-κB activation in infected cardiomyocytes.
FIGURE 4.

ROS and PARP-1 induce cytokine gene expression in infected cardiomyocytes. A, the mRNAs for TNF-α and IL-1β are increased in infected cells. Total RNA was isolated from cardiomyocytes at 0–24 h pi, and real-time RT-PCR performed as described under “Experimental Procedures.” After normalization to β-actin mRNA, relative increase in TNF-α and IL-1β mRNA levels in infected cardiomyocytes was plotted in comparison to normal controls. Data are presented as mean ± S.D. (p < 0.01; *, normal versus T. cruzi-infected). B and C, ROS, PARP-1, and NF-κB inhibition attenuated the enhanced cytokine mRNA level in infected cardiomyocytes. Cardiomyocytes (normal and infected) were incubated for 12 h in the presence or absence of 10 μm PJ34, 50 μm H2O2, 500 μm PBN, or 50 μg/ml emodin (inhibits NF-κB activation), and a real-time RT-PCR was performed to measure the TNF-α and IL-1β mRNA levels. Data (mean ± S.D.) are representative of triplicate experiments (p < 0.01; *, normal versus T. cruzi-infected; #, infected/PBN-treated versus infected/untreated).
We performed a dual luciferase reporter assay to evaluate whether NF-κB signaling of gene expression is indeed induced in T. cruzi-infected cardiomyocytes. AC16 cells transiently transfected with luciferase reporter plasmid pNF-κB-luc with 5×NF-κB binding site and pREP7-Rluc (expresses Renilla luciferase) were infected with T. cruzi for 12 h, and NF-κB-dependent luciferase activity was monitored. We observed that infected cardiomyocytes had a >2-fold increase in luciferase activity (normalized to Renilla luciferase) (Fig. 5A). Treatment with PBN and PJ34 resulted in a significant decline in luciferase activity (Fig. 5A), suggesting that ROS and PARP-1 signaled NF-κB-dependent gene expression in cardiomyocytes infected by T. cruzi. We considered that intracellular ROS facilitate nuclear translocation of activated NF-κB, and PARP-1 (nuclear protein) facilitates interaction of NF-κB with other factors of the transcription complex. RelA (p65) is an important subunit of activated NF-κB dimers (e.g. p50/p65, p65/p65, and p65/c-Rel). To investigate whether RelA (p65) translocation to nucleus occurs in an ROS-dependent manner, AC16 cells infected with T. cruzi were treated with PBN, and cellular and nuclear fractions were used for immunoblotting with anti-p65 antibody. Our results showed >2.5-fold increase in nuclear translocation of RelA (p65) that correlated with a similar level of decline in cytosolic RelA (p65) of infected cardiomyocytes (Fig. 5B). PBN blocked the nuclear translocation of RelA (p65) in infected cardiomyocytes, thus validating that ROS signal nuclear translocation of RelA (p65) in infected cardiomyocytes. It was, however, surprising to find that PJ34 treatment of infected cardiomyocytes also blocked the nuclear translocation of RelA (p65) (Fig. 5B), as PARP-1 is localized in nucleus, and thus, was not expected to interact with cytosolic NF-κB.
FIGURE 5.
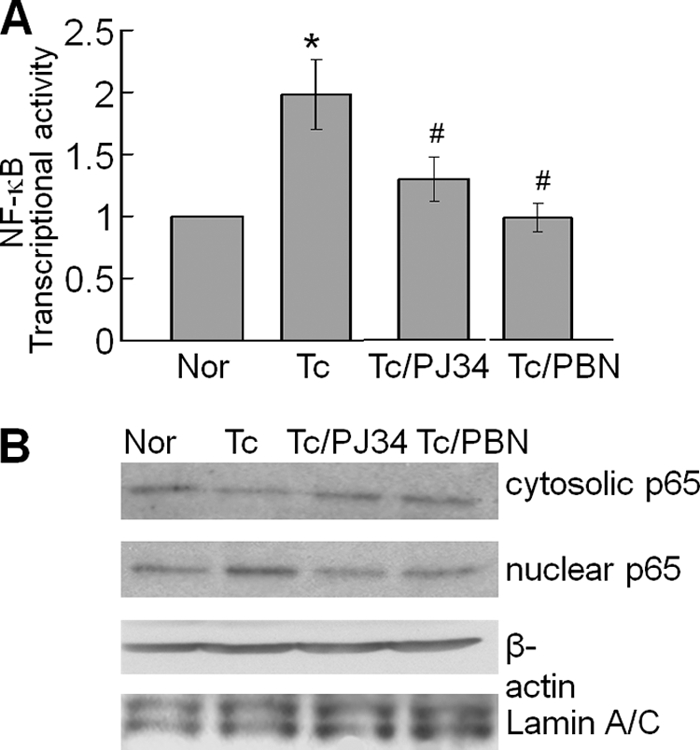
A, ROS and PARP-1 signal NF-κB activation in T. cruzi-infected cardiomyocytes. AC16 cells were transiently transfected with pNF-κB-luc (with 5× NF-κB binding site). Cells were infected with T. cruzi and incubated in the presence or absence of 10 μm PJ34 or 500 μm PBN for 12 h. The relative NF-κB transcriptional activity was measured by firefly luciferase activity normalized to Renilla luciferase activity. The transcriptional activity of NF-κB in normal cells was considered as baseline and valued 1 or 100%. Data (mean ± S.D.) are representative of three independent experiments (p < 0.01; *, normal versus T. cruzi-infected; #, infected/PBN-treated versus infected/untreated). B, PARP-1 and ROS inhibitors decreased the nuclear translocation of RelA (p65) in infected cardiomyocytes. AC16 cells (wt and infected, ± PJ34 or PBN) were incubated for 12 h, and cytosolic and nuclear fractions were prepared as described under “Experimental Procedures.” The p65 expression level was detected by immunoblotting with specific antibody. Membranes were re-probed with antibody to β-actin and Lamin A/C to normalize the cytosolic and nuclear fractions, respectively.
To verify if PARP-1 binds with RelA (p65), facilitating its nuclear translocation and NF-κB-dependent gene transcription, we utilized cytosolic and nuclear fractions of normal and infected cardiomyocytes, and performed immunoprecipitation with anti-PARP-1 or anti-p65 antibodies followed by immunoblotting with specific antibodies. Repeated attempts with various protein lysates concentration, antibody dilution, and incubation time to co-precipitate PARP-1 with p65 were unsuccessful. These results suggested that PARP-1 does not directly bind with p65 to facilitate its translocation or interaction with transcription complex in nuclei of cardiomyocytes infected by T. cruzi (data not shown). Next, we examined whether NF-κB-dependent gene expression is facilitated by PARP-1 activity via PAR modification of RelA (p65) or other interacting partners of the transcription complex. For this, cell lysates were subjected to immunoprecipitation with p65-specific antibody, and immunoblotting was performed with anti-PAR antibody. We found no detectable level of PARylation of RelA (p65) in infected cardiomyocytes (Fig. 6). Instead, we observed enhanced PAR modification of several proteins that co-precipitated with RelA (p65) (Fig. 6). The enhanced PARylation of RelA (p65)-binding proteins was decreased when infected cardiomyocytes were treated with PJ34 or PBN. These data suggested that PAR modification of RelA (p65)-binding subunits facilitated the activation of NF-κB-dependent gene transcription of infected cardiomyocytes.
FIGURE 6.
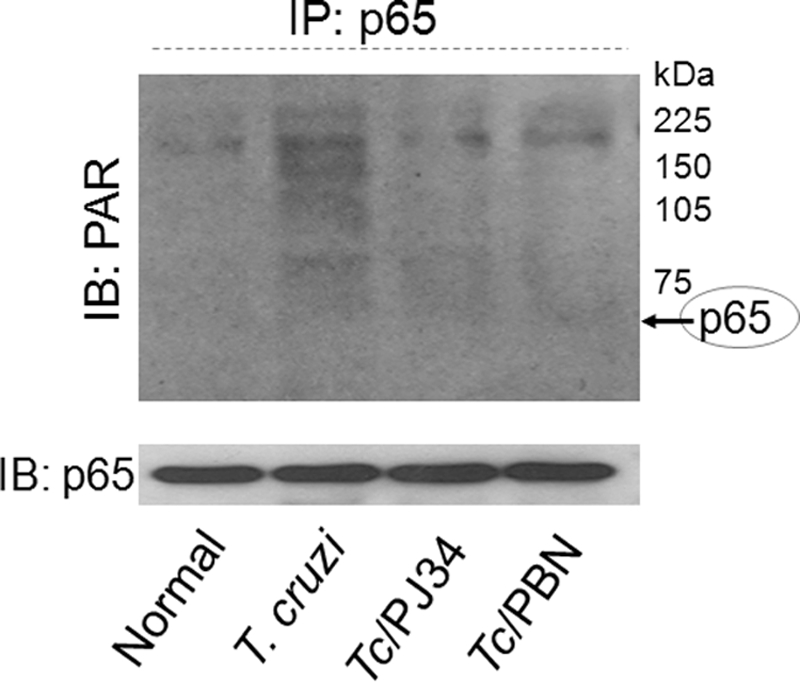
PAR modification of p65-associated proteins was enhanced in an ROS-dependent manner. AC16 cells (normal and infected) were incubated in the presence of 10 μm PJ34 or 500 μm PBN for 12 h. Immunoprecipitation was performed with RelA (p65)-specific antibody. Immune complexes were resolved on 10% SDS-PAGE, and immunoblotting was performed using antibodies specific for PAR polymer and RelA (p65) subunit of the NF-κB complex.
We considered the possibility that PAR modification of mitochondrial membranes signals ROS-induced NF-κB activation in infected cardiomyocytes. Dual labeling with anti-PAR antibody and MitoTracker Red demonstrated a significant increase in PAR-specific fluorescence in mitochondria of infected cardiomyocytes that was inhibited by PJ34 treatment (Fig. 7A). To determine if PARylation affected mitochondrial membrane potential (Δψm), cells were stained with JC-1. JC-1 accumulates in coupled, healthy mitochondria as red aggregates and remains in a monomeric green form if ψm is collapsed. Normal cardiomyocytes exhibited a high red aggregates/green monomers ratio. Comparatively, we noted a marked decline in red aggregates with a corresponding increase in green monomers in infected cardiomyocytes that was prevented by PJ34 treatment (Fig. 7B). The PJ34-dependent normalization of Δψm was associated with control of mtROS formation evidenced by a significant decline in MitoSOX Red (detects mitochondrial O2˙̄) and DHE (detects intracellular ROS) fluorescence in infected cardiomyocytes (Fig. 7C). These data, along with data presented in Figs. 1–3, implied that T. cruzi initiated mtROS production and PARP-1 activation in infected cardiomyocytes, and PARP-1 activation provided a secondary signal for intracellular ROS formation through PAR modification of mitochondrial membranes. Thus, PARP-1 enhanced nuclear translocation of RelA (p65) via providing a secondary stimulus for ROS production in infected cardiomyocytes.
FIGURE 7.

PARP-1 activation contributes to a loss of mitochondrial membrane potential and enhanced ROS production in infected cardiomyocytes. AC16 cells (normal and infected) were incubated in presence or absence of 10 μm PJ34 for 12 h. A, shown are the representative fluorescence micrographs of normal (panels a, d, and g), T. cruzi-infected (panels b, e, and h), and infected/PJ34-treated (panels c, f, and i) cardiomyocytes, stained with anti-PAR antibody (panels a–c, green) and MitoTracker Red (localizes to mitochondria, panels d–f). Overlay images (panels g–i, yellow) demonstrate mitochondrial localization of PAR in infected cardiomyocytes. B, shown are the representative fluorescence micrographs of JC-1-stained wt and infected cardiomyocytes (±PJ34). Note the accumulation of green monomers in infected cardiomyocytes (panel e) was inhibited by PJ34 (panel f). Overlay images (panels g–i) show that a majority of mitochondria fluoresced red in normal (panel g) and PJ34-treated/infected cardiomyocytes (panel i), whereas mitochondria in infected cardiomyocytes fluoresced green (panels h). C, shown are fluorescent micrographs of wt and infected cardiomyocytes (±PJ34) stained with MitoSOX Red (detects mitochondrial O2˙̄, panels a–c), and DHE (detects intracellular/cytosolic ROS, panels d–f, red) probes. Note that mitochondrial and cytosolic ROS were decreased by PJ34 treatment of infected cardiomyocytes.
To confirm the role of PARP-1, we further applied an RNAi approach to inhibit the PARP-1 expression. The PARP-1 RNAi (iPARP-1), but not scrambled RNAi (iCTR, negative control), resulted in a 2-fold decline in PARP-1 expression (Fig. 8A). The iPARP-1 rescued the collapse of Δψm (Fig. 8B) and reduced the production of mitochondrial ROS (Fig. 8C) in T. cruzi-infected cardiomyocytes. In addition, iPARP-1 blocked the nuclear translocation of p65 (Fig. 8, D and E) and the expression of inflammatory cytokines (Fig. 8F) in infected cardiomyocytes. These data confirmed the role of PARP-1 in mitochondrial dysfunction and inflammatory cytokine production in cardiomyocytes infected by T. cruzi.
FIGURE 8.

Genetic interference of PARP-1 rescued mitochondrial membrane potential, reduced mitochondrial ROS, and decreased NF-κB-mediated transcription of inflammatory genes in infected cardiomyocytes. A, AC16 cells were transfected with small RNAi of PARP-1 (iPARP-1) or negative control (iCTR) for 48 h, and immunoblotting was performed with anti-PARP-1 and anti-β-actin (loading control) antibodies. B–F, cells were transfected with PARP-1 or control RNAi (as above) and infected with T. cruzi for 12-h (B) JC-1 staining. Note the accumulation of green monomers and the loss of red aggregates in infected cardiomyocytes (panels b, f, and j) was inhibited by PARP-1 RNAi (panels c, g, and k) but not control RNAi (panels d, h, and g). C, MitoSOX Red staining. Note the mitochondrial ROS in infected cardiomyocytes (panel b) was inhibited by PARP-1 RNAi (panel c) but not control RNAi (panel d). D and E, the cytosolic and nuclear fractions of wt and infected cardiomyocytes (±PARP-1 or control RNAi) were prepared as described under “Experimental Procedures.” The p65 expression level was detected by immunoblotting with specific antibody. Membranes were re-probed with antibody to β-actin and Lamin A/C. Densitometric analysis of cytosolic and nuclear p65, normalized to β-actin and Lamin A/C signal, respectively, is shown in panel E. F, a real-time RT-PCR was performed to measure the TNF-α and IL-1β mRNA levels in wt and infected cardiomyocytes (±PARP-1 or control RNAi). Data (mean ± S.D.) are representative of triplicate experiments (p < 0.01; *, normal versus T. cruzi-infected; #, infected/PARP-1-interfered versus infected/untreated).
DISCUSSION
T. cruzi infects a wide range of host tissues, and inflammatory infiltrate and oxidative stress-induced injuries are a common finding in chagasic myocardium (reviewed in Refs. 2, 8). We have documented previously that mitochondria are a major source of O2˙̄ production in murine cardiomyocytes and myocardium infected by T. cruzi (15, 16). In this study, we demonstrate that mtROS diffuse to cytosol and elicit DNA damage and PARP-1 activation in human cardiomyocytes infected by T. cruzi. ROS, either through direct modification of cytosolic NF-κB, or via PARP-1-dependent PAR modification of RelA (p65)-interacting nuclear proteins, signaled the pro-inflammatory cytokine gene expression in infected cardiomyocytes. Further studies demonstrated that PAR polymers altered the mitochondrial membrane potential and provided a secondary signal for sustained mtROS production, PARP-1 activation, and NF-κB-dependent cytokine gene expression in cardiomyocytes infected by T. cruzi. This, to the best of our knowledge, is the first study demonstrating a link between ROS and inflammatory responses in cardiomyocytes infected by T. cruzi and providing a clue to the pathomechanism of sustained inflammation in Chagas disease.
The heart is dependent on mitochondria for ∼90% of the cellular energy required for its contractile and other metabolic activities. Due to high energy demand, mitochondria represent 30% of the total volume of cardiomyocytes and produce energy through the oxidative phosphorylation pathway (23), in which respiratory electron flow in the inner membrane of the mitochondria is coupled to the FoF1 ATP synthase and produces ATP. During this process, as much as 2–4% of the reducing equivalents escape the respiratory chain, leading to the formation of free radicals. Considering the high rate of O2 consumption in cardiomyocytes, it is inferred that, even in normal conditions, heart tissue is maximally exposed to ROS of mitochondrial origin (24). We have documented that respiratory chain efficiency is compromised in chagasic hearts, and the resultant increase in electron leakage and O2˙̄ production leads to sustained oxidative stress during the course of T. cruzi infection and disease development (15, 25). In this study, dual labeling with mitochondria- and O2˙̄-specific fluorescent probes validated that mitochondrial ROS production is indeed increased in cardiomyocytes within 3 h of infection by T. cruzi (Fig. 1). The ROS response was not a bystander effect caused by the presence of parasite antigens, and active invasion by T. cruzi was essential to elicit ROS production in cardiomyocytes (16). The diffusion of mtROS to cytosol was evidenced by a significant increase in DHE fluorescence in infected cardiomyocytes. DHE is oxidized by cytosolic ROS, and the latter binds to nuclear DNA, leading to nuclear red fluorescence, observed during 3–12 h pi (Fig. 1D).
DNA is sensitive to ROS, and nuclear enzyme PARP-1 is a sensor of damaged DNA. The basal level activation of PARP-1 by mild genotoxic stimuli causes PARylation of histone proteins (e.g. H1 and H2B) that mediates relaxation of the chromatin superstructure and recruitment of DNA-break repair enzymes (18, 19), resulting in DNA repair and cell survival (reviewed in Ref. 26). The hyperactivation of PARP-1, however, is harmful, because it may utilize NAD+ at a very high rate and result in excessive formation of PAR polymers that bind to cellular proteins and signal various pathological stimuli (27). Accumulated evidence suggests that PARP-1 activation plays a role in various forms of heart failure, cardiomyopathies, and myocardial hypertrophy, and inhibitors of PARP-1 or PARP-1 gene deletion ameliorate the features in these diseases (28). In this study, we observed a significant increase in DNA oxidative damage evidenced by nuclear 8-oxoG lesions and DNA Comet tails (indicator of DNA fragmentation) (Fig. 2), and PARP-1 activation, evidenced by the detection of PARylation of PARP-1 (Fig. 3D) and other cellular proteins (Fig. 3A) in cardiomyocytes infected by T. cruzi. Treatment of infected cardiomyocytes with ROS scavenger (PBN) and PARP-1 inhibitor (PJ34) attenuated the mtROS formation, and subsequently DNA damage and PARP-1 activation were decreased. These data allow us to surmise that T. cruzi induced an increase in mitochondrial ROS-signaled PARP-1 activation, which resulted in post-translational PAR modification of PARP-1 and other proteins in cardiomyocytes.
The plasma level of TNF-α and other pro-inflammatory cytokines is elevated in chagasic patients (3, 10). The enhanced expression of TNF-α, IL-1β, and IL-6 at both mRNA and protein levels has also been noted in the myocardium of T. cruzi-infected experimental models (7, 29, 30). It is generally accepted that macrophages and neutrophils that constitute the inflammatory infiltrate in the heart are the major source of IL-1β and TNF-α cytokines (31, 32), and CD4+ and CD8+ T cells contribute to interferon-γ production (33) in chagasic myocardium. Our finding of increased TNF-α and IL-1β mRNAs in infected cardiomyocytes (Fig. 4) suggested that cardiomyocytes also respond to T. cruzi by inflammatory cytokine production. ROS appeared to be important regulators of inflammatory cytokines, because treatment with PBN resulted in a significant decline in cytokine expression in infected cardiomyocytes (Fig. 4). The cytokine gene expression was also inhibited by emodin (NF-κB inhibitor) suggesting that ROS signals cytokine gene expression through NF-κB activation in infected cardiomyocytes. NF-κB is a known redox-sensitive, inducible transcription factor (34), and has been shown to regulate the production of pro-inflammatory cytokines and chemokines in a variety of in vitro and in vivo models (reviewed in Ref. 35). Accordingly, scavenging of ROS by PBN treatment blocked the nuclear translocation of RelA (p65), an important subunit of activated NF-κB dimers (p50/p65, p65/p65, and p65/c-Rel), and NF-κB transcriptional activity, validated by the dual luciferase reporter assay (Fig. 5). Treatment of infected cardiomyocytes with enbrel (TNF-α receptor inhibitor) had no effect on NF-κB activation and downstream events (data not shown). Together, these observations allow us to conclude that T. cruzi-induced intracellular ROS signals the NF-κB transcriptional activation and pro-inflammatory cytokine gene expression in cardiomyocytes.
Surprisingly, PJ34, the inhibitor of PARP-1 activation, blocked the nuclear translocation and activation of NF-κB and cytokine gene expression in infected cardiomyocytes. A few in vitro and in vivo studies have reported that PARP-1 acts as a co-activator of NF-κB, and inhibition of PARP-1 attenuated the pro-inflammatory cytokine response (36–38), although additional studies have debated against this notion (39, 40). Others have shown that PARP-1 and NF-κB form a stable, immunoprecipitable nuclear complex (41). Hassa et al. generated a PARP-1 mutant lacking the enzymatic and DNA-binding activity and showed that this mutant interacted, comparably to the wild-type PARP-1, with p65 or p50 subunits of NF-κB, thus, concluding that PARP-1 enzymatic activity is not essential for its interaction with NF-κB and DNA binding (40). We did not anticipate that PARP-1, a nuclear protein, would reach the cytosolic NF-κB to facilitate its translocation to the nucleus for the formation of transcription complex and cytokine gene expression. Our notion was validated by the fact that we observed no increase in PARP-1 protein level in response to T. cruzi infection, and repeated attempts to co-precipitate PARP-1 and NF-κB from the cytosolic and nuclear fractions of infected cardiomyocytes were not successful. We, therefore, surmised that PARP-1 does not facilitate NF-κB translocation to nucleus and transcription complex formation via direct binding to RelA (p65) in cardiomyocytes infected by T. cruzi. Chang and Alvarez-Gonzalez showed that DNA binding by NF-κB-p50 was inefficient in the absence of NAD and blocked in the presence of pharmacologic inhibitors of PARP-1, and suggested that NF-κB-p50 DNA binding and transcription complex formation are perhaps regulated in a protein-PARylation-dependent manner (39). Indeed our data provide support to this notion. We noted no PARylation of RelA (p65), however, proteins co-precipitated by anti-p65 antibody were PAR-modified in infected cardiomyocytes. PJ34 arrested the PARP-1 activity and PARylation of p65-binding proteins, and, subsequently, NF-κB activation and cytokine gene expression were blocked in PJ34-treated/infected cardiomyocytes. Together, these observations allow us to conclude that ROS-mediated PARP-1 activation induces PAR modification of p65-binding nuclear proteins and NF-κB activation that leads to cytokine gene expression in T. cruzi-infected cardiomyocytes.
PARP activation is suggested to play a crucial role in cross-talk between the nucleus and the mitochondria, although how PARP transfers information to the mitochondria is unclear. Some studies have indicated that PARP-1 is present in the mitochondria (42), and besides playing a role in repairing the mitochondrial DNA (43), it may also PAR-modify mitochondrial proteins, including the members of the Krebs cycle, the urea cycle, and the electron transport chain (44). In our study, dual labeling with PAR-specific antibody and mitochondria-targeted fluorescent probes, as well as JC-1 staining, showed a significant increase in mitochondrial PAR deposition associated with a loss in membrane potential (Δψm) in cardiomyocytes infected by T. cruzi (Figs. 7 and 8). Alterations in ψm affect several mitochondrial functions. One is the dissipation of protonmotive force that in itself serves as a negative modulator of respiratory chain activity in infected cardiomyocytes (16). We, thus, considered that T. cruzi-induced PARP-1 activation provides a secondary signal for PAR-dependent mitochondrial electron transport chain dysfunction and ROS formation in infected cardiomyocytes. This notion was validated by the observation that pharmacological treatment (PJ34) or genetic interference (PARP-1 RNAi) of infected cardiomyocytes blocked the PARP-1-dependent PARylation of mitochondrial membranes and loss of mitochondrial ψm, and subsequently, ROS production was decreased in pharmacologically treated or genetically interfered cardiomyocytes (Figs. 7 and 8). Thus, we conclude that PAR modification of mitochondrial membranes contributes to Δψm and enhanced ROS production in infected cardiomyocytes. These data also clarify the role of PARP-1 in NF-κB activation and suggest that PARP-1/PAR-mtROS provides a secondary signal for NF-κB nuclear translocation and activation.
In summary, we have shown that cross-talk between nuclei and mitochondria contribute to ROS-dependent cytokine gene expression in cardiomyocytes infected by T. cruzi (Fig. 9). Our data demonstrate that cardiomyocytes respond to T. cruzi infection by an exponential increase in mtROS production, some of which are diffused to cytosol and nucleus. The ROS-induced DNA oxidative damage elicited PARP-1 activation and PAR formation, and PARylation of mitochondrial membranes provided a secondary signal resulting in a positive feedback cycle of mtROS production, DNA damage, and PARP-1/PAR activation. ROS, either through direct modulation of cytosolic NF-κB or via PARP-1-dependent PAR modification of RelA (p65) interacting nuclear proteins, contributed to NF-κB activation and cytokine gene expression. The ROS-PARP-1-RelA signaling pathway may contribute to injurious inflammatory cytokine production and infiltration of inflammatory infiltrate in chronic chagasic hearts, to be investigated in future studies.
FIGURE 9.

Potential mechanism of ROS-PARP-1-RelA signaling of cytokine gene expression in T. cruzi-infected cardiomyocytes. ROS induced by T. cruzi infection elicit nuclear DNA damage and PARP-1 activation that then sustains an autocatalytic cycle of mitochondrial and intracellular ROS production, PARP-1 activation, and PAR formation. ROS (produced in response to T. cruzi infection) and by PARP-1/PAR-dependent mitochondrial alterations provide primary and secondary stimuli, respectively, for nuclear translocation of NF-κB, NF-κB activation, and cytokine gene expression. PARP-1 activation and enhanced PAR modification of p65-associated proteins may also signal optimal transcriptional activation of NF-κB-dependent pro-inflammatory gene expression.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-HL094802 from NHLBI and RO1-AI054578 from NIAID (to N. J. G.).
- ROS
- reactive oxygen species
- DHE
- dihydroethidium
- H2DCF-DA
- dichlorodihydrofluorescein diacetate
- IL-1β
- interleukin-1β
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide
- Δψm
- membrane potential
- O2˙̄
- superoxide
- PBN
- phenyl-α-tert-butyl nitrone
- PARP-1
- poly(ADP-ribose) polymerase-1
- PAR
- poly(ADP-ribose)
- PARylation
- poly(ADP-ribosyl)ation
- PARylated
- poly(ADP-ribosyl)ated
- mtROS
- mitochondrial ROS
- TNF-α
- tumor necrosis factor-α
- wt
- wild type
- RNAi
- RNA interference
- RT
- reverse transcription
- 8-oxoG
- 8-oxoguanine
- pi
- post-infection.
REFERENCES
- 1.World Health Organization (2006) Report of the Scientific Working Group on Chagas Disease, UNDP/World Bank/WHO [Google Scholar]
- 2.Tanowitz H. B., Machado F. S., Jelicks L. A., Shirani J., de Carvalho A. C., Spray D. C., Factor S. M., Kirchhoff L. V., Weiss L. M. (2009) Prog. Cardiovasc. Dis. 51, 524–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talvani A., Rocha M. O., Barcelos L. S., Gomes Y. M., Ribeiro A. L., Teixeira M. M. (2004) Clin. Infect. Dis. 38, 943–950 [DOI] [PubMed] [Google Scholar]
- 4.Machado F. S., Souto J. T., Rossi M. A., Esper L., Tanowitz H. B., Aliberti J., Silva J. S. (2008) Microbes Infect. 10, 1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossi M. A., Ramos S. G., Bestetti R. B. (2003) Front. Biosci. 8, 94–109 [DOI] [PubMed] [Google Scholar]
- 6.Zacks M. A., Wen J. J., Vyatkina G., Bhatia V., Garg N. J. (2005) An. Acad. Bras. Cienc. 77, 695–715 [DOI] [PubMed] [Google Scholar]
- 7.Machado F. S., Martins G. A., Aliberti J. C., Mestriner F. L., Cunha F. Q., Silva J. S. (2000) Circulation 102, 3003–3008 [DOI] [PubMed] [Google Scholar]
- 8.Gupta S., Wen J. J., Garg N. J. (2009) Interdiscip. Perspect. Infect. Dis. 2009, 190354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen J. J., Vyatkina G., Garg N. J. (2004) Free Radic. Biol. Med. 37, 1821–1833 [DOI] [PubMed] [Google Scholar]
- 10.Pérez-Fuentes R., Guégan J. F., Barnabé C., Lopez-Colombo A., Salgado-Rosas H., Torres-Rasgado E., Briones B., Romero-Díaz M., Ramos-Jiménez J., Sánchez-Guillén, Mdel C. (2003) Int. J. Parasitol. 33, 293–299 [DOI] [PubMed] [Google Scholar]
- 11.Wen J. J., Yachelini P. C., Sembaj A., Manzur R. E., Garg N. J. (2006) Free Radic. Biol. Med. 41, 270–276 [DOI] [PubMed] [Google Scholar]
- 12.de Oliveira T. B., Pedrosa R. C., Filho D. W. (2007) Int. J. Cardiol. 116, 357–363 [DOI] [PubMed] [Google Scholar]
- 13.Cardoni R. L., Rottenberg M. E., Segura E. L. (1990) Cell. Immunol. 128, 11–21 [DOI] [PubMed] [Google Scholar]
- 14.Cardoni R. L., Antunez M. I., Morales C., Nantes I. R. (1997) Am. J. Trop. Med. Hyg. 56, 329–334 [DOI] [PubMed] [Google Scholar]
- 15.Wen J. J., Garg N. J. (2008) J. Bioenerg. Biomembr. 40, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta S., Bhatia V., Wen J. J., Wu Y., Huang M. H., Garg N. J. (2009) Free Radic. Biol. Med. 47, 1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodhouse B. C., Dianov G. L. (2008) DNA Repair 7, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 18.El-Khamisy S. F., Masutani M., Suzuki H., Caldecott K. W. (2003) Nucleic Acids Res. 31, 5526–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde M. L., Hazra T. K., Mitra S. (2008) Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davidson M. M., Nesti C., Palenzuela L., Walker W. F., Hernandez E., Protas L., Hirano M., Isaac N. D. (2005) J. Mol. Cell. Cardiol. 39, 133–147 [DOI] [PubMed] [Google Scholar]
- 21.Dyer R. B., Herzog N. K. (1995) BioTechniques 19, 192–195 [PubMed] [Google Scholar]
- 22.Wen J. J., Bhatia V., Popov V. L., Garg N. J. (2006) Am. J. Pathol. 169, 1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheeran F. L., Pepe S. (2006) Biochim. Biophys. Acta 1757, 543–552 [DOI] [PubMed] [Google Scholar]
- 24.Genova M. L., Pich M. M., Bernacchia A., Bianchi C., Biondi A., Bovina C., Falasca A. I., Formiggini G., Castelli G. P., Lenaz G. (2004) Ann. N. Y. Acad. Sci. 1011, 86–100 [DOI] [PubMed] [Google Scholar]
- 25.Wen J. J., Garg N. J. (2010) Antiox. Redox Signal. 12, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouchard V. J., Rouleau M., Poirier G. G. (2003) Exp. Hematol. 31, 446–454 [DOI] [PubMed] [Google Scholar]
- 27.Andrabi S. A., Kim N. S., Yu S. W., Wang H., Koh D. W., Sasaki M., Klaus J. A., Otsuka T., Zhang Z., Koehler R. C., Hurn P. D., Poirier G. G., Dawson V. L., Dawson T. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18308–18313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacher P., Szabó C. (2007) Cardiovasc. Drug Rev. 25, 235–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandrasekar B., Melby P. C., Troyer D. A., Colston J. T., Freeman G. L. (1998) Am. J. Pathol. 152, 925–934 [PMC free article] [PubMed] [Google Scholar]
- 30.Talvani A., Ribeiro C. S., Aliberti J. C., Michailowsky V., Santos P. V., Murta S. M., Romanha A. J., Almeida I. C., Farber J., Lannes-Vieira J., Silva J. S., Gazzinelli R. T. (2000) Microbes. Infect. 2, 851–866 [DOI] [PubMed] [Google Scholar]
- 31.Muñoz-Fernández M. A., Fernández M. A., Fresno M. (1992) Immunol. Lett. 33, 35–40 [DOI] [PubMed] [Google Scholar]
- 32.Lima E. C., Garcia I., Vicentelli M. H., Vassalli P., Minoprio P. (1997) Infect. Immunol. 65, 457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardoni R. L., Antúnez M. I., Abrami A. A. (1999) Medicina 59, 84–90 [PubMed] [Google Scholar]
- 34.Kamata H., Hirata H. (1999) Cell. Signal. 11, 1–14 [DOI] [PubMed] [Google Scholar]
- 35.Saklatvala J., Dean J., Clark A. (2003) Biochem. Soc. Symp. 70, 95–106 [DOI] [PubMed] [Google Scholar]
- 36.Mabley J. G., Jagtap P., Perretti M., Getting S. J., Salzman A. L., Virág L., Szabó E., Soriano F. G., Liaudet L., Abdelkarim G. E., Haskó G., Marton A., Southan G. J., Szabó C. (2001) Inflamm. Res. 50, 561–569 [DOI] [PubMed] [Google Scholar]
- 37.Liaudet L., Pacher P., Mabley J. G., Virág L., Soriano F. G., Haskó G., Szabó C. (2002) Am. J. Respir. Crit. Care Med. 165, 372–377 [DOI] [PubMed] [Google Scholar]
- 38.Boulares A. H., Zoltoski A. J., Sherif Z. A., Jolly P., Massaro D., Smulson M. E. (2003) Am. J. Respir. Cell Mol. Biol. 28, 322–329 [DOI] [PubMed] [Google Scholar]
- 39.Chang W. J., Alvarez-Gonzalez R. (2001) J. Biol. Chem. 276, 47664–47670 [DOI] [PubMed] [Google Scholar]
- 40.Hassa P. O., Covic M., Hasan S., Imhof R., Hottiger M. O. (2001) J. Biol. Chem. 276, 45588–45597 [DOI] [PubMed] [Google Scholar]
- 41.Hassa P. O., Hottiger M. O. (1999) Biol. Chem. 380, 953–959 [DOI] [PubMed] [Google Scholar]
- 42.Du L., Zhang X., Han Y. Y., Burke N. A., Kochanek P. M., Watkins S. C., Graham S. H., Carcillo J. A., Szabó C., Clark R. S. (2003) J. Biol. Chem. 278, 18426–18433 [DOI] [PubMed] [Google Scholar]
- 43.Druzhyna N., Smulson M. E., LeDoux S. P., Wilson G. L. (2000) Diabetes 49, 1849–1855 [DOI] [PubMed] [Google Scholar]
- 44.Pankotai E., Lacza Z., Murányi M., Szabó C. (2009) Mitochondrion 9, 159–164 [DOI] [PubMed] [Google Scholar]