

Abstract
Human immunodeficiency virus-1 (HIV-1) impairs tumor necrosis factor-α (TNF-α)-mediated macrophage apoptosis induced by Mycobacterium tuberculosis (Mtb). HIV Nef protein plays an important role in the pathogenesis of AIDS. We have tested the hypothesis that exogenous Nef is a factor that inhibits TNF-α production/apoptosis in macrophages infected with Mtb. We demonstrate that Mtb and Nef individually trigger TNF-α production in macrophages. However, TNF-α production is dampened when the two are present simultaneously, probably through cross-regulation of the individual signaling pathways leading to activation of the TNF-α promoter. Mtb-induced TNF-α production is abrogated upon mutation of the Ets, Egr, Sp1, CRE, or AP1 binding sites on the TNF-α promoter, whereas Nef-mediated promoter activation depends only on the CRE and AP1 binding sites, pointing to differences in the mechanisms of activation of the promoter. Mtb-dependent promoter activation depends on the mitogen-activated kinase (MAPK) kinase kinase ASK1 and on MEK/ERK signaling. Nef inhibits ASK1/p38 MAPK-dependent Mtb-induced TNF-α production probably by inhibiting binding of ATF2 to the TNF-α promoter. It also inhibits MEK/ERK-dependent Mtb-induced binding of FosB to the promoter. Nef-driven TNF-α production occurs in an ASK1-independent, Rac1/PAK1/p38 MAPK-dependent, and MEK/ERK-independent manner. The signaling pathways used by Mtb and Nef to trigger TNF-α production are therefore distinctly different. In addition to attenuating Mtb-dependent TNF-α promoter activation, Nef also reduces Mtb-dependent TNF-α mRNA stability probably through its ability to inhibit ASK1/p38 MAPK signaling. These results provide new insight into how HIV Nef probably exacerbates tuberculosis infection by virtue of its ability to dampen Mtb-induced TNF-α production.
Keywords: Apoptosis, ERK, HIV, p38 MAPK, Tumor Necrosis Factor (TNF), Mycobacterium tuberculosis, ASK1
Introduction
The development and progression of AIDS is intimately associated with loss of normal immunological functions. Nef is a 27-kDa protein expressed by HIV-1/23 early during infection from multiple spliced viral mRNAs (1). It is considered to be a factor involved in the progression to AIDS (2–5). There is a large body of literature establishing the role of Nef in lymphocyte signaling. Nef increases viral replication in lymphocytes and down-regulates the cell surface expression of CD4, major histocompatibility complex class I A and B but not C (6–11), and several other receptors, including CD28 (12), CD80, CD86 (13), and CCR5 (14). Major histocompatibility complex class I down-regulation protects infected cells from killing by major histocompatibility complex class I A or B restricted cytotoxic T cells (15) and avoids killing by natural killer cells (16). Nef also prevents apoptosis of HIV-1-infected T cells (17). It deregulates cofilin in a PAK2-dependent manner, thereby restricting migration of T cells (18). It interacts with and regulates the activity of Src family kinases (19), class I phosphatidylinositol 3-kinase, guanine nucleotide exchange factor Vav, and calmodulin (20–22). Very recently, it has been established that Nef activates bidirectional membrane trafficking in T cells, which promotes transfer of Nef from infected to bystander cells (23). This offers an explanation for the detrimental effects observed in bystander cells in HIV infection.
Less is known about the role of Nef on signaling in macrophages. Nef reduces the expression of the mannose receptor on the macrophage cell surface by ∼50% (24). This is likely to contribute to crippling the host immune response. Nef-expressing macrophages attract CD4+ T cells thereby promoting productive HIV infection (25).
Considering that extracellular Nef has been detected in supernatants from HIV-1-infected cell cultures and in the serum of AIDS patients (26), signals delivered by exogenous Nef to immune cells are likely to be relevant to disease progression. Exogenous Nef activates the IκB kinase complex and the MAPKs JNK, ERK, and p38 in macrophages (27). Nef was detected inside B cells in vivo and shown to hamper B cell responses (28). Exogenous Nef reportedly activates NF-κB, AP-1, and c-Jun N-terminal kinase (JNK) in promonocytic cells (29). Exogenous Nef enters CD4+ T cells and primary macrophages by adsorptive endocytosis and activates signal transducers and activators of transcription 1 in macrophages (30). Although the stimulatory effects of exogenous Nef on NF-κB, AP-1, and JNK have been observed in U937 cells, Ma et al. (31) have reported that intracellular Nef, expressed through transduction of primary monocytes and promonocytic THP1 cells with retroviral nef gene, inhibits lipopolysaccharide-induced interleukin-12 p40 transcription by inhibiting JNK. The effects of Nef clearly appear to be context-specific. HIV-infected macrophages transfer Nef to B cells through long cellular protrusions, resulting in inhibition of immunoglobulin class switching (32).
Mycobacterium tuberculosis (Mtb) co-infection occurs in a large number of HIV-positive subjects, and mortality rates are very high (33). Alveolar macrophages serve as reservoirs of Mtb. Apoptosis of macrophages is believed to be essential for efficient control of Mtb infection, and increased apoptosis is a hallmark of bronchoalveolar lavage and lung tissue specimens of Mtb-infected patients (34, 35). In vitro HIV infection of alveolar macrophages has been reported to reduce both macrophage apoptosis as well as release of the apoptosis-inducing cytokine TNF-α in response to challenge with Mtb (36). Taken together with the observation that Nef attenuates HIV-induced macrophage apoptosis (37), this provided the motivation to test whether Nef could modulate apoptosis in Mtb-infected macrophages.
The viral burden is high in tissue compartments such as lymph nodes, even during the clinically latent stage of the disease (38). There is close interaction between infected lymphocytes and macrophages, in these compartments, raising the possibility that exogenous Nef could influence signaling in bystander macrophages. In this report we have tested the effects of exogenous Nef on signaling in Mtb-infected macrophages. We provide evidence that Nef attenuates apoptosis of Mtb-infected macrophages. This is most likely due to an attenuation of TNF-α production by Nef. Nef and Mtb both elicit TNF-α production. However, there is a net inhibitory effect when macrophages are challenged simultaneously with Nef and Mtb, probably due to mutual cross-regulation of signaling pathways.
EXPERIMENTAL PROCEDURES
Bacterial Strains
Escherichia coli BL21 (DE3) and E. coli DH5α strains were grown in Luria-Bertani (LB) Miller media. M. tuberculosis H37Rv was grown in Middlebrook 7H9 broth or Middlebrook 7H10 solid medium supplemented with albumin-dextrose-catalase and 0.05% Tween 80.
Molecular Biological Procedures
Standard procedures were used for cloning and analysis of DNA, PCR, and transformation. Enzymes used to manipulate DNA were from Roche Applied Science. All constructs made by PCR were sequenced to verify their integrity.
Antibodies
Antibodies against ASK1, Sp1, FosB, and ATF2 were from Santa Cruz Biotechnology (Santa Cruz, CA); Rac1, PAK1, ERK1/2, p38 MAPK, phospho-ERK1/2, phospho p38 MAPK, and phospho PAK1 antibodies were from Cell Signaling Technology (Beverly, MA).
Expression and Purification of SF2 Nef
N-terminal His-tagged SF2 Nef in pET 15b was a gift from Dr. Yong Hui Zheng (Michigan State University). SF2 Nef in pET15b was transformed in E. coli BL21 (DE3). Expression was carried out in the presence of 0.1 mm isopropyl-β-d-thiogalactopyranoside at 37 °C for 4 h. Cells were disrupted by sonication in 10 mm Tris-HCl (pH 8.0) containing 1 mm MgCl2, 1 mm Pefabloc, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Protein was purified from the post-sonicate supernatant by chromatography on nickel-nitrilotriacetic acid-agarose.
Plasmids
HA-tagged wild-type ASK1 and a catalytically inactive mutant (K709M) of ASK1(ASK1, KM) were obtained from Prof. Hidenori Ichijo, University of Tokyo. The dominant negative mutant of p38 MAPK (p38(agf)) was obtained from Dr. Roger Davis, University of Massachusetts Medical School, Worcester, MA. Dominant-negative MEK1 was a gift from Dr. D. J. Templeton, Case Western Reserve University, Cleveland, OH. Kinase-dead pCMV-PAK1 (K299R) (PAK1-KD) was a gift from Dr. Jeffrey Frost, University of Texas Southwestern Medical Center, Dallas, TX.
Using genomic DNA from RAW264.7 cells as template, the TNF-α promoter region (−185 to +69) was amplified by PCR using the sense and antisense primers 5′-ATAGGTACCCCCCAACTTTCCAAACCC-3′ and 5′-AAAGATCTAGCTATTTCCAAGATGTTC-3′, respectively. The resulting TNF-α promoter region (wild type) was cloned into the vector pGL3-basic (Promega) harboring the promoter-less luciferase gene, using asymmetric KpnI and BglII sites (underlined). CRE, Sp1, AP1, and Ets binding sites were mutated by overlap extension PCR.
Cell Culture and Treatments of Cells
THP-1 cells were obtained from the National Center for Cell Science, Pune, India, cultured and differentiated with phorbol 12-myristate 13-acetate as described by Maiti et al. (39). Adherent cells were 95% viable as determined by trypan blue dye exclusion. Differentiated THP-1 cells were either left untreated or treated with recombinant Nef (0.1–1 μg/ml) or with M. tuberculosis H37Rv (multiplicity of infection of 10) or with Nef and M. tuberculosis for 2 h or for the indicated periods of time. Unless otherwise stated, Nef was used at 1 μg/ml (the concentration at which the inhibitory effect of Nef was maximal). Cells were washed once, fresh medium was added, and incubations were carried out for the indicated time periods.
Cell Death Measurement
For the detection of histone by ELISA, cells (6 × 104) were plated in 96-well plates. After treatments, cell death was measured by the detection of histones in the cell supernatant using the Cell Death ELISA Plus kit (Roche Applied Science) according to the manufacturer's protocol.
ASK1 Kinase Assay
Cells were lysed after treatment in buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% (v/v) Triton X-100, 1% sodium deoxycholate, 0.5 mm sodium pyrophosphate, 1 mm sodium β-glycerophosphate, 1 mm Na3VO4, and 1 μg/ml leupeptin. The supernatant (equivalent to 200 μg of protein) was incubated overnight at 4 °C with rabbit polyclonal ASK1 antibody. Protein A/G Plus-agarose was added and incubated at 4 °C for an additional 3 h. The beads were washed twice with lysis buffer and twice with kinase buffer (25 mm Tris-HCl, pH 7.5, containing 5 mm sodium β-glycerophosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, and 10 mm MgCl2). The pellet was washed once with kinase buffer without protease inhibitors. The beads were then incubated in 20 μl of kinase buffer in the presence of 2.5 μCi of [γ-32P]ATP (specific activity, 6000 Ci/mmol) with 1 μg of myelin basic protein as substrate at 30 °C for 15 min. The reaction was stopped by adding protein gel denaturing buffer. After SDS-PAGE, gels were dried and subjected to autoradiography.
Rac1 Activation Assay
To assess the activation of Rac1 and formation of Rac1-GTP in response to stimulation with Nef, affinity precipitation was performed with a GST fusion protein corresponding to the p21-binding domain (PBD) of PAK1 (GST-PBD) that specifically binds to and precipitates Rac-GTP from cell lysates (40). The presence of Rac1 in the precipitate was assessed using Rac1 antibody.
Immunoprecipitation and Immunoblotting
For immunoprecipitations, cells were lysed, clarified by centrifugation, and then immunoprecipitated using appropriate antibody. Proteins were separated by SDS-PAGE and then transferred electrophoretically to polyvinylidene difluoride membranes. Blots after blocking were incubated overnight at 4 °C with primary antibody in Tris-buffered saline-Tween 20 (1%, v/v) with 5% (w/v) bovine serum albumin. After washing, the blots were incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) in blocking buffer for 1 h at room temperature. Blots were developed by chemiluminescence.
TransFactor Assays
TransFactor assays were carried out as described previously (41). Briefly, nuclear extracts from cells were prepared using the TransFactor extraction kit (Clontech). After centrifugation for 5 min at 20,000 × g at 4 °C, supernatants were assayed for the presence of the respective transcription factors by addition of equal amounts of lysates to wells precoated with DNA-binding consensus sequences. The presence of any particular transcription factor in the nucleus was then assessed by using the Mercury TransFactor kit (Clontech) according to the manufacturer's instructions. Plates were read at 655 nm.
Luciferase Reporter Assays
Cells were transfected with luciferase reporter plasmids. After treatments, cells were washed once with phosphate-buffered saline and scraped into luciferase lysis buffer (50 mm Tris-HCl, pH 8, 70 mm K2HPO4, 0.1% Nonidet P-40, 2 mm MgCl2, 1 mm dithiothreitol, 20 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml leupeptin). The lysates were rapidly mixed, and insoluble material was pelleted by centrifugation at 4 °C. The supernatant was removed and stored at −80 °C. For promoter activation analysis, luciferase activity assays were performed in a luminometer, and the results were normalized for transfection efficiencies by assay of β-galactosidase activity.
ChIP Assay
Chromatin immunoprecipitation (ChIP) assays were carried out using the Upstate Biotechnology ChIP assay kit. Briefly, cells after treatment were fixed by addition of formaldehyde (1%) to the culture medium for 10 min at 37 °C, washed in phosphate-buffered saline, scraped, lysed in lysis buffer (10 mm Tris-HCl, pH 8.0, 1% SDS, 0.5 mm Pefabloc, 2 μg/ml pepstatin A, and 2 μg/ml aprotinin) for 10 min at 4 °C, and sonicated to generate DNA fragments with an average size of 1 kb. The debris was removed by centrifugation. One-third of the lysate was used as DNA input control. The remaining two-thirds of the lysate were diluted 10-fold with a dilution buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.01% SDS, 1% Triton X-100, 1 mm EDTA), precleared with salmon sperm DNA/protein A/G-agarose slurry, followed by incubation of the supernatant with appropriate antibody (1.5–2 μg) overnight at 4 °C. Immunoprecipitated complexes were collected by pulldown assay with salmon sperm DNA/protein A/G-agarose beads. The precipitates were extensively washed and incubated in elution buffer (25 mm Tris-HCl, pH 8, 10 mm EDTA, 0.5% SDS) at 60 °C for 15 min. Cross-linking of protein-DNA complexes was reversed at 65 °C for 4 h, followed by ethanol precipitation overnight and centrifugation at 14,000 rpm for 20 min at 4 °C. The pellet was air-dried and reconstituted in Tris-EDTA buffer followed by treatment with 100 μg/ml proteinase K in PK buffer (50 mm Tris-HCl, pH 8, 25 mm EDTA, 1.25% SDS, and glycogen) for 1 h at 45 °C. DNA was extracted twice with phenol/chloroform and precipitated with ethanol. Pellets were resuspended in Tris-EDTA buffer and subjected to PCR amplification using TNF promoter-specific primers 5′-ATAGGTACCCCCCAACTTTCCAAACCC-3′ and 5′-AAAGATCTAGCTATTTCCAAGATGTTC-3′.
TNF-α mRNA Stability
TNF-α mRNA stability was assessed 16 h after treatment of cells with either M. tuberculosis, or M. tuberculosis and Nef by the addition of actinomycin D (5 μg/ml) for different periods of time. Total RNA was extracted using an RNeasy kit (Qiagen), and cDNA was prepared using first strand synthesis kit (Roche Applied Science) and quantification of TNF-α mRNA was carried out by quantitative real-time PCR on an ABI 7500 Fast detection system using SYBR green PCR master mix (Applied Biosystems). The sense and antisense primers used for TNF-α were 5′-GAGTGACAAGCCTGTAGCCCATGTTGTAGC-3′ and 5′-CTGGGAGTAGATGAGGTACAGGCCCTCTGA-3′, respectively. The sense and antisense primers used for glyceraldehyde-3-phosphate dehydrogenase were 5′-GATGGGATTTCCATTGATGACA-3′ and 5′-CCACCCATGGCAAATTCC-3′, respectively.
ELISA for TNF-α
Conditioned medium was removed 24 h after treatments with M. tuberculosis or Nef or a combination of the two, and assayed for TNF-α by ELISA using the TNF-α assay kit (BD Biosciences).
Statistical Analysis
Data are represented as means ± S.D. of three separate experiments. Student's t test was performed to test statistical significance.
RESULTS
Inhibition of Apoptosis and TNF-α Release in Macrophages Challenged with a Combination of Nef and M. tuberculosis
Exogenous Nef was able to inhibit apoptosis induced in differentiated THP-1 cells by M. tuberculosis as measured by histone ELISA (Fig. 1A). M. tuberculosis-induced macrophage apoptosis is known to depend on TNF-α, with HIV being able to dampen this process (36). In view of this, we tested the effect of Nef on M. tuberculosis-induced TNF-α release. TNF-α was released from differentiated THP-1 cells challenged with either M. tuberculosis or with recombinant Nef (Fig. 1B) in conformity with earlier reports on the effect of Nef or M. tuberculosis on macrophages (42, 43). However, when macrophages were challenged simultaneously with M. tuberculosis and Nef, there was significant decrease in TNF-α production (Fig. 1B). This suggested that M. tuberculosis and Nef were perhaps cross-inhibiting the respective signaling pathways associated with TNF-α production.
FIGURE 1.

Nef inhibits M. tuberculosis-mediated induction of TNF-α. A, THP-1 cells (in 96-well plates) were left untreated (−) or treated with either M. tuberculosis (Mtb) or Nef or both (Mtb + Nef) for 24 h. Cells were washed and lysed, and cell death was measured using the cell death ELISA kit according to the manufacturer's instructions. B, THP-1 cells (in 96-well plates) were left untreated (−) or treated with either M. tuberculosis (Mtb) or Nef or both (Mtb + Nef). In a separate set of experiments, Nef was pretreated with polymyxin B (PB-Nef) or heated at 95 °C for 10 min (heat-treated) and then used alone or in combination with M. tuberculosis as described above. The release of TNF-α in the supernatant was quantitated by TNF-α ELISA according to the manufacturer's instructions, 24 h after infection. THP-1 cells were transfected with a TNF-α promoter luciferase reporter construct (wt) (C and E) or with mutants devoid of the indicated transcription factor binding sites (E), along with a β-galactosidase expression vector. Cells were then left untreated (− in C), or incubated with either Mtb or Nef or with both (C and E) as indicated. Cells were lysed, and luciferase activities were determined 14 h after infection. The activities were normalized with β-galactosidase activity. Data represent means ± S.D. for three different experiments. D is a diagrammatic representation of the TNF-α promoter indicating the different transcription factor binding sites (underlined). The sequence in the bottom line indicates the mutation by replacement of the corresponding bases on the upper line.
Inhibition of TNF-α Promoter Activity in Macrophages Challenged with a Combination of Nef and M. tuberculosis
Production of TNF-α in cells of the monocytic lineage in response to stimulus, is regulated at the transcriptional and post-transcriptional level depending both on cell type and stimulus (44–46). Among the several possible steps at which Nef and M. tuberculosis could exert cross-inhibitory effects, we tested the possibility that this could be at the level of activation of the TNF-α promoter. It was observed that exogenous Nef and M. tuberculosis could individually activate TNF-α promoter-driven luciferase expression (Fig. 1C). However, in macrophages challenged simultaneously with Nef and M. tuberculosis, there was a significant decrease in TNF-α promoter-driven luciferase expression, compared with either entity used alone (Fig. 1C). Heat treatment abrogated the ability of Nef to induce TNF-α (Fig. 1, B and C), suggesting that the effect was attributable specifically to the protein, rather than any contaminant. The ability of Nef to down-regulate Mtb-dependent TNF-α production was not attributable to endotoxin contamination, because pretreatment of Nef with polymyxin B did not alter this activity (Fig. 1, B and C).
We next reasoned that, in the event of a cross-inhibitory mechanism being at work, M. tuberculosis and Nef were likely to operate through different mechanisms to activate the TNF-α promoter. Sequences in the proximal 200 bp and the distal (−627 to −487 bp) of the TNF-α promoter are remarkably conserved in mouse and human and encompass binding sites for multiple transcription factors (46–50). Previous studies have shown that the TNF-α gene is regulated in a cell type-specific manner (51). In addition, within the same cell type, the TNF-α gene is regulated in a stimulus-specific manner (52) through the action of distinct sets of transcription factors. We therefore tested the hypothesis that M. tuberculosis and Nef each induce TNF-α transcription through different sets of transcription factors. Both the human and the murine proximal TNF-α promoter contain, among other sites, multiple NFAT/ETS, Egr, Sp1, CRE, and AP1 binding sites. We mutated the −180 Ets, −117 Ets, −84 Ets, −76 Ets, Egr, Sp1, CRE, and AP1 sites (Fig. 1D) on the TNF-α promoter to evaluate the contributions of these binding sites in M. tuberculosis or Nef-induced TNF-α activation. M. tuberculosis-driven TNF-α promoter activation was abrogated when any one of the Ets or Egr or Sp1 or CRE or AP1 binding sites was mutated (Fig. 1E), suggesting a cooperative effect of these transcription factors on M. tuberculosis-induced TNF-α promoter activation as demonstrated by Barthel et al. (43). On the other hand, Nef-mediated promoter activation was affected only when the CRE or AP1 binding site was mutated. These results argued in favor of distinctly different signals and mechanisms driving TNF-α gene expression by Nef and by M. tuberculosis.
Role of ASK1 and p38 MAPK in TNF-α Release Driven by M. tuberculosis or Nef
Because the above results suggested that different signals were likely involved in M. tuberculosis or Nef-driven TNF-α promoter activation, we sought to identify at least some of these differences. Transfection of cells with a kinase-dead mutant of ASK1 (ASK1(KM)) or with dominant-negative ATF2 (ATF2(dn)) led to attenuation of M. tuberculosis-driven TNF-α release, but not to Nef-driven TNF-α release (Fig. 2A). ASK1 is known to activate p38 MAPK (53). Interestingly, transfection of cells with dominant-negative p38 MAPK (p38(agf)) attenuated M. tuberculosis- as well as Nef-driven TNF-α production (Fig. 2A), suggesting a role of p38 MAPK in the signaling pathway leading to TNF-α production triggered by both stimuli. This result also pointed to the fact that, whereas M. tuberculosis-induced TNF-α production depends on ASK1/p38 MAPK signaling, Nef-induced TNF-α production occurs in a p38 MAPK-dependent, but ASK1-independent manner. We tested activation of p38 MAPK induced by M. tuberculosis and Nef. Both M. tuberculosis and Nef activated p38 MAPK (Fig. 2B). However, M. tuberculosis-induced p38 MAPK activation was inhibited in cells transfected with ASK1(KM), whereas Nef-mediated p38 MAPK activation was ASK1-independent (Fig. 2B). This suggested that Nef-driven p38 MAPK activation was dependent on an MAP3K other than ASK1. We next tested whether Nef inhibits M. tuberculosis-driven ASK1 activation in macrophages. In vitro ASK1 kinase assays using ASK1 immunoprecipitates from cells treated with Nef and/or M. tuberculosis showed that Nef inhibits M. tuberculosis-driven ASK1 activation (Fig. 2C). This suggested that Nef inhibits M. tuberculosis-driven TNF-α production at least in part by inhibiting ASK1. In our search for activators of p38 MAPK in Nef-treated cells, we evaluated the role of Rac1 and PAK1 in p38 MAPK activation. Transfection of cells with dn-Rac1 or kinase-dead PAK1 prior to challenge with Nef led to the abrogation of p38 MAPK activation (Fig. 2D). On the other hand, M. tuberculosis-mediated p38 MAPK activation was independent of Rac1 or PAK1. In addition, we also observed activation of both Rac1 and PAK1 in macrophages challenged with Nef (Fig. 2, E and F).
FIGURE 2.

Nef inhibits M. tuberculosis-mediated activation of p38 MAPK and ASK1. A, THP-1 cells were transfected with vector alone or with a kinase-dead mutant of ASK1 (ASK1(KM)) or dominant-negative ATF2 (ATF2(dn)) or dominant-negative p38 MAPK (p38(agf)). Transfected cells were treated with M. tuberculosis (Mtb) or Nef separately, and release of TNF-α was measured as described under Fig. 1A. Data represent the means ± S.D. of three separate experiments. B–D, THP-1 cells transfected with either empty vector or ASK1 (KM) (B) or Rac1 (dn) (D) or PAK1-KD (D) were left untreated or treated with Mtb or with Nef separately for 90 (B and D) or 60 (C) min. Cell lysates were either immunoblotted with phospho-p38 MAPK antibody and reprobed with p38 MAPK antibody (B and D) or immunoprecipitated with ASK1 antibody, and the immunoprecipitate was used to study the phosphorylation of myelin basic protein using [γ-32P]ATP followed by autoradiography (C). Actin in the cell lysate was blotted to confirm equal amounts of proteins in cell lysates (bottom panel of C). E, activation of Rac1 in cellular extracts was assessed by affinity precipitation of the Rac1-GTP complex from whole cell lysates using PAK1-PBD followed by Western blotting using anti-Rac1 antibody as described under “Experimental Procedures.” F, cell extracts were prepared, and phosphorylation of PAK1 was assessed by Western blotting using anti-phospho-PAK1 antibody followed by reprobing with PAK1 antibody, respectively. The data in panels B–F are representative of those obtained in three different experiments.
Activation of Transcription Factors by M. tuberculosis or Nef and Binding to the TNF-α Promoter
To gain further insight into the differences between M. tuberculosis and Nef-driven TNF-α gene expression, we evaluated the activation of a set of transcription factors using a TransFactor ELISA kit (Clontech). We observed that M. tuberculosis activated ATF2, c-Jun, and Sp1 (Fig. 3A). Barthel et al. (43) had earlier reported recruitment of ATF2, c-Jun, and Sp1 to the TNF-α promoter. Nef activated c-Jun, but not Sp1 or ATF 2 (Fig. 3B). Nef did not attenuate M. tuberculosis-driven c-Jun or Sp1 activation (Fig. 3C). Interestingly, Nef inhibited M. tuberculosis-induced ATF2 activation (Fig. 3C). We performed ChIP assays of ATF2 binding to the TNF-α promoter. M. tuberculosis stimulated the binding of ATF2 to the TNF-α promoter, whereas Nef did not (Fig. 3D). Expectedly, the M. tuberculosis-dependent binding of ATF2 was inhibited by the p38 MAPK inhibitor SB203580 but not by the MEK inhibitor U0126. In addition, M. tuberculosis-driven binding of ATF2 to the TNF-α promoter was inhibited by Nef (Fig. 3D). This suggested that the inhibition of M. tuberculosis-induced p38 MAPK activation by Nef likely compromises binding of ATF2 to the TNF-α promoter, contributing in part to the process by which Nef inhibits M. tuberculosis-driven TNF-α production. On the other hand, M. tuberculosis-induced binding of Sp1 to the TNF-α promoter could not be inhibited by Nef, although the binding could be inhibited by U0126 as well as SB203580 (Fig. 3E). These observations led us to conclude that the signaling pathway operating upstream of Sp1 activation is distinct from that involved in ATF2 activation.
FIGURE 3.

Activation of transcription factor by M. tuberculosis and Nef. A and B, THP-1 cells were left untreated or treated with Mtb (A) or Nef (B), and the activation of c-Jun, Sp1, and ATF2 was quantified using the TransFactor ELISA kit (Clontech) according to the manufacturer's protocol. C, THP-1 cells were treated with Mtb alone or with Mtb and Nef, and the activation of c-Jun, Sp1, and ATF2 was determined using the TransFactor ELISA kit as described above. Data in A–C represent means ± S.D. of three separate determinations. D and E, THP-1 cells were left untreated (−) or preincubated with the inhibitors U0126 (U) or SB203580 (SB) prior to treatment with Mtb. In a separate set of experiments cells were left untreated or treated with either Mtb or Nef or with both. Chromatin was prepared, and ChIP analysis was carried out with primers specific for the AP1- (D) or Sp1- (E) binding site of the TNF-α promoter after immunoprecipitation (IP) with anti-ATF2 (D) or anti-Sp1 (E) antibody. The input panel shows the PCR product obtained when no immunoprecipitation was performed.
M. tuberculosis-induced Activation of the TNF-α Promoter Depends on MEK-ERK Signaling
M. tuberculosis-driven activation of the TNF-α promoter was inhibited in cells transfected with dominant-negative MEK (MEK(dn)) (Fig. 4A). On the other hand, Nef-driven TNF-α promoter activation was independent of MEK-ERK signaling. M. tuberculosis-induced ERK activation was inhibited in the presence of Nef (Fig. 4B). Taken together, we concluded that the ability of Nef to inhibit M. tuberculosis-induced ERK activation contributes another arm of the signaling pathway exploited by Nef to dampen M. tuberculosis-driven TNF-α production. c-Jun, JunD, FosB, and ATF2 are the classic AP1 transcription factors that have been identified in association with the TNF-α promoter (54). We observed in TransFactor assays that M. tuberculosis activated FosB, whereas Nef did not (Fig. 4C). ChIP analysis confirmed that FosB is associated in with the AP1-binding site of the TNF-α promoter in M. tuberculosis, but not in Nef-treated cells (Fig. 4D). This association was inhibited by U0126. Further, Nef inhibited the M. tuberculosis-driven binding of FosB to the TNF-α promoter (Fig. 4D). The inhibition of ERK activity by Nef therefore likely culminates in the attenuation of M. tuberculosis-induced FosB binding to the TNF-α promoter.
FIGURE 4.

Role of MAPKs in the activation of TNF-α. A, THP-1 cells were transfected with empty vector or dominant-negative MEK (MEK(dn)) along with the TNF-α promoter luciferase reporter construct. Transfected cells were left untreated or treated with Mtb or with Nef, and luciferase activity was measured as described previously. B, THP-1 cells were left untreated or treated with Mtb in the absence or presence of Nef for different periods of time. Cell lysates were immunoblotted with phospho-ERK antibody, and blots were reprobed with ERK antibody. C, THP-1 cells were left untreated or treated with Mtb or Nef for the indicated periods of time (in hours), and the activation of FosB was quantified using the TransFactor ELISA kit (Clontech) according to the manufacturer's protocol. D, THP-1 cells were left untreated (−) or preincubated with the inhibitor U0126 prior to treatment with Mtb. In a separate set of experiments, cells were treated with either Mtb or Nef or with both. After treatments, ChIP analysis was carried out with primers specific for the AP1-binding site of the TNF-α promoter after immunoprecipitation (IP) with anti-FosB antibody. The input panel shows the PCR product obtained when no immunoprecipitation was performed.
Nef Destabilizes M. tuberculosis-induced TNF-α mRNA
TNF-α production is dependent on the stability of TNF-α mRNA (55–57). To test whether Nef affects the stability of TNF-α mRNA induced by M. tuberculosis, cells were challenged with either M. tuberculosis or Nef or a combination of M. tuberculosis and Nef, followed by addition of actinomycin D, a transcriptional inhibitor and time-dependent monitoring of TNF-α mRNA by quantitative real-time PCR. It was observed that TNF-α mRNA decreased <10%, 90 min after actinomycin D treatment (compared with the amount of mRNA at the point of actinomycin D addition) in cells treated with M. tuberculosis alone. When cells were treated simultaneously with Nef and M. tuberculosis, >40% decrease of TNF-α mRNA was observed, 90 min after actinomycin D treatment (Fig. 5). These results suggested that the inhibitory effect of Nef, on M. tuberculosis-driven TNF-α production in macrophages, extends to destabilization of TNF-α mRNA. It is well established that p38 MAPK signaling is required for stabilization of TNF-α mRNA (58). It is therefore likely that the inhibition of M. tuberculosis-induced p38 MAPK by Nef, impacts TNF-α mRNA stability.
FIGURE 5.
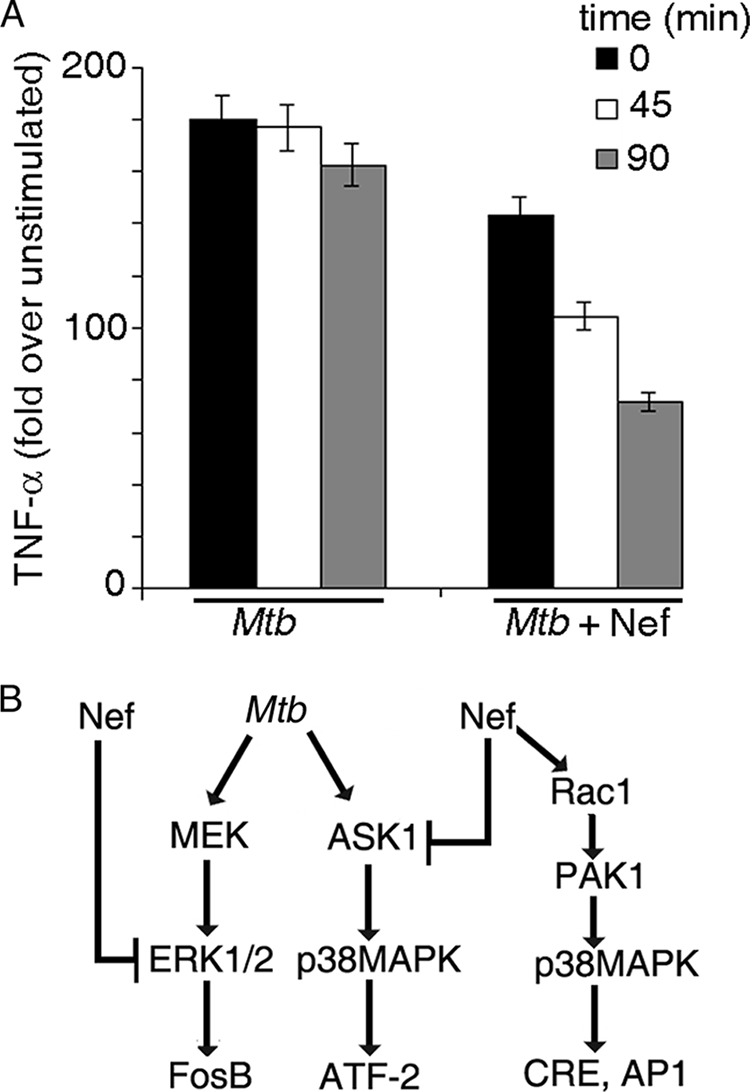
Role of Nef in TNF-α mRNA stability. A, THP-1 cells were treated with Mtb in the absence or in the presence of Nef for 14 h. Actinomycin D (5 μg/ml) was added, total RNA was harvested, and TNF-α mRNA was estimated by quantitative real-time PCR at 0, 45, and 90 min. Data represents % mRNA remaining at the indicated time points after actinomycin D addition. 100% represents the amount of mRNA at the zero time point. B, schematic diagram of modulation of Mtb-induced activation of TNF-α promoter by Nef.
DISCUSSION
The significance of exogenous Nef in the pathogenesis of HIV remains unclear. Exogenous Nef has been reported to activate NF-κB and AP-1 in U937 cells (14). HIV-TB co-infections present major challenges in terms of disease management. Several reports show that HIV exacerbates the course of TB infection (59). One of the mechanisms associated with this is likely to be attenuation of macrophage apoptosis. This benefits the bacterium by preservation of its intracellular niche. In this report we have tested the role of exogenous Nef in modulating the signaling events associated with infection of macrophages with M. tuberculosis. We report that M. tuberculosis or exogenous Nef individually triggers signaling pathways associated with macrophage apoptosis. However, their combined effect is an attenuated macrophage response. Several studies have established that autocrine TNF-α signaling is one of the major contributors toward macrophage apoptosis (60, 61). We have dissected the differences in signaling pathways triggered by either M. tuberculosis or exogenous Nef, culminating in TNF-α production. M. tuberculosis-dependent TNF-α production was abrogated when any one of the Ets or Egr or Sp1 or CRE binding sites on the TNF-promoter is mutated, whereas Nef-dependent TNF-α production is dependent on CRE and AP-1 sites only. This suggests that distinctly different mechanisms of activation of the TNF-α promoter operate in the case of M. tuberculosis and Nef. Considering that transcription factor activation depends on upstream MAPK signaling, we tested the role of the ASK1/p38 MAPK pathway in TNF-α production. Interestingly, although both Nef- and M. tuberculosis-induced TNF-α production depends on p38 MAPK, only M. tuberculosis, but not Nef-induced TNF-α production depends on ASK1, one of the MAP3Ks that signals upstream of p38. Nef inhibits M. tuberculosis-dependent ASK1 activation. This finding points to at least one signaling node where Nef exerts its inhibitory function. Nef has previously been reported to inhibit ASK1 in T cells by virtue of its ability to interact directly with ASK1 (16). It appears likely that a similar mechanism prevails in this instance as well, once Nef traverses the membrane and enters cells. In macrophages, Rac1/PAK1 signaling is associated with activation of p38 MAPK (62). Here we establish that Nef, but not M. tuberculosis, activates p38 MAPK in a Rac1/PAK1-dependent manner. These findings establish the distinctly different pathways leading to p38 MAPK activation in response to challenge with M. tuberculosis or Nef. Nef also inhibits M. tuberculosis-mediated ERK activation. We demonstrate that M. tuberculosis induces p38 MAPK-dependent binding of ATF2 and ERK-dependent binding of FosB to the TNF-α promoter (Fig. 5B). Interestingly, neither ATF2 nor FosB binds to the TNF-α promoter in Nef-stimulated cells. Putting these facts together, we conclude that, by virtue of inhibiting M. tuberculosis-driven ERK and p38 MAPK activation, Nef inhibits the binding of FosB and ATF2 to the TNF-α promoter, thereby attenuating M. tuberculosis-induced TNF-α production. On the other hand, M. tuberculosis-induced binding of Sp1 to the TNF-α promoter, although dependent on p38 MAPK, is not inhibited by Nef, suggesting that a MAP3K other than ASK1 likely drives Sp1 activation.
Finally, we observed that exogenous Nef attenuates the stability of TNF-α mRNA induced by M. tuberculosis. Considering the well established role of p38 MAPK in positively regulating mRNA stability, it appears likely that the effect of Nef is due to its ability to inhibit p38 MAPK.
The above findings provide important mechanistic insight into how exogenous Nef sourced from bystander cells could modulate the fate of M. tuberculosis-infected macrophages. These studies extend the observations of Patel et al. (36) that HIV infection is associated with attenuation of macrophage apoptosis upon exposure to virulent M. tuberculosis. We propose that this is attributable at least in part to the fact that the release of TNF-α is attenuated by the HIV protein Nef. The findings reported here support our contention that M. tuberculosis and Nef cross-regulate distinct signaling pathways employed by these two entities to drive the production of at least one cytokine, TNF-α. In this report we have focused on how Nef attenuates M. tuberculosis-induced signaling pathways. How M. tuberculosis attenuates Nef-driven pathways for TNF-α production, awaits clarification. What emerges from these studies is a glimpse into how HIV-TB co-infection leads to a dampening of the innate immune response, a process in which Nef plays a critical role.
This work was supported by a grant from the Swedish Research Council (to J. B. and A.-L. S.).
- HIV-1/2
- human immunodeficiency virus, types 1 and 2
- TNF
- tumor necrosis factor
- ASK1
- apoptosis signal-regulating kinase 1
- ChIP
- chromatin immunoprecipitation
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal regulated kinase
- MEK1
- MAPK/ERK kinase 1
- JNK
- c-Jun N-terminal kinase
- Mtb
- M. tuberculosis
- ELISA
- enzyme-linked immunosorbent assay
- GST
- glutathione S-transferase
- MAP3K
- MAPK kinase kinase
- KD
- kinase-dead
- KM
- kinase-dead mutant
- dn
- dominant negative.
REFERENCES
- 1.Kim S. Y., Byrn R., Groopman J., Baltimore D. (1989) J. Virol. 63, 3708–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greene W. C., Peterlin B. M. (2002) Nat. Med. 8, 673–680 [DOI] [PubMed] [Google Scholar]
- 3.Brambilla A., Turchetto L., Gatti A., Bovolenta C., Veglia F., Santagostino E., Gringeri A., Clementi M., Poli G., Bagnarelli P., Vicenzi E. (1999) Virology 259, 349–368 [DOI] [PubMed] [Google Scholar]
- 4.Miller M. D., Warmerdam M. T., Gaston I., Greene W. C., Feinberg M. B. (1994) J. Exp. Med. 179, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spina C. A., Kwoh T. J., Chowers M. Y., Guatelli J. C., Richman D. D. (1994) J. Exp. Med. 179, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lama J., Mangasarian A., Trono D. (1999) Curr. Biol. 9, 622–631 [DOI] [PubMed] [Google Scholar]
- 7.Ross T. M., Oran A. E., Cullen B. R. (1999) Curr. Biol. 9, 613–621 [DOI] [PubMed] [Google Scholar]
- 8.Schwartz O., Maréchal V., Le Gall S., Lemonnier F., Heard J. M. (1996) Nat. Med. 2, 338–342 [DOI] [PubMed] [Google Scholar]
- 9.Schindler M., Würfl S., Benaroch P., Greenough T. C., Daniels R., Easterbrook P., Brenner M., Münch J., Kirchhoff F. (2003) J. Virol. 77, 10548–10556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen G. B., Gandhi R. T., Davis D. M., Mandelboim O., Chen B. K., Strominger J. L., Baltimore D. (1999) Immunity 10, 661–671 [DOI] [PubMed] [Google Scholar]
- 11.Hung C. H., Thomas L., Ruby C. E., Atkins K. M., Morris N. P., Knight Z. A., Scholz I., Barklis E., Weinberg A. D., Shokat K. M., Thomas G. (2007) Cell Host Microbe 1, 121–133 [DOI] [PubMed] [Google Scholar]
- 12.Swigut T., Shohdy N., Skowronski J. (2001) EMBO J. 20, 1593–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhry A., Das S. R., Hussain A., Mayor S., George A., Bal V., Jameel S., Rath S. (2005) J. Immunol. 175, 4566–4574 [DOI] [PubMed] [Google Scholar]
- 14.Michel N., Allespach I., Venzke S., Fackler O. T., Keppler O. T. (2005) Curr. Biol. 15, 714–723 [DOI] [PubMed] [Google Scholar]
- 15.Collins K. L., Chen B. K., Kalams S. A., Walker B. D., Baltimore D. (1998) Nature 391, 397–401 [DOI] [PubMed] [Google Scholar]
- 16.Geleziunas R., Xu W., Takeda K., Ichijo H., Greene W. C. (2001) Nature 410, 834–838 [DOI] [PubMed] [Google Scholar]
- 17.Wolf D., Witte V., Laffert B., Blume K., Stromer E., Trapp S., d'Aloja P., Schürmann A., Baur A. S. (2001) Nat. Med. 7, 1217–1224 [DOI] [PubMed] [Google Scholar]
- 18.Stolp B., Reichman-Fried M., Abraham L., Pan X., Giese S. I., Hannemann S., Goulimari P., Raz E., Grosse R., Fackler O. T. (2009) Cell Host Microbe 6, 174–186 [DOI] [PubMed] [Google Scholar]
- 19.Trible R. P., Emert-Sedlak L., Smithgall T. E. (2006) J. Biol. Chem. 281, 27029–27038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenway A. L., Holloway G., McPhee D. A., Ellis P., Cornall A., Lidman M. (2003) J. Biosci. 28, 323–335 [DOI] [PubMed] [Google Scholar]
- 21.Hayashi N., Matsubara M., Jinbo Y., Titani K., Izumi Y., Matsushima N. (2002) Protein Sci. 11, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renkema G. H., Saksela K. (2000) Front. Biosci. 5, D268–D283 [DOI] [PubMed] [Google Scholar]
- 23.Muratori C., Cavallin L. E., Krätzel K., Tinari A., De Milito A., Fais S., D'Aloja P., Federico M., Vullo V., Fomina A., Mesri E. A., Superti F., Baur A. S. (2009) Cell Host Microbe 6, 218–230 [DOI] [PubMed] [Google Scholar]
- 24.Vigerust D. L., Egan B. S., Shepherd V. L. (2005) J. Leukocyte Biol. 77, 522–534 [DOI] [PubMed] [Google Scholar]
- 25.Mahlknecht U., Herbein G. (2001) Trends Immunol. 22, 256–260 [DOI] [PubMed] [Google Scholar]
- 26.Fujii Y., Otake K., Tashiro M., Adachi A. (1996) FEBS Lett. 393, 93–96 [DOI] [PubMed] [Google Scholar]
- 27.Mangino G., Percario Z. A., Fiorucci G., Vaccari G., Manrique S., Romeo G., Federico M., Geyer M., Affabris E. (2007) J. Virol. 81, 2777–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao X., He B., Chiu A., Knowles D. M., Chadburn A., Cerutti A. (2006) Nat. Immunol. 7, 302–310 [DOI] [PubMed] [Google Scholar]
- 29.Varin A., Manna S. K., Quivy V., Decrion A. Z., Van Lint C., Herbein G., Aggarwal B. B. (2003) J. Biol. Chem. 278, 2219–2227 [DOI] [PubMed] [Google Scholar]
- 30.Federico M., Percario Z., Olivetta E., Fiorucci G., Muratori C., Micheli A., Romeo G., Affabris E. (2001) Blood 98, 2752–2761 [DOI] [PubMed] [Google Scholar]
- 31.Ma W., Mishra S., Gajanayaka N., Angel J. B., Kumar A. (2009) J. Biol. Chem. 284, 7578–7587 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Xu W., Santini P. A., Sullivan J. S., He B., Shan M., Ball S. C., Dyer W. B., Ketas T. J., Chadburn A., Cohen-Gould L., Knowles D. M., Chiu A., Sanders R. W., Chen K., Cerutti A. (2009) Nat. Immunol. 10, 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nunn P., Williams B., Floyd K., Dye G., Elzinga G., Raviglione M. (2005) Nat. Rev. Immunol. 5, 819–826 [DOI] [PubMed] [Google Scholar]
- 34.Klingler K., Tchou-Wong K. M., Brändli O., Aston C., Kim R., Chi C., Rom W. N. (1997) Infect. Immun. 65, 5272–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keane J., Balcewicz-Sablinska M. K., Remold H. G., Chupp G. L., Meek B. B., Fenton M. J., Kornfeld H. (1997) Infect. Immun. 65, 298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel N. R., Zhu J., Tachado S. D., Zhang J., Wan Z., Saukkonen J., Koziel H. (2007) J. Immunol. 179, 6973–6980 [DOI] [PubMed] [Google Scholar]
- 37.Olivetta E., Federico M. (2006) Exp. Cell Res. 312, 890–900 [DOI] [PubMed] [Google Scholar]
- 38.Pantaleo G., Graziosi C., Demarest J. F., Butini L., Montroni M., Fox C. H., Orenstein J. M., Kotler D. P., Fauci A. S. (1993) Nature 362, 355–358 [DOI] [PubMed] [Google Scholar]
- 39.Maiti D., Bhattacharyya A., Basu J. (2001) J. Biol. Chem. 276, 329–333 [DOI] [PubMed] [Google Scholar]
- 40.Benard V., Bohl B. P., Bokoch G. M. (1999) J. Biol. Chem. 274, 13198–13204 [DOI] [PubMed] [Google Scholar]
- 41.Pathak S. K., Basu S., Bhattacharyya A., Pathak S., Kundu M., Basu J. (2005) J. Biol. Chem. 280, 42794–42800 [DOI] [PubMed] [Google Scholar]
- 42.Olivetta E., Percario Z., Fiorucci G., Mattia G., Schiavoni I., Dennis C., Jäger J., Harris M., Romeo G., Affabris E., Federico M. (2003) J. Immunol. 170, 1716–1727 [DOI] [PubMed] [Google Scholar]
- 43.Barthel R., Tsytsykova A. V., Barczak A. K., Tsai E. Y., Dascher C. C., Brenner M. B., Goldfeld A. E. (2003) Mol. Cell. Biol. 23, 526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swantek J. L., Cobb M. H., Geppert T. D. (1997) Mol. Cell. Biol. 17, 6274–6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raabe T., Bukrinsky M., Currie R. A. (1998) J. Biol. Chem. 273, 974–980 [DOI] [PubMed] [Google Scholar]
- 46.Udalova I. A., Knight J. C., Vidal V., Nedospasov S. A., Kwiatkowski D. (1998) J. Biol. Chem. 273, 21178–21186 [DOI] [PubMed] [Google Scholar]
- 47.Yao J., Mackman N., Edgington T. S., Fan S. T. (1997) J. Biol. Chem. 272, 17795–17801 [DOI] [PubMed] [Google Scholar]
- 48.Newell C. L., Deisseroth A. B., Lopez-Berestein G. (1994) J. Leukocyte Biol. 56, 27–35 [DOI] [PubMed] [Google Scholar]
- 49.Pope R. M., Leutz A., Ness S. A. (1994) J. Clin. Invest. 94, 1449–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geist L. J., Hopkins H. A., Dai L. Y., He B., Monick M. M., Hunninghake G. W. (1997) Am. J. Respir. Cell Mol. Biol. 16, 31–37 [DOI] [PubMed] [Google Scholar]
- 51.Tsai E. Y., Yie J., Thanos D., Goldfeld A. E. (1996) Mol. Cell. Biol. 16, 5232–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falvo J. V., Uglialoro A. M., Brinkman B. N., Merika M., Parekh B. S., Tsai E. Y., King H. C., Morielli A. D., Peralta E. G., Maniatis T., Thanos D., Goldfeld A. E. (2000) Mol. Cell. Biol. 20, 2239–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsukawa J., Matsuzawa A., Takeda K., Ichijo H. (2004) J. Biochem. 136, 261–265 [DOI] [PubMed] [Google Scholar]
- 54.Novotny V., Prieschl E. E., Csonga R., Fabjani G., Baumruker T. (1998) Nucleic Acids Res. 26, 5480–5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carballo E., Lai W. S., Blackshear P. J. (1998) Science 281, 1001–1005 [DOI] [PubMed] [Google Scholar]
- 56.Brook M., Sully G., Clark A. R., Saklatvala J. (2000) FEBS Lett. 483, 57–61 [DOI] [PubMed] [Google Scholar]
- 57.Rajasingh J., Bord E., Luedemann C., Asai J., Hamada H., Thorne T., Qin G., Goukassian D., Zhu Y., Losordo D. W., Kishore R. (2006) FASEB J. 20, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 58.Dean J. L., Sarsfield S. J., Tsounakou E., Saklatvala J. (2003) J. Biol. Chem. 278, 39470–39476 [DOI] [PubMed] [Google Scholar]
- 59.de Jong B. C., Israelski D. M., Corbett E. L., Small P. M. (2004) Annu. Rev. Med. 55, 283–301 [DOI] [PubMed] [Google Scholar]
- 60.Hsu H., Shu H. B., Pan M. G., Goeddel D. V. (1996) Cell 84, 299–308 [DOI] [PubMed] [Google Scholar]
- 61.Kundu M., Pathak S. K., Kumawat K., Basu S., Chatterjee G., Pathak S., Noguchi T., Takeda K., Ichijo H., Thien C. B., Langdon W. Y., Basu J. (2009) Nat. Immunol. 10, 918–926 [DOI] [PubMed] [Google Scholar]
- 62.Basak C., Pathak S. K., Bhattacharyya A., Mandal D., Pathak S., Kundu M. (2005) J. Biol. Chem. 280, 4279–4288 [DOI] [PubMed] [Google Scholar]